

1. INTRODUCTION
Tachypnoea and exercise dyspnea are children's most common signs of respiratory failure. In the long‐term presence of these symptoms, it is necessary to consider chronic lung diseases in the differential diagnosis and to establish personalized treatment specific to the patient after diagnosis. In this case report, we would like to share the successful clinical improvement of a rare chronic lung disease with a rarely used treatment modality in children.
2. CASE PRESENTATION
A 15‐year‐old male patient presented to our clinic with fatigue and shortness of breath for 2 years. He had a 5 kg weight loss over the last 3 months, dry cough, and tachypnea, which increased with effort and continued at rest. The patient had two hospitalizations for pneumonia at the ages of 4 and 8 years, which did not require respiratory support. The parents were not consanguineous, and he had no known family history of chronic respiratory disease or other conditions (Podcast 1).
On physical examination, the patient's growth parameters were normal. Although there was no fever, he had tachypnea (respiratory rate: 42/min) and dyspnea with jugular retractions at rest. His peripheral oxygen saturation (SpO2) was 91% in room air (RA). Pectus excavatum and clubbing of the fingers and toes were present. No abnormal breath sounds were heard on auscultation.
Pulmonary function tests (PFTs) were found to be abnormal (expressed as % predicted). The spirometry test revealed the following results: forced expiratory volume in 1 s (FEV1) 45%, forced vital capacity (FVC) 48%, FEV1/FVC 94%, and carbon monoxide diffusion capacity (DLCO): 53%. The body plethysmography test results were as follows: total lung capacity (TLC) 66%, residual volume (RV) 94%, RV/TLC 42,4. In the 6‐min walk test (6‐MWT), his SpO2 was found to be 84%, intermittent coughing started, dyspnea increased, and the test was discontinued at a distance of 400 m. His arterial blood gas test at rest showed borderline hypercapnia and hypoxemia (pCO2: 48.6 mmHg, pO2: 66.9 mmHg). The partial alveolar‐arterial pressure gradient (P(A‐a)) was found to be increased to 80 mmHg (normal < 15 mmHg). The serum lactate dehydrogenase (LDH) level was found to be 328 U/L (elevated).
2.1. Challenge point
A 15‐year‐old male with a 3‐month history of tachypnea, fatigue, and weight loss presented to the pediatric pulmonology clinic. Pectus excavatum, clubbing, restrictive pattern in spirometry, decreased DLCO, increased P(A‐a), and elevated LDH were detected.
2.2. Learner reflection
-
(1)
What is the most likely diagnosis in this patient (leading and differential)?
-
(2)
What are the next steps based on history and clinical findings?
Listen to Podcast 2 to hear the consortium's decision‐making progress.
2.3. Case progression
Chest X‐ray showed bilateral diffuse infiltrates sparing the left lower lobe (Figure 1A), and thorax computed tomography (CT) showed a crazy paving pattern, diffuse ground‐glass opacity, and thickening of secondary lobules. The Haller index was found to be 3.6 1 (Figure 1B).
Figure 1.
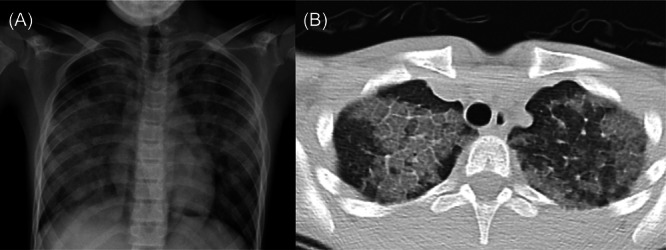
Pretreatment images (A) chest X‐ray; bilateral ground‐glass opacity, batwing pattern. (B) The chest computed tomography; crazy paving pattern, thickening of secondary lobules, and septal thickening.
