

Abstract
患儿,女,11 d,因生后呼吸困难11 d入院,临床主要表现为吸气性呼吸困难、喂养困难,伴特殊面容(小下颌、高腭弓、腭裂、舌后坠和口角向右侧歪斜),初步诊断皮罗序列征。治疗上给予呼吸机辅助通气、营养、手术结扎动脉导管等治疗后无明显好转。家系全外显子组测序示CHD7基因存在杂合变异c.3082A>G(p.Ile1028 Val),系CHARGE综合征的致病性变异。患儿最终诊断为CHARGE综合征,家属考虑预后不良而放弃治疗。该文报道1例新生儿期起病的CHD7基因变异所致CHARGE综合征病例,以及对该疾病的多学科诊疗,有助于早期疾病识别和指导临床决策。
Keywords: CHARGE综合征, CHD7基因, 皮罗序列征, 新生儿
Abstract
A female infant, aged 11 days, was admitted due to dyspnea for 11 days after birth, with the main clinical manifestations of inspiratory dyspnea, feeding difficulties, and unusual facies (micrognathia, high palatal arch, cleft palate, glossoptosis, and oblique mouth to the right), and the preliminary diagnosis was Pierre-Robin syndrome. There was no marked improvement after treatment such as ventilator-assisted ventilation, nutrition, and surgical ligation of patent ductus arteriosus. Whole-exome sequencing of the family showed a heterozygous mutation of c.3082A>G (p.Ile1028 Val) in the CHD7 gene, which was a pathogenic mutation of CHARGE syndrome. The neonate was ultimately diagnosed with CHARGE syndrome, and the family decided to withdraw treatment due to concerns about poor prognosis. This article reports a case of CHARGE syndrome caused by a mutation in the CHD7 gene and the multidisciplinary diagnosis and treatment of this disease, in order to provide help for early disease identification and guide clinical decision-making.
Keywords: CHARGE syndrome, CHD7 gene, Pierre-Robin syndrome, Neonate
1. 前言
CHARGE综合征(CHARGE syndrome, CS)是一种罕见的主要由染色质域解旋酶DNA结合蛋白7(chromodomain helicase DNA-binding protein 7, CHD7)基因突变所致的单基因常染色体显性遗传病,临床表现为多器官受累的先天性发育异常,发病率为1/15 000~1/10 000[1],预后视病情而定。该病临床表现多种多样,1979年首次被Hall[2]报道,1981年Pagon等[3]根据临床上一系列症状的英文首字母将其命名为“CHARGE syndrome”,包括眼器官先天裂开与脑神经缺损(coloboma)、心脏缺损(heart defects)、后鼻孔闭锁(atresia of the choanae)、生长与发育迟缓(retardation of growth and development)、生殖泌尿道系统异常(genital and urinary abnormalities)、耳朵异常或听力丧失(ear abnormalities and/or hearing loss)。