

ABSTRACT
Jumping translocations (JT) are rare cytogenetic abnormalities associated with progression in myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Typically, a tri–tetra‐somic 1q chromosome is translocated to two or more recipient chromosomes. In multiple myeloma JT were shown to originate after DNA demethylation and decondensation. Using epigenomics, we investigated sequential samples in an SRSF2‐mutated MDS and AML cohort with normal karyotype at diagnosis and 1qJT at disease evolution after 5′‐azacytidine (AZA). 1qJT breakpoints fell within repetitive DNA at both 1q12 and the translocation partners, namely acrocentrics n. 14, 15, 21, and 22, chromosome 16, and chromosome Y. The global methylome at diagnosis showed hypermethylation at 61% of the differentially methylated regions (DMRs), followed by hypomethylation at 80% of DMRs under AZA, mostly affecting pathways related to immune system, chromatin organization, chromosome condensation, telomere maintenance, rRNA, and DNA repair. At disease evolution, a shift toward hypermethylation, intronic enhancers enrichment and epigenetic involvement of the PI3K/AKT and MAPK signaling emerged. In particular, AKT1 phosphorylation behaved as a hallmark of the progression. Overall, we provided new insights on the characterization of 1qJT in SRSF2‐mutated myeloid neoplasms and first showed that epigenetics is a powerful tool to investigate the molecular landscape of repetitive DNA rearrangements.
Keywords: 5‐azacytidine, DNA methylation, jumping 1q, MDS/AML
1. Introduction
Heterochromatin is composed of distinct families of repetitive elements, well characterized in terms of sequence content and epigenetic marks [1]. Historically, it was defined as functionally useless “junk” DNA, whereas it is now clear that constitutive heterochromatin has an important role in maintaining genome stability and that it acts on neighboring genes by a cis/trans effect [2]. In contrast to deep knowledge on the molecular counterparts of chromosomal changes involving gene sequences, information on the molecular consequences of chromosome recombinations at repetitive heterochromatic sequences are still scarce.
Jumping translocations (JT) are cytogenetic abnormalities in which a donor chromosome, most frequently a trisomic or tetrasomic long (q) arm of chromosome n. 1, recombines with two or more recipient chromosomes [3, 4]. In aggressive multiple myeloma (MM), 1q12 JT have been related to DNA decondensation and hypomethylation [5, 6]. Evidence for a role of hypomethylation in the origin of 1qJT has been also provided by the rare autosomal recessive ICF (immunodeficiency, centromeric instability, and facial anomalies, OMIM#242860, OMIM#614069, OMIM#616910, and OMIM#616911) syndrome, in which hypomethylation at 1q12 underlies recombinations between pericentromeric regions, frequently involving chromosomes n. 1, 9, and 16 [7, 8, 9, 10].
In myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML), 1qJT have been associated with poor clinical outcome [11]. However, in‐depth molecular studies are still limited.
In this study, we used an integrated approach of fluorescence in situ hybridization (FISH), targeted resequencing, and global DNA methylation to investigate 1q12 JTs in AML.
2. Materials and Methods
2.1. Samples
Samples were retrospectively collected from the Laboratory of Cytogenetics and Molecular Genetics at the Hematology Department of the University of Perugia, Italy. The study was conducted according to the Helsinki Declaration and approved by the Institutional Bioethics Committee (Prot.1.X.2011). We collected nine longitudinal bone marrow (BM) samples from three MDS and AML cases, each at three timepoints. Hematological and cytogenetic features of all patients are listed in Table 1. As controls, we included three sex/age‐matched BM from cases with non‐neoplastic cytopenias and normal BM morphology and cytogenetics (Supporting Information: Methods).
TABLE 1.
Hematological and cytogenetic features of all the samples included in the study.
| Sample | S/A | Type | Karyotype | I‐FISH | SNPa | Mutation(s) | |
|---|---|---|---|---|---|---|---|
|
Case 1 (UPN660) |
Diagnosis (T1) | M/61 | MDS‐IB1 | 46,XY[20] | 0.7% | Normal |
SRSF2 (40.6%) IDH2 (41.6%) |
| Remission, C (T2) | After 5′‐azacytidine treatment | 46,XY[20] | 20% |
Gain: 1q21.1q44(145644984_249224684) Loss: 15q21.3(56899523_57874494) Loss: 16q11.2q24.3(46463673_90155062) |
SRSF2 (40.6%) IDH2 (36.9%) RUNX1 (19.7%) |
||
| Progression (T3) | AML |
46,XY,+1,+1,der(1;14)(q10;q10), der(1;15)(q10;q10)[7]/ 46,XY,+1,der(1;16)(q10;p10)[3]/ 46,XY[11] |
40.5% |
Gain: 1q21.1q44(145495568_249224684) Loss: 15q21.3(56899523_57874019) Loss: 16q11.2q24.3(46503193_90155062) |
SRSF2 (38.9%) IDH2 (39.1%) RUNX1 (12.8%) |
||
|
Case 2 (UPN661) |
Diagnosis (T1) | M/60 | AML | 46,XY[20] | 1.2% | LOH: 14q23.1q32.33(58237922_107285437) |
SRSF2 (32.6%) IDH2 (31%) ASXL1 (33.7%) |
| Remission, C (T2) |
After daunorubicin + cytarabine 5′‐azacytidine Venetoclax treatment |
46,XY[20] | 0% | Not available |
SRSF2 (26%) IDH2 (25.1%) ASXL1 (29.4%) KRAS (22.1%) |
||
| Progression (T3) | AML |
46,XY,+1,der(1;16)(q10;p10)[11]/ 46,X,der(Y)t(Y;1)(q12;q10)[5]/ 46,XY[4] |
50% |
Gain: 1q21.1q44(145624380_249224684) Loss: 16q11.2q24.3(46716436_90155062) LOH: 14q23.1q32.33(58237922_107285437) |
SRSF2 (35.4%) IDH2 (39.1%) ASXL1 (36.4%) KRAS (31.6%) RUNX1 (14.1%) |
||
|
Case 3 (UPN662) |
Diagnosis (T1) | F/60 | MDS‐IB1 | 46,XX[20] | 1.7% | Normal | SRSF2 (36.7%) |
| Remission, P (T2) | After 5′‐azacytidine treatment | 46,XX[20] | 2% | Not available | SRSF2 (29.8%) | ||
| Progression (T3) | AML |
46,XX,+1,der(1;21)(q10;q10)[9]/ 46,XX,+1,der(1;22)(q10;q10)[2]/ 46,XX,del(7)(q22q34)[7]/ 46,XX[9] |
16% |
Gain: 1q21.1q44(143932350_249224684) Loss: 6q12q27(65892047_170919482) Loss: 7q31.1q36.3(109368903_155826004) |
SRSF2 (29.9%) KRAS (8.8%) |
Note: For SNPa, numbers within brackets indicate start and end coordinates for the identified events (refers to GRCh37). For mutations, the variant allele frequency (VAF) is indicated between brackets. In bold are reported the derivative chromosomes resulting from the 1qJT and the common SRSF2 mutation found since diagnosis.
