

Graphical abstract

Overview of the study. IL: interleukin; TSLP: thymic stromal lymphopoietin; SLPI: secretory leukocyte protease inhibitor.
Abstract
Aim
To elucidate the important cellular and molecular drivers of pulmonary long COVID, we generated a single-cell transcriptomic map of the airway mucosa using bronchial brushings from patients with long COVID who reported persistent pulmonary symptoms.
Method
Adults with and without long COVID were recruited from the general community in Greater Vancouver, Canada. The cohort was divided into those with pulmonary long COVID, which was defined as persons with new or worsening respiratory symptoms following ≥12 weeks from their initial acute severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (n=9); and control subjects defined as SARS-CoV-2 infected persons whose acute respiratory symptoms had fully resolved or individuals who had no history of acute coronavirus disease 2019 (COVID-19) (n=9). These participants underwent bronchoscopy from which a single cell suspension was created from bronchial brush samples and then sequenced.
Results
A total of 56 906 cells were recovered for the downstream analysis, with 34 840 cells belonging to the pulmonary long COVID group, which strikingly showed a unique cluster of neutrophils in the pulmonary long COVID group (p<0.05). Ingenuity Pathway Analysis revealed that the neutrophil degranulation pathway was enriched across epithelial cell clusters. Differential gene expression analysis between the pulmonary long COVID and control groups demonstrated upregulation of inflammatory chemokines and epithelial barrier dysfunction across epithelial cell clusters, as well as over-expression of mucin genes across secretory cell clusters.
Conclusion
A single-cell transcriptomic landscape of the small airways suggest that neutrophils may play a significant role in mediating the chronic small airway inflammation driving pulmonary symptoms of long COVID.
Shareable abstract
Single-cell profiling shows infiltration of neutrophils with upregulation of inflammatory chemokines and mucin genes in the airway mucosa of patients with pulmonary long COVID, indicating persistent small airway inflammation in pulmonary long COVID https://bit.ly/3XieXQN
Introduction
The coronavirus disease 2019 (COVID-19) pandemic has infected >771 million people, accounting for >6.9 million deaths worldwide [1]. As we transition into the post-pandemic era, a new clinical entity has emerged: post-acute sequelae of severe acute respiratory coronavirus 2 (SARS-CoV-2) infection, commonly known as long COVID [2]. Approximately 10% of all infected adult survivors experience long COVID, with almost half reporting persistent symptoms beyond 1 year post-infection [3, 4]. However, long COVID is clinically complicated, as it is associated with >200 different symptoms involving numerous organs in the body [3, 5]. Among persistent symptoms, respiratory complaints are common [6–8], with 38.5% of all adult Canadians with long COVID experiencing dyspnoea and 39.3% experiencing cough [8]. Despite this, the pathophysiology of pulmonary long COVID remains obscure. To elucidate cellular and molecular drivers of pulmonary long COVID, we generated a single-cell transcriptomic map of the airway mucosa using bronchial brushings from patients with long COVID and with persistent pulmonary symptoms for >10 months following acute SARS-CoV-2 infection.
Methods
Study population
Adults (aged ≥19 years) with and without long COVID were recruited from a tertiary care clinic at St Paul's Hospital as well as from the general community in greater Vancouver, Canada, through advertising. Following informed consent (University of British Columbia/Providence Health Care research ethics board approval H21–02149 and H19–02222), all participants answered a series of questionnaires including the St George's Respiratory Questionnaire (SGRQ), and underwent a pulmonary function test (PFT), low-dose chest computed tomography (CT), and phlebotomy for complete blood count. Bronchoscopy was also performed in a subset of these participants who consented to a research bronchoscopy, the details of which have been published previously [9].
A priori, the cohort was divided into three groups: 1) patients with pulmonary long COVID [10], defined as individuals who remained persistently symptomatic >12 weeks post-infection, with a new onset or worsening of respiratory symptoms including cough or dyspnoea, in conjunction with SGRQ total score of >10 in the absence of known chronic lung conditions, such as bronchiectasis, pulmonary fibrosis or COPD; 2) individuals without any lingering respiratory symptoms post-COVID-19 infection (i.e. recovered COVID-19 patients); and 3) subjects who did not have any history of COVID infection at the time of enrolment (never COVID-19). As the demographic and clinical features of groups 2 and 3 were similar, in this study, we merged them as one control group (table 1). We excluded any participants with pre-existing chronic respiratory disorders, as well as participants who were heavily smoking cigarettes, cannabis or e-cigarettes at the time of recruitment.
TABLE 1.
Baseline characteristics of study participants in the control group
| Recovered COVID-19 | Never COVID-19 | p-value | |
|---|---|---|---|
| Participants | 4 | 5 | |
| Age, years | 48 (32–61) | 26 (24–65) | 1.00 |
| Male | 2 (50) | 2 (40) | 1.00 |
| BMI kg·m−2 | 25.6 (21.9–287.7) | 22.9 (22.4–24.6) | 0.71 |
| Cigarette smoking status | 0.17 | ||
| Former | 2 (50) | 0 (0) | |
| Never | 2 (50) | 5 (100) | |
| Cannabis smoking status | 1.00 | ||
| Current | 1 (25) | 1 (20) | |
| Never | 3 (75) | 4 (80) | |
| E-cigarette smoking status | 1.00 | ||
| Current | 0 (0) | 1 (20) | |
| Never | 4 (100) | 4 (80) | |
| SGRQ | 3.1 (1.9–4.9) | 1 (0.8–5.7) | 0.54 |
| Symptoms | 2.6 (0–6) | 6.7 (5.7–23.2) | 0.26 |
| Activity | 9.2 (4.7–13.7) | 0 (0–0) | 0.23 |
| Impact | 0 (0–0) | 0 (0–0) | 0.50 |
| Pulmonary function testing | |||
| FVC L | 4.1 (3.9–4.4) | 3.9 (3.8–4.8) | 0.90 |
| FVC % predicted | 122.2 (113.4–127.5) | 116.5 (106.6–122.3) | 0.39 |
| FEV1 L | 3.5 (3.3–3.8) | 3.4 (3–4.2) | 1.00 |
| FEV1 % predicted | 123.1 (117.5–125.4) | 116.9 (104.6–117.7) | 0.11 |
| FEV1/FVC % | 80.8 (80.3–83.9) | 81 (78.4–86.9) | 0.66 |
| DLCO % predicted | 108.2 (105.3–111.6) | 108.1 (99.4–112.2) | 0.71 |
| Blood cell counts | |||
| White blood cells ×109 cells·L−1 | 5.2 (4.9–5.5) | 4.5 (3.9–4.6) | 0.11 |
| Neutrophils % | 58.3 (54.6–61.8) | 55.7 (46.8–57.1) | 0.71 |
| Lymphocytes % | 29.7 (28–31.3) | 34.9 (34.6–36.5) | 0.18 |
| Monocytes % | 4.5 (4.1–5.2) | 4.3 (4.2–4.4) | 0.90 |
| Eosinophils % | 3.1 (1.8–4.6) | 2 (1.9–2.5) | 1.00 |
| Basophils % | 0.5 (0.4–0.5) | 0.7 (0.6–1) | 0.10 |
| BAL differential cell count# | |||
| Macrophage % | 90 (82.3–94.8) | 79 (77–91) | 0.54 |
| Lymphocytes % | 8.5 (5–14) | 8 (4–16) | 1.00 |
| Neutrophils % | 1.5 (0.8–3) | 5 (2–5) | 0.39 |
| Eosinophils % | 0 (0–0.3) | 0 (0–0) | 0.37 |
Data are presented as n, median (interquartile range) or n (%), unless otherwise stated. COVID-19: coronavirus disease 2019; BMI: body mass index; SGRQ: St George's Respiratory Questionnaire; FVC: forced vital capacity; FEV1: forced expiratory volume in 1 s; DLCO: diffusion capacity for carbon monoxide; BAL: bronchoalveolar lavage. #: the percentages of each type of leukocyte present in the BAL fluid.
The present cohort study included 24 participants who underwent bronchoscopy between January 2021 and August 2023. All participants were vaccinated against SARS-CoV-2 at the time of enrolment. Based on history, the predominant strains at the time of the patient's initial SARS-CoV-2 infection were alpha, beta, gamma and delta variants. Bronchoscopies were performed when the subjects were clinically stable for >2 months. Among 13 subjects with pulmonary long COVID, four were excluded due to poor quality of samples or technical issues in sample processing or library preparation. Among six COVID-19 patients without pulmonary symptoms, two were also excluded for these reasons. The final number of subjects with pulmonary long COVID in the analysis was nine. The final number of control subjects was also nine, with five who had experienced acute COVID-19, but were now free of significant pulmonary symptoms, and four without a prior history of acute COVID-19 (figure 1).
FIGURE 1.

A flow diagram summarising the participants who were enrolled between January 2021 and August 2023 and underwent bronchoscopy. 13 subjects were identified as having persistent respiratory symptoms ≥3 months post-COVID-19 (coronavirus disease 2019) infection and scored >10 in the St George's Respiratory Questionnaire (pulmonary long COVID). Nine subjects were selected as controls due to either no history of COVID-19 infection or lack of persistent respiratory symptoms post-infection. The final number of subjects used for analysis was nine in the pulmonary long COVID group and nine in the control group. #: subjects with pre-existing chronic respiratory disorders and participants who were heavily smoking tobacco cigarettes or using cannabis or e-cigarettes at the time of recruitment were excluded.
Bronchoscopy and single-cell suspension preparation
Under conscious sedation, a fibreoptic bronchoscope (Olympus Corporation, Tokyo, Japan) was passed through the mouth of participants and into the trachea. With the bronchoscope positioned in one of the subsegmental bronchi of the right or left upper lobe, a cytological brush was inserted through a bronchoscope channel into a sixth- to eighth-generation airway from where bronchial brush samples were collected. A cytological brush was then withdrawn from the bronchoscope, and using a pair of stainless-steel scissors, the brush was cut into a microcentrifuge tube containing 1000 μL medium and kept on ice until further processing. A single-cell suspension was then created according to our established protocol [11]. In brief, the single-cell suspension resulted from using Accutase dissociation agent (Stemcell Technologies, Vancouver, BC, Canada), and multiple washing steps using Pneumacult-Ex (Stemcell Technologies) media. During the sample collections or throughout the processing, if visible blood was noted on the brush or cell pellets, the sample was deemed contaminated and was not submitted for sequencing.