Bronchoalveolar lavage (BAL) fluid was serous and transparent, and the cell differential showed 82% macrophages, 3% neutrophils, and 15% lymphocytes. A video‐assisted thoracoscopic lung biopsy was performed. Histological evaluation showed alveolar wall distortion, epithelial hyperplasia, and wall thickening in the alveolar spaces (Figure 2A). periodic acid‐Schiff (PAS) staining was positive and demonstrated intra‐alveolar accumulation of abundant lipoproteinous surfactant material (Figure 2B). The biopsy was complicated by a postoperative pneumothorax on the right side, which was treated by chest tube drainage.
Figure 2.
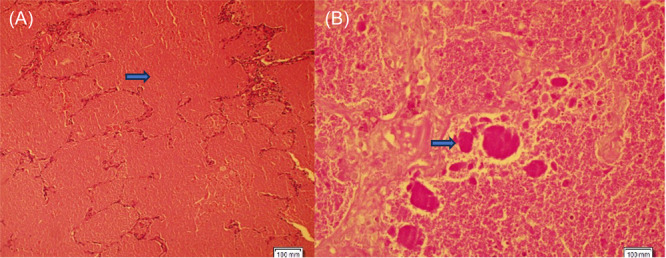
The lung biopsy material was stained with the hematoxylin–eosin (the first arrow) and periodic acid‐Schiff (PAS) (the second arrow). The histopathological section and histochemical analysis showed eosinophilic proteinaceous material filling the space of the alveolar space. (A): hemotoxylin–eosin stain, magnification ×100; (B): PAS stain+, magnification ×100).
2.4. Challenge point
In a 15‐year‐old male patient, bilateral diffuse infiltration was observed on chest radiography, crazy paving pattern was detected on thorax CT, and intra‐alveolar PAS+ staining material was detected on lung biopsy.
2.5. Learner reflection
-
(1)
What is the most likely diagnosis and differential diagnosis?
-
(2)
What do you do next and why?
Listen to Podcast 3 to hear the consortium's decision‐making progress.
Genetic analysis was performed to assess for congenital pulmonary alveolar proteinosis (PAP) caused by mutations of the surfactant proteins or granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) receptor subunits, and no pathology was detected. Metabolic, immunologic, and infectious etiologies were investigated, and no pathology was found. Anti‐GM‐CSF antibody was tested, and a level of 8.14 μg/mL (pathological >7 μg/mL) was obtained, confirming the diagnosis of autoimmune PAP (aPAP).
After careful discussion of the treatment options within the team and together with the patient and his family, we decided to start off‐label treatment with inhaled recombinant GM‐CSF (rGM‐CSF). An insurance claim was filed, and a positive response was received 2 weeks later.
As there was no need for an immediate improvement in the respiratory condition, inhaled rGM‐CSF (Leukine® [Sargramostim], used with a jet nebulizer) was started without performing a whole lung lavage (WLL).
2.6. Challenge point
A 15‐year‐old male was diagnosed with PAP following a lung biopsy, which also detected the anti‐GM‐CSF antibody. He was subsequently started on inhaled rGM‐CSF treatment.
2.7. Learner reflection
-
(1)
What is most likely happening to this patient?
-
(2)
How would you monitor this patient for treatment response and improvement?
Listen to Podcast 4 to hear the consortium's decision‐making progress.
The patient underwent inhalation therapy with rGM‐CSF in cycles of 14 days, following a total treatment schedule of 6 months. The patient received 125 μg inhaled rGM‐CSF twice daily during the first half of the 6 months and once daily during the latter half. 2 Please see the Supporting Information S1: Table S1 for the specific treatment regimen.
