


Abstract
Enhancers play a critical role in dynamically regulating spatial-temporal gene expression and establishing cell identity, underscoring the significance of designing them with specific properties for applications in biosynthetic engineering and gene therapy. Despite numerous high-throughput methods facilitating genome-wide enhancer identification, deciphering the sequence determinants of their activity remains challenging. Here, we present the DREAM (DNA cis-Regulatory Elements with controllable Activity design platforM) framework, a novel deep learning-based approach for synthetic enhancer design. Proficient in uncovering subtle and intricate patterns within extensive enhancer screening data, DREAM achieves cutting-edge sequence-based enhancer activity prediction and highlights critical sequence features implicating strong enhancer activity. Leveraging DREAM, we have engineered enhancers that surpass the potency of the strongest enhancer within the Drosophila genome by approximately 3.6-fold. Remarkably, these synthetic enhancers exhibited conserved functionality across species that have diverged more than billion years, indicating that DREAM was able to learn highly conserved enhancer regulatory grammar. Additionally, we designed silencers and cell line-specific enhancers using DREAM, demonstrating its versatility. Overall, our study not only introduces an interpretable approach for enhancer design but also lays out a general framework applicable to the design of other types of cis-regulatory elements.
Graphical Abstract
Graphical Abstract.

Introduction
The temporal and spatial pattern of gene expression is primarily orchestrated by cis-regulatory elements (CREs), such as promoters and enhancers, which play a critical role in establishing and maintaining the identity and function of tissues or organs. Due to their remarkable capacity in driving the transcription of any coding sequence in an expression cassette, synthetic CREs are widely used in nucleic acid-based therapeutics to obtain the appropriate expression of pharmaceutic products, and in cell-based bioreactors to increase production yield of valuable biologics. Recently, CRE engineering has also emerged as a valuable tool in genomic breeding, playing a pivotal role in advancing sustainable agricultural development (1–3). To identify CREs residing in the genome, a plethora of high-throughput based functional genomics technologies has been exploited and applied in various organisms, including biochemical marker-based epigenomic profiling assays (4–6) and massively parallel reporter assays (MPRAs) that directly measure CRE activity (7–10). Despite the wealth of natural CREs identified from existing genomes, they may not always align with the expression objectives required for diverse application scenarios. This discrepancy underscores the necessity for designing de novo synthetic CREs (11).
The rational CRE design relies heavily on a comprehensive understanding of the intrinsic cis-regulatory code governing the activity of CREs. Over past decades, researchers have unveiled numerous combinations of DNA sequence features, notably transcription factor binding sites (TFBSs), that constitute this regulatory code (12–14). Therefore, conventional approaches to crafting CREs with specific attributes predominantly involve leveraging known functional sequence motifs and employing iterative mutagenesis. For example, the manipulation of the orientation, number and spacing of well-established key functional motifs within existing or native CREs has proven instrumental in artificial promoter design (2,15). Additionally, the introduction of random mutations in a functional screening library has led to the identification of several novel promoters with higher regulatory activity compared to their native counterparts (16). However, progress in CRE design has been sluggish due to the intricate specificity of spatio-temporal patterns and the nuanced flexibility of regulatory grammar, particularly in promoter-distal CREs. The incomplete understanding of regulatory code largely limits the number of lexicons available in the CRE design toolbox. In addition, the exponential expansion of the sequence search space (4L for a CRE, where L represents its sequence length) and the inherent complexity of the weak grammar of the regulatory code, such as the impact from the flanking sequences of motifs, further hinders the rational CRE design (17). Owing to the collective effects of these factors, conventional CRE design strategies, i.e. rational CRE design strategies, suffer from lack of efficiency, demanding a profound understanding from seasoned experts to craft the backbone sequence, and series of time-consuming and labor-intensive experiments to evolve sequence design (18).
In recent years, the incorporation of deep neural networks (DNNs) has propelled genomics research forward, particularly in predicting TF (DNA-binding protein) binding sites (19), CRE activity (20–23) and alternative splicing events (24–26). This progress extends to forecasting the activity of CREs. Notably, the Enformer, employing a deep learning architecture that assimilates information from up to 100 kb away in the genome, stands out for its capacity to precisely predict numerous epigenetic and transcriptional profiles using only the DNA sequence as input (23). The efficacy of DNNs in these domains lies in their ability to leverage extensive and highly heterogeneous datasets, autonomously revealing hidden predictive patterns within sequence data (27,28). Based on its superior prediction accuracy, DNNs have been integrated with evolutionary optimization algorithms (EOAs) in the application of de novo CRE design. By constructing a DNN model that precisely recapitulates the sequence–activity relationship of CREs, EOA can be employed to emulate the natural evolutionary process. This algorithm navigates an expansive sequence space in search of mutations that enhance fitness, which aligns with the target of CRE optimization. Specifically, the procedure involves systematically introducing mutations into an initial population. At each iteration, the DNN model’s predictive prowess serves as the fitness function, assessing the activity of the evolved sequence. This evaluation guides the selection of mutations that confer the desired CRE function, facilitating the in silico evolution of CRE sequences through multiple mutational steps (29). The integration of DNNs and EOAs offers a powerful approach to streamline and enhance the CRE design process. However, existing work mainly focus on promoters, untranslated regions (UTRs), or other promoter-proximal CREs (29–32). The promoter-distal CREs, such as enhancers, present distinct sequence features and more flexible organizational principles (33,34), thereby intensifying both theoretical and practical challenges in de novo CRE design. A notable advancement in this domain is the development of DeepSTARR, a model designed to predict enhancers. This model was trained using data from self-transcribing active regulatory region sequencing (STARR-seq), which measured genome-wide enhancer activities in Drosophila S2 cells. In the following phase of enhancer design, the researchers chose to predict the regulatory activity of one billion random sequences using the model. Unlike an EOA approach, this random in silico screening only yielded synthetic sequences with activity comparable to natural enhancers (35). The question remains open as to whether unexplored DNA sequences exist that could demonstrate activity surpassing that observed in natural enhancers.
Another strategy of DNN-based CRE design is built on the deep generative adversarial networks (GANs). GANs operate by engaging in a minimax adversarial game between the generator and discriminator neural networks, enabling the generation of novel molecules from the latent space (36). Remarkable success has been demonstrated by GANs in tasks such as promoter design and protein engineering (37–41). However, GANs suffer from poor interpretability. Specifically, the origins and organization of semantics or functional sequence motifs in the latent space remain unclear. GANs are known for challenges such as mode collapse, non-convergence and instability during training, particularly when faced with inappropriate network structures and parameter initializations. Furthermore, to obtain CREs with specific properties, pre-trained classifier or regression DNNs are often required, which further increases the computational burden (38).
Here, we developed DREAM (DNA cis-Regulatory Elements with controllable Activity design platforM), an efficient, scalable and explainable computational framework to design CREs from scratch. DREAM can learn a repertoire of the regulatory lexicon related to the regulatory activity and accurately predicts the regulatory activity of enhancers. The enhancer regulatory activity prediction module within DREAM exhibits superior performance compared to the DeepSTARR model (35), representing state-of-the-art performance. Using this framework, we emulated the optimization trajectory of developmental and housekeeping enhancers within the sequence space, obtaining synthetic enhancers that exhibit approximately 3.6-fold higher activity than the strongest natural enhancer in the Drosophila genome. Surprisingly, the function of these enhancers optimized in Drosophila S2 cells are conserved across a diverse range of species and exhibit strong ability to stimulate the transcription of the luciferase reporter gene. We suggest that DREAM could find broad applications in designing various classes of CREs and provide valuable biological insights into their underlying regulatory grammar.
Materials and methods
UMI-STARR-seq data collection and processing
The genome-wide high-resolution Drosophila developmental and housekeeping enhancer UMI-STARR-seq (Unique Molecular Identifiers-STARR-seq) dataset was retrieved from the GEO database (accession number GSE183939) (35). The RNA and DNA input reads were mapped to the Drosophila genome (dm3) using Bowtie2 with default parameters. For paired-end RNA reads that mapped to the same positions, only paired-end RNA reads with different UMIs were retained. Enhancer activity was quantified as the log2 fold change of RNA reads count mapped to the genomic region over the input DNA read counts. To maintain comparability with the DeepSTARR model, we utilized an identical training, validation and hold-out chromosome dataset for training and evaluating the multitask deep learning model. Briefly, the Drosophila dm3 genome was divided into 249 bp windows with a stride of 100 bp. To ensure high-fidelity regulatory activity, the dataset only includes the bins with more than five reads in the DNA library and at least one read in the RNA library. To increase the sequences diversity in the dataset, three type potential enhancer sequences were introduced into the dataset: (i) 20 000 randomly sampled sequences overlapping the chromatin accessible regions in Drosophila S2 (7580), kc167 (7175) and OSC (5245) cell types (42,43); (ii) 8842 enhancers from Drosophila OSC (4640) and BG3 (4202) cell types (44); (iii) 1778 inducible enhancers in Drosophila S2 cells for ecdysone (1593) and Wnt (185) signaling (45,46). Additionally, we included 11 658 developmental and 7062 housekeeping enhancers, as well as 21 0686 random windows with a range of enhancer activity levels. The dataset was augmented by adding the reverse complement of each sequence with the same regulatory activity. Ultimately, the validation dataset comprised 40 570 sequences, and the testing dataset comprised 4 1186 sequences, all derived from the first and second halves of chr2R, respectively.
The architecture of SENet
The Squeeze-and-Excitation (SE) attention mechanism represents a channel-wise attention mechanism widely employed in computer vision and deep learning. SE blocks dynamically recalibrate channel-wise feature responses by explicitly modeling interdependencies between convolutional feature channels, thereby enhancing the representational power of conventional convolutional neural networks (47). In this study, we constructed a multi-task convolutional neural network that only takes one-hot encoded DNA sequences as the input to predict both developmental and housekeeping enhancer regulatory activities by incorporating the novel SE block.
First, the DNA sequences were transformed by the 1D convolutional layer (filters = 512, kernel_size = 7, strides = 1) followed by the batch normalization, the non-linearity activation function and average pooling (size = 5, strides = 2). Previous study has shown that the convolutional neural networks utilizing an exponential activation function in the first layer filters consistently lead to interpretable and robust representations of DNA motifs (48). To enhance the model interoperability, the exponential activation was utilized as the non-linearity activation function. Subsequently, the DNA feature maps were further transformed by four SE-ResNet modules (filters = [256, 256, 512, 512], blocks = [2, 2, 2, 2]). Finally, there are flatten layer followed by two fully connected layers with 512 and 256 neurons, respectively. The output of the fully connected layer was activated by a Rectified Linear Unit (ReLU) non-linear activation function, followed by a dropout layer with a dropout rate set to 0.2.
The SE block involves three computational operations: the squeeze operation, excitation operation, and scale operation. The SE computational block can build on any given transformation Ftr, e.g. a convolution, mapping the input 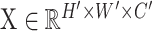 to the feature maps
to the feature maps 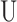 where
where 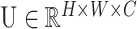 to perform feature recalibration. The features U are first passed through a squeeze operation, which produces a channel descriptor by aggregating feature maps across their spatial dimensions (H × W). This descriptor aims to create an embedding of the global distribution of channel-wise feature responses, enabling information from the global receptive field of the network to be utilized by all its layers. The aggregation is followed by an excitation operation, which takes the form of a simple self-gating mechanism that takes the embedding as input and produces a collection of per-channel modulation weights. To mitigate the vanishing/exploding gradient problem in our deeper convolutional neural network, we integrate SE blocks with the ResNet by using the SE block transformation Ftr is taken to be the non-identity branch of a residual module. SE both act before summation with the identity branch. The detailed architecture of our model is plotted in Supplementary Figure S1.
to perform feature recalibration. The features U are first passed through a squeeze operation, which produces a channel descriptor by aggregating feature maps across their spatial dimensions (H × W). This descriptor aims to create an embedding of the global distribution of channel-wise feature responses, enabling information from the global receptive field of the network to be utilized by all its layers. The aggregation is followed by an excitation operation, which takes the form of a simple self-gating mechanism that takes the embedding as input and produces a collection of per-channel modulation weights. To mitigate the vanishing/exploding gradient problem in our deeper convolutional neural network, we integrate SE blocks with the ResNet by using the SE block transformation Ftr is taken to be the non-identity branch of a residual module. SE both act before summation with the identity branch. The detailed architecture of our model is plotted in Supplementary Figure S1.