Single-cell sequencing
The created single-cell suspension was transferred on ice to a sequencing facility where the samples were loaded onto the Chromium Controller using the Chromium Next GEM Single Cell 3′ Kit v3.1 and Chip G (10x Genomics, CA, USA). The sequenced libraries were prepared in accordance with a previously published Chromium Single Cell 3′ Reagent Kits User Guide [12]. The integrity of the ccomplementary DNA libraries was examined using a 2100 Bioanalyzer instrument with a High Sensitivity DNA Kit (Agilent Technologies, CA, USA). Sequencing of the final libraries was performed at a loading concentration of 650 pM with a 2% PhiX spike-in on the NextSeq 2000 (Illumina, CA, USA) as recommended by 10x Genomics. The final depth of sequenced samples was targeted to reach 60 000 reads per cell. The Fastq files containing the data were then inputted to the Cell Ranger v6.01 (10x Genomics) and the reads were aligned to the human reference genome (hg19).
Bioinformatics and statistical analysis
Single-cell analysis pipeline
Correction for ambient RNA was done using the R package SoupX (version 1.5.2) [13] prior to performing further quality control tasks and downstream analysis. Following ambient RNA correction, using the Pegasus package (version 1.8.1) in Python, we performed additional quality control steps, in which cells with high mitochondrial genes (>20%) or high haemoglobin genes (>1%) were filtered out. To ensure the integrity of the data, we also filtered out cells that expressed <150 or >8000 genes, as very low or high gene counts could be the result of contamination or a technical artifact. We then removed mitochondrial genes, ribosomal genes and MALAT1 to reduce the impact of the aforementioned genes on downstream analysis and to improve the accuracy of gene expression profiles. The data were then processed for normalisation, logarithmic transformation, principal component analysis, and batch effect correction using the Scanorama algorithm (version 1.7.3). After nearest neighbour calculation, the Leiden algorithm was used to cluster the data [14]. Cell type annotation was then performed using a curated gene list from the Pegasus human lung and human immune legacy markers, which was complemented by other resources including the Protein Atlas (www.proteinatlas.org), PanglaoDB [15] and the markers highlighted in publications such as Hewitt et al. [16] and Zaragosi et al. [17] (supplementary table S1) [16–19]. Next, during annotation, within each cluster, we grouped cells into subclusters based on the differential gene expression of canonical markers. For example, within the basal cell cluster, we subdivided the cells into basal and basal-2 subclusters based on differences in the gene expression of ≥20% for TP63, a known canonical gene marker for basal cells.
Wilcoxon rank-sum tests were used to determine if there were significant cell proportion differences between the pulmonary long COVID group and controls. In addition, we performed differential gene expression between the two groups within each cluster, and significantly expressed genes were identified at a false discovery rate (FDR) of <0.05. Differential gene expression analysis among pooled cell clusters (e.g. epithelial) was performed using NEBULA [20], a negative binomial mixed model to conduct association analysis of multisubjects between the pulmonary long COVID group versus the control group. Pre-ranked gene set enrichment analysis was performed using log fold change (logFC) ranks from differential expression results with the Bioconductor R package, fgsea. Cell trajectory and pseudotime analysis were carried out using Velocyto 0.17.17, and ScVelo 0.2.5 packages in Python. All analyses were performed in R (4.3.1) and Python (3.9.12).
Ingenuity Pathway Analysis
All cluster-specific genes, in addition to their respective log2FC and FDR values, were uploaded to Ingenuity Pathway Analysis (IPA; Qiagen, Hilden, Germany), excluding cell clusters that comprised <2% of immune or epithelial cells in disease or control groups. Any clusters, such as suprabasal, that were considered to be cells in transition, were further excluded even if they had >2% presence in both disease and control groups to minimise the potential for false positive results due to their transition state. For each cluster-specific differentially expressed gene (DEG) set (FDR<0.05 and a |log2FC|>0.5), IPA core analysis functions generated p-values (threshold [–log (adjusted p-value)>1.3]) and predicted activation or inhibition status (positive or negative z-scores) for representative canonical pathways and upstream regulators (including associated downstream genes), the latter of which were used to identify chemical and biological drug targets predicted to modulate each DEG set. Core-analysis results from all epithelial clusters were also inputted for comparison analysis, with top upstream regulators and canonical pathways ranked based on the sum of absolute z-score across all compared cell types. Bubble plots for these analyses were then generated using the R package Plotly (version 4.10.4).
Results
Study subjects
There were no significant differences in age, sex, smoking status or PFT measures between subjects with pulmonary long COVID (n=9) and controls (n=9). However, the median (interquartile range) body mass index was significantly higher in subjects with pulmonary long COVID than in control subjects (28 (27–31) kg·m−2 versus 23 (22–26) kg·m−2, p=0.03). Similarly, the SGRQ total score and all three domains of symptoms, activity and impact were also significantly higher in subjects with pulmonary long COVID than in control subjects (45.0 (33.1–69.1) versus 2.1 (1.0–5.7), p<0.001 for SGRQ total score; 42.7 (40.8–44.1) versus 5.7 (0–8.8), p<0.001 for the symptoms domain; 60.4 (48.3–86.5) versus 0 (0–12.2), p<0.001 for the activity domain; and 32.5 (21.0–63.6) versus 0 (0–0), p<0.001 for the impact domain) (table 2) There were no significant differences in blood leukocytes or their differentials including neutrophils, lymphocytes, monocytes, eosinophils and basophils between the two groups. In bronchoalveolar lavage fluid, the cell counts for alveolar macrophages, lymphocytes, neutrophils and eosinophils were similar between the two groups and there was no evidence of a microbiological infection (table 2). The median (range) follow-up from the acute COVID-19 infection to bronchoscopy was 23.7 (11.2–30.6) months and 18.5 (10–24.8) months for pulmonary long COVID group and the control group that recovered from COVID-19, respectively. On a low-dose chest CT scan, there were no abnormal findings in the control group and there were minimal abnormal findings in the pulmonary long COVID group, which included mosaic attenuation (n=1), ground-glass opacity (n=1) and reticulation (n=3) without emphysema or honeycombing (table 3).
TABLE 2.
Baseline characteristics of study participants in pulmonary long COVID and control groups
| Pulmonary long COVID | Control | p-value | |
|---|---|---|---|
| Participants | 9 | 9 | |
| Age years | 54 (44–60) | 35 (24–62) | 0.63 |
| Male | 5 (55.6) | 4 (44.4) | 1.00 |
| BMI kg·m−2 | 28 (27–31) | 23 (22–26) | 0.03 |
| Cigarette smoking status | 1.00 | ||
| Former | 1 (11.1) | 2 (22.2) | |
| Never | 8 (88.9) | 7 (77.8) | |
| Cannabis smoking status | 0.47 | ||
| Current | 0 | 2 (22.2) | |
| Never | 7 (100) | 7 (77.8) | |
| E-cigarette smoking status | 1.00 | ||
| Current | 0 | 1 (11.1) | |
| Never | 7 (100) | 8 (88.9) | |
| SGRQ, total | 45.0 (33.1–69.1) | 2.1 (1.0–5.7) | <0.001 |
| Symptoms | 42.7 (40.8–44.1) | 5.7 (0–8.8) | <0.001 |
| Activity | 60.4 (48.3–86.5) | 0 (0–12.2) | <0.001 |
| Impact | 32.5 (21.0–63.6) | 0 (0–0) | <0.001 |
| Pulmonary function testing | |||
| FVC L | 4.0 (3.0–5.4) | 4.0 (3.8–4.8) | 1.00 |
| FVC % predicted | 108.6 (101.6–114.6) | 117.8 (106.6–126.5) | 0.19 |
| FEV1 L | 3.0 (2.4–4.2) | 3.4 (3.0–4.1) | 0.48 |
| FEV1 % predicted | 110.7 (94.7–117.1) | 117.7 (106.3–125.0) | 0.11 |
| FEV1/FVC % | 80.6 (76.8–84.4) | 81.0 (79.8–86.9) | 0.66 |
| DLCO % predicted | 96.9 (83.8–102.5) | 108.1 (100.7–112.2) | 0.13 |
| Blood cell counts | |||
| White blood cells ×109 cells·L−1 | 5.3 (4.7–6.5) | 4.6 (4.5–5.1) | 0.31 |
| Neutrophils % | 56.4 (52.1–58.5) | 55.7 (53.9–61.7) | 1.00 |
| Lymphocytes % | 32.5 (30.4–38.5) | 32.3 (28.4–34.9) | 0.60 |
| Monocytes % | 5.1 (4.0–6.4) | 4.3 (4.1–4.9) | 0.60 |
| Eosinophils % | 2.2 (1.6–3.1) | 2.0 (1.9–4.2) | 0.49 |
| Basophils % | 0.6 (0.5–0.9) | 0.5 (0.4–0.7) | 0.59 |
| BAL differential cell count# | |||
| Macrophage % | 79 (76–95) | 86 (77–93) | 0.79 |
| Lymphocytes % | 18 (4–20) | 8 (4–16) | 0.59 |
| Neutrophils % | 1 (0–3) | 2 (1–5) | 0.37 |
| Eosinophils % | 0 (0–0) | 0 (0–0) | 0.37 |
| Days from COVID-19 infection to bronchoscopy | 711 (444–832) | 556 (374–703)¶ | 0.35 |
Data are presented as n, median (interquartile range) or n (%), unless otherwise stated. BMI: body mass index; SGRQ: St George's Respiratory Questionnaire; FVC: forced vital capacity; FEV1: forced expiratory volume in 1 s; DLCO: diffusion capacity of the lung for carbon monoxide; BAL: bronchoalveolar lavage; COVID-19: coronavirus disease 2019. #: the percentages of each type of leukocyte present in the BAL fluid; ¶: the time from infection to bronchoscopy for individuals in the control group who are post-COVID, but do not report any persistent pulmonary symptoms (n=4).
TABLE 3.
Chest computed tomography findings
| Pulmonary long COVID | Control | |
|---|---|---|
| Participants | 9 | 9 |
| Emphysema | 0 (0) | 0 (0) |
| Mosaic attenuation | 1 (11) | 0 (0) |
| Ground-glass opacity | 1 (11) | 0 (0) |
| Reticulation | 3 (33) | 0 (0) |
| Honeycombing | 0 (0) | 0 (0) |
Data are presented as n or n (%).
Profiling the airway mucosal landscape using single-cell RNA sequencing
A total of 56 906 cells were recovered for downstream analysis, with 34 840 cells belonging to the pulmonary long COVID group. Using uniform manifold approximation and projection, we generated a dimensionality reduction plot, which revealed several clusters. These clusters were then labelled using legacy markers and manual curation (figure 2). With this approach, we identified a cluster predominantly derived from bronchial brush samples of the pulmonary long COVID patients (figure 2a), annotated as neutrophils. Overall, we found 16 clusters of epithelial origin and 15 clusters of immune cells (figure 2b). A composition plot (figure 3) illustrates the distribution of all established clusters.