Clinical improvements such as tachypnoea, retractions, dry cough, and increasing SpO2 in RA to 96% were observed within 1 month; however, objective parameters such as PFT did not improve adequately, and the second treatment period was administered, resulting in further improvements: FEV1 increased to 69%, DLCO was 53%, and the 6‐MWT distance increased to 702 m (Figure 3). Chest X‐rays showed reduced bilateral ground‐glass opacity, and the P(A‐a) decreased to 14 mmHg (Figure 4). Although PFT improved, they plateaued at subnormal values, leading to a third treatment period, during which significant PFT improvements were observed (FEV1: 86%, FVC: 83%, DLCO: 88%, TLC: 82%, RV: 73%). The improvement trend in the patient's PFT, body mass index, SpO2, and 6‐MWT is shown in Figure 3. His respiratory rate was 22/min.
Figure 3.
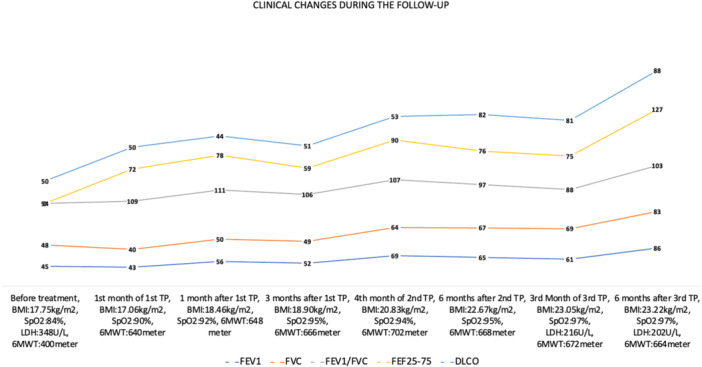
Changes in spirometry, carbon monoxide diffusing capacity (DLco) (all data expressed as %‐predicted), body mass index (BMI), oxygen saturation in room air, serum lactate dehydrogenase (LDH) level, and 6‐min walking test (6‐MWT) results during follow‐up. FEF25–75, forced expiratory flow between 25% and 75% of expired vital capacity; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; SpO2, peripheral oxygen saturation; TP, treatment period.
Figure 4.
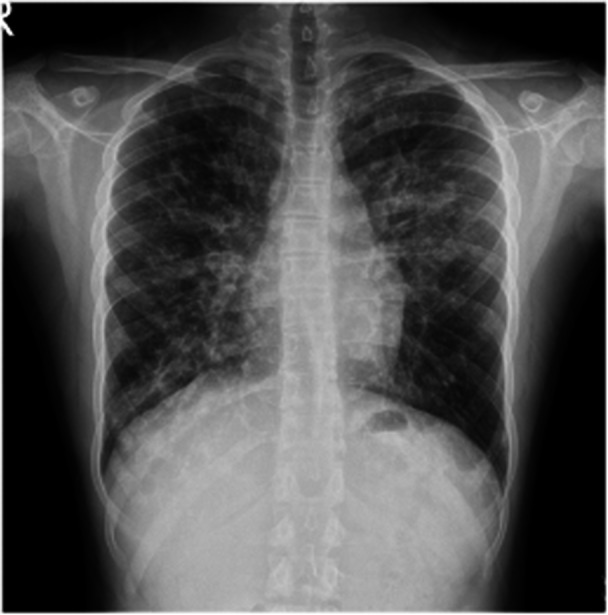
The posttreatment chest X‐ray (after second treatment period).
His quality of life has improved; he prefers stairs to the lift, goes to school regularly, and rides a bicycle. No treatment‐related side effects were observed during this period. The patient has been followed without medication for more than 1 year.