CS的诊断标准包括2个主要表现+任一次要表现[4],主要表现:(1)眼器官先天裂开与脑神经缺损;(2)后鼻道闭锁或唇/腭裂;(3)外耳/中耳/内耳的缺陷(包括半规管异常);(4)致病性CHD7基因变异。次要表现:(1)颅神经功能障碍(包括听力损伤);(2)吞咽困难/喂养困难;(3)大脑结构异常;(4)发育迟缓/孤独症谱系障碍/智力迟缓;(5)下丘脑-垂体功能不全(性激素/生长激素缺乏)和性腺异常;(6)心脏或食管畸形;(7)肾脏异常;(8)骨骼或指端畸形。本例患儿存在致病性CHD7基因变异和腭裂等主要表现,以及吞咽困难/喂养困难、大脑结构异常、心脏畸形等多个次要表现,故明确诊断为CS。
2. 病例介绍
现病史:患儿,女,11 d,因生后呼吸困难11 d转入我院。患儿系第1胎第1产,经阴道分娩出生,胎龄36+6周,出生体重2 750 g,出生时羊水、脐带、胎盘未见异常,Apgar评分均为10分。生后2 h出现呼吸困难,体格检查示小下颌、高腭弓、腭裂、舌后坠,伴有口角向右侧歪斜,收住当地医院给予低流量吸氧、经验性抗感染、雾化、吞咽功能锻炼等治疗11 d后无好转,考虑病情复杂联系我院转运组出车接回,转运途中给予高流量吸氧(8 L/min,吸入气氧浓度30%),生命体征平稳。
入院体格检查:呼吸机辅助通气下体温36.5 ℃,心率158次/min,呼吸56次/min,血压68/51 mmHg,体重2.96 kg,身长48 cm,头围32 cm。神志清楚,精神反应正常,小下颌、腭弓高、舌后坠、腭裂伴口角向右侧歪斜,哭闹时加重,双肺呼吸音粗,可闻及吸气性喉鸣音及痰鸣音;心音有力,心前区可闻及连续性杂音;腹平软,未触及包块,肝脾不大;肠鸣音正常,生殖器无畸形,四肢肌张力正常。肢端温暖,毛细血管充盈时间2 s。
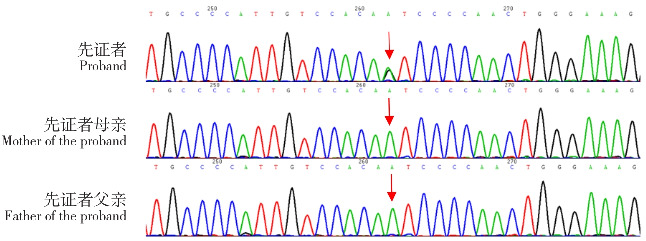
入院辅助检查:血气分析示酸碱度7.34(参考范围:7.35~7.45),碳酸氢根29.7 mmol/L(参考范围:22~27 mmol/L)。甲功三项示促甲状腺激素 20.252 μIU/mL(参考范围:0~5 μIU/mL),其余未见异常。血常规、C反应蛋白、肝肾功能、凝血功能、尿有机酸分析、血酰基肉碱分析、血浆氨基酸分析均未见明显异常。胸部X线片提示心影增大,双肺纹理增粗。胸部+颌面部CT+气道三维重建示小下颌、腭裂、舌后坠;颈7椎体水平气管、右主支气管起始段管腔变窄(图1)。纤维支气管镜示喉软化,左主支气管轻度狭窄(外压)(图2)。心脏超声示全心增大,动脉导管未闭(patent ductus arteriosus, PDA)(4.2 mm,左向右分流);卵圆孔未闭(双孔4.3 mm、4.8 mm,左向右分流);永存左上腔静脉。头颅磁共振成像示存在颅内发育性静脉畸形及双侧顶叶脑室旁白质少许点状异常信号(图3)。24 h脑电图示背景活动成熟度延迟(图4)。眼底及泌尿生殖系统超声未见明显异常,听力检测未完善。患儿家系全外显子组测序示CHD7基因存在新发杂合错义变异c.3082A>G(p.Ile1028 Val)(NM_017780.4:chr8:61735186)(图5)。根据美国医学遗传学与基因组学学会的遗传变异分类标准和指南[5]:查阅相关文献[6]及数据库,该变异类型在dbSNP数据库收录ID为rs121434338,对应的氨基酸残基位于基因编码蛋白质的Helicase ATP-binding结构域;该变异在多例CS患者中报道(PMID:15300250、25472840、32914532、35904121等);该变异在ClinVar数据库中注释为致病性(variation ID:2022);在HGMD数据库中收录ID为CM042321;综合家系分析,患儿为杂合变异,其父母未检出该变异,提示该变异为新发变异(不排除父母存在生殖腺或体细胞嵌合)。综上该变异判定为致病性变异。
图1. 患儿胸部+颌面部CT+气道三维重建结果 A~B:气道三维重建,右主支气管起始段变窄;C~D:胸部CT,颈7椎体水平气管、右主支气管起始段变窄;E:颌面部CT,下颌相对短小,后缩;F:硬腭后部中央见部分骨质连续性中断,裂隙宽处约0.4 mm,舌根后坠。箭头所示为病变位置。.