Training the SENet
First, the DNA sequences were converted to the one-hot encoding, where A, C, G and T are encoded as [1, 0, 0, 0], [0, 1, 0, 0], [0, 0, 1, 0] and [0, 0, 0, 1] respectively. Specifically, the parameters of SENet were learnt as follows. The observed regulatory activities of developmental and housekeeping enhancers were denoted as Y. To train the regression SENet model, the mean squared error loss (MSE) was selected as the loss function. Therefore, the goal of the training process is to find the Θ such that:
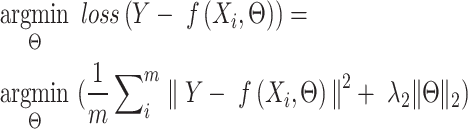 |
where Θ represents all learnable parameters of the SENet, f is the whole neural network, 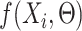 is the predicted regulatory activity vector of developmental and housekeeping enhancers,
is the predicted regulatory activity vector of developmental and housekeeping enhancers, 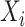 indicate the one-hot encoded DNA sequence, and
indicate the one-hot encoded DNA sequence, and 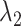 is the weight-decay hyper-parameter penalty for large model weights quantified by the L2 norm. The parameters in each layer of SENet were initialized with the Xavier Glorot's initialization method (49). The stochastic gradient descent (SGD) algorithm was used to train the parameters, i.e. the Θ was updated as follows:
is the weight-decay hyper-parameter penalty for large model weights quantified by the L2 norm. The parameters in each layer of SENet were initialized with the Xavier Glorot's initialization method (49). The stochastic gradient descent (SGD) algorithm was used to train the parameters, i.e. the Θ was updated as follows:
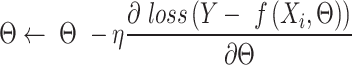 |
where 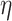 is the learning rate, and the optimizer Adaptive Moment Estimation (ADAM) to train our model (50). To prevent overfitting, an early stopping strategy is applied to the validation set (patience = 10). In each SE module, the dropout rate was set to 0.1, and after the flatten layer, it was set to 0.4.
is the learning rate, and the optimizer Adaptive Moment Estimation (ADAM) to train our model (50). To prevent overfitting, an early stopping strategy is applied to the validation set (patience = 10). In each SE module, the dropout rate was set to 0.1, and after the flatten layer, it was set to 0.4.
The model was implemented and trained with the Keras (version: 2.8.0, https://keras.io/) (with TensorFlow version: 2.8.0) The architecture and hyper-parameters of the SENet were tuning with the Bayesian optimization algorithm on the validation set.
Evaluation of prediction accuracy and model comparison
The performance of the model was evaluated separately for developmental and housekeeping predictions on the held-out test dataset. We used the Pearson correlation coefficient (PCC) between the observed and predicted enhancer functional activity to evaluate the predictive performance of our model. We also evaluate the model performance on bins that are not overlapping with repeats and locate within the Drosophila genome chromatin accessible regions, respectively. The repeat annotation for Drosophila dm3 genome was downloaded from the RepeatMasker database (https://www.repeatmasker.org/genomes/dm3/RepeatMasker-rm405-db20140131/dm3.fa.out.gz), and the ATAC-seq data for the Drosophila S2 and kc167 cell line were obtained from the GEO database (accession number GSE119708) (42). To provide a comprehensive and systematic comparison of SENet with other current mainstream models, the architecture and implementation of the DeepSTARR (https://github.com/bernardo-de-almeida/DeepSTARR) (35), DanQ (https://github.com/uci-cbcl/DanQ) (51), DNABERT (https://github.com/jerryji1993/DNABERT) (52), DenseLSTM (https://github.com/WangLabTHU/deepseed) (53), AttnBiLSTM (https://github.com/1edv/evolution) (29), Basset (https://github.com/davek44/Basset) (21), DeepATT (54), DeepMEL (https://github.com/aertslab/DeepMEL) (55,56), DeepSEA (https://deepsea.princeton.edu/) (57) and DeepSTARR2 (https://github.com/bernardo-de-almeida/DeepSTARR_embryo) (58) models were obtained from their respective code repositories. We modified the activation function of the output layer (from sigmoid to linear) and the loss function (from cross entropy to MSE) of these classification models to adapt them for enhancer activity prediction (regression), while keeping the other hyperparameters unchanged. For the DNABERT model, we obtained the pre-trained model following the instructions in the documentation at https://github.com/jerryji1993/DNABERT and performed fine-tuning. Using Bayesian optimization algorithms within the Optuna framework (version 4.0, https://optuna.org/), we optimized the learning rate and warm-up percentage parameters for the DNABERT model across 30 trials, achieving an optimized learning rate of 0.0002 and a warm-up percentage of 0.07. To account for the variance caused by the random initialization of deep learning model parameters, we repeated the training for each deep learning model 100 times. The mean and variance of the model performance on the hold-out chromosome dataset were calculated. We employed four metrics, i.e. PCC, Spearman correlation coefficient (SCC), coefficient of determination (R2) and MSE to compare the predictive performance between our model and DeepSTARR on the hold-out chromosome dataset.
Motif visualization
We used the method established in the previous study (48,21) to extract the DNA representation of the enhancer regulatory activity. Specifically, for each filter in the first convolution layer of SENet, the sequences which can activate the filter to more half of its maximum value were identified and extracted to construct the position weight matrix (PWM). The nucleotide occurrences in these sequences was counted and transformed to the probabilistic PWM. To identify the PWM which is likely corresponding to the known TFBS, we align these PWMs to the motifs in the JASPAR database (59) using the Tomtom search tool (60) with the threshold of FDR (False Discovery Rate) q-value < 0.1. The information content (IC) for each PWM was calculated as:
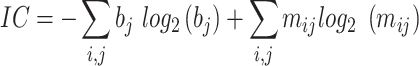 |
where m is the matrix of nucleotide probabilities for the motif, and b is the array of background Drosophila dm3 nucleotide probabilities. TFBSs within the Drosophila enhancer region was identified using the FIMO (Find Individual Motif Occurrences) (61) with the threshold of P-value 1e-5.
We calculated the contribution scores of all nucleotides in the sequence to the activity of developmental or housekeeping enhancers using DeepExplainer (62), an implementation of DeepLIFT (63) within DeepSHAP (version: 0.46.0). Background sequences were set as 1000 dinucleotide-shuffled sequences of the target sequence, serving as reference sequences. For each sequence, the nucleotide contribution scores were obtained by multiplying the importance scores calculated by DeepSHAP with the one-hot encoding matrix of the sequence. The visualization of these scores was performed using the ggseqlogo function from the R package ggseqlogo (version: 0.124).
Motif importance and motif co-occurrence analysis
To quantify the importance of filters in the first layer of the SENet, we used two metrics devised in the previous study, i.e. the activity (occurrence frequency) and the influence on the predictions of model (64). Specifically, the activity of the filter f for a set of sequences within a certain genomic context was computed as follows:
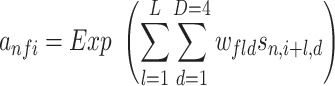 |
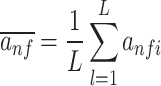 |
where 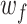 are the learnable parameters or weights of convolutional filter f of length L, D indicates the dimension of the one-hot encoded DNA matrix
are the learnable parameters or weights of convolutional filter f of length L, D indicates the dimension of the one-hot encoded DNA matrix 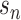 ,
, 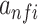 is the exponential activation for the convolutional filter f at position i of the input sequence, and
is the exponential activation for the convolutional filter f at position i of the input sequence, and 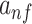 is the average of mean sequence activities. The exponential activation function always greater than zero, such that
is the average of mean sequence activities. The exponential activation function always greater than zero, such that 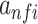 can be considered as the evidence that the motif represented by
can be considered as the evidence that the motif represented by 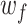 occurs at position i. The influence of filter f on the predicted developmental and housekeeping enhancer activity
occurs at position i. The influence of filter f on the predicted developmental and housekeeping enhancer activity 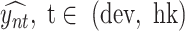 was computed as the Pearson correlation
was computed as the Pearson correlation 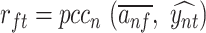 over a set of input sequences n.
over a set of input sequences n.
The co-occurrence of filters was visualized using principal component analysis (PCA) on the mean activations  on input sequences (Figure 3) and the pairwise correlations between mean sequence activations.
on input sequences (Figure 3) and the pairwise correlations between mean sequence activations.
Figure 3.

The SENet component of DREAM framework identifies sequence motifs associated with enhancer activity. (A–F) The filters in the first convolutional layer of SENet can recover the motifs associated with enhancer activity, including GATAd (A), Strat93E (B), kay (C), Dref (D), sna (E), and Ets21C (F) motifs. (G) Clustering of 512 motifs discovered by DREAM framework. The plot shows are the first two principal components of the motif occurrence frequencies in sequence windows (activity). Triangles represent the de novo motifs and dots denote motifs with significant (FDR < 0.1) similarity to the annotated motifs in the JASPAR databases. Marker size indicates the average activity; the estimated motif effect on the developmental enhancer activity (association) is shown by color.
In silico enhancer syntax analysis
(i) In silico motif position effect analysis: We employed two methods to calculate the positional effects of motifs: (a) Random backbone sequences: The consensus sequence of the target TF motif was embedded into 50 000 random 249 bp DNA backbone sequences. The motif’s position within the backbone was denoted as p, and the predicted enhancer activity of the sequence was denoted as Ap. The length of the TF motif was denoted as l. Backbone sequences were generated by sampling each base with equal probability, and their predicted enhancer activity was denoted as Arnd. The positional effect of the TF motif at position p was calculated as mean (log2 (Ap/Arnd)). By varying p from 1 to 249 − l, we obtained the positional effect profile of the TF motif. (b) Natural enhancers: Natural enhancers containing only one instance of the target TF motif were selected. The predicted enhancer activity of these enhancers was denoted as Aenh. Using the function swap (enh, p, motif), we swapped the motif instance with the sequence (enh) at position p: (p+ l), and the predicted enhancer activity of the resulting sequence was denoted as Ap. The positional effect of the TF motif at position p was calculated as mean (log2 (Ap/Aenh)). By varying p from 1 to 249 − l, we obtained the positional effect profile of the TF motif. (ii) In silico motif epistasis-distance analysis: The consensus sequence of TF motifA was embedded in the center of 50 000 random 249 bp DNA backbone sequences. TF motifB was then embedded at a distance d upstream or downstream of motifA. The enhancer activities of the following sequences were predicted using SENet: (a) random backbone sequence (Arnd), (b) sequence with only motifA embedded (AmotifA), (c) sequence with only motifB embedded (AmotifB), (d) sequence with both motifA and motifB embedded (AmotifAB). The epistasis between motifA and motifB at distance d was defined as log2 (AmotifAB/(AmotifA+ AmotifB− Arnd)) (35). A value of 1 indicates an additive effect, while a value >1 indicates positive synergy. For three TF motifs, motifA and motifB were fixed at their optimal relative distance in the random backbone, and motifC was moved. The rest of the calculation remained the same. (iii) In silico three-order TF combination effect analysis: The consensus sequences of motifA, motifB and motifC were embedded in the center of 50 000 random backbone sequences, maintaining the optimal relative distances between motifs. The predicted enhancer activity of these sequences was denoted as A. The predicted enhancer activity of the backbone sequences was denoted as Arnd. The higher-order TF combination effect was defined as mean (log2 (A/Arnd)). (iv) Multivariate linear regression model based on key TF motif features: TF motifs within the Drosophila enhancer regions were identified using FIMO (61) with a P-value threshold of 1 × 10−5. The DNA shape features of the sequences flanking the TF motifs (10 bp on each side) were estimated using the DNAshapeR package (version:1.32.0, https://bioconductor.org/packages/DNAshapeR/) (65). These features included minor groove width (MGW), roll (Roll), propeller twist (ProT) and helix twist (HelT). For both developmental and housekeeping enhancers, a multivariate linear regression model was constructed using features related to key TF motifs. These features included the number of motif instances, the distance of the motif from the center of the enhancer sequence, the binding strength of the TF (motif core, -log (binding probability) as a proxy), the DNA shape scores of the flanking sequences and the relative distances between key motifs. Only motif instances starting after position 10 and ending before position 239 of the 249 bp oligos were used to ensure the retrieval of their 10 bp flanking sequences. For motif distance analysis, only non-overlapping motif pairs were considered. The P-values of the features in the multivariate linear model were used to assess the significance of their contribution to enhancer activity.
Sequences property analysis
The TF motifs within the Drosophila enhancer region and designed sequences were identified using FIMO (61) with a threshold P-value of 1e-5. The number of motifs, distance between motifs, GC content of motifs, GC content of sequences and k-mer frequency of sequences were calculated using custom R scripts. Levenshtein distance and Hamming distance between sequences were calculated using the stringdist R package (version 0.9.12). The DNA shape features of the sequences flanking the TF motifs (10 bp on each side) were estimated using the DNAshapeR package (65). Nucleotide diversity was calculated as the average number of nucleotide differences per site between two DNA sequences in all possible pairs in the sample population (66). Entropy was computed using the formula: 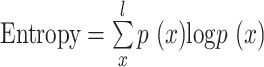 , where p (x) is the frequency of the motif x in the sequence, and l represents all TF motifs in the sequence. We used the -log (binding probability) as a proxy for TF binding affinity, determined using the FIMO software (61).
, where p (x) is the frequency of the motif x in the sequence, and l represents all TF motifs in the sequence. We used the -log (binding probability) as a proxy for TF binding affinity, determined using the FIMO software (61).