3. DISCUSSION
PAP is a rare condition characterized by an excessive build‐up of proteinaceous surfactant in the alveoli, impairing gas exchange, and it can lead to death if left untreated. 3 , 4 , 5 PAP is conventionally classified into two main types: congenital and acquired. The acquired form is further subdivided into autoimmune, reported in fewer than 20 cases among children to date, 4 , 5 , 6 and secondary forms associated with an underlying disorder. 4 Genetic causes that are predominant among children have been identified as disorders of surfactant protein metabolism, GM‐CSF receptor gene mutations, and other genetic disorders (methionyl transfer RNA synthetase [MARS], stimulator of interferon [STING], non‐clathrin‐coated vesicular coat protein [COPA], and GATA‐binding protein 2 [GATA2] mutations). 4 Other causes of PAP include metabolic diseases (lysinuric protein intolerance, Niemann–Pick disease), immunodeficiencies, malignancies, connective tissue diseases, congenital heart diseases, and infectious diseases. 4 aPAP is the most common type in adults, caused by autoantibodies targeting GM‐CSF. 5 , 6
Regardless of the etiology, the symptoms of patients with PAP are nonspecific. 5 , 7 Chronic dry cough and progressive dyspnea may occur after exercise or at rest; PAP may present with fatigue, weight loss, and chest pain; it may sometimes be asymptomatic and may not be diagnosed for months. 5 , 7 Physical examination is also nonspecific. 5 , 7 Clubbing occurs in up to 25% of individuals. 7 Rales can sometimes be heard. 7
The literature on aPAP in childhood consists of case reports. 5 , 6 , 8 , 9 , 10 In one case report, chronic dyspnea, hypoxemia findings, “crazy paving” findings on CT, a milky appearance on BAL, and high anti‐GM‐CSF antibody levels were common findings. 5 The patient's findings were nonspecific but indicated chronic hypoxemia. 5 The significant standard features in cases of PAP include a decrease in DLCO, an increase in P(A‐a), and a restrictive type of respiratory disorder indicating impaired alveolar‐arterial gas exchange. 7 A confirmed correlation exists between disease severity, P(A‐a), and DLCO. 7 The patient's elevated P(A‐a) improved during follow‐up, while spirometry and DLCO values continued to show a restrictive pattern. There is no specific biomarker to be monitored in PAP. However, an increase in serum LDH can be detected. 11 LDH levels may return to normal after inhaled rGM‐CSF therapy, as in the presented case. 11 The “Crazy paving” appearance on CT is a significant indicator of PAP. 12 It should be kept in mind that the discrepancy between the severity of thorax CT findings and the patient's symptoms may be an important feature of PAP. 6 The presence of a widespread “crazy paving pattern” on thorax CT helped guide us diagnostically.
In some cases, BAL may be transparent 13 ; a lung biopsy is required for diagnosis. 6 , 13 In our patient, BAL effluent was macroscopically normal, and a lung biopsy was required for diagnosis. After establishing the diagnosis of PAP, its etiology needs to be differentiated. Genetic analysis was performed to exclude rare congenital forms. Serum autoantibody levels are highly sensitive and specific for aPAP, nearly approaching 100%, and their measurement could prevent delays in diagnosis. 14 GM‐CSF autoantibody values were found to be elevated. Secondary etiological causes of PAP were ruled out.
Due to insufficient knowledge of aPAP in children, it is challenging to predict the disease's prognosis. Treatment options depend on the underlying etiology of PAP. 4 For some cases, a single procedure using WLL, the widely accepted treatment approach for PAP, may result in long‐term symptom improvement, while others may require multiple procedures over several years. 4 Hospitalization is required for WLL, along with an experienced team and general anesthesia. 15 WLL improves symptoms and oxygenation in approximately 95% of PAP patients. 4 Complications are rare and may include hydrothorax, pneumothorax, and intraoperative hypoxia. 4 Some forms of PAP respond well to WLL, while aPAP responds to rGM‐CSF. 4 In our patient, due to the clinical condition being stable and following detailed discussions with the patient's family and our team, it was decided to administer inhaled rGM‐CSF therapy.
