


Abstract
High-throughput technologies have led to the rapid generation of large-scale datasets about genes and gene products. These technologies have also shifted our research focus from ‘single genes’ to ‘gene sets’. We have developed a web-based integrated data mining system, WebGestalt (http://genereg.ornl.gov/webgestalt/), to help biologists in exploring large sets of genes. WebGestalt is composed of four modules: gene set management, information retrieval, organization/visualization, and statistics. The management module uploads, saves, retrieves and deletes gene sets, as well as performs Boolean operations to generate the unions, intersections or differences between different gene sets. The information retrieval module currently retrieves information for up to 20 attributes for all genes in a gene set. The organization/visualization module organizes and visualizes gene sets in various biological contexts, including Gene Ontology, tissue expression pattern, chromosome distribution, metabolic and signaling pathways, protein domain information and publications. The statistics module recommends and performs statistical tests to suggest biological areas that are important to a gene set and warrant further investigation. In order to demonstrate the use of WebGestalt, we have generated 48 gene sets with genes over-represented in various human tissue types. Exploration of all the 48 gene sets using WebGestalt is available for the public at http://genereg.ornl.gov/webgestalt/wg_enrich.php.
INTRODUCTION
The development of high-throughput methodologies, as epitomized by microarray technologies, has led to the rapid generation of large-scale datasets about RNA transcripts or proteins. While in the past biologists studied single genes at a time, now we can use high-throughput technologies to analyze tens of thousands of genes simultaneously. The nature of high throughput technologies requires that bioinformatics tools focus on ‘gene sets’ instead of ‘single genes’. For example, microarray and proteome technologies are producing sets of genes and proteins that are differentially expressed under certain conditions, or sets of genes and proteins that are co-expressed under varying conditions. Other studies such as quantitative trait analysis, large-scale mutagenesis studies, and other large-scale genetic studies are also producing sets of interesting genes. Translating the identified gene sets into a better understanding of the underlying biological processes constitutes a huge challenge for today's biologists. Even retrieving the associated functional information for large gene sets can be time-consuming. Further manipulating, visualizing, and statistically analyzing the interrelated data can involve complex processes for an average biologist. Without the assistance of appropriate bioinformatics tools, exploring the gene sets to discover important patterns is not a trivial task for biologists.
Traditional resources that are available for retrieving functional information, such as the LocusLink from NCBI (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/LocusLink/), are typically displayed in a one-gene-at-a-time format (1). A newer generation of resources has been created to facilitate batch information retrieval for sets of genes (2–4). One such example is ENSMART (http://www.ensembl.org/Multi/martview), in which the users can perform a genome information search and retrieval for sets of genes in human and several other eukaryotic species (3). ENSMART covers a broad spectrum of functional information pertaining to gene- and protein-specific attributes as well as disease, expression, sequence variation and cross-species attributes. Despite being an excellent batch information retrieval tool, ENSMART does not help biologists in efficiently exploring the abundant information associated with a gene set.
One way to help biologists in exploring large gene sets is to organize the genes based on common functional features, such as Gene Ontology (GO) (5) categories or biochemical pathways. Several bioinformatics tools have been developed for organizing sets of genes based on GO (6–10). Most of these tools have also implemented statistical tests to identify enriched GO categories and to suggest the most important biological areas associated with a given gene set. Although the use of ontological methods to structure biological knowledge is an active area of research and development, the body of biological knowledge associated with any gene set extends far beyond GO. In addition to organizing gene sets within the context of GO, MAPPFinder (11), DAVID (12) and GFINDer (13) provide the option of organizing and visualizing gene sets within the context of KEGG (Kyoto Encyclopedia of Genes and Genomes, http://www.genome.ad.jp/kegg) biochemical pathways (14). DAVID and GFINDer can also organize gene sets based on protein domain information. Other features, such as chromosome location, tissue expression pattern and association in publication, could also be used to organize a gene set. However, these features are not implemented in the current gene set analysis tools.
Although methods of gene organization help biologists explore large gene sets, they frequently generate complex results with hundreds of categories. Information visualization enables people to deal with the overwhelming amount of information associated with a gene set by taking advantage of our innate visual perception capabilities. Visual methods are useful in displaying data in ways that capitalize upon the particular strengths of human pattern processing abilities (15). Information visualization techniques have been successfully used in many areas of bioinformatics, including molecular structures, expression profile, genome and sequence annotation, sequence analysis, molecular pathway, ontology, taxonomy and phylogeny (16). Application of information visualization techniques in gene set analysis will not only help the visualization of large amount of information, but also facilitate data mining by aiding recognition of patterns and trends.