Abbreviations: AML, acute myeloid leukemia; F, female; IB1, MDS with increased blasts; I‐FISH, interphase nuclei fluorescent in situ hybridization using pUC1.77 probe for 1q12 heterochromatin: cut‐off values upper limits was setting at 2%–3% for both trisomy and tetrasomy by using 10 peripheral blood from healthy donors; LOH, loss of heterozygosity; M, male; P/C, partial or complete; S/A, sex/age; SNPa, single nucleotide polymorphism array.
2.2. Conventional and Molecular Cytogenetics
G‐banded karyotypes were described according to the ISCN [12]. FISH was performed following standard procedures (Table 2 and Supporting Information: Methods). Single‐nucleotide polymorphism array (SNPa) was performed using a CytoScan HD Affymetrix platform following manufacturer's instructions (Thermo Fisher Scientific, Whaltam, Massachusetts, USA) (Supporting Information: Methods).
TABLE 2.
Table showing the probes used in FISH analysis for the breakpoint characterization.






Note: In green are listed the centromeric probes; in red, the specific ones for satellite‐II and ‐III sequences; in black, the probes that identified NORs; in blue the probes for euchromatic regions.
Abbreviations: Chr, chromosome; NORs, nucleolar organizer regions.
2.3. Targeted Next Generation Sequencing
Genomic DNAs were longitudinally analyzed using the Myeloid panel by SOPHiA GENETICS (SOPHiA Genetics, Saint‐Sulpice, Switzerland) and the Custom Hereditary Hematological Disorders gene panel (CHHD_A_v2) for germline variants, following manufacturer's instructions (Table S1 and Supporting Information: Methods). Pooled libraries were sequenced on the Miseq platform (Illumina, San Diego, California, USA) and analyzed with SOPHiA DDM version 5 using the hg19 reference genome (SOPHiA Genetics) (Supporting Information: Methods). The identified variants from targeted next generation sequencing (t‐NGS) were confirmed by Sanger sequencing (3500 Genetic Analyzer, Applied Biosystems, Waltham, Massachusetts, USA), using specific primer pairs (Table S2).
2.4. Multiplex Enhanced Reduced Representation Bisulfite Sequencing
Libraries were prepared as previously described [18, 19] and sequenced on Illumina HiSeq 2500 using the manufacturer's recommended protocol for 50 bp stranded single‐end read runs (Illumina). Reads were aligned against the bisulfite‐converted hg19 genome using bismark with bowtie2 [20]. Downstream analysis was performed using MethylKit [21] and MethylSig [22] R packages [23] (Supporting Information: Methods). Differentially methylated regions (DMRs, FDR ≤ 0.1 and cut‐off of 25% methylation difference) were annotated to the RefSeq genes (NCBI) using ChIP‐Enrich annotation (locus.def = “nearest_tss”) [24]. For enhancers annotation we used histone profiles dataset from hematopoietic stem cells, defining active enhancers as non‐promoter regions marked by both H3K4me1 > H3K4me3 and the presence of active chromatin histone mark H3K27ac [25]. Annotation to repetitive sequences was performed using UCSC hg19 track of RepeatMasker Repbase Library [26]. Functional pathway analysis was performed on DMRs annotated genes using ChIP‐Enrich and EnrichR [24, 27].
2.5. Western Blot and Quantitative Real‐Time PCR
The phospho‐AKT(pAKT1)/AKT1 ratio was assessed with a rabbit monoclonal Ab recognizing the phosphorylation at serine residue 473 (clone D9E, Cell Signaling, Danvers, Massachusetts, USA) and at threonine residue 308 (Clone 244F9, Cell Signaling), followed by the detection of total AKT1 (Cell Signaling). The pPDK1/PDK1 ratio was measured by immunoblot with a rabbit monoclonal Ab recognizing the phosphorylation of PDK1 at serine 241 (Cell signaling), followed by the assessment of total PDK1 (Cell signaling). Specific Abs recognizing PTEN (Clone 138G6, Cell signaling) and SHIP (Clone P1C1, Santa Cruz Biotechnology, Dallas, Texas, USA) were also used with mouse monoclonal Ab against human β‐actin (SigmaAldrich, St. Louis, Missouri, USA), as normalizer (Supporting Information: Methods).
RNA was retrotranscribed using Superscript IV with esa‐random primers (Invitrogen, Waltham, Massachusetts, USA). Real‐time reactions were performed in triplicate (Light Cycler 480 Roche, Basilea, Swizerland); fluorescence data were analyzed with the software version 1.5 and second derivative maximum method; gene expression was expressed as Crossing point (Cp) values, and statistical significance was tested by Mann–Whitney test (*p < 0.05) on GraphPad prism (Supporting Information: Methods).
3. Results
3.1. Samples
BM samples from three AML cases were analyzed at three disease timepoints: timepoint 1 (T1) corresponding to first diagnosis of MDS or AML; timepoint 2 (T2) corresponding to partial or complete remission after 5′‐azacytidine treatment (AZA); timepoint 3 (T3) corresponding to AML as disease progression or relapse (Table 1).
The first case was a 61‐year‐old male with diagnosis of MDS with increased blasts 1 (MDS‐IB1) and normal karyotype (T1). Remission was achieved by six AZA cycles (T2), continued till 18 cycles, when the disease evolved to AML (T3) and 1q JT involving n. 14, 15, and 16 as recipient chromosomes appeared at karyotype (Table 1).
The second case was a 60‐year‐old man first diagnosed as AML (T1) and treated by the daunorubicin/cytarabine scheme. At first relapse, AZA and venetoclax were administered. Hematological remission was obtained after 6 cycles (T2), and treatment was continued up to 16 cycles of AZA and 15 of venetoclax. At that time, T3, a second relapse occurred with abnormal karyotype showing 1q translocations to chromosomes 16 and Y (Table 1).
The third case was a 60‐years‐old female with MDS‐IB1 and normal karyotype (T1). Eight AZA cycles induced partial remission (T2), which was maintained up to 9 cycles, when BM evaluation revealed progression (T3) to a cytogenetically abnormal AML with one clone with 1qJT involving chromosome 21 as recipient, a second clone with 1qJT involving chromosome 22, and an additional clone with interstitial 7q deletion (Table 1).