GM‐CSF autoantibodies may neutralize endogenous GM‐CSF, reduce the uptake of surfactant by alveolar macrophages, and cause surfactant accumulation in the alveoli. 5 , 16 Treatment with rGM‐CSF via inhalation or subcutaneous administration restores alveolar macrophage function and contributes to surfactant clearance. 15 Inhaled rGM‐CSF may be an effective treatment for idiopathic PAP. 17 In addition to WLL, which is the primary treatment approach independent of the condition's etiology, therapies restoring GM‐CSF stimulation by inhaled rGM‐CSF or systemic substitution of rGM‐CSF are being improved for patients with aPAP. 18 In their case series, Sirin Kose et al. stated that inhaled GM‐CSF alone or combined with WLL may be the future approach for aPAP patients who are unresponsive to WLL treatment. 19 The inhaled form exhibited is 71%–89% more effective with fewer side effects compared to the subcutaneous form. 20 Significant benefits in gas exchange are expected from inhaled rGM‐CSF. 17 Although the exact contraindication of inhaled rGM‐CSF therapy is not clearly defined, side effects such as respiratory tract infections, gastrointestinal disorders, and exacerbation of pre‐existing respiratory conditions can rarely be seen. 21 However, studies are still needed to clarify the optimal dose and treatment duration. 15 Monitoring patients closely and conducting thorough assessments to minimize side effects is crucial. In an adult study, 18 aPAP patients who received inhaled rGM‐CSF after initial WLL had a significantly reduced need for recurrent WLL, and clinical outcomes were better in the rGM‐CSF group. 15 In another study by Munsif et al., 22 following treatment with inhaled rGM‐CSF, significant improvements were observed in clinical symptoms, dyspnea scores, pulmonary function, gas exchange, and radiological findings, with no reported severe adverse effects. 22 In the case series by Griese et al., 5 it was observed that a gradual response was achieved solely by administering inhaled GM‐CSF without performing WLL in one child; the requirement for WLL decreased after rGM‐CSF. Complete remission of respiratory failure at rest was observed at the end of the first month of treatment, with improvement in thorax CT at month four and an increase in spirometry. 5 In our patient treated solely with inhaled GM‐CSF without undergoing any WLL, clinical improvement and resolution of RA desaturation were observed in the first month of follow‐up. At 6 months, an increase in DLCO, a decrease in P(A‐a) O2 gradient, an improvement in the 6‐MWT, and an increase in lung capacity were noted.
4. CONCLUSION
aPAP is rare in children, and there is currently no consensus on treatment. Therefore, registry systems can guide diagnosing and treating rare diseases such as aPAP. In the presented case of aPAP, we observed that inhaled rGM‐CSF was easily applicable, efficient, and safe and thus might have a high therapeutic potential in children. Considering the logistic needs and experience of WLL treatment, inhaled rGM‐CSF treatment may be viewed as an alternative therapy in children with aPAP. We reviewed the aPAP literature relevant to this encouraging case and described the importance of individualized management of rare diseases.
AUTHOR CONTRIBUTIONS
Sinem Can Oksay: Conceptualization; data curation; formal analysis; visualization; writing—original draft; investigation; methodology; project administration; writing—review and editing; validation; funding acquisition; resources; software. Zeynep R. Onay: Conceptualization; data curation; visualization; investigation; writing—review and editing; validation; software. Gulay Bilgin: Conceptualization; data curation; visualization; investigation; writing—review and editing; validation; software. Deniz Mavi Tortop: Conceptualization; data curation; visualization; investigation; writing—review and editing; validation; software. Sabriye Gülçin Bozbeyoglu: Data curation; investigation; validation. Ayşe Nur Toksöz Yildirim: Data curation; visualization; investigation; validation. Matthias Griese: Conceptualization; data curation; formal analysis; visualization; supervision; investigation; methodology; project administration; writing—review and editing; validation. Saniye Girit: Conceptualization; data curation; formal analysis; visualization; writing—original draft; supervision; investigation; writing—review and editing; validation; methodology; project administration.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
The case report was written in accordance with the Declaration of Helsinki. In cases where written informed consent is obtained from the patient's parents and consent appropriate to the age of the patient is obtained, there is an ethics committee exemption for the case report at the ethics committee of Medeniyet University. Informed consent was obtained from the patient's family.