FIGURE 2.

Uniform manifold approximation (UMAP) of 56 906 of profiled cells from both pulmonary long COVID and control groups, which were annotated using legacy markers. Final annotation of the cluster revealed 16 clusters that were of epithelial origin and 15 clusters that were immune cells. a) A dimensionality reduction plot of pulmonary long COVID versus control reveals distribution of clusters across both categories. It prominently highlights a specific cluster derived from the pulmonary long COVID group. b) Various clusters of cells originating from both epithelial and immune cell lineages were observed. As anticipated, the diversity among these identified cell clusters serves as a representation of the cellular heterogeneity within the airway mucosa. Additionally, distinct neutrophil cluster was highlighted. MAIT: mucosal-associated invariant T-cell; TCM: central memory T-cell; NK: natural killer.
FIGURE 3.

A four-part composition plot, each representing the distribution of epithelial and immune cells identified in the sample. TCM: central memory T-cell; MAIT: mucosal-associated invariant T-cell; NK: natural killer.
A Wilcoxon rank-sum test comparing the composition of clusters between the pulmonary long COVID and control groups revealed that the abundance of neutrophils was significantly increased in pulmonary long COVID participants (p<0.05) (supplementary figure S1a and b). To validate this observation, we prepared cytospin slides from randomly selected bronchial brushes and confirmed the presence of neutrophils in pulmonary long COVID group samples (n=3). In contrast, we did not observe any neutrophils in the control samples (n=3) (supplementary figure S2a and b).
Trajectory analysis
The RNA velocity dynamical model in conjunction with a pseudotime analysis revealed that the majority of cells in our dataset were relatively transcriptomically stable and thus probably in a quiescent state. This was confirmed by evaluating the Antigen Kiel 67 (MKI67) expression levels across all cell types (supplementary figure S3a and b). The calculated coherence of the vector field provided us confidence of accuracy for the calculated RNA velocity (supplementary figure S3c).
Differential gene expression analysis of epithelial and immune cells
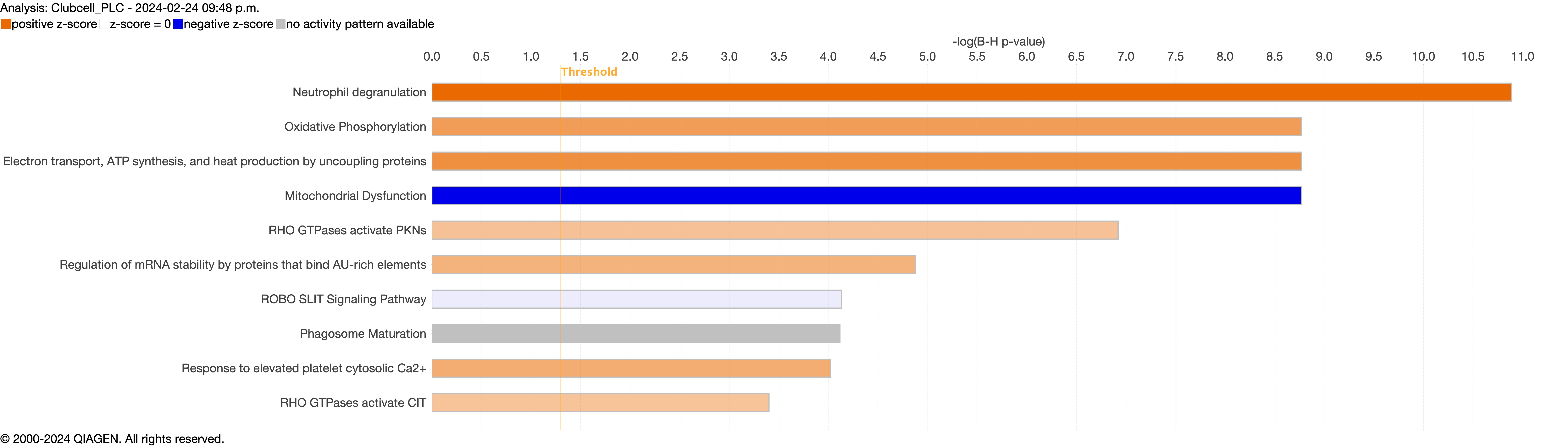
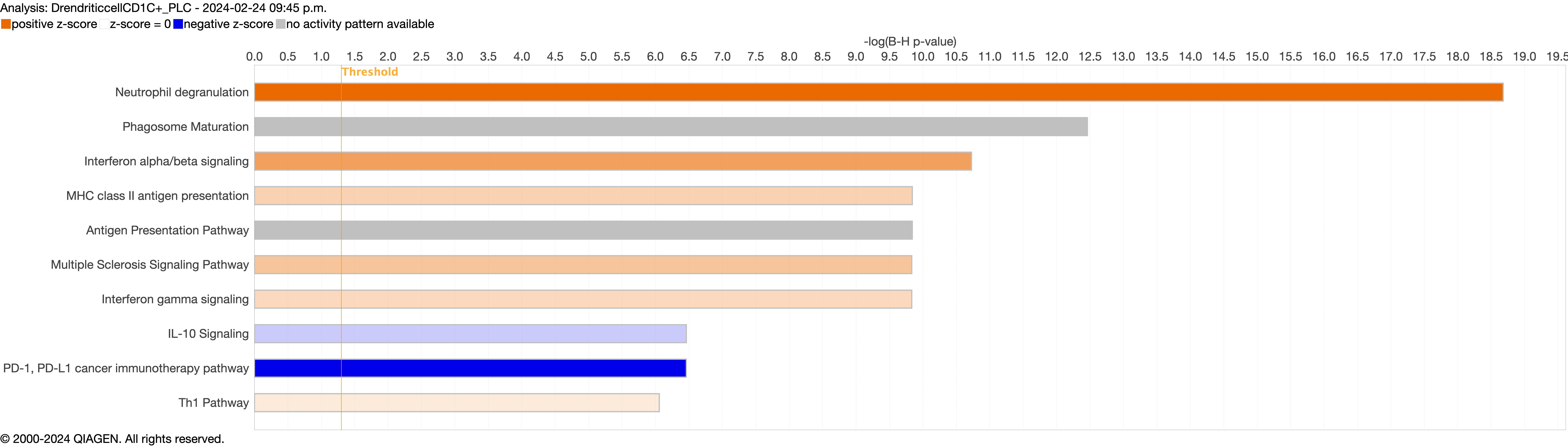
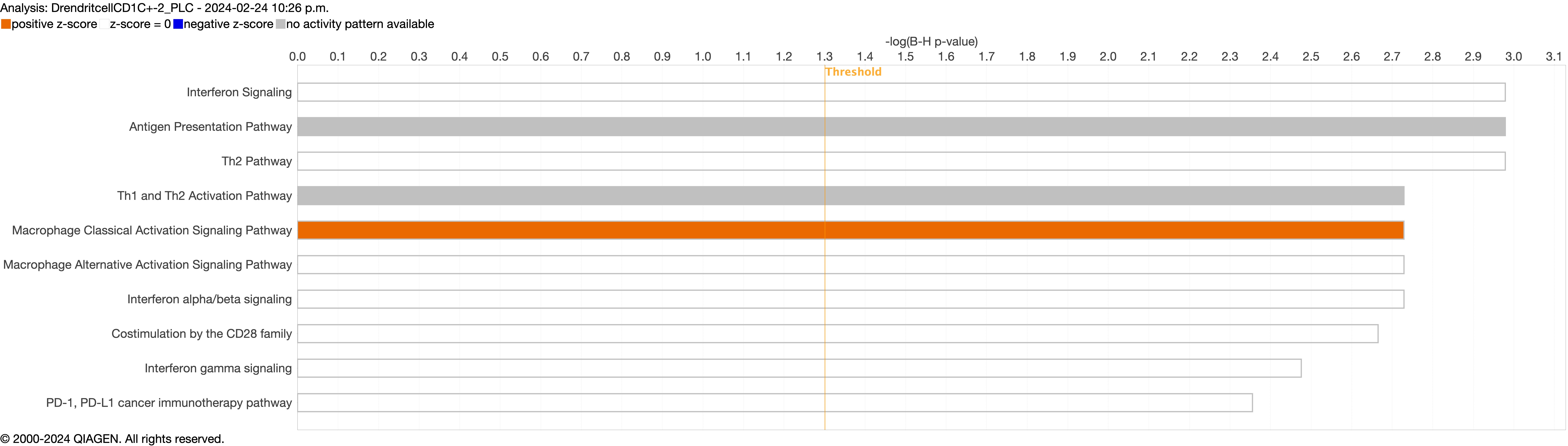
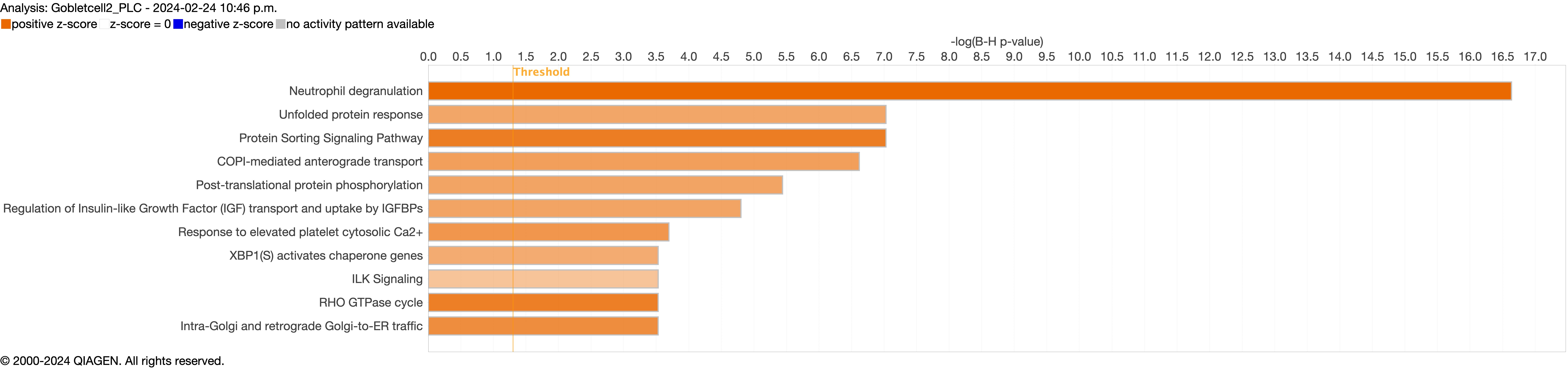
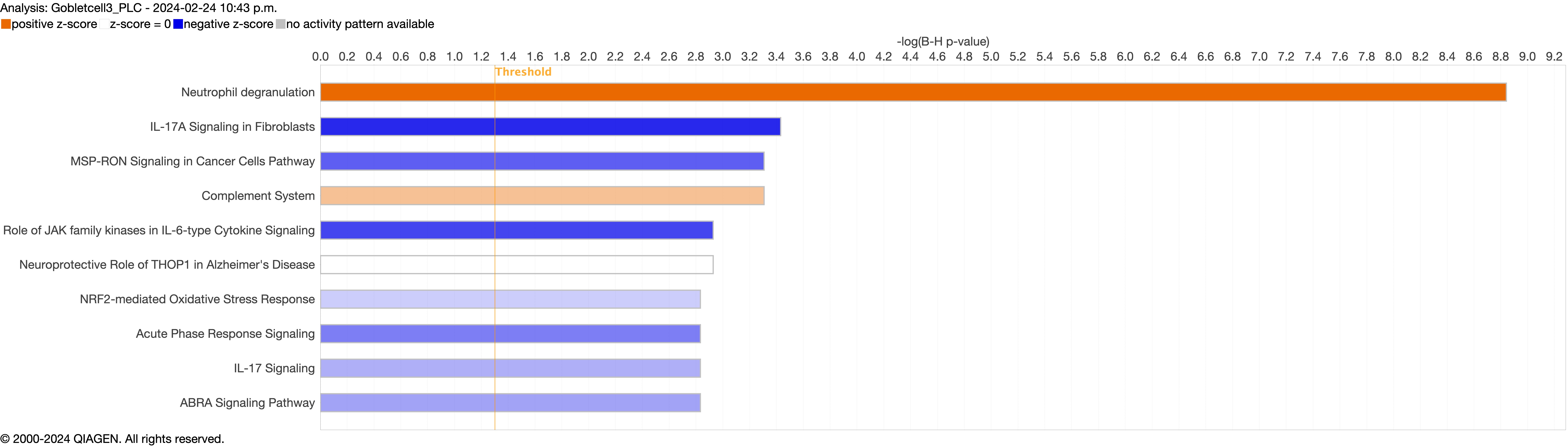
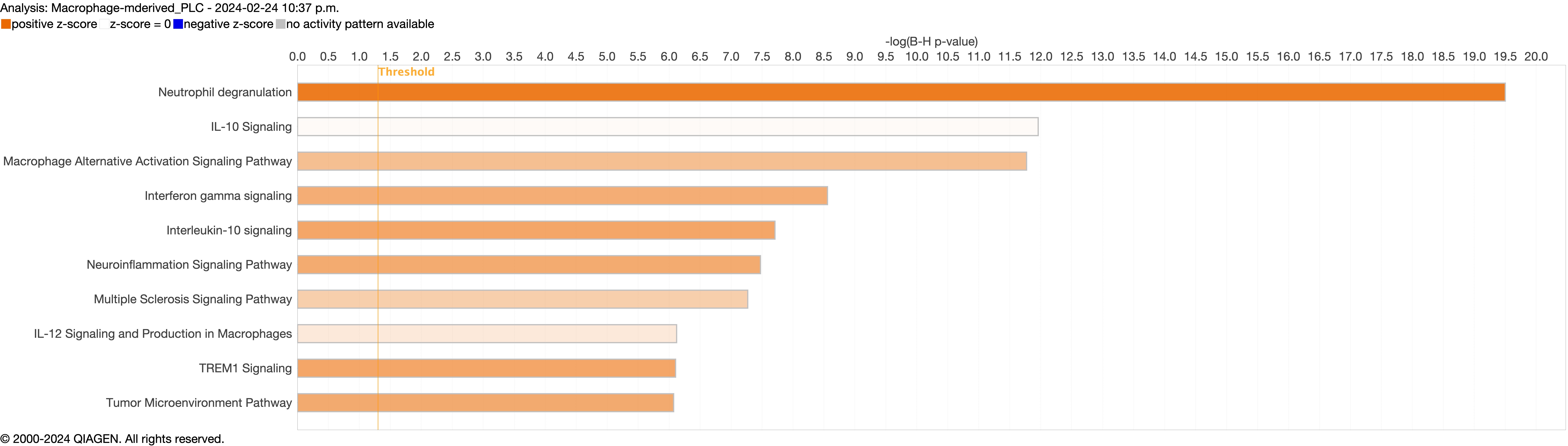
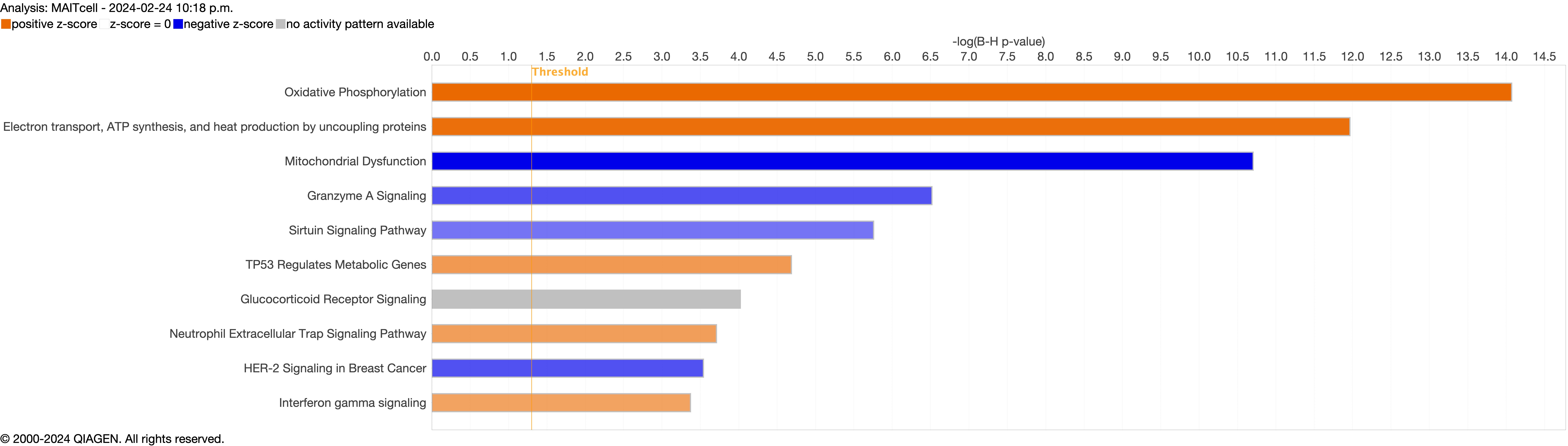
Next, we explored the transcriptional profiling differences in the airway mucosa of pulmonary long COVID compared to control groups. Here, the DEGs between pulmonary long COVID and control participants were subjected to canonical pathway analysis in IPA (supplementary figure S4a–s). Among all epithelial cells, consistent enrichment in pathways associated with neutrophil activation was observed. These pathways included neutrophil degranulation in basal and secretory cell clusters, neutrophil extracellular trap signalling in basal cell clusters, oxidative phosphorylation in basal and ciliated cell clusters, the Ras homology (Rho)-GTPase cycle pathway in secretory cell clusters, and interferon-γ signalling in ciliated cell clusters (figure 4a–c).
FIGURE 4.