Sequences optimization
To generate novel enhancers with the high regulatory activity, we implemented a genetic algorithm using the parallelized Python DEAP package (version 1.3.3, available at https://github.com/deap/deap) with a distributed evolutionary approach. Initially, we set the population size to 100 000 individuals, initializing them with the nucleotide frequencies similar to the Drosophila reference genome. The mutation probability and two-point crossover probability were both set to 0.1, with a selection tournament size of 3. The genetic algorithm comprised 90 generations, each aimed at maximizing the regulatory activity of developmental and housekeeping enhancers, respectively. Eight enhancers generated from the intermediate steps of the iterative optimization process and two final optimized enhancers were synthesized to measure their activity experimentally. We also extended DREAM by designing different fitness functions to meet various enhancer or silencer design requirements as follows: (i) ‘AT rich + strong activity’ enhancers: fitness = f (sequence)/gc (sequence); (ii) strong housekeeping silencers: fitness = -f (sequence); (iii) strong housekeeping enhancers: fitness = f (sequence); (iv) enhancers with user-specified activity: fitness = -|T - f (sequence)|; (v) enhancers specific to the human A549 cell line: fitness = gA549 (sequence) - max (gHCT116 (sequence), gMCF7 (sequence); where f (sequence) represents the predicted enhancer activity in Drosophila S2 cells, gc (sequence) represents the GC content, T represents the user-specified enhancer activity, and gA549 (sequence), gHCT116 (sequence) and gMCF7 (sequence) represent the predicted enhancer activity in human A549, HCT116 and MCF7 cells, respectively. The optimization target in the DREAM framework’s genetic algorithm is to maximize the corresponding fitness function. Additionally, during the GA operations, the sequences were fixed with three restriction enzyme sites (RESs) at positions 50, 150 and 200 bp (AgeI = ‘ACCGGT’, SalI = ‘GTCGAC’, HindIII = ‘AAGCTT’), while continuing to optimize the corresponding fitness function to obtain the ‘with 3 fixed RESs’ enhancers.
Cell line and transfection
A diverse array of cell lines across seven species, namely Drosophila S2, Spodoptera frugiperda SF9, chicken DF1, fish (Cynoglossus semilaevis) spermatogonium, Pichia pastoris, human 293T, A549, HEPG2, K562 cell, mouse 3T3, C2C12, CHO, ARPE-19 cell, pig PK15, PSKM cell and sheep MSC cell, was employed to examine the regulatory activity of candidate enhancers. Comprehensive details outlining the cell culture and transfection methodologies are provided in the supplementary notes.
Plasmid construction and extraction
The Plasmid used in this study was constructed as follows (the details about plasmids are provided in the supplementary notes):
E-pGL3_DSCP_luc Plasmid Construction: The synthesis of the E-pGL3_DSCP_luc plasmid, designed to initiate luciferase reporter gene expression with the DSCP promoter, was facilitated in collaboration with Sangon Biotech, China. In this construct, an enhancer was strategically inserted upstream of the DSCP promoter in E-pGL3_DSCP_luc, augmenting its regulatory capacity. The CMV enhancer sequence (Addgene, #171379) sourced from the literature (67–69).
pGL3_DSCP_Rluc Plasmid Construction: Using the pT-TK Plasmid (Addgene, #31549) as a template, PCR amplification was performed to obtain the Rluc expression cassette. The Rluc cassette was then inserted downstream of the DSCP promoter in the pGL3_DSCP_luc plasmid after removing the luciferase gene, resulting in the pGL3_DSCP_Rluc Plasmid.
E-CAG-luc Plasmid Construction: The CAG promoter was obtained through PCR amplification of the VB220421-1515nsc plasmid (kindly provided by VectorBuilder). Subsequent steps involved excising the SV40 promoter and the Renilla luciferase reporter plasmid from pmirGLO using restriction endonuclease. The CAG promoter was then ligated onto the modified pmirGLO plasmid through Gibson cloning (NEB, E2611L). Notably, the CMV sequence within CAG was substituted with the enhancer sequence derived from the E-pGL3_DSCP_luc plasmid on the transformed plasmid, yielding the final E-CAG-luc plasmid.
E-HIS-Rluc-luc Plasmid Construction: Using the pLyGo-Kp-1 Plasmid (Addgene, #163143) as a template, PCR amplification of Pphis4 was performed to obtain the His tag. The His tag was then inserted downstream of the SV40 polyadenylation signal in the E-pGL3_DSCP_luc plasmid using Gibson assembly, resulting in the E-HIS-Luc plasmid. Subsequently, using the pT-TK Plasmid (Addgene, #31549) as a template, PCR amplification was carried out to obtain the Rluc expression cassette driven by the DSCP promoter. This cassette was then inserted upstream of the pause site in the E-HIS-Luc plasmid using Gibson assembly, resulting in the final E-HIS-Rluc-luc plasmid for dual luciferase reporter experiments in Pichia pastoris.
Plasmid extraction was executed using the Endo-Free Plasmid Maxi Kit (Omega, D6926-03).
Luciferase reporter assays and data analysis
The Dual-Luciferase® Reporter 1000 Assay System (Promega, E1960) was employed to conduct the dual luciferase reporter assay. Following a 24-h transfection period, the cellular medium was discarded, and PBS was used for cell washing. Subsequently, 200 μl of lysate, diluted to a 1 × concentration, was administered to the cells. After 10-min cell lysis, 20 μl of the resultant cell lysate was carefully transferred to a 1.5 ml centrifuge tube (Selection, MCT-001–150). This was followed by the addition of 100 μl of Luciferase Assay Reagent II to the lysate. The luminometric reading was acquired using a GLOMAX20/20 instrument (Promega E5311). To terminate the reaction, 100 μl of Stop & Glo was introduced, and the centrifuge tube was repositioned in the GLOMAX20/20 instrument (Promega, E5311) for the final reading, with meticulous record-keeping of the results. Three independent biological replicates were performed for each sequence. We normalized all firefly luciferase signals to the signal of Renilla luciferase to control for transfection efficiency and cell number (the relative luciferase signal).
Results
The computational framework of DREAM in CRE design
To de novo design transcription start site (TSS) distal CREs with desired regulatory activity, we developed an innovative framework named DREAM. This framework integrates a state-of-the-art SE CNN (SENet) model and a genetic algorithm, featuring two interconnected modules: the sequence-function module and the evolutionary sequence optimization module (Figure 1, ‘Materials and Methods’ section).
Figure 1.

Overview of the DREAM framework. DREAM comprises two integral modules: the state-of-the-art SENet that models enhancer activity using DNA sequences as the input (A) and the evolutionary optimization module of DNA sequences (B, ‘Genetic Algorithm’). The SENet was trained using UMI-STARR-seq data to learn the DNA regulatory lexicon underlying enhancer activity and was subsequently used to predict the regulatory activity of diverse DNA sequences. In tandem, the evolutionary optimization module employs a genetic algorithm, iteratively maximizing regulatory activity as predicted by the SENet-derived model. This iterative process ensures precise and targeted enhancement of enhancer functionality, thus facilitating the design of tailored sequences for specific regulatory tasks.
To be specific, SENet was first employed and trained to predict the activity of CREs ab initio solely using DNA sequences as model input (Figure 1A). SENet can explicitly model the dynamical non-linear dependencies between convolutional feature channels and enhance the representation ability of vanilla CNNs by using the SE attention mechanism (47). The SE attention mechanism has been applied in predicting TF-DNA binding (70). By scanning the input CRE sequences with a moving sliding window, the first convolutional layer of SENet is responsible for extracting and recovering a spectrum of potential DNA motifs related to regulatory activity. This parallels the mechanism by which TFs identify specific DNA motifs within regulatory regions, activating unique transcriptional programs that play a pivotal role in defining cell identity and fate. The subsequent convolutional layers, hierarchically and adaptively, recapitulate the dynamic interplay and spatial relationships among the filters (analogous to TFs biologically) and aptly epitomize it into a high-dimensional representation space. Second, utilizing the trained SENet as the fitness function, we employed a genetic algorithm to iteratively improve individual fitness within a randomly initialized sequence population, working toward a predefined design objective (Figure 1B). Briefly, at the lower level, in each generation, the new candidate CREs (offspring) are introduced by iteratively applying mutation, recombination and selection operators to the selected CREs (parents) from the preceding generation. At the higher level, the fitness of each candidate CRE is measured by the well-trained SENet, and strong selection mechanisms determine which offspring advance to the next generation. Importantly, as mentioned earlier, leveraging the regulatory lexicon learned by the filters in the first convolutional layer of SENet, DREAM inherently enables visualization of the preferred regulatory lexicons during CRE optimization. Therefore, the whole trajectory of CRE design and optimization is transparent and biologically interpretable.
DREAM can accurately recapitulate the enhancer activity
To obtain training data for DREAM, we downloaded UMI-STARR-seq data in Drosophila melanogaster S2 cells from a previous publication (35). Specifically, the dataset used by de Almeida et al. utilized UMI-STARR-seq to measure enhancer activity toward two distinct transcriptional programs defined by their representative promoters: a synthetic core promoter (DSCP) derived from the even-skipped TF for developmental enhancers, and the core promoter of Ribosomal protein gene 12 (Rps12) for housekeeping enhancers, in D. melanogaster S2 cells (71). These cells, derived from late-stage embryos, are widely used in gene expression and regulation studies. This dataset, characterized by high resolution and fidelity, offers a quantitative assessment of enhancer activity across the Drosophila genome (35). The dataset screened a total of 242 026 sequences for potential enhancer activity. Among these sequences, 11 658 and 7062 were identified as boosting transcription initiated from a developmental and housekeeping a promoter, respectively. Unlike other epigenomic assays based on the enrichment of specific biochemical marks (such as H3K27ac) to infer enhancer activity, STARR-seq directly measures the intrinsic enhancer activity of DNA sequences in a high-throughput manner (17,35,43,72). Therefore, deep learning models trained with STARR-seq datasets can directly predict enhancer activity, rather than relying on proxy measurements such as H3K27ac signal.
When it comes to predicting enhancer activity solely from DNA sequences, DeepSTARR (35) has set a notable benchmark with its state-of-the-art performance employing a CNN-based model. In order to gauge our approach against this benchmark, we directed the same training, validation and hold-out chromosome dataset to the multitask SENet component of DREAM (‘Materials and Methods’ section). Using the hold-out chromosome data as independent testing data, SENet achieved Pearson correlation coefficients (PCC) of 0.71 (P-value < 2.2e-16) and 0.80 (P-value < 2.2e-16) for developmental enhancers and housekeeping enhancers, respectively (‘Materials and Methods’ section; Figure 2A and B). SENet also accurately predicted the difference in enhancer activity between the developmental and housekeeping promoters on the hold-out chromosome (PCC = 0.92, P-value < 2.2e-16; Figure 2C). The predictive performance of SENet are robust on the non-repeat regions (developmental enhancer: PCC = 0.71, P-value < 2.2e-16; housekeeping enhancer: PCC = 0.80, P-value < 2.2e-16; Supplementary Figure S2) and the open chromatin regions (S2 cell and kc167 cell developmental enhancer: PCC = 0.75, P-value < 2.2e-16; housekeeping enhancer: PCC = 0.85, P-value < 2.2e-16; Supplementary Figure S3). To further validate the generalizability of SENet, we subjected it to 249 randomly generated synthetic enhancer sequences spanning a broad spectrum of activity levels (experimentally measured by de Almeida et al. (35)). We found that, remarkably, SENet also demonstrated excellent predictive performance in these exogenous sequences which are not present in the Drosophila genome (‘Materials and Methods’ section, PCC = 0.65, P-value < 2.2e-16; Supplementary Figure S4).
Figure 2.

The sequence-activity model of DREAM framework can quantitatively and accurately predict enhancer activity solely from DNA sequences. (A and B) 2D kernel density plots showing the correlation between the observed and the predicted regulatory activity for developmental enhancers (A) and housekeeping enhancers (B) on the held-out chromosome, respectively. (C) The SENet accurately captures the activity difference between enhancers coupled to developmental and housekeeping promoters. (D) Four metrics—PCC, SCC, R² and MSE—were utilized to assess the predictive performance of the SENet and other ten models on developmental enhancers and housekeeping enhancers. The evaluation encompassed diverse datasets, including the kc167 (kc167 ATAC) and S2 cell (S2 ATAC) accessible regions, 249 synthetic developmental enhancers designed by de Almeida et al. (Synth), non-repetitive regions on the hold-out chromosome (Non-repeats), and the entirety of the hold-out chromosome dataset (Testset).
To provide a comprehensive and systematic comparison of SENet with other current mainstream models, we included ten deep learning models capable of predicting enhancer activity (including the DeepSTARR model (35)): DanQ (51), DNABERT (52), DenseLSTM (53), AttnBiLSTM (29), Basset (21), DeepATT (54), DeepMEL (55,56), DeepSEA (57) and DeepSTARR2 (58) (see ‘Materials and Methods’ section). We evaluated the performance of these models on a hold-out chromosome dataset using four metrics: PCC, SCC, R² and MSE. The results demonstrated that SENet outperformed all the other models. Specifically, compared to the second-best performing model, DNABERT, SENet achieved average improvements of 3.51% in PCC (housekeeping enhancer: 3.11%, developmental enhancer: 3.91%), 3.02% in SCC (housekeeping enhancer: 3.02%, developmental enhancer: 3.01%), 6.80% in R² (housekeeping enhancer: 6.41%, developmental enhancer: 7.19%) and 9.06% in MSE (housekeeping enhancer: 10.28%, developmental enhancer: 7.83%) (Figure 2D). The results of 10-fold cross-validation also indicate that SENet has the best performance in predicting enhancer activity (Supplementary Figure S5). In contrast to the DeepSTARR model, the novel SE block proposed in SENet can explicitly model the non-linear interactions between the filters (corresponding to DNA motifs). This capability enhances the representational power in modeling the regulatory grammar that governs enhancer activity.