Case‐Based Dynamic Learning of Inhaled treatment for dyspnea in a rare childhood disease
Podcast 1
(pulmonologist #1): In a patient presenting with dyspnea, fatigue, weight loss, and clubbing, these complaints may be signs of a chronic disease involving the respiratory tract. In addition, other causes, including cardiac causes and other organ system diseases, must be considered with these findings.
(pulmonologist#2): In the differential diagnosis of cardiac disease, we search for findings of pulmonary edema, elevated pulmonary artery or systemic blood pressure, and rhythm disorderfindings.
(pulmonologist#3): Tuberculosis, malignancy, and collagen tissue diseases are primarily includedin the differential diagnosis because of weight loss. The patient's sedimentation, C‐reactive protein (CRP), hemoglobin (Hb), and leucocyte values were within normal limits. However, tuberculosis, pulmonary involvement of collagen tissue diseases, or interstitial lung disease may be investigatedin cases where findings related to the respiratory system are at the forefront. PFT and imagingmethods may be the next step for differential diagnosis.
Podcast 2
(pulmonologist#1): Tachypnoea without fever, low SpO2 and normal CO2 indicate hypoxaemic respiratory disorder in the foreground; this is called type 1 respiratory insufficiency. Type 2 respiratory insufficiency occurs with additionally increased pCO2, assessed in blood gas analysis.
(pulmonologist#2): Chronic hypoxia is a major etiology of clubbing. Our patient's SpO2 was 84%, indicating type 1 respiratory failure. Differential diagnosis should include alveolar hypoventilation, ventilation‐perfusion mismatch, cardiac or pulmonary shunt formation, diffusion impairment, or reduced oxygen levels. To determine whether the respiratory failure is of pulmonary or extrapulmonary origin, the alveolar‐arterial O2 gradient should be measured for a differential diagnosis. Elevated alveolar‐arterial O2 gradient in hypoxemia indicates the presence of intrapulmonary shunt, impairing gas exchange in the lung parenchyma, such as in pneumonia, acute respiratory distress syndrome, and interstitial lung diseases.
(pulmonologist#3): Normal findings on auscultation do not exclude impaired PFTs. On spirometry, a low FEV1, low FVC, and a normal or increased FEV1/FVC ratio may indicate a restrictive respiratory problem. However, it is necessary to differentiate the cause further, which may include chest wall problems like scoliosis or previous operations on the chest cage and thoracic muscle weakness. In addition to the restrictive respiratory pattern on spirometry, a reduced DLco indicates that perfusion is also impaired. Radiological imaging may be helpful in the next step.
Podcast 3
(radiologist#1): The chest X‐ray shows bilateral diffuse parenchymal opacities. They appear more irregular in areas with air trapping in the upper lobes. Ground glass opacities without air bronchograms are dominant. A high‐resolution thorax CT is required for a detailed examination.
(pulmonologist#3): What would be expected in a CT scan?
(radiologist#1): Ground‐glass opacity is a term used to describe an area of increased density in the lung seen on CT scans, where bronchial and vascular markings remain visible. This finding is nonspecific and can be caused by various conditions such as infections, chronic interstitial lung diseases, and acute alveolar diseases.
(Pulmonologist#3): If inter and intralobular septal thickening are present in addition to groundglass opacity, the resulting pattern is called a crazy‐paving pattern. Together with the patient's signs and symptoms, PAP can be considered. For the differential diagnosis of PAP, it is necessary to demonstrate the accumulation of PAS staining material in the alveoli in BAL and/or tissue sampling.
(pathologist#1): The presence of a positive PAS staining of extracellular material in BAL demonstrates PAP. In rare cases, as in this case, BAL may not prove PAP and lung tissue biopsy is necessary for a definitive diagnosis. Our patient's lung biopsy demonstrated plenty of lumpy alveolar phospholipid and protein material, which stained positive for PAS.