3.2. Breakpoints Encompassed Repetitive Sequences at Both Chromosome 1 and All the Partners of Jumping Translocations
In case 1 at T3, double color FISH with the 1q pericentromeric probe pUC1.77 and the α‐satellite probes D14Z1, D15Z3, and D16Z2, generated fusion signals between the pericentromeric regions of chromosomes 1q and 14, 15, and 16 (Figure 1a–c). FISH with oligonucleotide S.2 revealed the presence of satellite‐II (sat‐II) from chromosome 1 in the derivatives, excluding the involvement of centromeric α‐satellite sequences from chromosome 1 (Figure 1a–c). Juxtapositions between NORs from both n. 14 and 15 and sat‐II from chromosome 1 were confirmed by fusion signals obtained with specific probes (Figure 1c). Additionally, in this case, there was a third clone with 1q moving to chromosome 16, in which the pHuR195 (D16Z3) probe identified the breakpoints within sat‐II. Identical results were seen in the der(1;16) also found in case 2 (Figure 1d). Moreover, in the last case double color FISH, using the pericentromeric probe for chromosome 1 sat‐II and the DYZ1 probe for the sat‐III at Yq12, generated a fusion signal, identifying sat‐III as the region of recombination on chromosome Y (Figure 1e). Finally, in case 3, NORs were involved at acrocentrics n. 21 and n. 22, as shown by double color FISH using the 1q pericentromeric pUC1.77 probe and the NOR‐specific probe RP5‐1174A5 (Figure 1f). Additionally, the small signal, clearly weaker than that corresponding to the normal homolog, generated by the specific RP5‐1174A5 NORs‐probe on der(21), was consistent with breakpoints falling within NOR regions (Figures 1f and S1).
FIGURE 1.
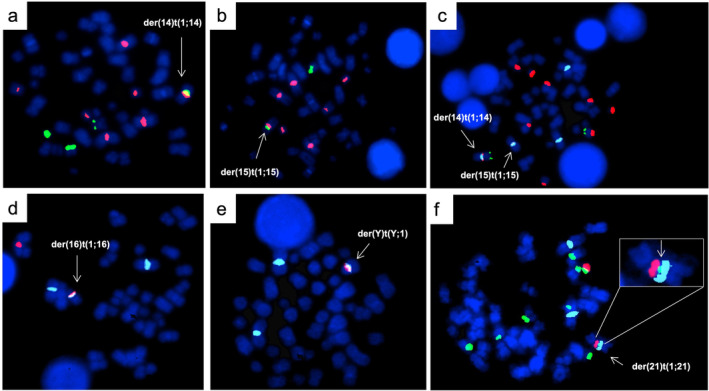
FISH characterization of the three patients. Case 1 (a–c). Hybridization of the Sat.2 oligo probe for 1q12 sat‐II region (orange) and D14Z1 centromeric probe for chromosome 14 (green) (panel a); and D15Z3 centromeric probe for chromosome 15 (green) (panel b); tricolor FISH showing der(14) and der(15) in the same metaphase with pUC1.77 for 1q12 heterochromatin (aqua), RP5‐1174A5 probe for NORs (orange) in acrocentrics and RP11‐431B1 probe for 14q32 (green), used as a reference for chromosome 14 (panel c). Case 2 (d and e). Hybridization of Sat.2 oligo probe for 1q12 sat‐II region (aqua) and D16Z3 probe for pericentromeric heterochromatin of chromosome 16 (orange) (panel d); and DYZ1 probe for sat‐III of chromosome Y (orange) (panel e). Case 3 (f). Hybridization of pUC1.77 for 1q12 heterochromatin (aqua), RP5‐1174A5 probe for NORs (green) in acrocentrics and whole chromosome paint probe for chromosome 21 (orange). On the top right the insert showing a reduction of the signal for RP5‐1174A5 probe for NORs (green) in the der(21).
SNPa analysis was available at all three timepoints in case 1. Interestingly, in this case SNPa identified both the 1q gain and 16q losses from T2, when the karyotype was still normal, suggesting that the affected cells were present, although non‐proliferating (Table 1). These results were further confirmed by interphase FISH with the pUC1.77 probe, which showed an abnormal tri/tetra‐somic clone already present in 20% of nuclei from T2 (Table 1). In addition, in the same case SNPa identified an acquired loss of 974.5 Kb at 15q encompassing ZNF280D, LOC145783, TCF12, LINC00926, LINC01413, and CGNL1 genes. In case 2, SNPa confirmed the copy number variations due to the t(1;16) JT at T3. In addition, SNPa detected a loss of heterozygosity of 49.04 Kb at 14q23.1q32.33 (Table 1). Finally, in case 3, SNPa at T3 showed the 1q trisomy and the del(7q) seen at karyotypic level and identified a cryptic del(6q) (Table 1). FISH analysis showed the JT, the del(7q), and the del(6q) marking three independent clones (data not shown).
Collectively, these results showed that the centromeres of all 1qJT belonged to the recipient chromosomes. Moreover, the sat‐II region of pericentromeric 1q heterochromatin was involved in all cases and alternatively recombined with NORs at acrocentrics, and with sat‐II and III at chromosome 16 and Y, respectively.
3.3. The P95_R102del Somatic SRSF2 Variant Was the Common Driver Mutation
The same SRSF2 deletion (p.P95_R102del) was identified by t‐NGS in all cases since diagnosis (Tables 1 and S3). Additional mutations, since T1, were the same p.(R140Q) IDH2 variant in case 1 and 2, and ASXL1 (p.R1068*) only in case 2. Moreover, variants at RUNX1 and/or KRAS characterized disease progression (Tables 1 and S3).
These results identified a common SRSF2 deletion (p.P95_R102del) in our 1qJT cohort from diagnosis and revealed that in all cases additional somatic variants contributed to the stepwise progression.