The SENet can reveal the regulatory lexicon and syntax of enhancers
Understanding and interpreting the underlying basis on which the models make decisions and predictions can provide profound insight into the biological mechanisms being studied. Previous research has established that the collaborative occupancy of TFs, along with the interplay between their motifs and genomic contexts, plays a pivotal role in fine-tuning enhancer activity (33–35,73,74). Here, to extract the enhancer cis-regulatory grammars learned by SENet, we employed a sequence-alignment-based approach. This approach sifted through sequences that robustly activated nodes in the first convolutional layer and subsequently aligned them for closer examination (19,31,21,64,75) (‘Materials and Methods’ section). We found that 21.29% (109/512) of the potential regulatory motifs learned by the convolutional filters significantly aligned with enhancer-activating TF DNA binding motifs (annotated motifs; ‘Materials and Methods’ section; q-value < 0.1), such as GATAd, Strat93E, kay, Dref, sna and Ets21C motifs (35,44,71) (Figure 3A–F). Some motifs were captured by different filters recurrently, implying that the filters were highly redundant (Supplementary Figure S6). Additionally, our observation also revealed that some filters responded to highly similar sequence patterns but displayed conspicuously diverse effects on the final predicted activity (Supplementary Figure S6). This implies that the motifs and their contextual relationships within enhancers are intricate and sophisticated, adding a layer of complexity to the understanding of their functional impact. Notably, some filters that can’t match any known motifs (de novo motifs) might recognize the low-level sequence features related to enhancers, such as higher GC content which is enriched in the chromatin accessible regions (76), or the sequence features determining DNA shape recognized by TF binding (77,78).
Moreover, two complementary metrics, i.e. (i) the occurrence frequency of the filters in each enhancer sequence (activation) and (ii) the influence of each filter on the final enhancer activity prediction (association), were employed to evaluate the contribution and importance of the motifs recovered by the filters (‘Materials and Methods’ section). The annotated motifs, which exhibit higher IC (P-value = 0.012, one-sided Wilcoxon rank-sum test) and activation (P-value = 0.046, one-sided Wilcoxon rank-sum test) in SENet (Supplementary Figure S7), exhibit a similar association with enhancer activity as the de novo motifs (Supplementary Figure S7D). We observed that the SENet-unveiled motifs were found to be as predictive, if not more so, of enhancer activity than known TF motifs (Supplementary Figure S8). Notably, the motif instances (matching the filters) exhibiting increased activation within housekeeping enhancers, which show greater evolutionary conservation (Spearman’s rho = –0.20, P-value = 9.13e-3). The TFs recovered by the filters with positive association on activity displayed significantly lower evolutionary rates than those captured by filters with negative association (housekeeping enhancer: P-value = 7.4e-3; developmental enhancer: P-value = 0.09; one-sided Wilcoxon rank-sum test; Supplementary Figure S9). Together, these observations affirm that, indeed, SENet has effectively learned the regulatory grammar underlying enhancer activity. To investigate the co-occurrence of motifs across sequences, we applied PCA (Figure 3G). We found that motifs with similar nucleotide composition tended to group together, with two major clusters associated with increased or decreased enhancer activity co-occurring within the same sequence. Hierarchical clustering of filter (motif) activation correlations reveals the presence of numerous co-activated motifs within enhancers (Supplementary Figure S10). These findings potentially indicate widespread TF–TF interactions within enhancers. Additionally, we observed that motifs with negative association on enhancer activity tended to be CG-rich.
We compared the motifs identified by SENet and DeepSTARR (Supplementary Figure S11) and found that only 13.6% of the motifs matched (q-value < 0.1, Supplementary Figure S11A). We speculate that one reason for this discrepancy is that some filters of the two models only capture a subset of TFBS motifs, leading to mismatches. Further analysis indicated that, compared to vanilla CNNs (i.e. DeepSTARR model), SENet captured more known TFBS motifs (observed at q-value < 0.1 and q-value < 0.05 thresholds, Supplementary Figure S11C). Additionally, the de novo filters and annotated filters in SENet exhibited higher IC and activation (both P-Value < 2.2e-16, one-sided Wilcoxon rank-sum test; Supplementary Figure S11D and E). We assessed the importance of filters by correlating their activation values with enhancer activity (PCC) and found that, in predicting developmental and housekeeping enhancers, SENet captured more important DNA motifs compared to the DeepSTARR model (filters with importance > 0.1, Supplementary Figure S11F). Finally, to further validate our findings, we used the motifs learned by SENet and DeepSTARR to construct random forest and XGBoost models for predicting the activity of developmental and housekeeping enhancers. The results showed that the DNA sequence features learned by SENet have higher predictive power (except for the non-significant prediction of developmental enhancer activity by the XGBoost model, with other P-values < 0.01, one-sided Wilcoxon rank-sum test; Supplementary Figure S11G). We also observed consistent conclusions when using the top 20, 50 and 100 ranked filters by importance as features (Supplementary Figure S11G).
To further explore the complexity and dynamics of enhancer regulatory syntax, we selected the top six important TF motifs learned by SENet (considered key motifs) and performed an in silico analysis of their positional effects, epistasis and higher-order motif interactions (Figure 4; Supplementary Figure S12 and Supplementary Figure S13). The key findings are as follows: (i) enhancers often display positional preferences for TF motifs, with instances of the same motif contributing differently to enhancer activity based on their positions. In developmental enhancers, the positional effect of TF motifs shows a unimodal distribution, where motifs at the center of the sequence contribute the most to enhancer activity (Figure 4B). In contrast, housekeeping enhancers exhibit a multimodal distribution, with significant contributions from motifs located not only at the center but also at the 5′ and 3′ ends of the sequence (Supplementary Figure S12A). (ii) Epistasis between TF motifs varies with their relative distance (Figure 4C and Supplementary Figure S12B). Different TF motifs show distinct patterns of interaction based on their relative distances, forming two categories: those with optimal distances of <25 bp and those >25 bp. When considering the interaction among three TF motifs (with two motifs fixed at their optimal interaction distance), the epistasis follows a similar pattern (Supplementary Figure S13A). (iii) The distance between some key motifs significantly influences enhancer activity. For instance, in developmental enhancers, the distance between key motifs such as GATAe motifs and grn motifs shows a notable impact on enhancer activity (Figure 4D and Supplementary Figure S12C). (iv) The DNA shape of the TF motifs flanking sequences also plays a crucial role in enhancer activity. By analyzing four main DNA shape features—MGW, roll (Roll), ProT and HelT—we found that the contribution of DNA shape to enhancer activity is more significant in housekeeping enhancers (Figure 4D and Supplementary Figure S12C). We hypothesize that these DNA shapes affect the binding affinity of TFs to DNA, thereby regulating enhancer activity. (v) Higher-order interactions among TF motifs (e.g. three motifs) exhibit complex patterns. Even when maintaining the optimal distance required for TF–TF interactions, only some combinations of three TF motifs tend to increase enhancer activity in developmental enhancers (Supplementary Figure S13B).
Figure 4.

In silico analysis reveals positional effects of key motifs in developmental enhancers, distance-dependent TF motif epistasis, and contributions of TF motif-related features to enhancer activity. (A) Schematic illustrating two computational strategies for assessing motif positional effects in silico: ‘Random Backbone Sequences’ and ‘Natural Enhancers’ (see ‘Materials and Methods’ section for details). (B) Positional effects of the top six TF motifs in developmental enhancers. Green lines represent the ‘Random Backbone Sequences’ strategy, orange lines indicate the ‘Natural Enhancers’ strategy, and blue lines denote the negative control ‘GGGCT’. Dashed line indicates an additive effect. (C) Epistasis effects between TF motifs as a function of the relative distance between motifs. The first motif in the title is fixed at the center of the backbone, while the second motif (motif B) is computationally moved (color-coded). The ‘GGGCT’ motif serves as a negative control (see ‘Materials and Methods’ section). (D) Contributions of TF motif-related features to developmental enhancer activity. For each TF motif (each row), multiple linear regression models were constructed using the number of motif instances, the distance from the enhancer center, the binding strength of the TF (motif core, with -log (binding probability) as a proxy), DNA shape scores of the flanking sequences, and the relative distances between key motifs. The P-value of each motif feature from these models indicate the significance of each motif feature's contribution to enhancer activity.
Optimize enhancers activity with the DREAM framework
Utilizing the highly predictive model for enhancer activity as the ‘fitness’ function for the genetic algorithms, we can design the CREs with prespecified regulatory activity, including enhancers exhibiting exceptional transcriptional-stimulating effect. To achieve this, we initialized 100 000 random sequences sampled from the sequence space with similar nucleotide frequencies as the Drosophila genome. These sequences were then subject to genetic algorithms which simulated optimization trajectories of 90 generations toward maximizing their enhancer activity (‘Materials and Methods’ section). We observed that enhancer activity of the developmental and housekeeping transcriptional programs approached saturation, reaching its peak after ∼70–80 generations. The activity levels of developmental and housekeeping enhancers derived from the final design using the DREAM framework were 6.8- and 5.3-fold (predicated by SENet), respectively, compared to the strongest enhancer identified in the Drosophila genome (35) (Figure 5A). We also found that increasing the initial population size improves the ability of genetic algorithms to find higher-activity enhancers but also significantly increases computational resource demands (Supplementary Figure S14). To gain insights into the putative TF binding motifs playing critical roles during the optimization of enhancer activity, we examined the activation value profiles during the in silico optimization trajectory, for the Top 10 and Bottom 10 filters found in the final optimal enhancers (filters with Top 10 and Bottom 10 activation values). Notably, we observed a gradual increase in activation values for the Top 10 filters. Particularly, the filter_288 capturing kay and Jra motifs exhibited the highest activation value. In contrast, the activation values of the Bottom 10 filters remained largely unchanged (Figure 5B). For a more comprehensive exploration of the properties of the artificial enhancers designed by DREAM, we visualized them in the DNA enhancer embedding space. This visualization included the DREAM-optimized developmental enhancers, natural sequences in the Drosophila genome (encompassing both developmental enhancers and random genomic sequences), random sequences with an identical nucleotide frequency distribution and synthetic developmental enhancers designed by de Almeida et al. (35)—serving as the control sequences (Figure 5C and D). The control sequences were located in regions with the lower enhancer activity, and the natural developmental enhancer samples clustered together. The natural sequences did not reach the optimal fitness (enhancer activity) peak, suggesting that natural enhancers may be subject to additional constraints beyond regulatory activity. In contrast, the DREAM-optimized enhancers scattered, and showed dramatically higher regulatory activity.
Figure 5.

DREAM framework leverages the most influential regulatory lexicon to optimize the regulatory activity of enhancers. (A) Distribution of predicted enhancer activity (y-axis) for developmental and housekeeping enhancers at mutational steps (x-axis) for the in silico optimization trajectories favoring high activity. Boxes demarcate the 25th, 50th and 75th percentile values, while whiskers indicate the outermost point with 1.5 times the interquartile range from the edges of the boxes. The blue and yellow dash lines represent the Drosophila developmental and housekeeping enhancer with the strongest activity measured in S2 cells, respectively. (B) The dynamic trajectories of activation values for filters ranked in the top 10 (dashed lines) and bottom 10 (solid lines) based on activation values within the final optimized enhancers during in silico evolution. The color indicates the filter influence on the predictions of developmental enhancer activity. (C and D) Unsupervised Clustering shows that the optimized developmental enhancers by the DREAM framework are conspicuously distinct from the Drosophila genome sequences (including developmental enhancers [11 658] and random genomic non-coding sequences [5000]), random sequences with the same nucleotide frequency distribution as the Drosophila genome (5000) and synthetic developmental enhancers designed by de Almeida et al. (249) (35). Those sequences were projected into the enhancer embedding space with the UMAP algorithm. The dot color represents the type of sequence type. The dot color represents the type of sequence type (C) and the predicted regulatory activity of developmental enhancers (D), respectively.