Besides information retrieval, organization, statistical analysis and visualization, management of large gene sets presents additional challenges for biologists. Bioinformatics tools are needed to create subsets of genes from a gene set based on different criteria, such as GO categories, biochemical pathways or chromosome location ranges. Tools are also needed to perform Boolean operations and generate the unions, intersections and differences between gene sets. Boolean operations could help to reveal the interrelationship among different gene sets.
In response to these challenges, we have developed WebGestalt (WEB-based GEne SeT AnaLysis Toolkit), an integrated data mining system for the management, information retrieval, organization, visualization and statistical analysis of large sets of genes.
METHODS
Database: GeneKeyDB
WebGestalt is based on an ORACLE relational database, GeneKeyDB. This database has used a strong gene and protein centric viewpoint. Gene and gene product information is primarily taken from NCBI LocusLink, Ensembl, Swiss-Prot, HomoloGene, Unigene, CGAP, UCSC, GO Consortium, KEGG, BioCarta and Affymetrix. As a consequence of the transition from LocusLink to Entrez Gene from NCBI, we are currently migrating from the LocusLink data to the Entrez Gene data. Updating of GeneKeyDB is automated by pre-prepared scripts. The Schema and dictionary of GeneKeyDB are available from http://genereg.ornl.gov/gkdb. More details of GeneKeyDB are available from (17).
WebGestalt modules
Figure 1 depicts the schematic overview of WebGestalt. WebGestalt is composed of four modules: gene set management, information retrieval, organization/visualization and statistics. The gene set management module receives gene sets submitted by the users. Received gene sets can be saved, retrieved and deleted. Boolean operations are also provided by this module to generate the unions, intersections or differences between gene sets. The information retrieval module currently retrieves information for up to 20 attributes through our local database GeneKeyDB for the received gene sets. The organization/visualization module helps the users to explore efficiently the retrieved information in various biological contexts, using eight sub-modules: GO Tree, KEGG Table and Maps, BioCarta Table and Maps, Protein Domain Table, Tissue Expression Bar Chart, Chromosome Distribution Chart, PubMed Table and GRIF Table. Subsets of genes based on the organization can be generated and saved as new gene sets. The statistics module currently provides two statistical tests (the hypergeometric test and the Fisher's exact test) to identify interesting patterns in the gene sets.
Figure 1.

Schematic overview of WebGestalt. WebGestalt is composed of four main modules: gene set management, information retrieval, organization/visualization and statistics. The gene set management module uploads, saves, retrieves and deletes gene sets, as well as performs Boolean operations to generate the unions, intersections and differences between gene sets. The uploading tool accepts datasets defined by experiment data, GO categories or chromosome location ranges. WebGestalt is flexible in the input identifier (Entrez Gene ID, Swiss-Prot ID, Ensembl ID, Unigene ID, gene symbol and Affymetrix Probe Set ID). The saving tool saves sub-sets of genes generated by the organization/visualization module. The information retrieval module currently retrieves information for up to 20 attributes for all genes in a gene set, including nomenclatures, various gene identifiers, map and functional information. Retrieved information can be exported to Microsoft Excel files. The organization/visualization module organizes and visualizes a gene set in figures or tables using eight sub-modules: GO Tree, Tissue Expression Bar Chart, Chromosome Distribution Chart, KEGG Table and Maps, BioCarta Table and Maps, Protein Domain Table, PubMed Table and GRIF Table. The statistics module provides two statistical tests, the hypergeometric test and Fisher's exact test and suggests important biological areas in a gene set.
Gene set management module
The gene set management module accepts gene sets submitted by files, by GO categories or by chromosome location ranges. The input file should be a plain text file, including the appropriate IDs (required) and corresponding microarray ratios or other values (optional), separated by tabs in the format of one ID per row. Gene identifiers that can be recognized are Entrez Gene IDs, Swiss-Prot IDs, Ensembl IDs, Unigene IDs, gene symbols and Affymetrix probe set IDs. WebGestalt works currently with human and mouse. More organisms will be added in the future. A unique analysis name is given for each gene set by the user and can be used to retrieve or delete the gene set in the future. Sub-sets of genes can be generated from an existing gene set through the organization/visualization module and saved as new gene sets through the management module. The management module also performs Boolean operations to generate the union, intersection and difference between two existing gene sets. Recursively applying these Boolean operations makes it possible to combine information from more than two sets of genes. Orthologs can be retrieved for a gene set using the management module. The orthologs are defined by HomoloGene from NCBI (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=homologene). Inclusion of orthologous information could assist in comparative genomics studies.
Information retrieval module
The information retrieval module provides rapid access to the existing information for all genes in a gene set. The attributes that can be retrieved include nomenclature, identifiers to different databases, map and functional information. Table 1 lists all of the 20 attributes, their sources and associated websites. Retrieved information for all genes in a gene set can be downloaded as a tab-delimited file or opened directly in the web browser using Microsoft Excel.
Table 1.