3.4. AZA‐Induced Global Hypomethylation That Reverted to Hypermethylation at Disease Progression
Unsupervised principal component analysis from multiplex enhanced reduced representation bisulfite sequencing (mERRBS) separated the leukemic samples from the normal controls (Figures 2a and 3). At diagnosis (T1) we found an hypermethylated signature, involving 3481 out of the 5753 DMRs (61%), significantly enriched at exons (binomial test, p value < 2.2−16) and intergenic regions (binomial test, p value < 2.2−16) (Figure 2b,c and Table S4). At T2, as an expected AZA‐effect, we detected a prevalent hypomethylated pattern affecting 29283/36403 (80%) DMRs, with enrichment at introns (binomial test, p value < 2.2−16) and intergenic regions (binomial test, p value < 2.2−16) (Figure 2b,c and Table S5). Of note, the T1 hypermethylated intergenic regions were only partially (384/1117, 34%) demethylated by AZA (Figure 2c and Tables S4 and S5). At T3 we found the highest number of DMRs, with a prevalent hypermethylation (56%; 14457/25823) and with significant enrichment at exons (binomial test, p value < 2.2−16) (Figure 2b,c and Table S6). Notably, hypermethylated enhancers were enriched (binomial test, p value = 1.061e−13) only at T3, suggesting their specific role during disease progression (Figure 2c). Enhancers were preferentially mapped at introns (202/326, 61%) and at distal intergenic regions (90/326, 20%), and annotated to 135 neighboring genes (Table S6). Since hypermethylated enhancers have been reported as a typical finding in IDH‐mutated AML [28, 29] and our cohort included two IDH‐mutated cases, we compared our epigenetic results with those published by Figueroa et al. [29] and found 45.7% of overlap. Additionally, methylation changes at enhancers significantly affected binding sites (FDR < 0.05) for 23 transcription factors (TFs), mostly belonging to helix‐turn‐helix (HTH) and zing finger (ZF) families (Figure S2).
FIGURE 2.
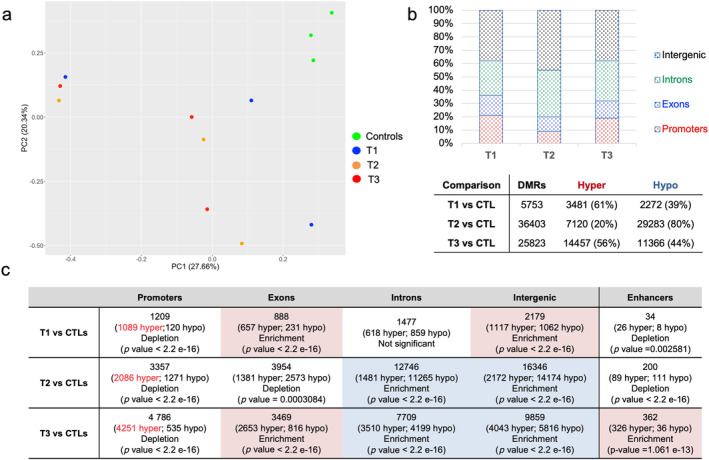
Summary of DMRs annotations. (a) Unsupervised principal component analysis (PCA) from mERRBS data separating leukemic samples from controls. (b) Upper, histograms showing DMRs distribution at each timepoint when compared to controls (FDR < 0.1; differential methylation > 25). Bottom, table reporting the total number DMRs with hyper‐ and hypo‐methylated DMRs at each timepoint. (c) Genomic localization of the identified DMRs. Statistical results for Binomial enrichment/depletion test were conduct on R, using 95% confidence interval and percentage of background annotation for each category (promoters, exons, introns, intergenic, and active enhancer) as true probability of success. As background, we used total identified DMRs without filters.
FIGURE 3.
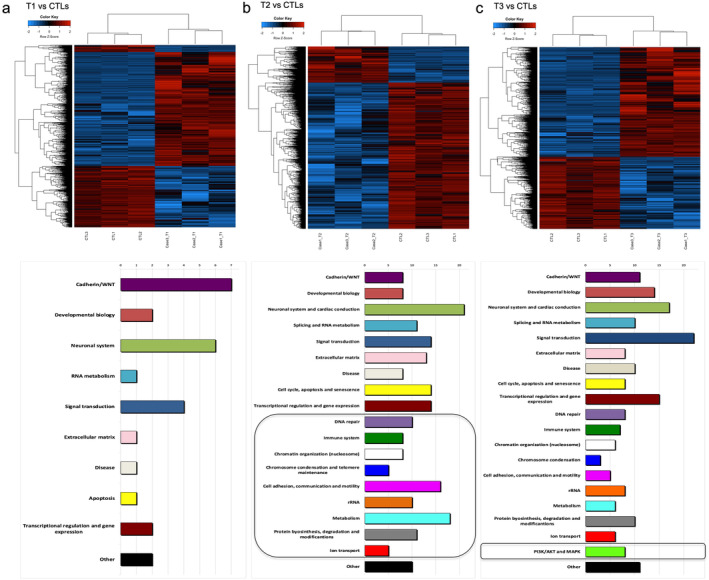
Summary of DNA methylation and pathways analysis. Supervised heatmaps (top) and enriched pathways (bottom) identified from DMRs annotation in the 1qJT cases analyzed versus controls at T1 (panel a), at T2 (panel b), and at T3 (panel c). Patients in the heatmaps are represented in the columns and CpGs in the rows. Blue and red heatmap colors refers to hypo‐ and hyper‐methylation, respectively as indicated in the top left color key legend. x‐axis on pathway histograms represented the number of pathways in each category, as reported by Arabic numbers on the top; black squares underline categories that emerged as new at each timepoint.
Notably, methylation changes did not significantly affect promoter regions at any times over this longitudinal study (Figure 2c).
Altogether, these epigenetic results underlined that the deep hypomethylating effect of AZA was only partially acting on the highly hypermethylated regions at diagnosis. Moreover, hypermethylation at exons and intronic enhancers marked the disease progression after AZA.
3.5. Longitudinal DMR Profiles Delineated Time Course‐Specific Pathways Deregulation
To investigate the biological effects of the identified epigenetic changes, we annotated DMRs to genes and analyzed the associated altered pathways. Our functional analysis on DMRs‐associated genes at T1 identified 27 enriched pathways (FDR < 0.1), mainly related to Cadherin/WNT signaling (Figure 3a and Table S7). At T2, according to the increased number of DMRs, a set of 211 enriched pathways emerged (Figure 3b and Table S8). In addition to all the altered pathways identified at T1, nine new functional categories with significant demethylation changes emerged at this timepoint (Figure 3b and Table S8). Intriguingly the new pathways related to DNA repair, chromatin organization, chromosome condensation and telomere maintenance, and rRNA (Figure 3b and Table S8) are mirroring known mechanisms in JT [5, 6, 30, 31].
Furthermore, after AZA treatment, we confirmed the hypomethylation of immune system related pathways and involvement of SINE/Alu, LTR, and LINE repetitive sequences, as previously reported [32] (Figure S3 and Table S8).