Bubble plots of top 15 canonical pathways from differentially expressed genes (false discovery rate <0.05 and log2 fold change >0.5) between pulmonary long COVID and control participants across a) basal, b) secretory and c) ciliated cell clusters. n: number of differentially expressed genes regulated per pathway, with colour denoting cell subclusters. HMOX: haem oxygenase; Ca2+: calcium; SNARE: soluble N-ethylmaleimide-sensitive factor attachment protein receptor; RHOGDI: Ras homology GDP-dissociation inhibitor; ER: endoplasmic reticulum; IGF: insulin-like growth factor; IGFBP: IGF binding protein; HSP: heat shock protein; IL: interleukin; TCR: T-cell receptor; IQGAP: Ras GTPase-activating-like protein; MSP-RON: macrophage stimulating protein receptor d'origine Nantais.
Given the enrichment of neutrophil-related pathways, we next explored epithelial gene expression changes related to neutrophil chemoattraction, as well as epithelial changes known to be altered by neutrophil accumulation (i.e. barrier dysfunction and mucin production) (figure 5a and b). CXCL6 and interleukin (IL)-8, known cytokines associated with neutrophil recruitment and activation, were upregulated in the pulmonary long COVID group (threshold: log2FC>|0.5| and FDR <0.05) (figure 5a). Using the same threshold, a similar upregulation pattern was also observed in genes for SLPI (secretory leukocyte protease inhibitor), IL-33, TRPV4 (transient receptor potential vanilloid-type 4) and TSLP (thymic stromal lymphopoietin), which could implicate epithelial barrier dysfunction (figure 5a). Looking at all known human mucin genes, compared to controls, the results indicated overall upregulation of MUC5AC, MUC4, MUC1 and MUC5B across pulmonary long COVID epithelial cells with secretory characteristics (figure 5b).
FIGURE 5.

The expression pattern of the commonly recognised neutrophil chemoattractant and markers of epithelial barrier dysfunction was assessed among the differentially expressed genes within our dataset. a) The cell level differential gene expression of the chemoattractant and markers of epithelial dysfunction across all epithelial cells that have >2% representation in the sample. b) The cell level differential gene expression of the all the known human mucin genes that we were able to identify across all epithelial cells that have >2% representation in the sample.
The top five significantly (FDR<0.05) identified enriched pathways among pooled epithelial cells, based on the reference Human Molecular Signatures Database (MSigDB), including Gene Ontology (GO), Reactom85, hallmark gene sets and the Kyoto Encyclopedia of Genes and Genomes (KEGG) (KEGG MEDICUS, KEGG legacy gene sets), were plotted (supplementary figure S5, supplementary table S2). The results also indicated enriched immune pathways in epithelial cells, similar to what we observed in the IPA analysis.
We grouped all the identified cell types in the samples based on their lineages (ciliated, secretory, dendritic, T-cells and myeloid lineage cells). Among the resulting top five most common cell type-specific DEGs (threshold: FDR <0.05 and log2FC>|0.5|), we noted 10 upregulated genes to be common across these five groups with no significant pathway enrichment. When looking at the enriched pathways for the two most abundant clusters (i.e. secretory and myeloid) using the R package WebGestaltR, we noted enrichment of GO pathways that were indicative of immune response activation (supplementary figure 6a–c).