Gene attributes that can be retrieved by WebGestalt
Ensembl ID, Ensembl gene stable ID; Cytogenetic, Cytogenetic map location; Physical, Physical map location; KEGG, KEGG pathway name; BioCarta, BioCarta pathway name.
Organization/visualization module
While the information retrieval module provides quick and easy information retrieval for large sets of genes and generates files that can be easily parsed and further utilized by other computational tools, it does not help biologists in exploring information associated with the gene sets. The organization/visualization module in WebGestalt is intended to assist biologists in exploring large gene sets by organizing and visualizing the genes in various biological contexts.
GO Tree. The GO Tree is based on our published tool GO Tree Machine, which was described in detail in (9). The GO Tree organizes a gene set based on the GO DAG (Directed Acyclic Graph), and has implemented several visualizations, including an expandable tree, a bar chart at selected annotation level and an enriched DAG. The expandable tree is suitable for exploring interactively the structure of GO. After exploring the expandable tree, the user may pick appropriate annotation levels to generate corresponding bar charts that are appropriate for publications and presentations. The enriched DAG is used for visualizing GO categories with enriched gene numbers as identified by the statistics module.
KEGG and BioCarta Tables and Maps. One of the most important tasks in the high-throughput experiments is to identify the pathways that are involved in the biological studies. Similarly to DAVID, MAPPFinder and GFINDer, WebGestalt can organize genes based on the KEGG biochemical pathways in a KEGG Table. The KEGG Table shows KEGG pathways associated with the gene set, the number of genes in each pathway and the Entrez Gene IDs for the genes. The KEGG table also provides P-values, indicating the significance of enrichment for each KEGG pathway. Each pathway name in the KEGG Table is hyperlinked to the KEGG Map, in which genes in the gene set are highlighted in red. WebGestalt can also organize genes based on another popular pathway database, BioCarta (http://www.biocarta.com), into a BioCarta Table. The BioCarta Table has the same structure as the KEGG Table. Each pathway name in the BioCarta Table is hyperlinked to the BioCarta Map.
Protein Domain Table. The Protein Domain Table organizes the genes based on the PFAM protein domains. The table shows the name of the PFAM domains associated with the gene set, the number of genes having each domain and the Entrez Gene IDs for the genes. The table also provides P-values, indicating the significance of enrichment for each domain. Each domain name is hyperlinked to the Conserved Domain Database of the NCBI, where the information of domain functions, structure and sequence is available. Each Entrez Gene ID is hyperlinked to the Conserved Domain Summary of the NCBI, where a graphical view of domains on the protein is available.
Tissue Expression Bar Chart. The Tissue Expression Bar Chart is designed to organize a gene set based on large-scale, publicly available gene expression data derived from a wild variety of tissue and organ types. The current version of WebGestalt uses the gene expression data from the CGAP-expressed sequence tag (EST) project (http://cgap.nci.nih.gov/Tissues) (18). It has been well accepted that the content of the EST pool for a given tissue type reflects the composition of original mRNA samples used for creation of the complementary DNA library (19). In the Tissue Expression Bar Chart, each tissue is represented by a bar. The height of the bars represents the number of genes that are in the active gene set, and also expressed in the tissue based on the CGAP data. For individual genes, WebGestalt evaluates the over/under-representation of the gene in individual tissue types using the statistics module.
Chromosome Distribution Chart. Chromosome distribution of the genes in a gene set is visualized using the Chromosome Distribution Chart. The chromosome location information comes from the UCSC genome annotation databases (ftp://hgdownload.cse.ucsc.edu/goldenPath/currentGenomes/). In this chart, each chromosome is represented by a vertical bar. Each gene is represented by a ‘red cross’ symbol and located on the chromosome based on its location. Clustered genes from a gene set can be easily visualized in the chart.
PubMed Table and GRIF Table. WebGestalt can organize genes according to their co-occurrence in publications, based on the gene-publication association information retrieved from in the LocusLink database. LocusLink provides two types of gene-publication indices. One is computed from the PubMed, the other is GRIF (1). WebGestalt organizes genes based on both indices and generates a PubMed Table or a GRIF Table. The PubMed Table shows PubMed IDs for the publications associated with the gene set, the number of genes in each publication and the Entrez Gene IDs for the genes. Each PubMed ID is hyperlinked to the corresponding PubMed record, where the abstract for the paper is available. The GRIF Table is similar to the PubMed Table, except for one additional column showing the GRIF comments.