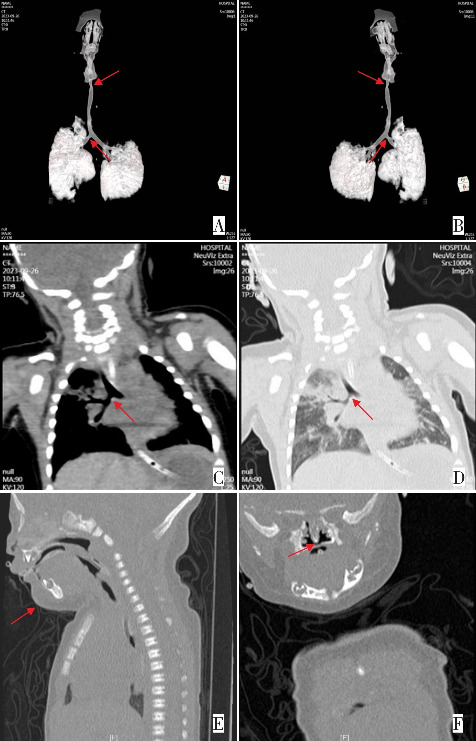
图2. 患儿纤维支气管镜结果 A:气道通畅;B:气管隆嵴;C:喉、杓状软骨内收,喉软化;D:左主支气管远端轻度狭窄。.
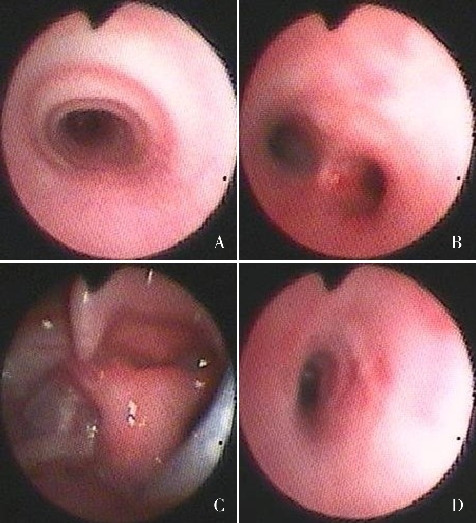
图3. 患儿头颅磁共振成像平扫结果 A:右侧顶叶白质内异常血管影,考虑发育性静脉畸形可能;B:双侧顶叶脑室旁白质少许点状异常信号。箭头所示为病变位置。.
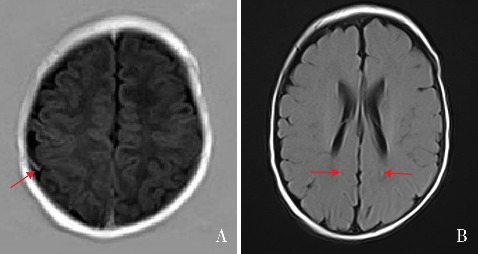
图4. 患儿24 h振幅整合脑电图结果 下边界稍低,下边界4~10 μV,上边界10~50 μV,可见睡眠觉醒周期,无典型发作图形。.
图5. 患儿及其父母家系全外显子组测序结果 患儿CHD基因存在新发杂合错义变异c.3082A>G(p.Ile1028 Val)(NM_017780.4,chr8:61735186),父母未检出该变异。箭头所示为变异位置。.