On the basis of upregulated genes and associated enriched pathways, we used the IPA database to explore potential therapeutics that could potentially reverse the transcriptomic signature across all epithelial cells (figure 6, supplementary table S3). For example, budesonide and fluticasone propionate were predicted to revert the signature of pulmonary long COVID airways to that resembling the control airway.
FIGURE 6.

Top 20 predicated biological and chemical drug targets from differentially expressed genes (false discovery rate <0.05 and log2 fold change >0.5) between the pulmonary long COVID group and control participants across epithelial cell clusters. Negative z-scores indicate that drug administration in pulmonary long COVID would reorient cell-cluster-specific differentially expressed genes to those of controls. n: number of differentially expressed genes regulated per target, with colour denoting cell subclusters. GABA: γ-aminobutyric acid; GnRH: gonadotropin hormone-releasing hormone.
Discussion
Here, we profiled the transcriptomic landscape of epithelial and airway immune cells in the small airways of patients with pulmonary long COVID at a single-cell resolution. The most striking feature was an increase in the number of neutrophils in the airway mucosa as well an upregulation in the neutrophil-associated activation signatures and increased expression of epithelial barrier dysfunction markers across all epithelial cells, alongside mucin genes in the secretory cells. in the pulmonary long COVID airways. We also observed an increase in MUC gene expression in the secretory cells of the pulmonary long COVID airways.
Our findings are in line with the important role that neutrophils play in the pathogenesis of severe COVID-19. During an acute infection, there is a significant increase in circulating neutrophils in peripheral blood of patients with severe COVID-19 infections [21, 22] and several studies using single cell RNA-sequencing (scRNA-seq) have reported an increase in dysfunctional circulating neutrophils in the blood of those with severe disease [23, 24], which may contribute to cytokine storm caused by uncontrolled innate immune system activation during active disease [25, 26].
However, the pathogenesis of long COVID-19 is largely unknown. A previous study indicated that individuals with interstitial lung changes at 3–6 months post-infection demonstrated upregulation in neutrophil-associated immune signatures including increased chemokines, proteases and markers of neutrophil extracellular traps in circulation compared to those who experienced a complete radiographic resolution at follow-up [27]. A recent study showed that long COVID patients display upregulation of certain inflammatory plasma proteins. Interestingly, the most enriched pathways (for these proteins) were those related to neutrophil degranulation [7]. We extend these data by showing that neutrophils and their associated signatures are increased in the small airway mucosa of patients with pulmonary long COVID, even among patients with no or minimal changes on chest imaging (by CT scan) or PFTs. Additionally, the IPA canonical pathway analysis indicated enrichment of neutrophilic degranulation pathways in both secretory and basal cells. Oxidative phosphorylation in the basal cells and ciliated cells clusters and Rho-GTPase cycle pathway in