Statistics module
While methods of gene organization provide an efficient way for biologists to explore large gene sets, these approaches, such as the GO Tree, frequently generate very complex results with hundreds of categories still requiring summarization. Statistical analysis is needed to guide biologists in finding the statistically significant categories that are associated with a gene set. In order to identify functional categories with significantly enriched gene numbers in a gene set we are interested in, we need to compare the gene set of interest to a reference gene set for the proportion of genes in the category. Suppose that we have n genes in the interesting gene set (A) and m genes in the reference gene set (B). Suppose further that there are k genes in A and j genes in B that are in a given category (C) (e.g. a GO category, a KEGG pathway, a BioCarta pathway etc.). Based on the reference gene set, the expected value of k would be ke = (n/m)* j. If k exceeds the above expected value, category C is said to be enriched, with a ratio of enrichment (r) given by r = k/ke. If B represents the population from which the genes in A are drawn, WebGestalt uses the hypergeometric test to evaluate the significance of enrichment for category C in gene set A,
If A and B are two independent gene sets, WebGestalt uses Fisher's exact test instead,
The users can select different significance levels for the statistical analysis. The users can also specify the minimum number of genes in a significant category. For example, categories with only one gene might be statistically enriched, but they might not be in the user's interest.
The hypergeometric test is also used for the evaluation of the over/under-representation of individual genes in a selected tissue type. Suppose that we have d EST sequences for a selected gene in all tissues and b EST sequences for all genes in all tissues. Suppose further that there are c EST sequences for the selected gene in a selected tissue and a EST sequences for all genes in the tissue. If c > (d/b)* a, we consider that the gene is over-represented in the tissue, and the P-value indicating the significance of over-representation is calculated by this formula:
If c < (d/b)*a, we consider that the gene is under-represented in the tissue, and the P-value indicating the significance of under-representation is calculated using this formula:
User interface
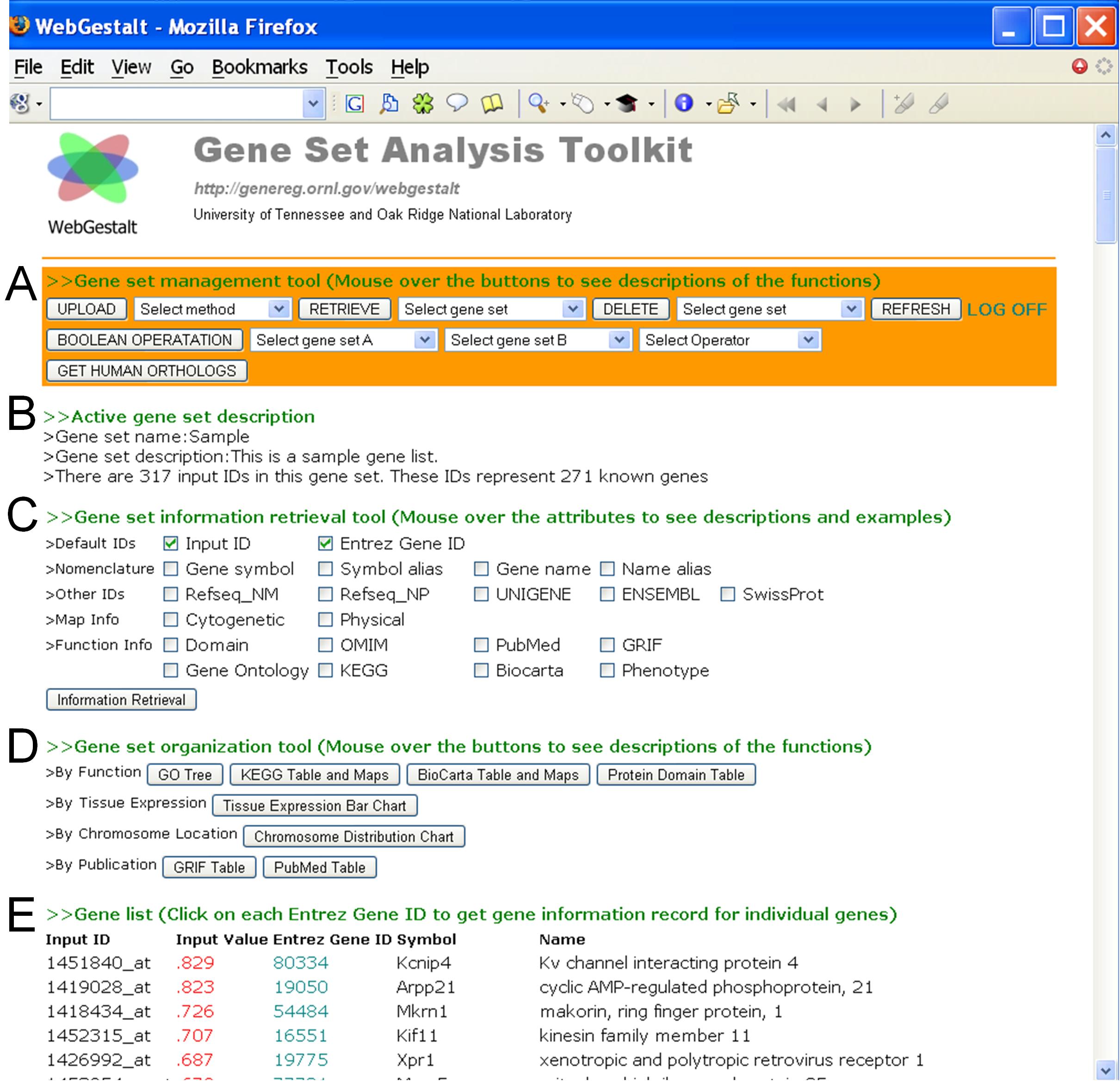
All of the above tools in WebGestalt can be accessed through a simple and intuitive user interface (Supplementary Figure S1). The interface can be divided into five areas. Area A provides gene set management tools for uploading, retrieving, deleting, performing Boolean operations and retrieving orthologs. Area B displays the name and description of the currently active gene set. Area C provides the gene set information retrieval tool, where the user can choose to retrieve information for up to 20 attributes. Area D provides the gene set organization and visualization tools that help users to explore large gene sets. Area E displays a table for the genes in the currently active gene set, including the ID used in the input file, the value provided in the input file, Entrez Gene ID, gene symbol and gene name. Each Entrez Gene ID is hyperlinked to a gene information record with detailed information retrieved from our local database GeneKeyDB. The values >0 are colored red, while those <0 are colored blue. Mouse-over descriptions are available for the buttons.