3. 多学科诊疗
3.1. 新生儿重症监护病房初诊
患儿生后即出现呼吸困难,伴有喂养困难,有特殊面容(下颌短小、高腭弓、腭裂、舌后坠、口角向右侧歪斜),经外院呼吸支持、经验性抗感染等常规治疗无效,考虑病情复杂转入我科。结合现有病史资料、临床表现、体格检查,初步诊断考虑皮罗序列征(Pierre-Robin syndrome, PRS),是否能用“一元论”来解释存在疑问,需全面评估患儿各系统情况后综合分析病因。首先患儿出生即出现呼吸困难,体格检查有上气道梗阻表现,应高度怀疑上气道梗阻性疾病,结合喂养困难及特殊面容,需完善胸部+颌面部CT+气道三维重建、电子喉镜、纤维支气管镜检查评估咽喉部、肺部及气道发育情况,必要时进一步完善胸部CT血管造影评估胸部大血管发育情况;其次体格检查可闻及心前区连续性杂音,需排除先天性心脏病,予完善心脏超声;患儿为晚期早产儿,出生后存在喂养困难,建议完善甲状腺功能、尿有机酸分析、血酰基肉碱分析、血浆氨基酸分析等代谢性疾病方面的检查。因患儿存在呼吸及喂养问题,伴多个器官先天发育异常,染色体基因病可能性较大,予完善家系全外显子组基因检测。积极联系多学科会诊讨论,协助评估病情明确病因。
3.2. 口腔颌面外科会诊
患儿因呼吸困难转入我院,初步诊断为PRS,临床表现及体格检查与之相符,同意新生儿科诊断。治疗上建议给予俯卧位通气,避免误吸及呛咳,目前有PRS手术适应证,但患儿合并症状性动脉导管未闭(symptomatic patent ductus arteriosus, sPDA),同台手术风险较大,且患儿术后护理难度大,建议先行PDA结扎术,待病情稳定后再考虑行下颌骨牵张成形术。PRS病因不明,可能与孕期营养不良、某些药物使用、接触放射性及有毒、有害物质损害及遗传因素有关。OMIM数据库目前已收录至少40种具有PRS表型的综合征,PRS更倾向于一种临床表型而非疾病诊断[7]。患儿同时存在下颌短小、腭裂、气道狭窄、喉软化、先天性心脏病、神经系统异常影像学等表现,建议对症治疗同时完善基因检测,进一步明确病因,帮助评估预后及指导治疗。
3.3. 心脏外科会诊
患儿入院体格检查发现心前区可闻及明显连续性杂音,心脏超声示全心增大,动脉导管4.2 mm,左向右分流,左房内径/主动脉根部内径比值1.57,胸部X线片示心影增大,同时临床上存在无法脱离呼吸支持且参数难以下调,符合sPDA诊断标准,有药物及手术干预指征。胸部CT平扫可见右主支气管起始段管腔变窄,但纤维支气管镜下未见明显狭窄,不排除与增大的心脏压迫相关。回顾病史及诊疗经过,考虑目前无法脱离呼吸支持的最主要因素为上气道问题,sPDA作为次要因素参与其中。sPDA药物关闭治疗无效,可考虑手术干预治疗,但获益未可知,需征求家属意见后决定。
3.4. 医学影像中心会诊
典型的PRS影像学表现为小下颌、舌后坠,大多数患儿合并腭裂,部分伴有喉软化或气道软化[7-8],该患儿存在上述所有表现,与PRS诊断相符。胸部CT平扫可见右主支气管起始段管腔变窄,但纤维支气管镜下未见明显气道狭窄,考虑为行胸部CT检查时患儿处于吸气相。头颅磁共振成像平扫提示右侧顶叶白质内可见一条异常信号影,T2加权信号呈低信号改变;双侧顶叶白质可见散在点状异常信号,T2加权信号呈低信号,T1加权信号呈略高信号。右顶叶异常信号影考虑发育性静脉畸形可能性大,双侧顶叶脑室旁白质存在局灶性损伤,具体情况需结合临床进一步评估。
3.5. 康复科会诊
患儿主要经胃管喂养,吸吮-吞咽-呼吸不协调,用奶嘴刺激后出现吸吮反应,新生儿口腔评估量表评分为4分,诊断为中度吞咽障碍,吸吮检查时主要表现为:(1)用奶嘴触及嘴唇无反应,晃动奶嘴刺激后出现反应;(2)持续出现与胎龄不相符的不成熟吸吮形态,考虑可能是呼吸困难、疲倦所致不能吸吮持续2 min;(3)吸吮-吞咽-呼吸不协调,出现鼻翼扇动、转头。建议行口周肌肉按摩、下颌被动运动、吸吮训练、口腔内感觉刺激、咽感觉刺激,双咬肌/舌骨上肌神经肌肉电刺激。
3.6. 胎儿医学及产前诊断中心会诊
患儿家系全外显子组测序结果提示CHD7基因杂合突变,CHD7基因位于8号染色体8q12上,全长195 kb,包含38个外显子,编码2 997个氨基酸,编码CHD7蛋白,该蛋白广泛表达于全身各组织,包括眼、耳、心脏、神经系统、生殖系统等均可受累[9]。CHD7蛋白在染色体重塑、细胞周期调节、细胞凋亡调控、转录调节和胚胎干细胞多样性中发挥作用,CHD7基因的突变导致产生无功能短CHD7蛋白,从而中断基因表达的调节,这些变化如果发生在胚胎期,就会导致基因多效性体征和症状的出现[10]。有学者统计,约90% CS的发生与CHD7基因突变相关[11],此外SME3A、SEMA3E、KMT2D、RERA、EP300、PUF60等基因突变也有案例报道[12-15]。本病例发现的CHD7基因变异为新发突变,父母均未检测到相关变异,变异外显子第3 082位点由腺嘌呤变为鸟嘌呤,为明确的致病变异位点。