To explore motif syntax underlying strong enhancer activity, we examined motif configuration changes during the in silico optimization trajectory. We found that during the optimization of enhancer activity based on DREAM, the occurrence of TF motifs with close proximity (motif distance <20 bp), the number of key motifs, TF binding affinity and the diversity of TF motifs gradually increased, while the average distance between motifs decreased, and sequence diversity reduced (Figure 6A and Supplementary Figure S15). Enhancers designed by DREAM exhibited significant differences in these sequence properties compared to natural Drosophila enhancers and those previously designed by DeepSTARR (35) and DeepSTARR2 (58) (all P-values < 2.22e-16, one-sided Wilcoxon rank-sum test; Figure 6A). Furthermore, enhancers designed by DREAM were closer to natural enhancers in terms of Hamming distance and Levenshtein distance (all P-values < 2.22e-16, one-sided Wilcoxon rank-sum test; Supplementary Figure S15B). Additionally, the designed enhancers showed differences from natural sequences in k-mer frequency (k = 5,6) and the DNA shape of motif flanking sequences (Supplementary Figure S15C and D). Additionally, during the optimization process, clear co-occurrence patterns emerged among functional motifs, such as the SREBP-kay, SREBP-Jra, Mitif-kay, etc., underscoring the interplay of these motifs is vital for increasing enhancer activity (Supplementary Figures S16 and S17). These observations suggest that, under intense selection pressure, populations rapidly converge towards the optimal fitness by strategically exploiting and reinforcing TF motifs and motifs syntax that substantially contribute to enhancer activity. The motif enrichment analysis further revealed that the optimized enhancers harbored a distinct set of overrepresented functional motifs (Figure 6B and C). These results indicated that the DREAM framework learned the enhancer grammar and applied it into the process of enhancer design, rather than merely copying the sequences of natural enhancers.
Figure 6.

The dynamic changes of known TF binding motifs during the in silico optimization trajectory. (A) This panel illustrates various characteristics, including the count of TF motif pairs with distances <20 bp (# (distance < 20)), the average number of (key) TF motifs (# (motifs)), TF binding affinity quantified by −log (binding probability), diversity of TF motifs evaluated through entropy, sequence diversity quantified by Levenshtein distance, the mean distance between TF motifs, and GC content of binding TF motifs (motif GC%) and sequences (sequence GC%) throughout the in silico optimization trajectory. The x-axis outlines the steps of the in silico optimization process, and the results are compared with natural enhancers in the Drosophila genome as well as synthetic enhancers designed by DeepSTARR (35) and DeepSTARR2 (58) (one-sided Wilcoxon rank-sum test). (B) TF motifs over-represented in optimized synthetic enhancers compared to natural developmental enhancers in the Drosophila genome. (C) TF motifs over-represented in developmental enhancers compared to genomic regions of non-enhancers (background). The x-axis represents the log2-transformed one-sided Fisher’s exact test odds ratio, and the y-axis represents the corresponding FDR-corrected significance.
To further demonstrate the scalability of the DREAM framework in designing CREs, we developed different fitness functions for various CREs design goals (‘Materials and Methods’ section, Supplementary Figure S18A and B): (i) enhancers with extremely high AT content and strong activity (‘AT rich + strong activity’ enhancers); (ii) enhancers with three user-specified restriction enzyme sites (RESs; with 3 fixed RESs, i.e. AgeI = ‘ACCGGT’, SalI = ‘GTCGAC’, HindIII = ‘AAGCTT’); (iii) CREs with strong activity (‘strong housekeeping silencers/enhancers’); (iv) developmental enhancers with user-specified activity levels (Supplementary Figure S20); (v) enhancers specific to the human A549 cell line (Figure 7). We observed that in these five different design scenarios, DREAM effectively optimized sequences for the predefined goals (Supplementary Figures S18 and S19). In the task of designing A549 high activity enhancers, DREAM produced sequences with significantly higher activity than natural A549 enhancers (human genomic A549 enhancers, Figure 7B and C). However, the increase in activity in A549 cells was accompanied by increased activity in HCT116 and MCF7 cells. TF motif analysis in two designed sequences revealed motifs primarily for TP53, TP63 and TP73. For A549 cell-type-specific enhancers, DREAM generated sequences with higher specificity than natural A549-specific enhancers (human genomic A549-specific enhancers, Figure 7D and E). TF motif analysis showed higher diversity in optimized A549-specific enhancers, including motifs for TP53, TP63, TP73, PATZ1, ZNF281 and ZBED4 (Figure 7D and E). This suggests an antagonistic relationship between enhancer activity and cell-type specificity, where achieving high specificity often comes at the cost of reduced activity. Furthermore, the motifs used differed between tasks, with cell-type-specific tasks incorporating motifs that enhance specificity alongside those that activate transcription.
Figure 7.

A549 high-activity and A549 specific enhancers designed Using DREAM. (A) Predictive performance of the DREAM enhancer activity prediction module across A549, HCT116 and MCF7 cell lines, evaluated on the hold-out chromosome 22 test dataset. (B and D) Optimization processes for designing high-activity and cell-type specific enhancers in A549 cells, respectively. These figures also compare the enhancer activities of designed sequences to natural enhancers. (C and E) Nucleotide contribution scores of the optimized enhancer sequences, derived from the DREAM enhancer activity prediction module using DeepExplainer (62). Additionally, instances of motifs identified using Vertebrate PWMs from the JASPAR2024 database are shown. Boxes demarcate the 25th, 50th and 75th percentile values, while whiskers indicate the outermost point with 1.5 times the interquartile range from the edges of the boxes.
DREAM-designed enhancers displayed extreme regulatory potential across multiple species
To validate the accuracy of the DREAM framework in designing enhancers, we employed the developmental enhancers (enhancers designed in the context of driving the developmental promoter) as a paradigmatic case study. Specifically, we selected an extensive and diverse set of enhancer sequences for luciferase reporter assays in S2 cell (Figure 8D). These sequences include (i) non-enhancer sequences in the Drosophila genome (non-enhancers), serving as the baseline control; (ii) the natural Drosophila developmental enhancers exhibiting the strongest and medium regulatory activity measured by STARR-seq; (iii) the five synthetic Drosophila developmental enhancers with the highest regulatory activity (Top 5) designed by de Almeida et al. (DeepSTARR did not target enhancer activity for directional optimization) (35); (iv) ten enhancers designed by the DREAM design framework, comprising eight enhancers generated from the intermediate steps of the iterative optimization process and two final optimized enhancers emerging upon algorithmic convergence. Notably, we found that the two final optimized enhancers designed by the DREAM framework exhibited a remarkable and significant 3.6-fold (average, one-sided Wilcoxon rank-sum test, P-value = 0.01) and a 3.9-fold (average, one-sided Wilcoxon rank-sum test, P-value = 1.84e-05) increase in regulatory activity, compared to the strongest Drosophila endogenous enhancer and the strongest artificial enhancer previously designed by de Almeida et al., respectively. Meanwhile, consistent with previous research, the strongest enhancer designed by de Almeida et al. exhibited regulatory activity comparable to the strongest natural Drosophila enhancer, reinforcing the robustness of our findings (35). Furthermore, the enhancer activities predicted by the DREAM framework exhibited a strong correlation with those measured by luciferase reporter assays (PCC = 0.80, P-value = 1.60e-05), which further underscores the exceptional performance of the DREAM framework for enhancer activity prediction.
Figure 8.

The final optimized enhancers displayed extreme regulatory activity and are functionally conserved across diverse species. (A) Nucleotide contribution scores for the optimized enhancers derived from the enhancer activity models using DeepExplainer. Instances of motifs identified by DREAM are emphasized, with known motifs indicated in black and de novo motifs marked in red. The number of known motifs (# (known motifs)) and the number of de novo motifs (# (de novo motifs)) are also marked. (B) The colored matrices illustrate the presence or absence of TF motifs (x-axis) in the corresponding taxonomy (y-axis), with blue indicating absence and orange indicating presence of the TF in the respective taxonomy. (C) Schematic representation of the luciferase reporter assays system for the test sequences, with the designed sequences positioned at the 5′ end of the DSCP promoter. (D) Comparing enhancer activity, as measured by luciferase reporter assays, with predictions generated by the DREAM framework in S2 cells. Five classes of sequences are shown in the plot, including (i) non-enhancer sequences in the Drosophila genome (non-enhancer), (ii) the Drosophila developmental enhancers exhibiting the strongest and medium regulatory activity, (iii) the top five synthetic developmental enhancers with the highest regulatory activity, as designed by de Almeida et al. (35), (iv) ten enhancers designed by the DREAM design framework and (v) the CMV enhancer and Hr5 enhancer. The firefly luciferase values were normalized with the signal of Renilla luciferase. Error bars: Standard error of the mean (n = 3 biological replicates). (E) Comparative analysis using luciferase reporter assays to assess the activities of enhancers optimized by the DREAM framework, enhancers designed by de Almeida et al. (35), and the CMV enhancer across diverse cell lines spanning nine species, including chicken, fish (Cynoglossus semilaevis), human, mouse, pig, sheep, cattle, insect (Spodoptera frugiperda) and yeast (Komagataella phaffii). The Hr5 (81) and UASE (82) enhancers served as controls for insect (Spodoptera frugiperda) and yeast (Komagataella phaffii) cell lines, respectively, while the CMV enhancer was used as a control for the remaining cell lines. The luciferase values are normalized against the signal of Renilla luciferase. Error bars: Standard error of the mean (n = 3 biological replicates).
Recent research has revealed that, despite low sequence conservation, the activity of enhancers across evolution could be conserved by retaining a collection of conserved TF DNA binding motifs with variations in ordering and spacing observed across different species (79,80). Despite the model of DREAM framework was trained with the Drosophila S2 cell dataset, we found that the 5.7% (29/512) and 7.2% (37/512) motifs recovered by the filters of SENet can align with fungi and vertebrate’s TF motifs retrieved from JASPAR database (q-value < 0.1). Therefore, we speculated that the function of these enhancers optimized in Drosophila S2 cells might be conserved across species. To test this hypothesis, we compared the activity of the designed enhancers and a 305-bp CMV enhancer frequently used in gene over-expression constructs. We performed luciferase reporter experiments on these sequences in diverse cell lines from nine species, spanning 1.275 billion years of evolutionary divergence, including chicken, fish (Cynoglossus semilaevis), human, mouse, pig, sheep, cattle, insect (Spodoptera frugiperda) and yeast (Komagataella phaffii, syn. Pichia pastoris) (Figure 8E). Intriguingly, we found that, comparing to CMV enhancer, which is one of the most potent CREs at activating transcription in mammalian cells, our designed enhancers presented an averaged ∼3.4-fold increase in transcription-stimulating activity. The advance of DREAM-designed enhancers is most prominent in non-mammalian cell lines, which are usually not a favored infection target of CMV virus. For example, in the SF9 cell of Spodoptera frugiperda, the regulatory activity of DREAM-designed enhancers surpassed that of the CMV enhancer by 209.5-fold and the Hr5 enhancer (commonly used in insects to over-express a gene) (81) by 15.7-fold. In spermatogonium cells of Cynoglossus semilaevis, the DREAM-designed sequence in exhibited regulatory activity 28.6-fold higher than that of the CMV enhancer. Additionally, in yeast, the DREAM-designed sequences demonstrated 9.1- and 3.9- fold higher activity than the UASE enhancer (82), and the most potent enhancer synthesized by de Almeida et al., respectively. Together, these results indicated that the extremely strong regulatory activity of optimized enhancers are conserved across various species and further demonstrated the ability of the DREAM framework to discern and comprehend the general regulatory grammar inherent to enhancers. The CMV (cytomegalovirus) immediate enhancer/β-actin (CAG) promoter has been shown to have strong ubiquitous activity in various cell types and is widely used in recombinant adeno-associated virus (rAAV) vectors as a versatile gene delivery platform for clinical gene therapy. Next, we sought to test whether the synthetic enhancers are able to further improve the expression efficiency of CAG promoter and CMV promoter in human K562 and A549 cells, Drosophila S2 cells. We found that DREAM-optimized enhancer displayed stronger ability to stimulate the expression of CAG and CMV promoter in all three cell lines (all P-values ≤ 0.05, one-sided Wilcoxon rank-sum test; Supplementary Figure S21). Finally, we individually selected the most potent designed sequences from S2 cells (Generation:90_individual:2) and human 293T cells (Generation:79_individual:2) to assess the regulatory activity of the enhancers designed in our study within an endogenous chromatin context. We generated luciferase transgenic Drosophila lines using the attB/attP site-specific recombination system. The result showed that, compared to the wild-type (utilizing the DSCP promoter), the optimized enhancer sequence increased luciferase expression by approximately 10 000-fold (‘Materials and Methods’ in Supplementary data, Supplementary Figure S22A). Additionally, we integrated the optimized enhancer into the genome of human 293T cells via recombinase-mediated integration and subsequently measured their activity. The result demonstrated that the optimized enhancer exhibited an activity 1.29-fold higher than that of the CMV enhancer (‘Materials and Methods’ in Supplementary data, P-values = 0.02, one-sided T-test; Supplementary Figure S22C). Collectively, these findings suggest that, beyond their role in episomal plasmids, synthetic enhancers could significantly amplify gene expression, and, in other words, induce specific molecular phenotypes within the endogenous chromatin context.
To validate DREAM’s accuracy in various CRE design tasks, including the design of strong housekeeping enhancers/silencers, we randomly selected 19 synthetic CREs (14 synthetic enhancers and 5 synthetic silencers) and validated their activity using luciferase reporter assays in Drosophila S2 cells (Supplementary Figure S18C–G): (i) 4 ‘AT rich + strong activity’ enhancers, (ii) 4 ‘with 3 fixed RESs’ enhancers, (iii) 11 ‘strong housekeeping silencers /(iv) enhancers’. The luciferase reporter assays results demonstrated that the designed enhancer sequences (Generation: 90_individual:1) had significantly higher regulatory activity compared to Hr5 enhancer. Specifically, the ‘AT rich + strong activity’ synthetic developmental and housekeeping enhancers were 1.7 and 3.1 times more active than Hr5, respectively. The ‘with 3 fixed RESs’ synthetic developmental and housekeeping enhancers showed 3.9 and 12.1 times higher activity than Hr5, respectively. The synthetic housekeeping enhancers were 6.6 times more active than Hr5. Notably, the strongest synthetic housekeeping silencers could reduce Rps12 promoter transcriptional activity by 43.8 times. Additionally, the designed housekeeping silencers also significantly inhibited PGK promoter transcriptional activity in human and mouse cell lines.