Concerning the T3, we identified 193 enriched pathways (Figure 3c and Table S9). The PI3K/AKT and MAPK signaling were the new altered pathways appearing at disease progression (Figure 3c and Table S9). In particular, the PI3K/AKT pathway consisted of 657 DMRs preferentially located at intergenic (n = 273) and intronic (n = 220) regions and annotated to 175 genes (Table S10). We focused on AKT as hub of this signaling and found AKT1 protein activation by phosphorylation (pAKT1) at T3, anticipated by a mild signal at T2 (Figures 4a and S4). With regards to activation mechanisms, by western blot and quantitative real‐time PCR (qRT‐PCR) on longitudinal samples, we first excluded the AKT1 mRNA upregulation (Figure S4). Moreover, positively (PDK1) or negatively (PTEN and SHIP) interacting regulator proteins were not significantly altered (Figure S4). Instead, the hypermethylation of the mir200c promoter, encoding for an AKT1 inhibitor [33], emerged as likely addressing the T3 activation (Figure 4b). Accordingly, we could document the mir200c downregulation by the hypermethylated promoter (Figure 4c).
FIGURE 4.
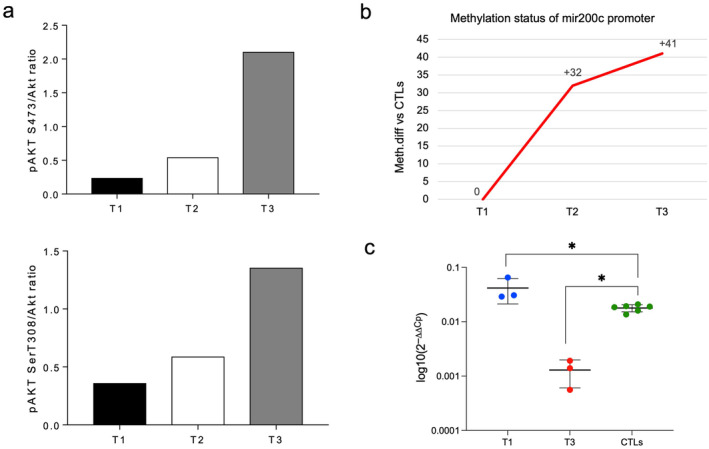
Results from AKT1 activation. (a) pAkt S473/Akt (upper) and pAkt T308/Akt (bottom) ratios of scanning densitometry from immunoblot analysis from case 1 at each timepoint (T1, T2, T3). (b) Methylation status of the mir200c promoter region at the three timepoints from mERRBS data. y‐axis indicates the methylation difference identified from mERRBS. (c) Significance for mir200c expression by qRT‐PCR (Mann–Whitney test, *p < 0.05); values are expressed as means ± SD.
Altogether, our results highlight the epigenetic involvement of the druggable PI3K/AKT signaling, in particular AKT1 activation behaved as a functional hallmark of disease progression in this cohort of AML with 1qJT after AZA treatment.
4. Discussion
In this study we combined cytogenetics, genomics, and epigenetics to characterize molecular events underlying 1qJT in AML.
Interestingly, common clinical–hematological and molecular features emerged in this selected cohort with 1qJT.
First, all cases had been previously diagnosed as MDS or AML with normal karyotypes. Since diagnosis, however, all cases shared a similar genotype characterized by a loss of function deletion at the SRSF2 gene, starting from proline at position 95 [34]. To our knowledge, previous studies investigating the impact of SRSF2 mutation on the response to therapy have shown controversial results. Persistence of mutations and inferior relapse‐free and overall survival have been reported by Rothenberg‐Thurley et al. in the 64% of SRSF2 mutated cases from a large series of AML [35]. Accordingly, Berton et al. [36] found the association of SRSF2 mutations with poor‐outcomes in AML cases treated with combination of azacytidine and venetoclax. Conversely, in a large series of AML published by Lachowiez et al. [37] SRSF2 mutations, alone or in combination with IDH2 m, were found in responders to azacytidine and venetoclax. Similarly, Hong et al. and Nannya et al. reported that the mutational status of the SRSF2 did not influence the response to decitabine or azacytidine in MDS [38, 39]. Our series is too small to draw conclusions on the role of SRSF2 mutations in the response to AZA. However, the allelic frequencies were virtually stable over the entire disease course in all our cases, suggesting that the affected cells were refractory to treatments. Moreover, 1qJT emerged after AZA in this genetic background. The p.P95_R102del variant emerged as the mutational hallmark in around 4.5% of cases in a large series of myeloid neoplasms [40]. In AML, Yoshimi et al. identified SRSF2, included the p.P95_R102del, as one of the most frequently mutated genes [41]. Of note, in the same cohort co‐existence of mutations at both SRSF2 and IDH2 genes emerged in the 47% of cases [41]. This cooperation was seen also in our study, in which the p.(R140Q) IDH2 variant was present as a recurrent additional event.
Thus, confirming data on the SRSF2 genotype as driver event in a subset of myeloid neoplasms, here we showed the non‐random association between the p.P95_R102del SRSF2 variant and 1qJT.
A pivotal common finding in this cohort was the appearance of 1q JT after treatment with the DNA methyltransferase inhibitor (DNMTi) AZA. Several in vitro studies proved the induction of chromatin decondensation, and 1q heterochromatin recombinations by DNMTi, involving 1q12 sat‐II repetitive sequences family [6, 42, 43]. Our data first showed these effects ex vivo in AML. Interestingly, Sawyer et al. [6, 30, 31] previously proved that an identical model applies to a subset of aggressive MM, suggesting that the chromosomal instability accompanying JT is triggering an uncontrolled malignant proliferation in both plasma cells and myeloid cells. Notably, in this study we found that RUNX1 and KRAS gene mutations were cooperating hits in AML evolution.
With respect to chromosome partners of 1qJT, our breakpoint narrowing showed that families of repetitive sequences at recipient regions were juxtaposed to 1q12 sat‐II in all cases. Specifically, we identified sat‐II at the pericentromeric region of chromosome 16, satellite III at chromosome Y, and NOR sequences at n. 14, 15, 21, and 22 acrocentric chromosomes [44], and epigenetics provided us with new insights to understand the molecular consequences of these heterochromatic recombinations.
An hypermethylated signature was present at diagnosis, whereas, as expected, the DNMTi treatment shifted the global methylome to hypomethylation, significantly involving pathways recapitulating the model of JT origin by DNA demethylation, chromatin decondensation, and chromosome instability [6, 30, 31, 42, 43, 45, 46, 47, 48, 49].