Podcast 4
(Pulmonologist #3): WLL is the initial treatment procedure for PAP. However, this is an invasive procedure that takes several hours and requires extensive practical experience, particularly in children and in patients with pneumothorax. Yes, I understand why the inhaled rGM‐CSF procedure was chosen for these reasons. And how would you measure clinical outcomes during his follow‐up?
(pulmonologist#4): Inhaled rGM‐CSF could be an alternative option, although it is still an experimental treatment approach among children. After a detailed discussion with our team and with his family, we put him on inhaled rGM‐CSF. Luckily, we experienced clinical improvement in a month. In the patient's follow‐up, it is necessary to continue regular check‐ups every 3‐6 months, including physical examination, detailed history, measurement of oxygen saturation in room air, spirometry, plethysmography, DLCO, and a 6‐minute walk test. Perhaps most importantly, the follow‐up should focus on rapidly and effectively managing potential pulmonary exacerbations. The patient should be closely monitored for the need for WLL.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
We acknowledge that we employed ChatGPT 3.5 and 4 to assist us in refining the clarity of our writing while developing the draft of this case report. We always maintained continuous human oversight (editing‐revising) and verified the artificial intelligence‐generated output. We never used AI to find, locate, or review the literature or resources, summarize the articles, analyze the selected articles, or synthesize the findings. The authors completed all analyses with higher‐level efforts.
Can Oksay S, Onay ZR, Bilgin G, et al. Inhaled treatment for dyspnea in a rare childhood disease. Pediatr Pulmonol. 2024;59:3692‐3698. 10.1002/ppul.27208
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Katrancioglu O, Ozgel M, Inceoglu F, Katrancioglu N, Sahin E. Is there a relationship between Haller Index and cardiopulmonary function in children with pectus excavatum? Turk Gogus Kalp Damar Cerrahisi Derg. 2023;31(3):367‐373. 10.5606/TGKDC.DERGISI.2023.24088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tazawa R, Trapnell BC, Inoue Y, et al. Inhaled granulocyte/macrophage‐colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am J Respir Crit Care Med. 2010;181(12):1345‐1354. 10.1164/RCCM.200906-0978OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Griese M. Pulmonary alveolar proteinosis: a comprehensive clinical perspective. Pediatrics. 2017;140(2):e20170610. 10.1542/PEDS.2017-0610 [DOI] [PubMed] [Google Scholar]
- 4. Bush A, Pabary R. Pulmonary alveolarproteinosis in children. Breathe (Sheff). 2020;16(2):200001. 10.1183/20734735.0001-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Griese M, Panagiotou P, Manali ED, et al. Autoimmune pulmonary alveolar proteinosis in children. ERJ Open Res. 2022;8(1):0701‐2021. 10.1183/23120541.00701-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alasiri AM, Alasbali RA, Alaqil MA, Alahmari AM, Alshamrani ND, Badri RN. Autoimmune pulmonary alveolar proteinosis successfully treated with lung lavage in an adolescent patient: a case report. J Med Case Rep. 2021;15(1):340. 10.1186/S13256-021-02906-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kumar A, Abdelmalak B, Inoue Y, Culver DA. Pulmonary alveolar proteinosis in adults: pathophysiology and clinical approach. Lancet Respir Med. 2018;6(7):554‐565. 10.1016/S2213-2600(18)30043-2 [DOI] [PubMed] [Google Scholar]