Discussion
Enhancers, pivotal genetic elements, play a crucial role in establishing and maintaining cell identity. Designing synthetic CREs with desired properties, for example, cell type specific or high-activity CREs, offers significant applications. Cell-type-specific enhancers can precisely control gene expression in targeted tissues or cells, essential for regenerative and personalized medicine. They enable therapies that activate genes only in specific cells, minimizing side effects and enhancing treatment efficacy. High-activity enhancers are valuable in industrial biotechnology, optimizing the production of biofuels, pharmaceuticals and other compounds by boosting the expression of key metabolic genes in microbes or plants. In this study, we present DREAM, an innovative framework for synthetic enhancer design and optimization, harnessing the power of deep learning. DREAM demonstrates state-of-the-art performance of enhancer activity prediction, surpassing its counterpart, the DeepSTARR model. Importantly, the enhancer design process within the DREAM framework is transparent and highly biologically interpretable, shedding light on cis-regulatory lexicon associated with enhancer activity. Leveraging learned motifs and DNA features, DREAM is able to design artificial enhancers with the predefined or highest regulatory activity. Notably, the final optimized enhancers exhibited a comparable sequence difference to natural enhancers, reinforcing the effectiveness of the DREAM framework in designing novel synthetic enhancers rather than merely memorizing natural enhancer sequences by rote. Moreover, these designed enhancers exhibit conserved functionality across a diverse range of species, including yeast, insects, avians and mammals.
Recent studies have leveraged deep learning models for the design of enhancer elements, highlighting the evolving landscape of computational approaches in this domain. Notably, de Almeida et al. developed the DeepSTARR model, a vanilla CNN trained on UMI-STARR-seq data, to predict the activity of 1 billion random DNA sequences, identifying sequences with varying enhancer activities, including enhancers with activity comparable to the strongest native Drosophila S2 developmental enhancers (35). In a follow-up study, they introduced the DeepSTARR2 model, which modified the convolutional layers of the original DeepSTARR model and was trained on ATAC-seq data (58). This approach enabled the prediction of the activity of 3 billion random DNA sequences and the identification of tissue-specific enhancers. However, the DeepSTARR series models employ a ‘random sampling and prediction’ design strategy that cannot guarantee the optimal target properties of enhancers within the DNA sequence space and is inefficient. Additionally, Taskiran et al. employed the DeepMEL model, trained on (sc)ATAC-seq data, and used a greedy algorithm to design cell type (line)-specific enhancer elements (56). However, the greedy search method used by Taskiran et al. is limited in exploring the DNA sequence space, making it prone to local optima, and ATAC-seq signals are only imperfect predictors and typically need to be complemented by methods that directly measure enhancer activities (83,84). In contrast to these methods, the DREAM framework we proposed demonstrates several notable advantages. First, the enhancer activity prediction module within the DREAM framework significantly outperforms the models used in the aforementioned studies, including DeepSTARR (35), DeepSTARR2 (58) and DeepMEL (55), in terms of predictive performance. Second, the DREAM framework offers enhanced interpretability, capturing more informative DNA sequence features for enhancer activity than the DeepSTARR model. Additionally, unlike the random sampling method employed by de Almeida et al., which is inefficient and lacks directional design capabilities, the DREAM framework can optimize from 0.1 million random DNA sequences to obtain enhancers with ∼3.6-fold higher activity than the strongest developmental enhancers in the Drosophila genome. Moreover, compared to Taskiran et al.’s method, the genetic algorithm used in the DREAM framework expands the search space effectively, mitigating the issue of local optima while maintaining design efficiency. The DREAM framework’s direct optimization of enhancer activity, facilitated by training on UMI-STARR-seq data, allows for the generation of enhancers with customized activity levels. This contrasts with models trained on (sc)ATAC-seq data, which cannot directly optimize enhancer activity due to the imperfect prediction of chromatin accessibility alone. Furthermore, the relatively short length of enhancers designed using the DREAM framework (249 bp) is advantageous for the miniaturization of AAV delivery vectors. Lastly, the DREAM framework allows users to customize the fitness function, enhancing its scalability and enabling the optimization of multiple enhancer properties simultaneously. For example, users can control cell specificity or simultaneously optimize sequence properties and enhancer activity.
The effectiveness of DREAM framework heavily relies on access to substantial amounts of high-quality enhancer sequence-activity datasets. Currently, there remains a scarcity of large-scale and highly reproducible datasets that directly measure the activity of enhancers. While the well-established high-throughput functional genomics assays, such as ChIP-seq and ATAC-seq, have provided valuable insights into enhancer function and regulation (85), it is important to note that they primarily generate signals indicative of some degree of correlation with enhancer activity rather than directly measuring enhancer activity itself. In contrast, cutting-edge techniques such as STARR-seq and other episomal MPRAs represent an advancement from the luciferase reporter assay, which has long been considered the gold standard for directly measuring enhancer activity. These methods bring enhancer quantification into an NGS-based format, enabling the direct measurement of enhancer activity in a high-throughput manner. Consequently, they stand out as ideal candidates for generating training dataset for the DREAM framework. In this study, we utilized the UMI-STARR-seq data from Drosophila melanogaster S2 cells to train the DREAM framework. UMI-STARR-seq incorporates unique molecular identifiers (UMIs) to accurately distinguish and remove PCR duplicates, which is crucial for increasing signal-to-nose ratio while avoiding false positives (84). Despite the effectiveness of STARR-seq and its derivatives, they are technically challenging and labor-intensive, involving >250 steps and relying on high transfection efficiency of the cells of interest (72). Consequently, high-quality STARR-seq data remains limited at present. In the future, with the ongoing standardization and optimization of STARR-seq technology protocols, coupled with the generation of high-quality datasets across various species, the current data constraints of the DREAM framework are poised to diminish. This progress will facilitate the design of enhancer sequences across a more diverse range of species.
Unraveling and cataloging DNA motifs linked to enhancer functionality will pave the way for understanding the molecular mechanisms of enhancer regulation. A notable advantage of our DREAM framework lies in its biological interpretability. It has the capacity to unveil and categorize DNA motifs linked to enhancer functionality, thus providing insights into the cis-regulatory grammar governing enhancer activity. In this study, we showcased the proficiency of DREAM in accurately recovering well-established enhancer-activating TF motifs, including kay and GATAd motifs. Additionally, DREAM also identified novel motifs that cannot align with any known ones, providing novel insights into enhancer regulation. Beyond providing important insights into the regulatory grammar of enhancers, the DREAM framework has the potential to simulate enhancer activity evolution in silico by designing proper fitness functions and population demographic history. The visualization of dynamic changes in the filter activation of SENet’s first layer allows us to understand how the DNA sequence space is explored during evolution and how the enhancer functional motifs are exploited in the in silico sequence design process. Intriguingly, we found a conspicuous co-occurrence pattern emerged among the functional motifs during the in silico enhancer evolution, such as the SREBP-kay, SREBP-Jra, which indicated the interactions between functional motifs are vital for manipulating the enhancer activity.
Utilizing natural CREs for fine-tuning gene expression faces limitations, particularly when there is a need to induce extremely high expression of a target gene. The constrained activity of natural enhancers may stem from the fact that natural selection does not explicitly optimize enhancer activity. The gene expression patterns within organisms are often intricately orchestrated by a network of multiple enhancers (74,86,87). In this scenario, natural selection may tend to optimize the overall module’s output and its internal interactions rather than solely elevating the activity of an individual regulatory element. Additionally, CREs with excessive activity might adversely impact the overall fitness of an individual, hindering their emergence in the process of natural evolution. In this work, utilizing the DEARM framework for synthetic enhancer design, we expanded the repertoire of CREs available in the genetic engineering toolbox beyond natural DNA sequences. Our strategy successfully designed enhancer elements with remarkably strong activity—exceeding approximately 3.6-fold the potency of the strongest natural enhancer in the Drosophila genome. Notably, despite the DEARM framework being trained on the Drosophila S2 cell dataset, the final optimized enhancers exhibited remarkably high regulatory activity across diverse species. This observation not only underscores the conservation of enhancer syntax across species but also implies that the designed enhancers harbor significant potential for application in various fields, including gene therapy, genomic breeding and industrial production from cell-based bioreactors.
While many studies evaluating enhancer activity often overlook specifying the associated promoter, emerging evidence suggests that the activity of an enhancer is influenced to some degree by its cognate promoter. Reports indicate that enhancer-promoter compatibility may be governed by interacting sets of TFs or cofactors (71,88–90). Hence, it is crucial to recognize that enhancers designed by the DREAM framework might not perform as anticipated when coupled with a promoter different from the one used in the training data. Conventional STARR-seq techniques typically assess enhancer activity with one promoter at a time, posing challenges in obtaining large-scale training datasets for promoter and enhancer interactions. A recent advancement, the ExP STARR-seq assay, allows simultaneous assessment of the activity of 1000 candidate enhancers on 1000 promoters (88). While commendable, these data may still fall short of meeting the training requirements for deep learning-based models. A promising direction for future research involves the development of innovative high-throughput technological approaches capable of efficiently and accurately providing information on promoter and enhancer compatibility. Once such datasets become available, the DREAM framework can incorporate both promoter and enhancer DNA sequences simultaneously for training and optimization, enabling the prediction of enhancer activity for a given sequence coupled with any specified promoter sequence.
DREAM framework can also be extended to efficiently design other CRE, such as promoters and silencers, with desirable properties with the corresponding training dataset. Recently, researchers proposed a new workflow that significantly enhances the capability of STARR-seq to efficiently and simultaneously measure the functional activity of both enhancers and silencers (91). This framework is poised to become a pivotal tool in the design of silencers. Additionally, the crafting of tissue-specific CREs can be accomplished by utilizing the corresponding high-throughput training dataset derived from diverse cell lines or tissues, which would necessitate only slight adjustments to the fitness function of DREAM.
To effectively harness and design specific phenotypes in plants and animals using synthetic CREs, it is essential to precisely design and regulate the activity of these CREs, including their spatiotemporal specificity. Moreover, a profound understanding of the underlying principles governing gene regulatory networks and their responses to environmental stimuli is crucial (92). These insights represent key future directions and research areas for applying synthetic biology in plant and animal breeding.
Supplementary Material
Acknowledgements
Author contribution: Z.L. and Y.L. conceived the project. Z.L. designed the project, performed the research, analyzed the data and wrote the manuscript. Z.Y. designed and organized validation experiments with the help from Q.S., Q.Z., C.C., Y.B., Y.C., B.L., Q.L., L.X., X.M., H.W., L.X., Y.Y., B.D., J.L., B.J.D., Y.T.C., J.W., Y.Z., Y.H, X.C., T.S. and T.L. R.L., Z.T., J.Z., E.Z., C.M., F.Z., C.S., G.W., N.W. and G.L. provided the cell lines and transfection protocols used in this study. P.B. and N.J. construct the luciferase transgenic Drosophila lines and validate the designed enhancers activity in vivo. Y.L. supervised this study. Z.L. and Y.L. revised the manuscript. All authors interpreted the results and approved the final paper.
We express our gratitude to Dr Ye Li and Dr Xiaoping Guo for their assistance in editing the figures. We also extend our sincere thanks to the five anonymous reviewers for their thorough review and invaluable suggestions.
Contributor Information
Zhaohong Li, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Key Laboratory of Livestock and Poultry Multi-Omics of MARA, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China; Innovation Group of Pig Genome Design and Breeding, Research Centre for Animal Genome, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Yuanyuan Zhang, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Key Laboratory of Livestock and Poultry Multi-Omics of MARA, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China; Innovation Group of Pig Genome Design and Breeding, Research Centre for Animal Genome, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Bo Peng, Gene Regulatory Lab, School of Basic Medical Sciences, Tsinghua University, NO. 30 Shuangqing road, Haidian district, Beijing 100084, China; State Key Laboratory of Molecular Oncology, Tsinghua University, NO. 30 Shuangqing road, Haidian district, Beijing 100084, China.
Shenghua Qin, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Key Laboratory of Livestock and Poultry Multi-Omics of MARA, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China; Innovation Group of Pig Genome Design and Breeding, Research Centre for Animal Genome, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Qian Zhang, State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, NO.1 Beichen West Road, Chaoyang District, Beijing 100101, China.
Yun Chen, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Key Laboratory of Livestock and Poultry Multi-Omics of MARA, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China; Innovation Group of Pig Genome Design and Breeding, Research Centre for Animal Genome, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Choulin Chen, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Key Laboratory of Livestock and Poultry Multi-Omics of MARA, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China; Innovation Group of Pig Genome Design and Breeding, Research Centre for Animal Genome, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Yongzhou Bao, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Key Laboratory of Livestock and Poultry Multi-Omics of MARA, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China; Innovation Group of Pig Genome Design and Breeding, Research Centre for Animal Genome, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Yuqi Zhu, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, NO. 7 Pengfei Road, Dapeng District, Shenzhen 518124, China.