Furthermore, at disease progression we found two specific epigenetic features, namely hypermethylation at intronic enhancers and deregulation of the leukemogenic PI3K/AKT and MAPK pathways. Regarding hypermethylated enhancers, an overlap emerged with those described in IDH‐mutated AML [29, 50], suggesting that the presence of IDH2 mutation in our cohort partially contributed to this epigenetic feature. In addition, enhancer enrichment in this series, only at disease progression, supports their involvement in a mechanism of drug resistance [51].
Concerning the PI3K/AKT and MAPK pathways, their activation has been reported in AML with poor response to therapies [52, 53, 54, 55, 56]. Interestingly, deregulation of PI3K/AKT and MAPK pathways has been related to resistance to chemotherapy in three AML cases who received induction chemotherapy [57] different than the 3 + 7 scheme used in our case n. 2 before azacytidine, suggesting that deregulation of these pathways may preferentially emerge after chemotherapy, included azacytidine.
Focusing on AKT1 as a hub in the PI3K/AKT signaling we identified its phosphorylation at disease progression after AZA, in keeping with in vitro experiments that showed AKT1 activation by AZA [58, 59]. Accordingly, increased AKT1 activation, has been reported in an MDS cell line with an AZA‐resistant phenotype [59]. Thus, monitoring of AKT1 phosphorylation under AZA treatment might be helpful to predict progression and to address the experimental use of PI3K/AKT inhibitors in myeloid neoplasms [53, 54, 55, 56, 59, 60].
At last, although it remains to be confirmed in additional cases, here epigenetics showed the hypermethylation at the promoter of the AKT inhibitor mir200c as a mechanism supporting AKT1 phosphorylation over disease evolution.
In conclusion, our results provided new insights to understand the origin and the molecular counterpart of 1q12JT in AML. Demethylation by AZA was the driver event generating 1qJT from recombinations between families of repetitive DNA sequences. Moreover, AKT1 phosphorylation additionally marked disease progression.
Author Contributions
A.G.L.F. and C.N. performed the methylation experiments, analysis, and drafted the article; V.P. and B.C. performed cytogenetic studies and FISH experiments; V.P. performed SNPa experiments and analysis; F.P. and C.Ma. performed and analyzed targeted NGS; M.C. and S.A. performed nucleic acids extraction and Sanger sequencing V.D.B. and M.Q. provided clinical – hematological data; G.M. and C.O. performed Western blot experiments and analysis; P.G. contributed to the analysis of qRT‐PCR and microRNA expression; C.M. conceived and designed the study, contributed to data interpretation, and wrote the article. All authors approved final version of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Data S1.
Table S4.
Table S5.
Table S6.
Table S7.
Table S8.
Table S9.
Table S10.
Acknowledgments
The authors would like to thank Dr. Silvia Romoli and Dr. Donatella Beacci for their technical assistance in preparing samples for cytogenetic experiments.
Funding: This work was supported by AIRC 5 × 1000 call “Metastatic disease: the key unmet need in oncology” to MYNERVA project, #21267 (MYeloid NEoplasms Research Venture Airc). A detailed description of the MYNERVA project is available at https://www.progettomynerva.it. This publication is based upon work from COST Action CA18233, supported by COST (European Cooperation in Science and Technology). CM Unit belongs to the Consorzio Interuniversitario per le Biotecnologie (C.I.B.). The Hematology Unit was supported by PNC, LSH‐TA (PNC‐E3‐2022‐23683269).
Data Availability Statement
The data that support the findings of this study are openly available in Gene Expression Omnibus (GEO) at http://www.ncbi.nlm.nih.gov/geo/, reference number GSE271912, GSE271912.
References
- 1. Allshire R. C. and Madhani H. D., “Ten Principles of Heterochromatin Formation and Function,” Nature Reviews Molecular Cell Biology 19, no. 4 (2018): 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Janssen A., Colmenares S. U., and Karpen G. H., “Heterochromatin: Guardian of the Genome,” Annual Review of Cell and Developmental Biology 34 (2018): 265–288. [DOI] [PubMed] [Google Scholar]
- 3. Couture T., Amato K., DiAdamo A., and Li P., “Jumping Translocations of 1q in Myelodysplastic Syndrome and Acute Myeloid Leukemia: Report of Three Cases and Review of Literature,” Case Reports in Genetics (2018): 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Borri D., Ommati L. V. M., and Velloso E. D. R., “Jumping Translocation: An Unusual Cytogenetic Finding in Myeloid Neoplasm,” Hematology, transfusion and Cell Therapy 44, no. 4 (2022): 612–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sawyer J. R., Tian E., J. D. Shaughnessy, Jr. , et al., “Hyperhaploidy Is a Novel High‐Risk Cytogenetic Subgroup in Multiple Myeloma,” Leukemia 31, no. 3 (2017): 637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sawyer J. R., Tian E., Heuck C. J., et al., “Evidence of an Epigenetic Origin for High‐Risk 1q21 Copy Number Aberrations in Multiple Myeloma,” Blood 125, no. 24 (2015): 3756–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tiepolo L., Maraschio P., Gimelli G., Cuoco C., Gargani G. F., and Romano C., “Multibranched Chromosomes 1, 9, and 16 in a Patient With Combined IgA and IgE Deficiency,” Human Genetics 51, no. 2 (1979): 127–137. [DOI] [PubMed] [Google Scholar]
- 8. Miniou P., Jeanpierre M., Bourc'his D., Barbosa A. C. C., Blanquet V., and Viegas‐Péquignot E., “α‐Satellite DNA Methylation in Normal Individuals and in ICF Patients: Heterogeneous Methylation of Constitutive Heterochromatin in Adult and Fetal Tissues,” Human Genetics 99, no. 6 (1997): 738–745. [DOI] [PubMed] [Google Scholar]