Implementation
WebGestalt is implemented in PHP. Gene set management, information retrieval and organization are mainly accomplished by querying the GeneKeyDB database. The expandable GO Tree is generated using the PHP Layers Menu System (http://phplayersmenu.sourceforge.net/). The bar chart for the GO organization, the tissue expression bar chart and the chromosome distribution chart are all generated by ChartDirector (http://www.advsofteng.com/index.html). The DAG for enriched GO categories is created using Graphviz (http://www.research.att.com/sw/tools/graphviz/). Genes on the KEGG map are highlighted using the KEGG Applications Programming Interface (API) (http://www.genome.ad.jp/kegg/soap/). WebGestalt is accessible through IE5.0 or higher, Netscape 7.0 or higher, Safari and Firefox from multiple platforms. WebGestalt can be accessed from the website http://genereg.ornl.gov/webgestalt/. A detailed manual can be downloaded from http://genereg.ornl.gov/webgestalt/WebGestalt_Manual.pdf.
RESULTS
In order to demonstrate the use of WebGestalt, we have generated 48 gene sets with genes over-represented in various human tissue types. These gene sets were generated based on the gene expression data from the publicly available human EST database (CGAP, http://cgap.nci.nih.gov/), the same data we used for creating the Tissue Expression Bar Chart. The tissue type is defined by CGAP. We did not separate different histological types. As described in the methods, we performed hypergeometric tests to identify tissue-enriched genes for each tissue type based on the EST representation profile. As we were doing multiple tests simultaneously, we considered a gene was over-represented in a select tissue if the P-value was <0.01 after the Bonferroni adjustment. To simplify, we will call these genes ‘tissue-enriched genes’. No tissue-enriched gene was found in adrenal medulla. It was probably due to the small number of available ESTs in this tissue type. An average of 190 tissue-enriched genes was identified for each of the other 48 tissue types, ranging from 6 in synovium to 817 in brain. All these 48 gene sets were uploaded to WebGestalt for exploration. Some sample results from the GO Tree analysis, KEGG pathway mapping and chromosome distribution analysis will be presented in this paper. Complete exploration of all the 48 gene sets using all available tools in WebGestalt is available for the public through this URL: http://genereg.ornl.gov/webgestalt/wg_enrich.php.
An example will be given for the GO Tree analysis using the set of 23 genes that are significantly over-represented in adrenal cortex. WebGestalt was able to found GO annotations for 21 out of the 23 genes. Nineteen GO categories were found to have enriched gene numbers using all genes in the human genome as a reference. Ten categories were under ‘biological process’, six were under ‘molecular function’ and three were under ‘cellular component’. Figure 2 is an enriched DAG for the 10 categories under ‘biological process’. An enriched DAG shows GO categories with enriched gene numbers (in red) and their non-enriched parents. Most of the enriched GO categories identified for this gene set were closely related to the function of adrenal cortex. The most significant category was ‘C21-steroid hormone biosyntheses’, which gives a P-value of 4.22 × 10−9. Similarly, GO Tree analysis was able to identify the most important functional areas for other tissue types. For example, the most significant category under ‘biological process’ for adipose is ‘lipid metabolism’ (P = 3.40 × 10−6), for cerebrum is ‘transmission of nerve impulse’ (P = 3.33 × 10−19), for ear is ‘perception of sound’ (P = 1.70 × 10−9), for heart is ‘muscle contraction’ (P = 2.00 × 10−22), for lymph node is ‘defense response’(P = 1.40 × 10−26), for retina is ‘sensory perception of light’ (P = 3.57 × 10−44) and for testis is ‘sexual reproduction’ (P = 1.35 × 10−21).