Yi Hong, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, NO. 7 Pengfei Road, Dapeng District, Shenzhen 518124, China.
Binghua Liu, State Key Laboratory of Maricultural Biobreeding and Sustainable Goods, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, NO.106 Nanjing Road, Shinan District, Qingdao, Shandong 266071, China.
Qian Liu, State Key Laboratory of Maricultural Biobreeding and Sustainable Goods, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, NO.106 Nanjing Road, Shinan District, Qingdao, Shandong 266071, China.
Lingna Xu, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Key Laboratory of Livestock and Poultry Multi-Omics of MARA, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China; Innovation Group of Pig Genome Design and Breeding, Research Centre for Animal Genome, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Xi Chen, Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture, Key Laboratory of Synthetic Biology, Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Xinhao Ma, College of Grassland Agriculture, National Beef Cattle Improvement Center, College of Animal Science and Technology, Northwest A&F University, NO. 3 Taicheng Road, Yangling District, Yangling, Shaanxi 712100, China.
Hongyan Wang, State Key Laboratory of Maricultural Biobreeding and Sustainable Goods, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, NO.106 Nanjing Road, Shinan District, Qingdao, Shandong 266071, China.
Long Xie, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Key Laboratory of Livestock and Poultry Multi-Omics of MARA, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Yilong Yao, Green Healthy Aquaculture Research Center, Kunpeng Institute of Modern Agriculture at Foshan, Chinese Academy of Agricultural Sciences, Building 26 Lihe Technology Park, Auxiliary Road of Xinxi Avenue South, Nanhai District, Foshan 528226, China.
Biao Deng, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Key Laboratory of Livestock and Poultry Multi-Omics of MARA, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China; Innovation Group of Pig Genome Design and Breeding, Research Centre for Animal Genome, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Jiaying Li, Department of Ophthalmology, Beijing Institute of Ophthalmology, Beijing Tongren Eye Center, Beijing Tongren Hospital, Capital Medical University, Dongjiaomin lane No1, Dongcheng District, Beijing 100101, China.
Baojun De, College of Life Sciences, Inner Mongolia Autonomous Region Key Laboratory of Biomanufacturing, Inner Mongolia Agricultural University, NO. 306 Zhaowuda Road, Saihan District, Hohhot 010018, China.
Yuting Chen, College of Life Sciences, Inner Mongolia Autonomous Region Key Laboratory of Biomanufacturing, Inner Mongolia Agricultural University, NO. 306 Zhaowuda Road, Saihan District, Hohhot 010018, China.
Jing Wang, Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture, Key Laboratory of Synthetic Biology, Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Tian Li, College of JUNCAO Science and Ecology, Haixia Institute of Science and Technology, National Engineering Research Center of JUNCAO, Fujian Agriculture and Forestry University (FAFU), NO.15 Shangxiadian Road, Cangshan District, Fuzhou 0350002, China.
Ranran Liu, Institute of Animal Science, Chinese Academy of Agricultural Sciences, Yuanmingyuan West Road NO. 2, Haidian District, Beijing 100193, China.
Zhonglin Tang, Green Healthy Aquaculture Research Center, Kunpeng Institute of Modern Agriculture at Foshan, Chinese Academy of Agricultural Sciences, Building 26 Lihe Technology Park, Auxiliary Road of Xinxi Avenue South, Nanhai District, Foshan 528226, China.
Junwei Cao, College of Life Sciences, Inner Mongolia Autonomous Region Key Laboratory of Biomanufacturing, Inner Mongolia Agricultural University, NO. 306 Zhaowuda Road, Saihan District, Hohhot 010018, China.
Erwei Zuo, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Key Laboratory of Livestock and Poultry Multi-Omics of MARA, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Chugang Mei, College of Grassland Agriculture, National Beef Cattle Improvement Center, College of Animal Science and Technology, Northwest A&F University, NO. 3 Taicheng Road, Yangling District, Yangling, Shaanxi 712100, China.
Fangjie Zhu, College of JUNCAO Science and Ecology, Haixia Institute of Science and Technology, National Engineering Research Center of JUNCAO, Fujian Agriculture and Forestry University (FAFU), NO.15 Shangxiadian Road, Cangshan District, Fuzhou 0350002, China.
Changwei Shao, State Key Laboratory of Maricultural Biobreeding and Sustainable Goods, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, NO.106 Nanjing Road, Shinan District, Qingdao, Shandong 266071, China.
Guirong Wang, Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture, Key Laboratory of Synthetic Biology, Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China.
Tongjun Sun, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, NO. 7 Pengfei Road, Dapeng District, Shenzhen 518124, China.
Ningli Wang, Department of Ophthalmology, Beijing Institute of Ophthalmology, Beijing Tongren Eye Center, Beijing Tongren Hospital, Capital Medical University, Dongjiaomin lane No1, Dongcheng District, Beijing 100101, China.
Gang Liu, State Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, NO.1 Beichen West Road, Chaoyang District, Beijing 100101, China.
Jian-Quan Ni, Gene Regulatory Lab, School of Basic Medical Sciences, Tsinghua University, NO. 30 Shuangqing road, Haidian district, Beijing 100084, China; State Key Laboratory of Molecular Oncology, Tsinghua University, NO. 30 Shuangqing road, Haidian district, Beijing 100084, China; SXMU-Tsinghua Collaborative Innovation Center for Frontier Medicine, Shanxi Medical University, NO. 56 Xinjian South Road, Yingze District, Taiyuan 030001, China.
Yuwen Liu, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Key Laboratory of Livestock and Poultry Multi-Omics of MARA, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China; Innovation Group of Pig Genome Design and Breeding, Research Centre for Animal Genome, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Buxin Road NO. 97, Dapeng District, Shenzhen 518124, China; Green Healthy Aquaculture Research Center, Kunpeng Institute of Modern Agriculture at Foshan, Chinese Academy of Agricultural Sciences, Building 26 Lihe Technology Park, Auxiliary Road of Xinxi Avenue South, Nanhai District, Foshan 528226, China.
Data availability
The source code used to implement, train and evaluate the SENet is available on GitHub (https://github.com/cisGrammar/DREAM) and Figshare (https://doi.org/10.6084/m9.figshare.27135255.v1). The source code for designing and optimizing the enhancers is also available from https://github.com/cisGrammar/DREAM/GA.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
China National Key R&D Program during the 14th Five-Year Plan Period [2021YFF1200500 to L.Y.]]; National Natural Science Foundation of China [32070595 to L.Y.]; Ministry of Science and Technology of the People’s Republic of China [20221250020]; National Natural Science Foundation of China [20181300988, 20201300797]. Funding for open access charge: National Key Research and Development Program of China [2021YFF1200500 to L.Y].
Conflict of interest statement. The enhancer design and optimization framework DREAM has filed a provisional patent application (2023114102122) related to the work described here.
References
- 1. Chen W.C.W., Gaidukov L., Lai Y., Wu M.-R., Cao J., Gutbrod M.J., Choi G.C.G., Utomo R.P., Chen Y.-C., Wroblewska L.et al.. A synthetic transcription platform for programmable gene expression in mammalian cells. Nat. Commun. 2022; 13:6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yasmeen E., Wang J., Riaz M., Zhang L., Zuo K.. Designing artificial synthetic promoters for accurate, smart, and versatile gene expression in plants. Plant Commun. 2023; 4:100558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Song X., Meng X., Guo H., Cheng Q., Jing Y., Chen M., Liu G., Wang B., Wang Y., Li J.et al.. Targeting a gene regulatory element enhances rice grain yield by decoupling panicle number and size. Nat. Biotechnol. 2022; 40:1403–1411. [DOI] [PubMed] [Google Scholar]
- 4. Grandi F.C., Modi H., Kampman L., Corces M.R.. Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 2022; 17:1518–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tsompana M., Buck M.J.. Chromatin accessibility: a window into the genome. Epigenetics Chromatin. 2014; 7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lareau C.A., Duarte F.M., Chew J.G., Kartha V.K., Burkett Z.D., Kohlway A.S., Pokholok D., Aryee M.J., Steemers F.J., Lebofsky R.et al.. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol. 2019; 37:916–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Arensbergen J., FitzPatrick V.D., de Haas M., Pagie L., Sluimer J., Bussemaker H.J., van Steensel B.. Genome-wide mapping of autonomous promoter activity in human cells. Nat. Biotechnol. 2017; 35:145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Melnikov A., Murugan A., Zhang X., Tesileanu T., Wang L., Rogov P., Feizi S., Gnirke A., Callan C.G., Kinney J.B.et al.. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012; 30:271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Boer C.G., Vaishnav E.D., Sadeh R., Abeyta E.L., Friedman N., Regev A.. Deciphering eukaryotic gene-regulatory logic with 100 million random promoters. Nat. Biotechnol. 2020; 38:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao S., Hong C.K.Y., Myers C.A., Granas D.M., White M.A., Corbo J.C., Cohen B.A.. A single-cell massively parallel reporter assay detects cell-type-specific gene regulation. Nat. Genet. 2023; 55:346–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Redden H., Alper H.S.. The development and characterization of synthetic minimal yeast promoters. Nat. Commun. 2015; 6:7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Neph S., Vierstra J., Stergachis A.B., Reynolds A.P., Haugen E., Vernot B., Thurman R.E., John S., Sandstrom R., Johnson A.Ket al.. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012; 489:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vierstra J., Lazar J., Sandstrom R., Halow J., Lee K., Bates D., Diegel M., Dunn D., Neri F., Haugen E.et al.. Global reference mapping of human transcription factor footprints. Nature. 2020; 583:729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., Kawli T., Davis C.A., Dobin A., Kaul R.et al.. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020; 583:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cai Y.-M., Kallam K., Tidd H., Gendarini G., Salzman A., Patron N.J.. Rational design of minimal synthetic promoters for plants. Nucleic Acids Res. 2020; 48:11845–11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guiziou S., Sauveplane V., Chang H.-J., Clerté C., Declerck N., Jules M., Bonnet J.. A part toolbox to tune genetic expression in Bacillus subtilis. Nucleic Acids Res. 2016; 44:7495–7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sahu B., Hartonen T., Pihlajamaa P., Wei B., Dave K., Zhu F., Kaasinen E., Lidschreiber K., Lidschreiber M., Daub C.O.et al.. Sequence determinants of human gene regulatory elements. Nat. Genet. 2022; 54:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Levo M., Segal E.. In pursuit of design principles of regulatory sequences. Nat. Rev. Genet. 2014; 15:453–468. [DOI] [PubMed] [Google Scholar]
- 19. Alipanahi B., Delong A., Weirauch M.T., Frey B.J.. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015; 33:831–838. [DOI] [PubMed] [Google Scholar]
- 20. Zhou J., Theesfeld C.L., Yao K., Chen K.M., Wong A.K., Troyanskaya O.G.. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 2018; 50:1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kelley D.R., Snoek J., Rinn J.L.. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016; 26:990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kelley D.R., Reshef Y.A., Bileschi M., Belanger D., McLean C.Y., Snoek J.. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 2018; 28:739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Avsec Ž., Agarwal V., Visentin D., Ledsam J.R., Grabska-Barwinska A., Taylor K.R., Assael Y., Jumper J., Kohli P., Kelley D.R.. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods. 2021; 18:1196–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jaganathan K., Kyriazopoulou Panagiotopoulou S., McRae J.F., Darbandi S.F., Knowles D., Li Y.I., Kosmicki J.A., Arbelaez J., Cui W., Schwartz G.B.et al.. Predicting splicing from primary sequence with deep learning. Cell. 2019; 176:535–548. [DOI] [PubMed] [Google Scholar]
- 25. Dawes R., Bournazos A.M., Bryen S.J., Bommireddipalli S., Marchant R.G., Joshi H., Cooper S.T.. SpliceVault predicts the precise nature of variant-associated mis-splicing. Nat. Genet. 2023; 55:324–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiong H.Y., Alipanahi B., Lee L.J., Bretschneider H., Merico D., Yuen R.K.C., Hua Y., Gueroussov S., Najafabadi H.S., Hughes T.R.et al.. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015; 347:1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eraslan G., Avsec Ž., Gagneur J., Theis F.J.. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 2019; 20:389–403. [DOI] [PubMed] [Google Scholar]