- 9. de Greef J. C., Wang J., Balog J., et al., “Mutations in ZBTB24 Are Associated With Immunodeficiency, Centromeric Instability, and Facial Anomalies Syndrome Type 2,” American Journal of Human Genetics 88, no. 6 (2011): 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thijssen P. E., Ito Y., Grillo G., et al., “Mutations in CDCA7 and HELLS Cause Immunodeficiency–Centromeric Instability–Facial Anomalies Syndrome,” Nature Communications 6, no. 1 (2015): 7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yeung C. C. S., Deeg H. J., Pritchard C., Wu D., and Fang M., “Jumping Translocations in Myelodysplastic Syndromes,” Cancer Genetics 209, no. 9 (2016): 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liehr T., “International System for Human Cytogenetic or Cytogenomic Nomenclature (ISCN): Some Thoughts,” Cytogenetic and Genome Research 161, no. 5 (2021): 223–224. [DOI] [PubMed] [Google Scholar]
- 13. Gosden J. R., Lawrie S. S., and Cooke H. J., “A Cloned Repeated DNA Sequence in Human Chromosome Heteromorphisms,” Cytogenetics and Cell Genetics 29, no. 1 (1981): 32–39. [DOI] [PubMed] [Google Scholar]
- 14. Tagarro I., Fernández‐Peralta A. M., and González‐Aguilera J. J., “Chromosomal Localization of Human Satellites 2 and 3 by a FISH Method Using Oligonucleotides as Probes,” Human Genetics 93, no. 4 (1994): 383–388. [DOI] [PubMed] [Google Scholar]
- 15. Finelli P., Sirchia S. M., Masciadri M., et al., “Juxtaposition of Heterochromatic and Euchromatic Regions by Chromosomal Translocation Mediates a Heterochromatic Long‐Range Position Effect Associated With a Severe Neurological Phenotype,” Molecular Cytogenetics 5 (2012): 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Knight S. J., Lese C. M., Precht K. S., et al., “An Optimized Set of Human Telomere Clones for Studying Telomere Integrity and Architecture,” The American Journal of Human Genetics 67, no. 2 (2000): 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moyzis R. K., Albright K. L., Bartholdi M. F., et al., “Human Chromosome‐Specific Repetitive DNA Sequences: Novel Markers for Genetic Analysis,” Chromosoma 95, no. 6 (1987): 375–386. [DOI] [PubMed] [Google Scholar]
- 18. Garrett‐Bakelman F. E., Sheridan C. K., Kacmarczyk T. J., et al., “Enhanced Reduced Representation Bisulfite Sequencing for Assessment of DNA Methylation at Base Pair Resolution,” Journal of Visualized Experiments 96 (2015): e52246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fernandez A. G. L., Crescenzi B., Pierini V., et al., “A Distinct Epigenetic Program Underlies the 1;7 Translocation in Myelodysplastic Syndromes,” Leukemia 33, no. 10 (2019): 2481–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krueger F. and Andrews S. R., “Bismark: A Flexible Aligner and Methy‐ Lation Caller for Bisulfite‐Seq Applications,” Bioinformatics 27, no. 11 (2011): 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Akalin A., Kormaksson M., Li S., et al., “methylKit: A Comprehensive R Package for the Analysis of Genome‐Wide DNA Methylation Profiles,” Genome Biology 13, no. 10 (2012): R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park Y., Figueroa M. E., Rozek L. S., and Sartor M. A., “MethylSig: A Whole Genome DNA Methylation Analysis Pipeline,” Bioinformatics 30, no. 17 (2014): 2414–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. R Core Team , R: A Language and Environment for Statistical Computing (Vienna, Austria: R Foundation for Statistical Computing, 2020), https://www.R‐project.org/. [Google Scholar]
- 24. Welch R. P., Lee C., Imbriano P. M., et al., “ChIP‐Enrich: Gene Set Enrichment Testing for ChIP‐Seq Data,” Nucleic Acids Research 42, no. 13 (2014): e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adelman E. R., Huang H. T., Roisman A., et al., “Aging Human Hematopoietic Stem Cells Manifest Profound Epigenetic Reprogramming of Enhancers That May Predispose to Leukemia,” Cancer Discovery 9 (2019): 1080–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bao W., Kojima K. K., and Kohany O., “Repbase Update, a Database of Repetitive Elements in Eukaryotic Genomes,” Mobile DNA 6 (2015): 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xie Z., Bailey A., Kuleshov M. V., et al., “Gene Set Knowledge Discovery With Enrichr,” Current Protocols 1, no. 3 (2021): e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Glass J. L., Hassane D., Wouters B. J., et al., “Epigenetic Identity in AML Depends on Disruption of Nonpromoter Regulatory Elements and Is Affected by Antagonistic Effects of Mutations in Epigenetic Modifiers,” Cancer Discovery 7, no. 8 (2017): 868–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Figueroa M. E., Abdel‐Wahab O., Lu C., et al., “Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation,” Cancer Cell 18, no. 6 (2010): 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sawyer J. R., Tian E., Walker B. A., et al., “An Acquired High‐Risk Chromosome Instability Phenotype in Multiple Myeloma: Jumping 1q Syndrome,” Blood Cancer Journal 9, no. 8 (2019): 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sawyer J. R., Tricot G., Mattox S., Jagannath S., and Barlogie B., “Jumping Translocations of Chromosome 1q in Multiple Myeloma: Evidence for a Mechanism Involving Decondensation of Pericentromeric Heterochromatin,” Blood 91, no. 5 (1998): 1732–1741. [PubMed] [Google Scholar]
- 32. Ohtani H., Ørskov A. D., Helbo A. S., et al., “Activation of a Subset of Evolutionarily Young Transposable Elements and Innate Immunity Are Linked to Clinical Responses to 5‐Azacytidine,” Cancer Research 80, no. 12 (2020): 2441–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao G.‐Z., Niu Y.‐Q., Li Z.‐M., Kou D., and Zhang L., “MiR‐200c Inhibits Proliferation and Promotes Apoptosis of Wilms Tumor Cells by Regulating Akt Signaling Pathway,” European Review for Medical and Pharmacological Sciences 24, no. 12 (2020): 6623–6631. [DOI] [PubMed] [Google Scholar]
- 34. Meggendorfer M., Roller A., Haferlach T., et al., “SRSF2 Mutations in 275 Cases With Chronic Myelomonocytic Leukemia (CMML),” Blood 120, no. 15 (2012): 3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rothenberg‐Thurley M., Amler S., Goerlich D., et al., “Persistence of Pre‐Leukemic Clones During First Remission and Risk of Relapse in Acute Myeloid Leukemia,” Leukemia 32, no. 7 (2018): 1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Berton G., Sedaki B., Collomb E., et al., “Poor Prognosis of SRSF2 Gene Mutations in Patients Treated With VEN‐AZA for Newly Diagnosed Acute Myeloid Leukemia,” Leukemia Research 141 (2024): 107500. [DOI] [PubMed] [Google Scholar]