Figure 2.

Enriched DAG under ‘biological process’ for a set of 23 genes that are significantly over-represented in adrenal cortex, using all genes in the human genome as a reference. The enriched GO categories are brought together and visualized as a DAG. Categories in red are enriched ones while those in black are non-enriched parents. Listed in the boxes are the name of the GO category, the number of genes in the category and the P-value indicating the significance of enrichment.
For the same gene set, the KEGG Table in WebGestalt reveals 18 KEGG pathways that involve genes over-represented in adrenal cortex. The ‘C21-Steroid Hormone Metabolism’ pathway was found to have enriched gene numbers (P = 3.09 × 10−8) using all genes in the human genome as a reference. This result is consistent with the GO Tree analysis. Three genes, CYP17A1, CYP21A2 and HSD3B2, were mapped to and highlighted on the ‘C21-Steroid Hormone Metabolism’ pathway using the KEGG Map in WebGestalt.
The Chromosome Distribution Chart was used to show the distribution of the set of 208 pancreas genes on the human chromosome. Several clusters of pancreas-enriched genes can be seen from the chart. For example, 18 out of the 208 genes are found to be located within 2.54M on chromosome 19, which is a >20 times of enrichment comparing with the background distribution (75 out of the 17 435 physically located genes were found in this region). Clustering of tissue-enriched genes on the chromosome was also found in other tissue types. Although the Chromosome Distribution Chart may help us to identify these important patterns, statistical analysis is needed to evaluate the significance. We are working on the statistically evaluation of local gene enrichment for a given gene set.
DISCUSSION
WebGestalt is designed for genomic, gene expression, proteomic and large-scale genetic studies from which high-throughput datasets are generated. Complementing and extending the functionality of similar data mining tools, WebGestalt provides a unique online resource for the management, information retrieval, organization, visualization and statistical analysis of sets of genes. The major advantages of WebGestalt compared with similar existing tools include: (i) the ability to retrieve more information for all genes in a gene set; (ii) more ways to organize a gene set; (iii) appropriate visualization for each organization; (iv) assistance in choosing appropriate statistical tests; (v) a simple and intuitive user interface; and (vi) Boolean operations on selected gene sets.
Functional features, such as GO (6–13), KEGG pathway (11–13) and PFAM domains (12,13), have been used to organize and help exploring gene sets. WebGestalt has added several new features for gene set organization, including tissue expression pattern, chromosome location and co-occurrence in publications. Their potential uses will be discussed below.
The Tissue Expression Bar Chart is especially useful in candidate gene identification for genetic experiments. For example, the critical interval identified from the QTL (Quantitative Trait Loci) analysis will be between 0.5 and 10 cM, with the number of genes anywhere between 5 and 300 (20). It has been shown that it is possible to identify plausible candidate genes for human multiple congenital anomaly syndromes by systematically using data on murine gene expression patterns (21). The tissue expression pattern of the genes in an interval can be easily analyzed and visualized using the Tissue Expression Bar Chart in WebGestalt. The sub-set of genes expressed in certain tissue types can be saved as new gene sets and analyzed by other modules in WebGestalt, such as the GO Tree to further prioritize the genes for mutation analyses. The current Tissue Expression Bar Chart is based on the gene expression data from the CGAP EST project (18). Microarray data on the tissue-specific pattern of mRNA expression are recently available for a panel of 79 human and 61 mouse tissues (22). Massively Parallel Signature Sequencing data on different mouse tissue types are also available from the Mouse Transcriptome Project (http://www.ncbi.nlm.nih.gov/genome/guide/mouse/MouseTranscriptome.html). We are considering adding these and other large datasets to WebGestalt.
The Chromosome Distribution Chart can help to identify clustered genes from a gene set. Tight clustering of co-expressed genes on the chromosomes is common in prokaryotes (23). In eukaryotes, it is typically assumed that genes are randomly distributed. Nonetheless, recent studies in yeast (24), worm (25), fly (19,26), mouse (27) and human (28,29) suggest that gene location might not be random. For example, among the 1661 testes-specific genes identified in Drosophila, one-third are clustered on chromosomes (19). Testis-specific clustering of genes on chromosomes has also been found in mouse (27). Although tissue-specific clustering of genes on chromosomes has not been found in human, Lercher et al. (29) have shown that housekeeping genes are strongly clustered in human. Since the Chromosome Distribution Chart organizes genes in a gene set based on their chromosome location, clustered genes can easily be visualized. Statistical methods are being developed and will be added in the statistics module for the identification of local gene enrichment on the chromosome.