- 28. Zou J., Huss M., Abid A., Mohammadi P., Torkamani A., Telenti A.. A primer on deep learning in genomics. Nat. Genet. 2019; 51:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vaishnav E.D., de Boer C.G., Molinet J., Yassour M., Fan L., Adiconis X., Thompson D.A., Levin J.Z., Cubillos F.A., Regev A.. The evolution, evolvability and engineering of gene regulatory DNA. Nature. 2022; 603:455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sample P.J., Wang B., Reid D.W., Presnyak V., McFadyen I.J., Morris D.R., Seelig G.. Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 2019; 37:803–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cuperus J.T., Groves B., Kuchina A., Rosenberg A.B., Jojic N., Fields S., Seelig G.. Deep learning of the regulatory grammar of yeast 5' untranslated regions from 500,000 random sequences. Genome Res. 2017; 27:2015–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jores T., Tonnies J., Wrightsman T., Buckler E.S., Cuperus J.T., Fields S., Queitsch C.. Synthetic promoter designs enabled by a comprehensive analysis of plant core promoters. Nat. Plants. 2021; 7:842–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reiter F., de Almeida B.P., Stark A.. Enhancers display constrained sequence flexibility and context-specific modulation of motif function. Genome Res. 2023; 33:346–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang M.G., Ling E., Cowley C.J., Greenberg M.E., Vierbuchen T.. Characterization of sequence determinants of enhancer function using natural genetic variation. eLife. 2022; 11:e76500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de Almeida B.P., Reiter F., Pagani M., Stark A.. DeepSTARR predicts enhancer activity from DNA sequence and enables the de novo design of synthetic enhancers. Nat. Genet. 2022; 54:613–624. [DOI] [PubMed] [Google Scholar]
- 36. Goodfellow I.J., Pouget-Abadie J., Mirza M., Xu B., Warde-Farley D., Ozair S., Courville A.C., Bengio Y.. Generative Adversarial Nets. Proceedings of the 27th International Conference on Neural Information Processing Systems. 2014; Montreal, Quebec, Canada: 2672–2680. [Google Scholar]
- 37. Wang Y., Wang H., Wei L., Li S., Liu L., Wang X.. Synthetic promoter design in Escherichia coli based on a deep generative network. Nucleic Acids Res. 2020; 48:6403–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zrimec J., Fu X., Muhammad A.S., Skrekas C., Jauniskis V., Speicher N.K., Börlin C.S., Verendel V., Chehreghani M.H., Dubhashi D.et al.. Controlling gene expression with deep generative design of regulatory DNA. Nat. Commun. 2022; 13:5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Repecka D., Jauniskis V., Karpus L., Rembeza E., Rokaitis I., Zrimec J., Poviloniene S., Laurynenas A., Viknander S., Abuajwa W.et al.. Expanding functional protein sequence spaces using generative adversarial networks. Nat. Machine Intelligence. 2021; 3:324–333. [Google Scholar]
- 40. Linder J., Bogard N., Rosenberg A.B., Seelig G.. A generative neural network for maximizing fitness and diversity of synthetic DNA and protein sequences. Cell Syst. 2020; 11:49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Strokach A., Kim P.M.. Deep generative modeling for protein design. Curr. Opin. Struct. Biol. 2022; 72:226–236. [DOI] [PubMed] [Google Scholar]
- 42. Albig C., Tikhonova E., Krause S., Maksimenko O., Regnard C., Becker P.B.. Factor cooperation for chromosome discrimination in Drosophila. Nucleic Acids Res. 2018; 47:1706–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arnold C.D., Gerlach D., Stelzer C., Boryń Ł.M., Rath M., Stark A.. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013; 339:1074–1077. [DOI] [PubMed] [Google Scholar]
- 44. Yáñez-Cuna J.O., Arnold C.D., Stampfel G., Boryń Ł.M., Gerlach D., Rath M., Stark A.. Dissection of thousands of cell type-specific enhancers identifies dinucleotide repeat motifs as general enhancer features. Genome Res. 2014; 24:1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shlyueva D., Stelzer C., Gerlach D., Yáñez-Cuna J.O., Rath M., Boryń Ł.u.M., Arnold C.D., Stark A.. Hormone-responsive enhancer-activity maps reveal predictive motifs, indirect repression, and targeting of closed chromatin. Mol. Cell. 2014; 54:180–192. [DOI] [PubMed] [Google Scholar]
- 46. Franz A., Shlyueva D., Brunner E., Stark A., Basler K.. Probing the canonicity of the Wnt/Wingless signaling pathway. PLoS Genet. 2017; 13:e1006700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu J., Shen L., Sun G.. Squeeze-and-Excitation Networks. 2018 IEEE/CVF Conference on Computer Vision and Pattern Recognition. 2017; Salt Lake City, UT, USA: 7132–7141. [Google Scholar]
- 48. Koo P.K., Ploenzke M.. Improving representations of genomic sequence motifs in convolutional networks with exponential activations. Nat. Machine Intelligence. 2021; 3:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Glorot X., Bengio Y.. Yee Whye T., Mike T.. Proceedings of the Thirteenth International Conference on Artificial Intelligence and Statistics. 2010; 9:PMLR, Proceedings of Machine Learning Research; 249–256. [Google Scholar]
- 50. Kingma D.P., Ba J.. Adam: a method for stochastic optimization. 2015 International Conference on Learning Representations (ICLR). 2014; San Diego, USA: 13–14. [Google Scholar]
- 51. Quang D., Xie X.. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 2016; 44:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ji Y., Zhou Z., Liu H., Davuluri R.V.. DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics. 2021; 37:2112–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang P., Wang H., Xu H., Wei L., Liu L., Hu Z., Wang X.. Deep flanking sequence engineering for efficient promoter design using DeepSEED. Nat. Commun. 2023; 14:6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li J., Pu Y., Tang J., Zou Q., Guo F.. DeepATT: a hybrid category attention neural network for identifying functional effects of DNA sequences. Brief. Bioinf. 2020; 22:bbaa159. [DOI] [PubMed] [Google Scholar]
- 55. Minnoye L., Taskiran I.I., Mauduit D., Fazio M., Van Aerschot L., Hulselmans G., Christiaens V., Makhzami S., Seltenhammer M., Karras P.et al.. Cross-species analysis of enhancer logic using deep learning. Genome Res. 2020; 30:1815–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Taskiran I.I., Spanier K.I., Dickmänken H., Kempynck N., Pančíková A., Ekşi E.C., Hulselmans G., Ismail J.N., Theunis K., Vandepoel R.et al.. Cell-type-directed design of synthetic enhancers. Nature. 2024; 626:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou J., Troyanskaya O.G.. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods. 2015; 12:931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. de Almeida B.P., Schaub C., Pagani M., Secchia S., Furlong E.E.M., Stark A.. Targeted design of synthetic enhancers for selected tissues in the Drosophila embryo. Nature. 2024; 626:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rauluseviciute I., Riudavets-Puig R., Blanc-Mathieu R., Castro-Mondragon J.A., Ferenc K., Kumar V., Lemma R.B., Lucas J., Chèneby J., Baranasic D.et al.. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2023; 52:D174–D182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S.. Quantifying similarity between motifs. Genome Biol. 2007; 8:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grant C.E., Bailey T.L., Noble W.S.. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011; 27:1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lundberg S.M., Erion G., Chen H., DeGrave A., Prutkin J.M., Nair B., Katz R., Himmelfarb J., Bansal N., Lee S.-I.. From local explanations to global understanding with explainable AI for trees. Nat. Machine Intelligence. 2020; 2:56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shrikumar A., Greenside P., Kundaje A.. Learning important features through propagating activation differences. Proceedings of the 34thInternational Conference on Machine Learning. 2017; Sydney, NSW, Australia: 3145–3153. [Google Scholar]
- 64. Angermueller C., Lee H.J., Reik W., Stegle O.. DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 2017; 18:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chiu T.-P., Comoglio F., Zhou T., Yang L., Paro R., Rohs R.. DNAshapeR: an R/Bioconductor package for DNA shape prediction and feature encoding. Bioinformatics. 2015; 32:1211–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nei M., Tajima F.. DNA polymorphism detectable by restriction endonucleases. Genetics. 1981; 97:145–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Boshart M., Weber F., Jahn G., Dorsch-H⇒ler K., Fleckenstein B., Schaffner W.. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell. 1985; 41:521–530. [DOI] [PubMed] [Google Scholar]
- 68. Xu C., Zhou Y., Xiao Q., He B., Geng G., Wang Z., Cao B., Dong X., Bai W., Wang Y.et al.. Programmable RNA editing with compact CRISPR–Cas13 systems from uncultivated microbes. Nat. Methods. 2021; 18:499–506. [DOI] [PubMed] [Google Scholar]
- 69. Gentili M., Kowal J., Tkach M., Satoh T., Lahaye X., Conrad C., Boyron M., Lombard B., Durand S., Kroemer G.et al.. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 2015; 349:1232–1236. [DOI] [PubMed] [Google Scholar]
- 70. Zhang Y., Wang Z., Liu Y., Lu L., Tan X., Zou Q.. 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2021; 594–599. [Google Scholar]
- 71. Zabidi M.A., Arnold C.D., Schernhuber K., Pagani M., Rath M., Frank O., Stark A.. Enhancer–core-promoter specificity separates developmental and housekeeping gene regulation. Nature. 2015; 518:556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Das M., Hossain A., Banerjee D., Praul C.A., Girirajan S.. Challenges and considerations for reproducibility of STARR-seq assays. Genome Res. 2023; 33:479–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Spitz F., Furlong E.E.M.. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 2012; 13:613–626. [DOI] [PubMed] [Google Scholar]
- 74. Panigrahi A., O’Malley B.W. Mechanisms of enhancer action: the known and the unknown. Genome Biol. 2021; 22:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bogard N., Linder J., Rosenberg A.B., Seelig G.. A deep neural network for predicting and engineering alternative polyadenylation. Cell. 2019; 178:91–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang J., Zhuang J., Iyer S., Lin X., Whitfield T.W., Greven M.C., Pierce B.G., Dong X., Kundaje A., Cheng Y.et al.. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012; 22:1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mathelier A., Xin B., Chiu T.-P., Yang L., Rohs R., Wasserman W.W.. DNA shape features improve transcription factor binding site predictions in vivo. Cell Syst. 2016; 3:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sielemann J., Wulf D., Schmidt R., Bräutigam A.. Local DNA shape is a general principle of transcription factor binding specificity in Arabidopsis thaliana. Nat. Commun. 2021; 12:6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wong E.S., Zheng D., Tan S.Z., Bower N.I., Garside V., Vanwalleghem G., Gaiti F., Scott E., Hogan B.M., Kikuchi K.et al.. Deep conservation of the enhancer regulatory code in animals. Science. 2020; 370:eaax8137. [DOI] [PubMed] [Google Scholar]
- 80. Snetkova V., Ypsilanti A.R., Akiyama J.A., Mannion B.J., Plajzer-Frick I., Novak C.S., Harrington A.N., Pham Q.T., Kato M., Zhu Y.et al.. Ultraconserved enhancer function does not require perfect sequence conservation. Nat. Genet. 2021; 53:521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Guarino L.A., Dong W.. Functional dissection of the Autographa californica nuclear polyhedrosis virus enhancer element hr5. Virology. 1994; 200:328–335. [DOI] [PubMed] [Google Scholar]
- 82. Lai J., Song L., Zhou Y., Zong H., Zhuge B., Lu X.. Fine-tuned gene expression elements from hybrid promoter libraries in Pichiapastoris. ACS Synth. Biol. 2024; 13:310–318. [DOI] [PubMed] [Google Scholar]
- 83. Catarino R.R., Stark A.. Assessing sufficiency and necessity of enhancer activities for gene expression and the mechanisms of transcription activation. Genes Dev. 2018; 32:202–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Neumayr C., Pagani M., Stark A., Arnold C.D.. STARR-seq and UMI-STARR-seq: assessing enhancer activities for genome-wide-, high-, and low-complexity candidate libraries. Curr. Protocol. Mol. Biol. 2019; 128:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Andersson R., Sandelin A.. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020; 21:71–87. [DOI] [PubMed] [Google Scholar]
- 86. Kvon E.Z., Waymack R., Elabd M.G., Wunderlich Z.. Enhancer redundancy in development and disease. Nat. Rev. Genet. 2021; 22:324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Osterwalder M., Barozzi I., Tissières V., Fukuda-Yuzawa Y., Mannion B.J., Afzal S.Y., Lee E.A., Zhu Y., Plajzer-Frick I., Pickle C.S.et al.. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature. 2018; 554:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bergman D.T., Jones T.R., Liu V., Ray J., Jagoda E., Siraj L., Kang H.Y., Nasser J., Kane M., Rios A.et al.. Compatibility rules of human enhancer and promoter sequences. Nature. 2022; 607:176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Martinez-Ara M., Comoglio F., van Arensbergen J., van Steensel B.. Systematic analysis of intrinsic enhancer-promoter compatibility in the mouse genome. Mol. Cell. 2022; 82:2519–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. van Arensbergen J., van Steensel B., Bussemaker H.J. In search of the determinants of enhancer–promoter interaction specificity. Trends Cell Biol. 2014; 24:695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hansen T.J., Hodges E.. ATAC-STARR-seq reveals transcription factor–bound activators and silencers within chromatin-accessible regions of the human genome. Genome Res. 2022; 32:1529–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Borowsky A.T., Bailey-Serres J.. Rewiring gene circuitry for plant improvement. Nat. Genet. 2024; 56:1574–1582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code used to implement, train and evaluate the SENet is available on GitHub (https://github.com/cisGrammar/DREAM) and Figshare (https://doi.org/10.6084/m9.figshare.27135255.v1). The source code for designing and optimizing the enhancers is also available from https://github.com/cisGrammar/DREAM/GA.