- 8. Meka SG, Mohr M, Nair GB, Salman BA. Autoimmune pulmonary alveolar proteinosis mimicking Mycoplasma pneumonia in an adolescent. Respir Med Case Rep. 2020;30:101100. 10.1016/J.RMCR.2020.101100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gajewska ME, Sritharan SS, Santoni‐Rugiu E, Bendstrup EM. Autoimmune pulmonary alveolar proteinosis in an adolescent successfully treated with inhaled rhGM‐CSF (molgramostim). Respir Med Case Rep. 2018;23:167‐169. 10.1016/J.RMCR.2018.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Price A, Manson D, Cutz E, Dell S. Pulmonary alveolar proteinosis associated with anti‐GM‐CSF antibodies in a child: successful treatment with inhaled GM‐CSF. Pediatr Pulmonol. 2006;41(4):367‐370. 10.1002/PPUL.20347 [DOI] [PubMed] [Google Scholar]
- 11. Shi S, Chen L, Qiu X, Zhao Q, Xiao Y, Yan X. Valuable serum markers in pulmonary alveolar proteinosis. Dis Markers. 2019;2019:1‐6. 10.1155/2019/9709531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Wever W, Meersschaert J, Coolen J, Verbeken E, Verschakelen JA. The crazy‐paving pattern: a radiological‐pathological correlation. Insights Imaging. 2011;2(2):117‐132. 10.1007/S13244-010-0060-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fujii K, Takeshima H, Nishimura T, et al. Autoimmune pulmonary alveolar proteinosis with features similar to nonspecific interstitial pneumonia. Respir Med Case Rep. 2022;36:101591. 10.1016/J.RMCR.2022.101591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suzuki T, Trapnell BC. Pulmonary alveolar proteinosis syndrome. Clin Chest Med. 2016;37(3):431‐440. 10.1016/J.CCM.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Campo I, Carey BC, Paracchini E, et al. Inhaled recombinant GM‐CSF reduces the need for whole lung lavage and improves gas exchange in autoimmune pulmonary alveolar proteinosis patients. Eur Respir J. 2024;63(1):2301233. 10.1183/13993003.01233-2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Borie R, Danel C, Debray MP, et al. Pulmonary alveolar proteinosis. Eur Respir Rev. 2011;20(120):98‐107. 10.1183/09059180.00001311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arai T, Hamano E, Inoue Y, et al. Serum neutralizing capacity of GM‐CSF reflects disease severity in a patient with pulmonary alveolar proteinosis successfully treated with inhaled GM‐CSF. Respir Med. 2004;98(12):1227‐1230. 10.1016/J.RMED.2004.08.011 [DOI] [PubMed] [Google Scholar]
- 18. Jehn LB, Bonella F. Pulmonary alveolar proteinosis—current and future therapeutical strategies. Curr Opin Pulm Med. 2023;29(5):465‐474. 10.1097/MCP.0000000000000982 [DOI] [PubMed] [Google Scholar]
- 19. Sirin Kose S, Asilsoy S, Uzuner N, Karaman O, Ozer E, Anal O. Pulmonary alveolar proteinosis in hereditary and autoimmune forms with 2 cases. Pediatr Emerg Care. 2020;36(8):e470‐e472. 10.1097/PEC.0000000000001536 [DOI] [PubMed] [Google Scholar]
- 20. Sheng G, Chen P, Wei Y, Chu J, Cao X, Zhang HL. Better approach for autoimmune pulmonary alveolar proteinosis treatment: inhaled or subcutaneous granulocyte‐macrophage colony‐stimulating factor: a meta‐analyses. Respir Res. 2018;19(1):163. 10.1186/S12931-018-0862-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tazawa R, Ueda T, Abe M, et al. Inhaled GM‐CSF for pulmonary alveolar proteinosis. N Engl J Med. 2019;381(10):923‐932. 10.1056/NEJMOA1816216 [DOI] [PubMed] [Google Scholar]
- 22. Munsif M, Sweeney D, Leong TL, Stirling RG. Nebulised granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) in autoimmune pulmonary alveolar proteinosis: a systematic review and meta‐analysis. Eur Respir Rev. 2023;32(170):230080. 10.1183/16000617.0080-2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.