本病例初诊为PRS,PRS本身并不是单一的综合征,而是一系列具有因果关系的疾病组成,目前病因尚不完全明确,与下颚发育相关的染色体或基因突变为最常见,也可能是环境问题而产生致畸。目前临床PRS更倾向于一种临床表型而非疾病诊断,可以作为独立的疾病存在,也可以作为全身其他系统疾病或综合征的症状之一,因此PRS和CS诊断并不冲突,最终通过基因检测结果修订诊断为“CHARGE综合征”。
CS产前超声筛查最常见异常为先天性心脏缺陷、后鼻孔闭锁和气管食管闭锁,内外耳异常、宫内生长受限及肾脏异常是超声诊断胎儿CHARGE综合征相对特征性表现[16-17],但超声筛查依赖于操作者水平,往往会出现漏诊。Polito等[18]的一项纳入1981—2022年943例CS患者案例的荟萃分析提示先天性心脏病的发病率为76.6%,其中PDA为26%,室间隔缺损为21%,房间隔缺损为18%,法洛四联症为11%,主动脉畸形为24%。目前无创产前监测为评估病情提供了新的替代方案,可以通过母亲血液检测到胎儿变异,但检测费用高昂且需排除母亲为低水平嵌合体携带者[19]。CS诊断需要详细的病史并结合产前、新生儿期表现,当在子宫内发现胎儿发育不良、宫内生长受限、后鼻孔闭锁、食管闭锁、肾脏发育异常、唇腭裂或先天性心脏缺陷时应予以考虑。
3.7. 新生儿重症监护病房诊断思路总结
该患儿生后即起病,入院后评估主要问题为吸气性呼吸困难、无法脱离呼吸支持、喂养困难及心脏明显杂音。(1)呼吸系统:通过外院病史及体格检查,入院初步诊断PRS,因PRS常常合并气道疾病及需评估手术指征,故入院后完善胸部+颌面部CT+气道三维重建、纤维支气管镜评估病情,影像学提示符合PRS表现,有喉软化、气道狭窄表现。(2)喂养困难:PRS患儿常常合并吞咽协调障碍,但需鉴别神经系统异常所致,故请康复科协助评估病情,同时完善头颅磁共振成像及脑电图进一步评估,头颅磁共振成像提示静脉发育性畸形,脑电背景活动成熟度延迟,无法完全解释存在喂养困难。(3)心脏疾病:入院心脏连续性杂音明显,结合心脏超声检查及临床诊断sPDA,经多学科讨论后予手术结扎PDA。上述异常临床表现及辅助检查无法用“一元论”来解释患儿病情,考虑目前病情以PRS为主,PRS病因不明,遗传性因素复杂,不同疾病预后及治疗方式各异,为进一步明确病因及指导后续诊疗,征得家属同意后予完善家系全外显子组基因测序,结果提示CHD7基因存在新发杂合突变,通过查阅文献及结合病史,最终确诊为CS。
4. 住院经过及转归
入院后根据病史及临床表现,初步诊断为PRS。治疗上继续予呼吸机辅助通气、加强营养、康复训练、口服布洛芬关闭sPDA(2个疗程均失败)等治疗。因PRS及sPDA均需手术治疗,经多学科会诊讨论综合评估后考虑同步手术风险大,决定先行PDA结扎术,术后再评估患儿呼吸困难改善情况,择期行PRS的手术治疗。患儿在全麻下行PDA结扎术,但术后仍无法脱离无创呼吸支持。家系全外显子组基因测序结果回报CHD7基因c.3082A>G(p.Ile1028Val)新发杂合致病性变异。结合患儿病史、辅助检查及基因检测结果,符合CS诊断。考虑到该病目前尚无特异性治疗手段,主要通过手术和序贯对症处理,远期预后差,家属放弃治疗,签字自动出院,出院后失访。
5. 小结
CS临床表现各异,特征性表现包括眼缺陷、后鼻道闭锁、耳部异常等均可见于其他疾病或综合征,典型CS诊断需2个主要表现+任一次要表现[4],往往难以识别,临床上评估如果用“一元论”无法解释,应考虑CS。CS治疗上以对症为主,需要多学科协作序贯开展、个体化定制,生后早期需要注意呼吸及摄食功能维护、全面脏器功能评估并进行手术干预。中期干预包括听觉、视觉及语言功能维护。后期需监测激素水平并进行适当的替代治疗,同时积极进行心理康复治疗[20-21]。这些治疗方案极大地提高了CS患儿的生存质量,对远期预后有获益。CS临床表现复杂多样,对于考虑该疾病患儿应尽早进行基因检测,明确其基因突变位点并进行遗传咨询,为下一步治疗提供依据。
利益冲突声明
所有作者声明无利益冲突。
作者贡献
高波负责整理数据及论文初稿撰写、论文修改;肖淑负责临床资料收集、参考文献查阅;陈晓文、李茹负责病例分析、指导文章的修改及审核;王玲负责多学科讨论组织以及文章的构思、设计和修改。
参 考 文 献
- 1. Hudson A, Trider CL, Blake K. CHARGE syndrome[J]. Pediatr Rev, 2017, 38(1): 56-59. DOI: 10.1542/pir.2016-0050. [DOI] [PubMed] [Google Scholar]