Bioinformatics tools based on literature profiling have been developed by a few groups to assist biologists in the interpretation of sets of interesting genes (30–32). Jenssen et al. (30) have constructed a gene network from the co-occurrence of gene symbols or short gene names in the title or the abstract of a common article record. They also demonstrated that literature co-occurrence associated biologically related genes, which suggests the value of organizing genes based on the co-occurrence in publications. In WebGestalt, instead of constructing a gene-publication index de novo, we used the indices available from the LocusLink database and organized the genes in a gene set using the PubMed Table and the GRIF Table. The PubMed table provides better coverage but with less specificity, while the GRIF table provides less coverage but better functional specificity.
Another feature of WebGestalt is the Boolean operations on existing gene sets. It will help to answer simple questions such as: ‘show me all genes identified through experiment A or experiment B’ (union), ‘show me the genes that are consistently up-regulated in both of two microarray experiments’ (intersection) or ‘show me the genes that are expressed in brain but not skin’ (difference). Recursively applying the Boolean operation makes it possible to combine information from any number of gene sets. Putting the organization module and the management module together, WebGestalt is able to answer complex questions such as ‘give me all genes in my gene set that are expressed in the brain or cerebellum, located on chromosome 5 and involved in signal transduction’.
WebGestalt incorporates information from different public resources, provides tools for the management, information retrieval, organization, visualization and statistical analysis of gene sets. The simple and intuitive, web-based interface provides experimental biologists easy access to the tool kit. Moreover, the modules in WebGestalt can be easily used by third-party applications. For example, WebGestalt has been implemented in WebQTL (http://www.webqtl.org), which is a unique service that allows biologists to rapidly identify and map genes and QTL (33). The WebGestalt modules are used to analyze sets of genes that are highly correlated with various phenotypes in WebQTL. We are working on an API to allow easy access of the WebGestalt modules from any third-party applications.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Supplementary Material
Acknowledgments
The authors thank Suzanne Baktash and Oakley Crawford for technical help in preparing the manuscript. This work was supported by the INIA project (NIH/NIAAA, U01-AA013532), the BISTI project (NIH/NIDA, P01-DA015027) and the ORNL LDRD project (DOE, AC05-00OR22725). Funding to pay the Open Access publication charges for this article was provided by the INIA project (NIH/NIAAA, U01-AA013532).
Conflict of interest statement. None declared.
REFERENCES
- 1.Pruitt K.D., Maglott D.R. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 2001;29:137–140. doi: 10.1093/nar/29.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsai J., Sultana R., Lee Y., Pertea G., Karamycheva S., Antonescu V., Cho J., Parvizi B., Cheung F., Quackenbush J. RESOURCERER: a database for annotating and linking microarray resources within and across species. Genome Biol. 2001;2:1–4. doi: 10.1186/gb-2001-2-11-software0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kasprzyk A., Keefe D., Smedley D., London D., Spooner W., Melsopp C., Hammond M., Rocca-Serra P., Cox T., Birney E. EnsMart: a generic system for fast and flexible access to biological data. Genome Res. 2004;14:160–169. doi: 10.1101/gr.1645104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diehn M., Sherlock G., Binkley G., Jin H., Matese J.C., Hernandez-Boussard T., Rees C.A., Cherry J.M., Botstein D., Brown P.O., et al. SOURCE: a unified genomic resource of functional annotations, ontologies, and gene expression data. Nucleic Acids Res. 2003;31:219–223. doi: 10.1093/nar/gkg014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology consortium. Nature Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herrero J., Al-Shahrour F., Diaz-Uriarte R., Mateos A., Vaquerizas J.M., Santoyo J., Dopazo J. GEPAS: a web-based resource for microarray gene expression data analysis. Nucleic Acids Res. 2003;31:3461–3467. doi: 10.1093/nar/gkg591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Draghici S., Khatri P., Martins R.P., Ostermeier G.C., Krawetz S.A. Global functional profiling of gene expression. Genomics. 2003;81:98–104. doi: 10.1016/s0888-7543(02)00021-6. [DOI] [PubMed] [Google Scholar]