- 37. Lachowiez C. A., Loghavi S., Furudate K., et al., “Impact of Splicing Mutations in Acute Myeloid Leukemia Treated With Hypomethylating Agents Combined With Venetoclax,” Blood Advances 5, no. 8 (2021): 2173–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hong J. Y., Seo J. Y., Kim S. H., et al., “Mutations in the Spliceosomal Machinery Genes SRSF2, U2AF1, and ZRSR2 and Response to Decitabine in Myelodysplastic Syndrome,” Anticancer Research 35, no. 5 (2015): 3081–3089. [PubMed] [Google Scholar]
- 39. Nannya Y., Tobiasson M., Sato S., et al., “Postazacitidine Clone Size Predicts Long‐Term Outcome of Patients With Myelodysplastic Syndromes and Related Myeloid Neoplasms,” Blood Advances 7, no. 14 (2023): 3624–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Todisco G., Creignou M., Gallì A., et al., “Co‐Mutation Pattern, Clonal Hierarchy, and Clone Size Concur to Determine Disease Phenotype of SRSF2P95‐Mutated Neoplasms,” Leukemia 35, no. 8 (2021): 2371–2381. [DOI] [PubMed] [Google Scholar]
- 41. Yoshimi A., Lin K. T., Wiseman D. H., et al., “Coordinated Alterations in RNA Splicing and Epigenetic Regulation Drive Leukaemogenesis,” Nature 574, no. 7777 (2019): 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Costa G., Barra V., Lentini L., Cilluffo D., and Di Leonardo A., “DNA Demethylation Caused by 5‐Aza‐2′‐Deoxycytidine Induces Mitotic Alterations and Aneuploidy,” Oncotarget 7, no. 4 (2016): 3726–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grövdal M., Karimi M., Tobiasson M., et al., “Azacitidine Induces Profound Genome‐Wide Hypomethylation in Primary Myelodysplastic Bone Marrow Cultures but May Also Reduce Histone Acetylation,” Leukemia 28, no. 2 (2014): 411–413. [DOI] [PubMed] [Google Scholar]
- 44. McStay B., “The p‐Arms of Human Acrocentric Chromosomes Play by a Different Set of Rules,” Annual Review of Genomics and Human Genetics 24, no. 1 (2023): 63–83. [DOI] [PubMed] [Google Scholar]
- 45. Aoki Y., Nojima M., Suzuki H., et al., “Genomic Vulnerability to LINE‐1 Hypomethylation Is a Potential Determinant of the Clinicogenetic Features of Multiple Myeloma,” Genome Medicine 4, no. 12 (2012): 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hagemann S., Heil O., Lyko F., and Brueckner B., “Azacytidine and Decitabine Induce Gene‐Specific and Non‐Random DNA Demethylation in Human Cancer Cell Lines,” PLoS One 6, no. 3 (2011): e17388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mund C., Hackanson B., Stresemann C., Lübbert M., and Lyko F., “Characterization of DNA Demethylation Effects Induced by 5‐Aza‐2′‐Deoxycytidine in Patients With Myelodysplastic Syndrome,” Cancer Research 65, no. 16 (2005): 7086–7090. [DOI] [PubMed] [Google Scholar]
- 48. Tobiasson M., Abdulkadir H., Lennartsson A., et al., “Comprehensive Mapping of the Effects of Azacitidine on DNA Methylation, Repressive/Permissive Histone Marks and Gene Expression in Primary Cells From Patients With MDS and MDS‐Related Disease,” Oncotarget 8, no. 17 (2017): 28812–28825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tran H. T. T., Kim H. N., Lee I. K., et al., “DNA Methylation Changes Following 5‐Azacitidine Treatment in Patients With Myelodysplastic Syndrome,” Journal of Korean Medical Science 26, no. 2 (2011): 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Akalin A., Garrett‐Bakelman F. E., Kormaksson M., et al., “Base‐Pair Resolution DNA Methylation Sequencing Reveals Profoundly Divergent Epigenetic Landscapes in Acute Myeloid Leukemia,” PLoS Genetics 8, no. 6 (2012): e1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Haertle L., Barrio S., Munawar U., et al., “Cereblon Enhancer Methylation and IMiD Resistance in Multiple Myeloma,” Blood 138, no. 18 (2021): 1721–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bosi C., Finelli C., Martelli A. M., et al., “Frequent Elevation of Akt Kinase Phosphorylation in Mononuclear Cells From High‐Risk Myelodysplastic Syndrome Patients,” Blood 106, no. 11 (2005): 3432. [DOI] [PubMed] [Google Scholar]
- 53. Nepstad I., Hatfield K. J., Grønningsæter I. S., and Reikvam H., “The PI3K‐Akt‐mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells,” International Journal of Molecular Sciences 21, no. 8 (2020): 2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Estruch M., Vittori C., Montesinos T. M., Reckzeh K., and Theilgaard‐Mönch K., “Targeting of PI3K/AKT Signaling and DNA Damage Response in Acute Myeloid Leukemia: A Novel Therapeutic Strategy to Boost Chemotherapy Response and Overcome Resistance,” Cancer Drug Resistance 4, no. 4 (2021): 984–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Platanias L. C., “Map Kinase Signaling Pathways and Hematologic Malignancies,” Blood 101, no. 12 (2003): 4667–4679. [DOI] [PubMed] [Google Scholar]
- 56. Murali I., Kasar S., Naeem A., et al., “Activation of the MAPK Pathway Mediates Resistance to PI3K Inhibitors in Chronic Lymphocytic Leukemia,” Blood 138, no. 1 (2021): 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Magi A., Mattei G., Mingrino A., et al., “High‐Resolution Nanopore Methylome‐Maps Reveal Random Hyper‐Methylation at CpG‐Poor Regions as Driver of Chemoresistance in Leukemias,” Communications Biology 6, no. 1 (2023): 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yu S. M. and Kim S. J., “DNA‐Hypomethylating Agent, 5′‐Azacytidine, Induces Cyclooxygenase‐2 Expression via the PI3‐Kinase/Akt and Extracellular Signal‐Regulated Kinase‐1/2 Pathways in Human HT1080 Fibrosarcoma Cells,” International Journal of Oncology 47, no. 4 (2015): 1469–1475. [DOI] [PubMed] [Google Scholar]
- 59. Minařík L., Pimková K., Kokavec J., et al., “Analysis of 5‐Azacytidine Resistance Models Reveals a Set of Targetable Pathways,” Cells 11, no. 2 (2022): 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. He Y., Sun M. M., Zhang G. G., et al., “Targeting PI3K/Akt Signal Transduction for Cancer Therapy,” Signal Transduction and Targeted Therapy 6, no. 1 (2021): 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Table S4.
Table S5.
Table S6.
Table S7.
Table S8.
Table S9.
Table S10.
Data Availability Statement
The data that support the findings of this study are openly available in Gene Expression Omnibus (GEO) at http://www.ncbi.nlm.nih.gov/geo/, reference number GSE271912, GSE271912.