secretory cells clusters were also enriched, indicating ongoing inflammation. Oxidative phosphorylation in mitochondria induces activation of innate immune response [28], suggesting a potential link between cellular energy metabolism and the inflammatory process. Rho-GTPases are associated with migration and function of innate immune cells during inflammation [29]. In addition, we observed an upregulation of inflammatory chemokines CXCL6 and IL-8 in epithelial cells. These chemokines are known for their role in recruiting and activating myeloid cells [30, 31], which can drive inflammation and immune responses. In line with these observations, we saw a significant upregulation of SLPI in epithelial cells, which neutralises neutrophil elastase and dampens extracellular trap signalling [32]. We also observed increased expression of MUC5AC and MUC5B genes, representing two major secreted airway mucins, in the airways of pulmonary long COVID compared with control subjects. Upregulation of these genes could result in increased mucus production in the airways [33, 34]. Together, these data suggest that even months and years following acute COVID, there may be “neutrophilic” inflammation in the small airways of long COVID patients, who have persistent pulmonary symptoms.
The upstream drivers of the inflammatory changes in the airway mucosa of pulmonary long COVID patients are obscure and largely speculative. Given that lung epithelial cells are the prime targets of entry and propagation of SARS-CoV-2, the enrichment of adaptive immune response signalling (T-cell receptor signalling) in ciliated cells raises the possibility of ongoing stimulation potentially by putative viral reservoirs [35, 36]. Another possibility for this ongoing inflammation in the small airways might be a loss of mucosal barrier integrity marked by upregulation of genes including SLPI, TRPV4 and TSLP, alongside subsequent release of cytokines, such as IL33, in epithelial cells [37–39].
Dissimilar to prior studies, which used peripheral blood samples for scRNA-seq, we used airway tissue samples obtained during research bronchoscopy. Notably, the patients in the present study had normal lung function measurements and normal or near-normal CT imaging. Moreover, we focused on patients with long-term COVID who had persistent pulmonary symptoms for >10 months post-acute infection, which enabled us to relate the scRNA-seq data in the small airways of these patients with their symptoms.
The present study has several limitations. First, this is a cross-sectional study, and thus longitudinal changes of involved cells and biological pathways for pulmonary long COVID could not be investigated. Secondly, due to the nature of single-cell transcriptomic analysis, the expression level of protein related to function could not be assessed. To validate the scRNA-seq data on neutrophils, we prepared cytospin slides and observed these cells in pulmonary long COVID, but not in control samples. However, it should be noted that the sample size for the cytospins was low and will require additional validation in the future. Thirdly, technical limitations of scRNA-seq such as dropout events of fragile cells and relatively low sequencing depth, leading to the loss of rare cell types or those with low RNA content could also have skewed our results, though any confounding or biases from these technical issues should have been nondifferential and affected both the pulmonary long COVID and control groups, resulting in a reduction in the signal. Finally, our primary recruitment relied on voluntary participation from the local community; thus, the results of our study may not be generalisable to different settings.