- 8.Zeeberg B.R., Feng W., Wang G., Wang M.D., Fojo A.T., Sunshine M., Narasimhan S., Kane D.W., Reinhold W.C., Lababidi S., et al. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang B., Schmoyer D., Kirov S., Snoddy J. GOTree Machine (GOTM): a web-based platform for interpreting sets of interesting genes using Gene Ontology hierarchies. BMC Bioinformatics. 2004;5:16. doi: 10.1186/1471-2105-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong S., Li C., Wong W.H. ChipInfo: software for extracting gene annotation and gene ontology information for microarray analysis. Nucleic Acids Res. 2003;31:3483–3486. doi: 10.1093/nar/gkg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doniger S.W., Salomonis N., Dahlquist K.D., Vranizan K., Lawlor S.C., Conklin B.R. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2003;4:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dennis G., Jr, Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated discovery. Genome Biol. 2003;4:R60. [PubMed] [Google Scholar]
- 13.Masseroli M., Martucci D., Pinciroli F. GFINDer: Genome Function INtegrated Discoverer through dynamic annotation, statistical analysis, and mining. Nucleic Acids Res. 2004;32:W293–W300. doi: 10.1093/nar/gkh432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanehisa M., Goto S., Kawashima S., Okuno Y., Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hand D., Mannila H., Smyth P. Principles of Data Mining. Cambridge, MA: The MIT Press; 2001. [Google Scholar]
- 16.Tao Y., Liu Y., Friedman C., Lussier Y.A. Information visualization techniques in bioinformatics during the postgenomic era. Biosilico. 2004;2:237–245. doi: 10.1016/S1741-8364(04)02423-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirov S.A., Peng X., Baker E., Schmoyer D., Zhang B., Snoddy J. GeneKeyDB: a lightweight, gene-centric, relational database to support data mining environments. BMC Bioinformatics. 2005;6:72. doi: 10.1186/1471-2105-6-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strausberg R.L. The Cancer Genome Anatomy Project: new resources for reading the molecular signatures of cancer. J. Pathol. 2001;195:31–40. doi: 10.1002/1096-9896(200109)195:1<31::AID-PATH920>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 19.Boutanaev A.M., Kalmykova A.I., Shevelyov Y.Y., Nurminsky D.I. Large clusters of co-expressed genes in the Drosophila genome. Nature. 2002;420:666–669. doi: 10.1038/nature01216. [DOI] [PubMed] [Google Scholar]
- 20.van Driel M.A., Cuelenaere K., Kemmeren P.P., Leunissen J.A., Brunner H.G. A new web-based data mining tool for the identification of candidate genes for human genetic disorders. Eur. J. Hum. Genet. 2003;11:57–63. doi: 10.1038/sj.ejhg.5200918. [DOI] [PubMed] [Google Scholar]
- 21.van Steensel M.A., Celli J., van Bokhoven J.H., Brunner H.G. Probing the gene expression database for candidate genes. Eur. J. Hum. Genet. 1999;7:910–919. doi: 10.1038/sj.ejhg.5200405. [DOI] [PubMed] [Google Scholar]
- 22.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dandekar T., Snel B., Huynen M., Bork P. Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem. Sci. 1998;23:324–328. doi: 10.1016/s0968-0004(98)01274-2. [DOI] [PubMed] [Google Scholar]
- 24.Cohen B.A., Mitra R.D., Hughes J.D., Church G.M. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nature Genet. 2000;26:183–186. doi: 10.1038/79896. [DOI] [PubMed] [Google Scholar]
- 25.Roy P.J., Stuart J.M., Lund J., Kim S.K. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature. 2002;418:975–979. doi: 10.1038/nature01012. [DOI] [PubMed] [Google Scholar]
- 26.Spellman P.T., Rubin G.M. Evidence for large domains of similarly expressed genes in the Drosophila genome. J. Biol. 2002;1:5. doi: 10.1186/1475-4924-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q., Lee B.T., Zhang L. Genome-scale analysis of positional clustering of mouse testis-specific genes. BMC Genomics. 2005;6:7. doi: 10.1186/1471-2164-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caron H., van Schaik B., van der Mee M., Baas F., Riggins G., van Sluis P., Hermus M.C., van Asperen R., Boon K., Voute P.A., et al. The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science. 2001;291:1289–1292. doi: 10.1126/science.1056794. [DOI] [PubMed] [Google Scholar]
- 29.Lercher M.J., Urrutia A.O., Hurst L.D. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nature Genet. 2002;31:180–183. doi: 10.1038/ng887. [DOI] [PubMed] [Google Scholar]
- 30.Jenssen T.K., Laegreid A., Komorowski J., Hovig E. A literature network of human genes for high-throughput analysis of gene expression. Nature Genet. 2001;28:21–28. doi: 10.1038/ng0501-21. [DOI] [PubMed] [Google Scholar]
- 31.Masys D.R., Welsh J.B., Lynn Fink J., Gribskov M., Klacansky I., Corbeil J. Use of keyword hierarchies to interpret gene expression patterns. Bioinformatics. 2001;17:319–326. doi: 10.1093/bioinformatics/17.4.319. [DOI] [PubMed] [Google Scholar]
- 32.Chaussabel D., Sher A. Mining microarray expression data by literature profiling. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-10-research0055. RESEARCH0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chesler E.J., Lu L., Wang J., Williams R.W., Manly K.F. WebQTL: rapid exploratory analysis of gene expression and genetic networks for brain and behavior. Nature Neurosci. 2004;7:485–486. doi: 10.1038/nn0504-485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}