- 2. Hall BD. Choanal atresia and associated multiple anomalies[J]. J Pediatr, 1979, 95(3): 395-398. DOI: 10.1016/s0022-3476(79)80513-2. [DOI] [PubMed] [Google Scholar]
- 3. Pagon RA, Graham JM, Zonana J, et al. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association[J]. J Pediatr, 1981, 99(2): 223-227. DOI: 10.1016/s0022-3476(81)80454-4. [DOI] [PubMed] [Google Scholar]
- 4. Hale CL, Niederriter AN, Green GE, et al. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria[J]. Am J Med Genet A, 2016, 170A(2): 344-354. DOI: 10.1002/ajmg.a.37435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424. DOI: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roux I, Fenollar-Ferrer C, Lee HJ, et al. CHD7 variants associated with hearing loss and enlargement of the vestibular aqueduct[J]. Hum Genet, 2023, 142(10): 1499-1517. DOI: 10.1007/s00439-023-02581-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karempelis P, Hagen M, Morrell N, et al. Associated syndromes in patients with Pierre Robin sequence[J]. Int J Pediatr Otorhinolaryngol, 2020, 131: 109842. DOI: 10.1016/j.ijporl.2019.109842. [DOI] [PubMed] [Google Scholar]
- 8. Hsieh ST, Woo AS. Pierre Robin sequence[J]. Clin Plast Surg, 2019, 46(2): 249-259. DOI: 10.1016/j.cps.2018.11.010. [DOI] [PubMed] [Google Scholar]
- 9. Bedeschi MF, Crippa BL, Colombo L, et al. A case series of CHARGE syndrome: identification of key features for a neonatal diagnosis[J]. Ital J Pediatr, 2020, 46(1): 53. DOI: 10.1186/s13052-020-0806-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. 吴逊, 宋洁, 唐幂. 新发CHD7基因突变致CHARGE综合征的诊断分析[J]. 临床小儿外科杂志, 2022, 21(9): 850-854. DOI: 10.3760/cma.j.cn101785-202205065-010. [DOI] [Google Scholar]
- 11. Zentner GE, Layman WS, Martin DM, et al. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome[J]. Am J Med Genet A, 2010, 152a(3): 674-686. DOI: 10.1002/ajmg.a.33323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lalani SR, Safiullah AM, Molinari LM, et al. SEMA3E mutation in a patient with CHARGE syndrome[J]. J Med Genet, 2004, 41(7): e94. DOI: 10.1136/jmg.2003.017640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sakata S, Okada S, Aoyama K, et al. Individual clinically diagnosed with CHARGE syndrome but with a mutation in KMT2D, a gene associated with Kabuki syndrome: a case report[J]. Front Genet, 2017, 8: 210. DOI: 10.3389/fgene.2017.