In conclusion, the single-cell transcriptomic landscape of the pulmonary long COVID airways in patients who were >10 months post-acute infection showed an increase in the number of neutrophils along with upregulation in the neutrophil-associated activation signature and its related inflammatory chemokines across clusters. An increased expression of epithelial barrier dysfunction markers across all epithelial cells and an increase in mucin gene expression in the secretory cells was also observed in pulmonary long COVID airways. Together, these changes may explain the persistent pulmonary symptoms of cough, sputum production and exertional dyspnoea, although additional studies will be required to fully validate this notion. Notwithstanding, these chronic inflammatory changes in the small airways provide new biologic insight on the pathogenesis of pulmonary long COVID and raise potential novel therapeutic investigations for these patients.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary table S1. Curated gene list used for annotating the identified clusters from the Pegasus human lung, human immune legacy markers, and other resources including the Protein Atlas, PanglaoDB and the markers highlighted in literatures such as Hewitt et al. and Zaragosi et al. ERJ-01947-2023.Table_S1 (33.1KB, pdf)
Supplementary table S2. Results of the pre-ranked GSEA analysis of the differentially expressed genes of the pooled epithelial cells based on the negative binomial mixed model identified enriched pathways based on the reference Human Molecular Signatures Database (MSigDB), including gene ontology (GO), Reactom85, hallmark gene sets, and KEGG (KEGG MEDICUS, KEGG legacy gene sets). ERJ-01947-2023.Table_S2 (57.4KB, xlsx)
Supplementary table S3. The list of the genes correspondent to the predicted therapeutics based on the IPA database that could potentially reverse the transcriptomic signature across all epithelial cells. ERJ-01947-2023.Table_S3 (15.9KB, csv)
Supplementary figure S1. The comparison of composition distribution in the clusters between the PLC and control groups. Wilcoxon rank-sum test of the composition of the clusters between the PLC and control groups revealed the neutrophil cluster to be of statistical significance (p<0.05). a) Epithelial cells. ERJ-01947-2023.Figure_S1a (202.6KB, png)
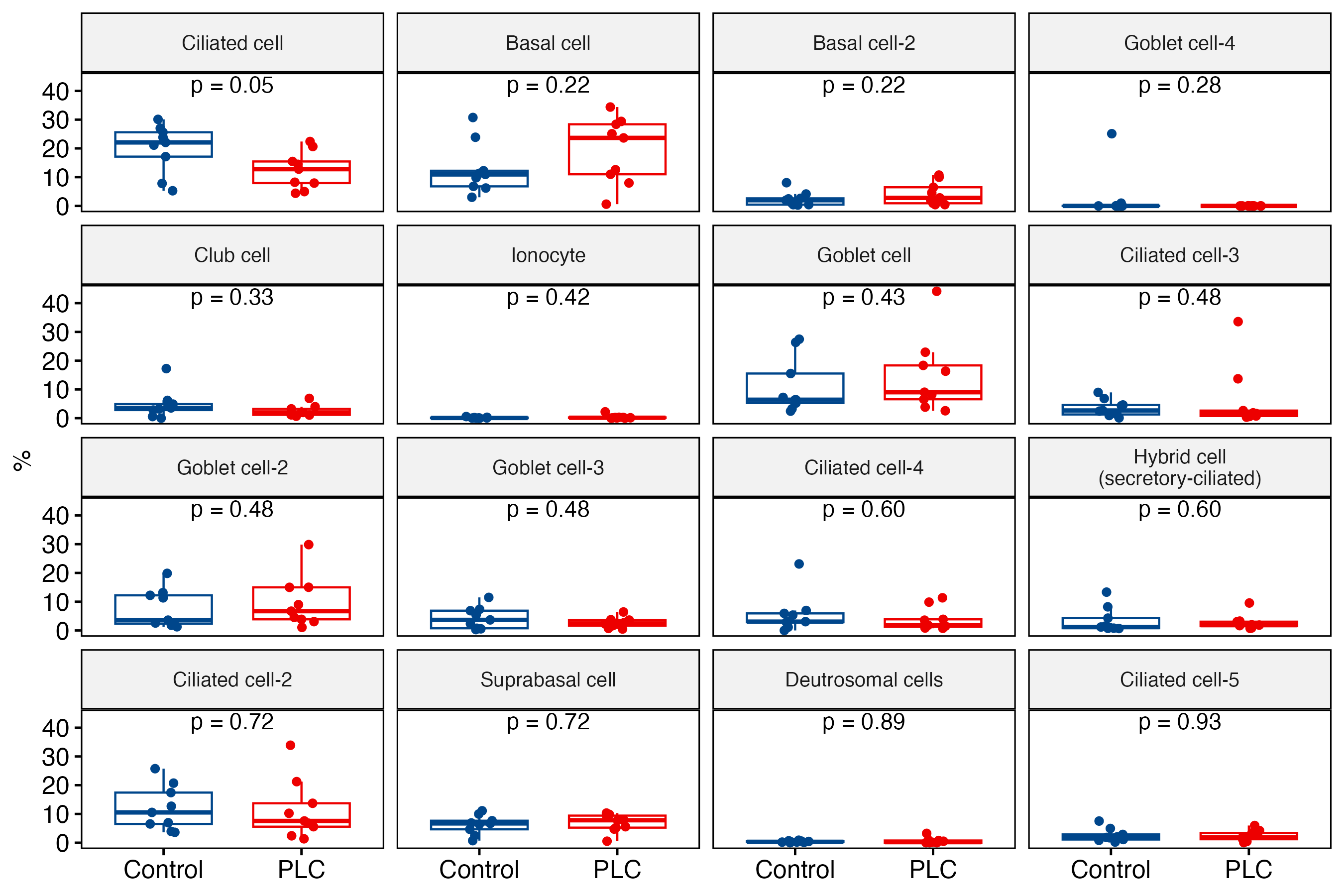{kind=link}
Supplementary figure S1. b) Immune cells. ERJ-01947-2023.Figure_S1b (208.5KB, png)
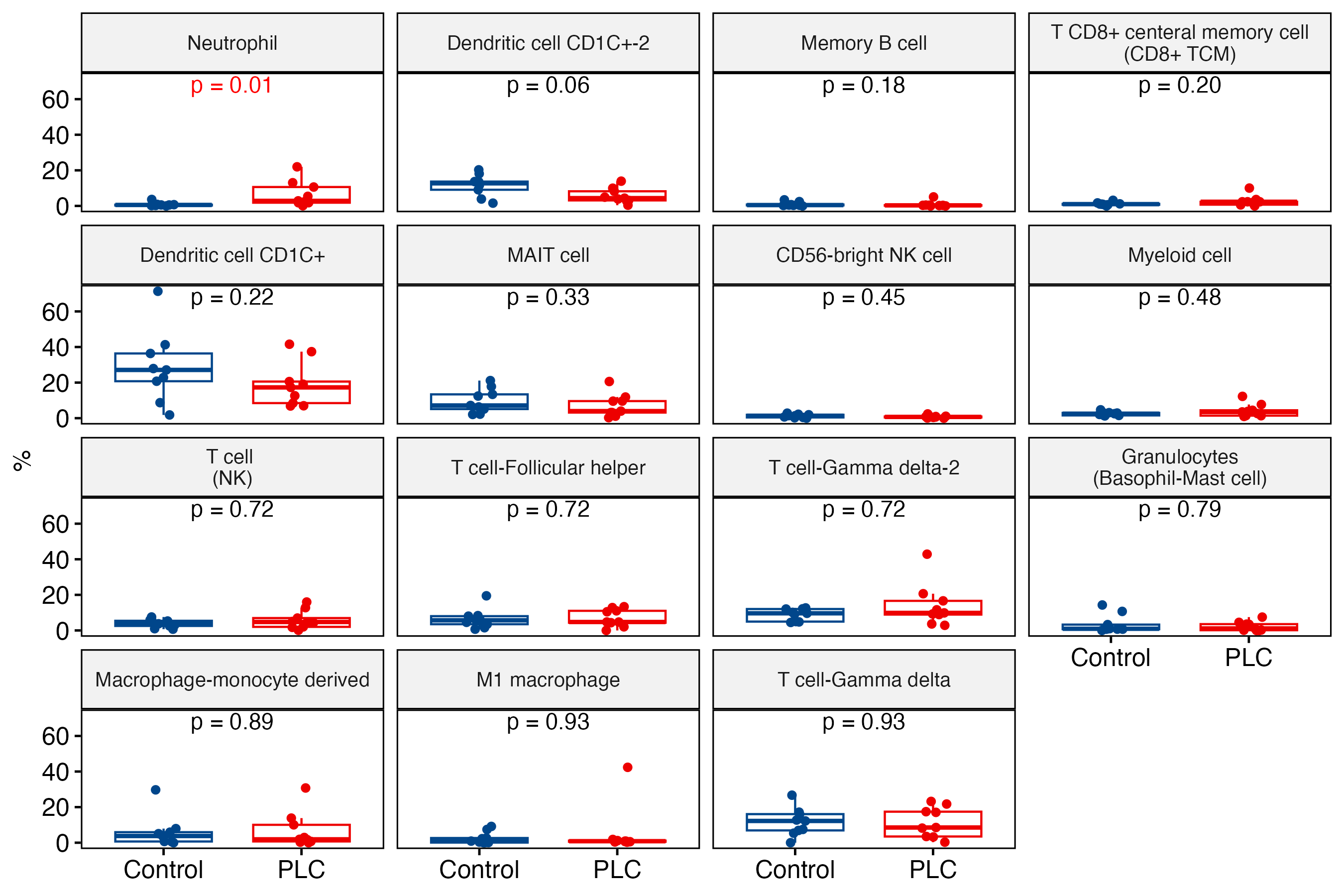{kind=link}
Supplementary figure S2. Cytospin slides in PLC and control samples. Haematoxylin and eosin staining, original magnification ×35. a) Image representative of a PLC sample showing neutrophil presence. Neutrophils have been highlighted using yellow arrow. ERJ-01947-2023.Figure_S2a (1.4MB, tif)
Supplementary figure S2. b) Image representative of a control sample, showing lack of neutrophil presence. ERJ-01947-2023.Figure_S2b (1.1MB, tif)