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moccia A, Srivastava A, Skidmore JM, et al. Genetic analysis of CHARGE syndrome identifies overlapping molecular biology[J]. Genet Med, 2018, 20(9): 1022-1029. DOI: 10.1038/gim.2017.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ufartes R, Schwenty-Lara J, Freese L, et al. Sema3a plays a role in the pathogenesis of CHARGE syndrome[J]. Hum Mol Genet, 2018, 27(8): 1343-1352. DOI: 10.1093/hmg/ddy045. [DOI] [PubMed] [Google Scholar]
- 16. Biard JM, Payrat S, Clapuyt P, et al. Antenatal diagnosis of CHARGE syndrome: prenatal ultrasound findings and crucial role of fetal dysmorphic signs. About a series of 10 cases and review of literature[J]. Eur J Med Genet, 2021, 64(4): 104189. DOI: 10.1016/j.ejmg.2021.104189. [DOI] [PubMed] [Google Scholar]
- 17. Traisrisilp K, Chankhunaphas W, Sittiwangkul R, et al. Prenatal sonographic features of CHARGE syndrome[J]. Diagnostics (Basel), 2021, 11(3): 415. DOI: 10.3390/diagnostics11030415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Polito MV, Ferraioli M, Nocilla A, et al. CHARGE syndrome and congenital heart diseases: systematic review of literature[J]. Monaldi Arch Chest Dis, 2023. Epub ahead of print. DOI: 10.4081/monaldi.2023.2661. [DOI] [PubMed] [Google Scholar]
- 19. Daley R, Hill M, Chitty LS. Non-invasive prenatal diagnosis: progress and potential[J]. Arch Dis Child Fetal Neonatal Ed, 2014, 99(5): F426-F430. DOI: 10.1136/archdischild-2013-304828. [DOI] [PubMed] [Google Scholar]
- 20. 杨国珺, 蒋海越. CHARGE综合征研究进展[J]. 中华整形外科杂志, 2018, 34(7): 582-585. DOI: 10.3760/cma.j.issn.1009-4598.2018.07.022. [DOI] [Google Scholar]
- 21. 张贝贝, 巩纯秀. CHARGE综合征诊疗新进展[J]. 中华实用儿科临床杂志, 2019, 34(14): 1116-1120. DOI: 10.3760/cma.j.issn.2095-428X.2019.14.019. [DOI] [Google Scholar]