Supplementary figure S3. RNA velocity and pseudotime analysis show majority of cells being in quiescent state. a) The RNA velocity dynamical map showing the full transcriptional dynamics of splicing kinetic estimating the parameters of reaction rates and latent cell-specific variables across all the identified clusters. b) The RNA velocity pseudotime based on the distance measures on the velocity graph, as well as expression of MKI67 proliferation marker across all the identified clusters in both PLC and control. Majority of the cells appears to be non-proliferating and at the quiescent state. c) Velocity confidence, calculated based on the rate of differentiation on the basis of the length of the velocity vector. It is providing a measure of confidence, as it showcases if the velocity direction is indeterminate. ERJ-01947-2023.Figure_S3 (515.3KB, tif)
Supplementary figure S4a. Figure S4 shows the top 10-13 IPA canonical pathways across all identified clusters that had >2% presence in the analysis. Excluding cells in transition. ERJ-01947-2023.Figure_S4a (1MB, jpg)
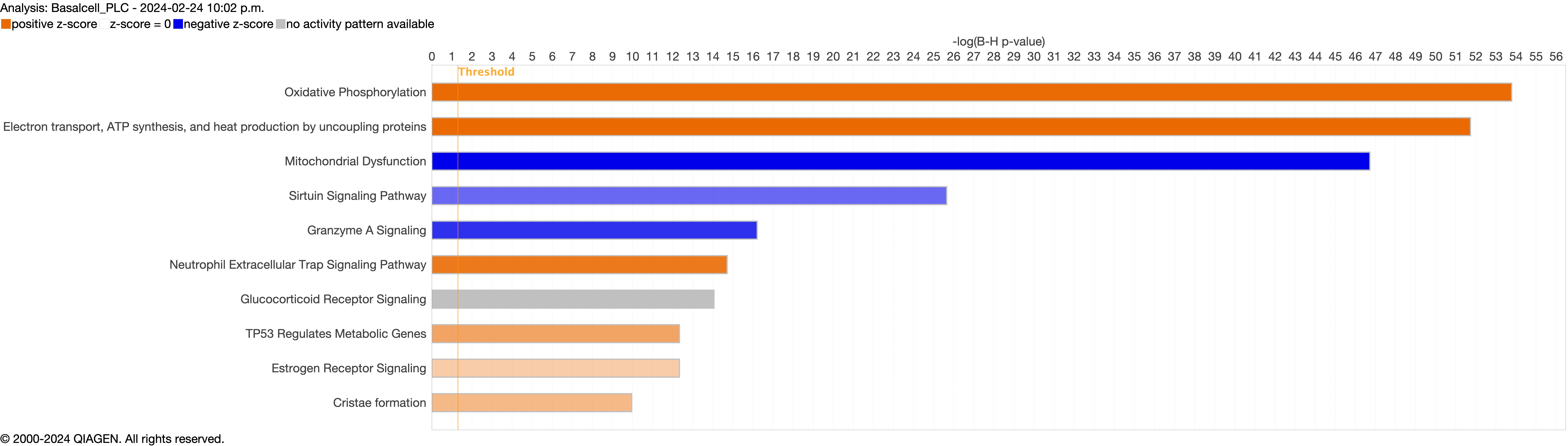{kind=link}
Supplementary figure S4b. ERJ-01947-2023.Figure_S4b (1MB, jpg)
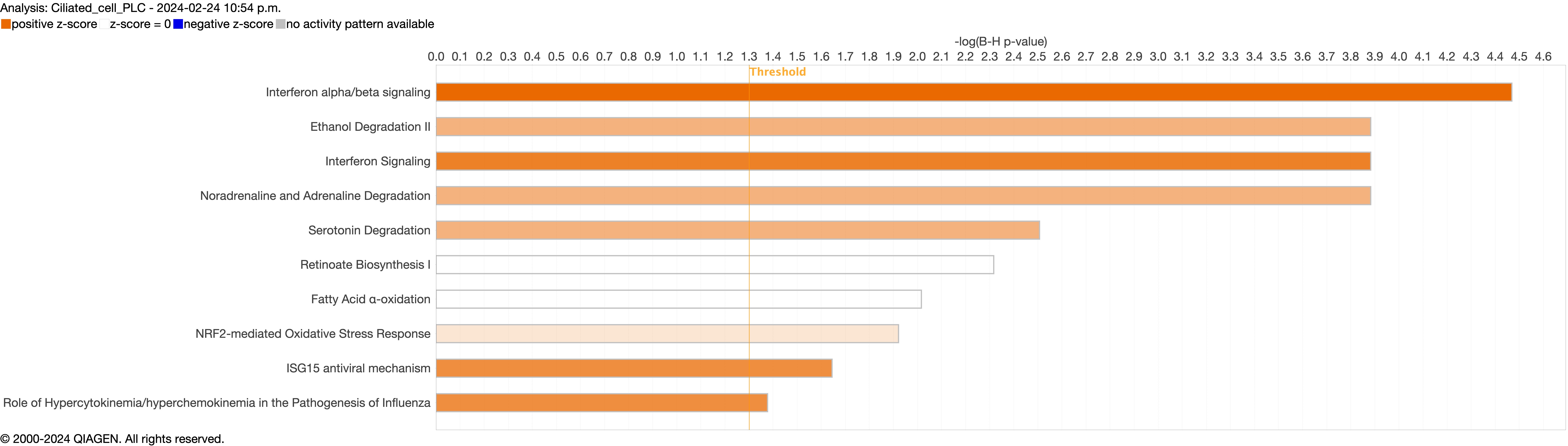{kind=link}
Supplementary figure S4c. ERJ-01947-2023.Figure_S4c (1.1MB, jpg)
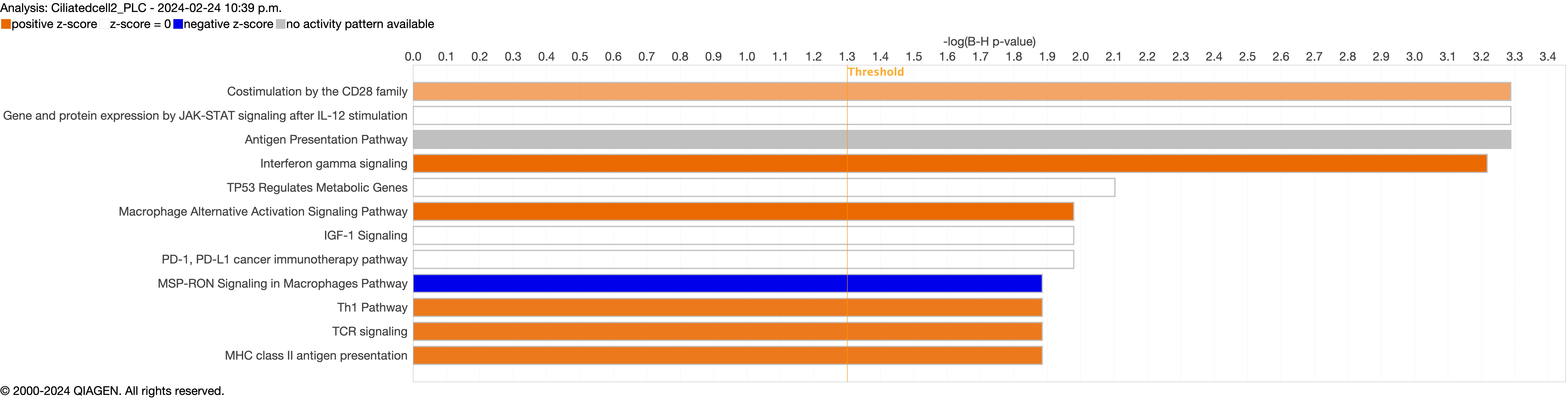{kind=link}
Supplementary figure S4d. ERJ-01947-2023.Figure_S4d (1.2MB, jpg)
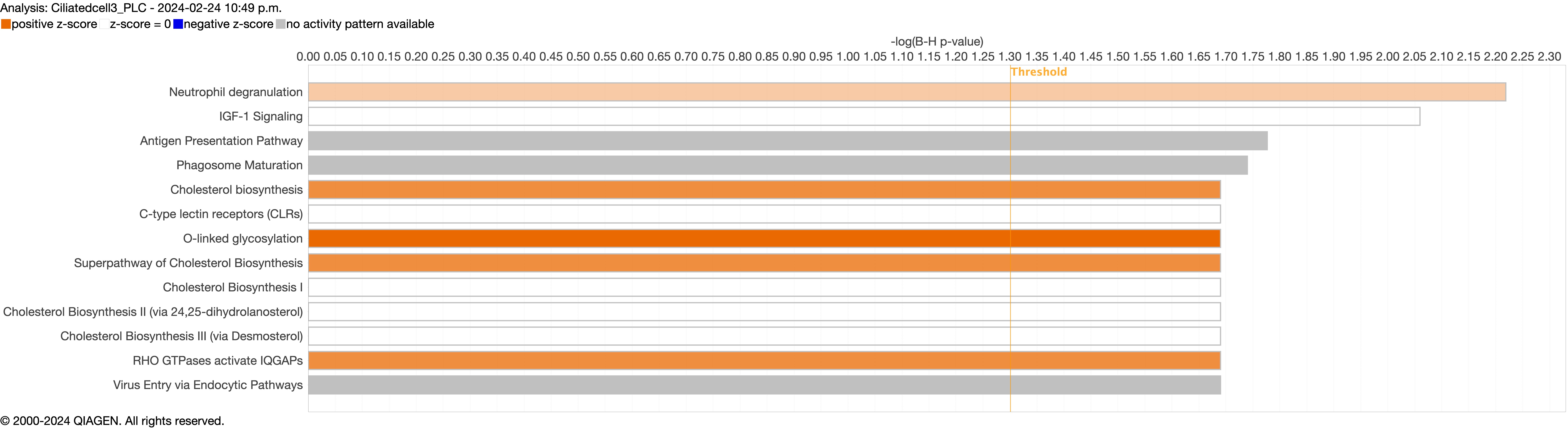{kind=link}
Supplementary figure S4e. ERJ-01947-2023.Figure_S4e (909.9KB, jpg)
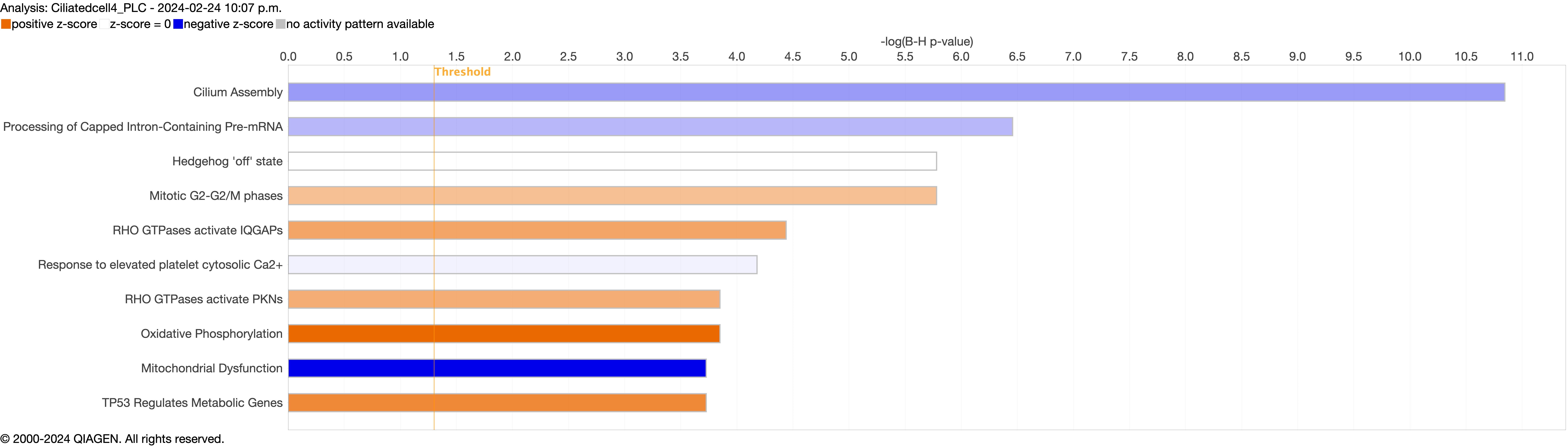{kind=link}
Supplementary figure S4f. ERJ-01947-2023.Figure_S4f (964.3KB, jpg)
{kind=link}
Supplementary figure S4g. ERJ-01947-2023.Figure_S4g (1,002.9KB, jpg)
{kind=link}
Supplementary figure S4h. ERJ-01947-2023.Figure_S4h (1.1MB, jpg)
{kind=link}
Supplementary figure S4i. ERJ-01947-2023.Figure_S4i (1.1MB, jpg)
{kind=link}
Supplementary figure S4j. ERJ-01947-2023.Figure_S4j (914.9KB, jpg)
{kind=link}
Supplementary figure S4k. ERJ-01947-2023.Figure_S4k (1MB, jpg)
{kind=link}
Supplementary figure S4l. ERJ-01947-2023.Figure_S4l (1.1MB, jpg)
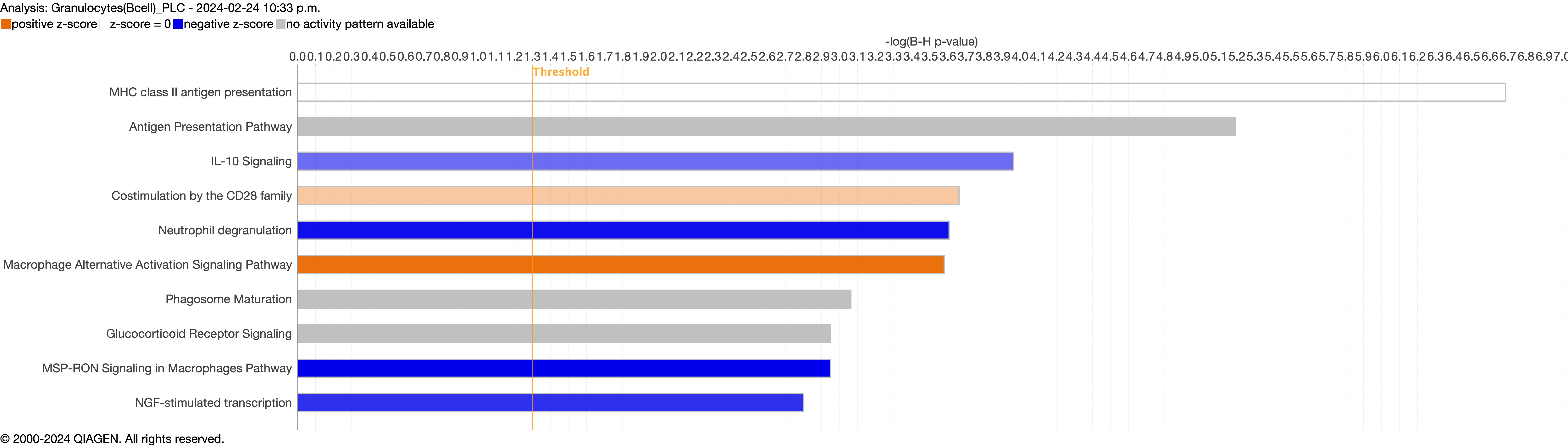{kind=link}
Supplementary figure S4m. ERJ-01947-2023.Figure_S4m (994.8KB, jpg)
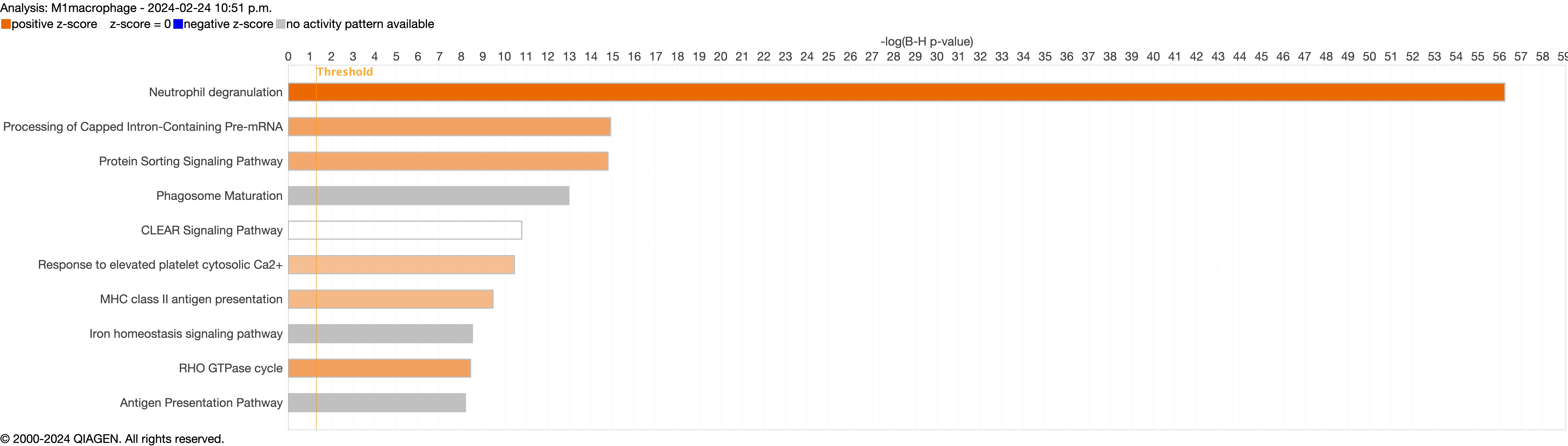{kind=link}
Supplementary figure S4n. ERJ-01947-2023.Figure_S4n (1,019.6KB, jpg)
{kind=link}
Supplementary figure S4o. ERJ-01947-2023.Figure_S4o (965.2KB, jpg)
{kind=link}
Supplementary figure S4p. ERJ-01947-2023.Figure_S4p (1.1MB, jpg)
{kind=link}
Supplementary figure S4q. ERJ-01947-2023.Figure_S4q (1MB, jpg)
{kind=link}
Supplementary figure S4r. ERJ-01947-2023.Figure_S4r (1.1MB, jpg)
{kind=link}
Supplementary figure S4s. ERJ-01947-2023.Figure_S4s (1.1MB, jpg)
{kind=link}
Supplementary figure S5. Enrichment of the epithelial cells from the differentially expressed genes based on the mixed effect model. ERJ-01947-2023.Figure_S5 (387.6KB, png)
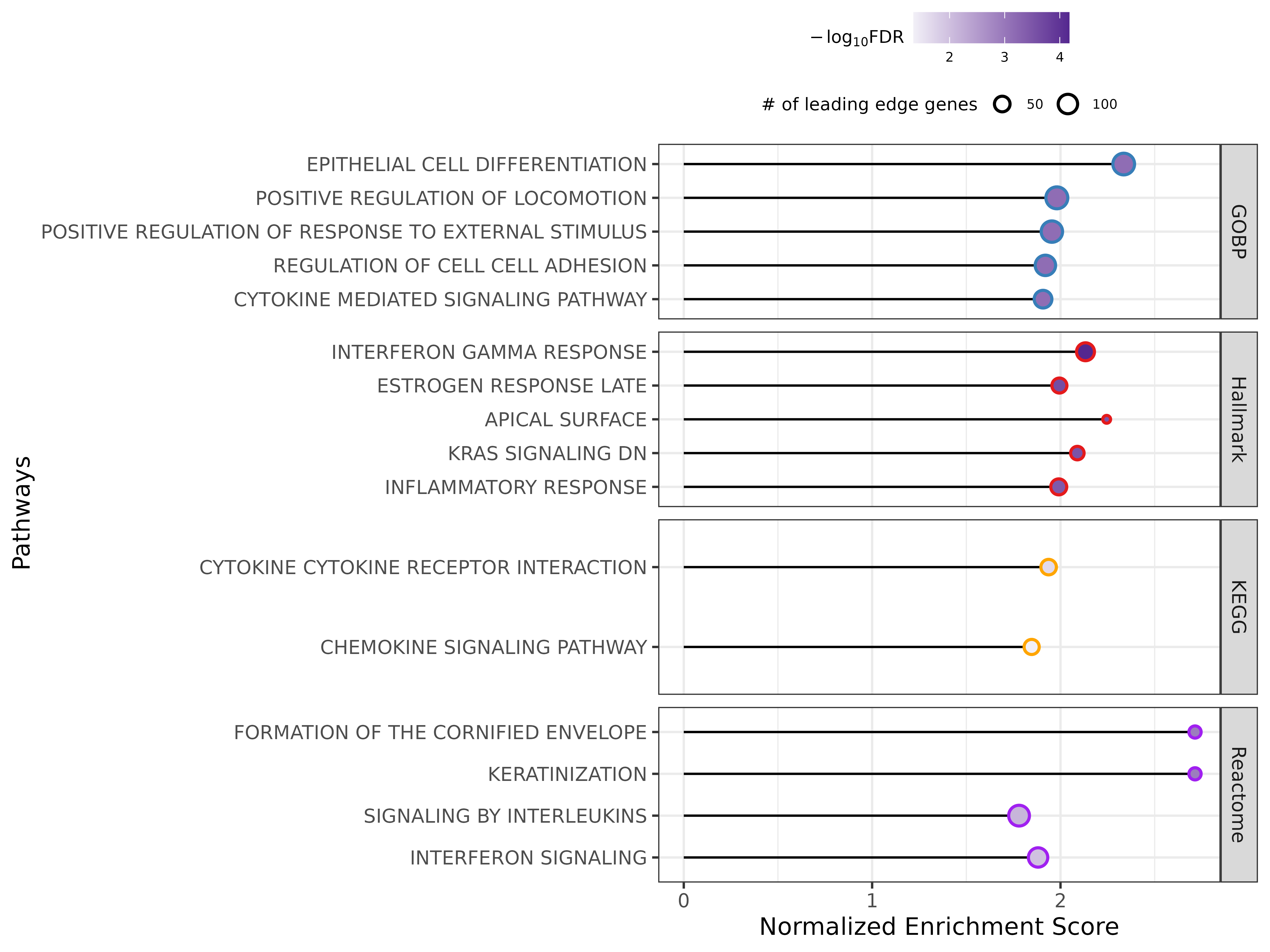{kind=link}
Supplementary figure S6a. Venn diagram of the most abundant cell types upregulated genes. After applying cut off threshold of <0.05 and log2FC >0.5 we identified 10 overlapping upregulated genes between them. ERJ-01947-2023.Figure_S6a (2.3MB, png)
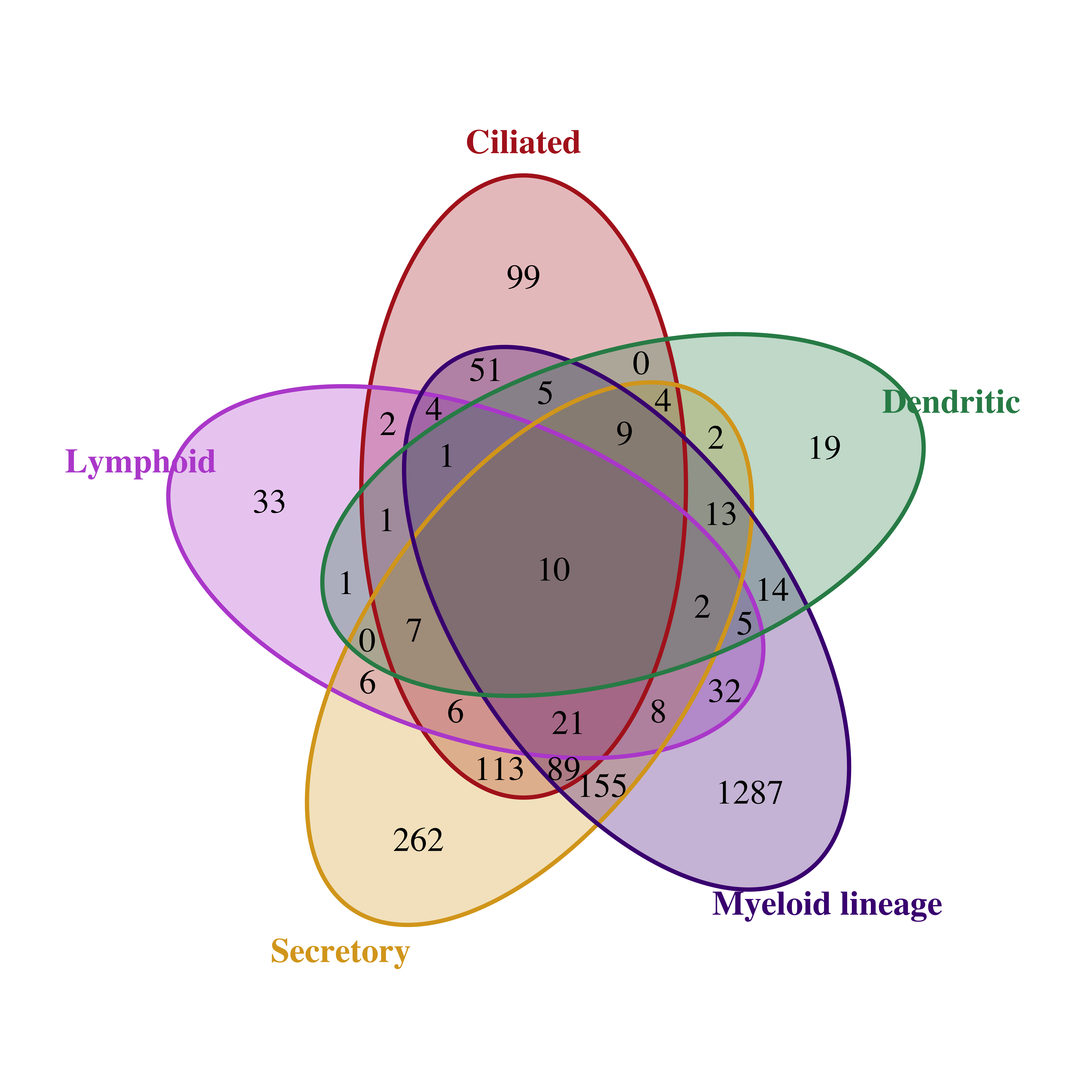{kind=link}
Supplementary figure S6b. GO pathway showing top 50 enriched pathways in the secretory cells including neutrophil mediated immunity. ERJ-01947-2023.Figure_S6b (327.3KB, png)
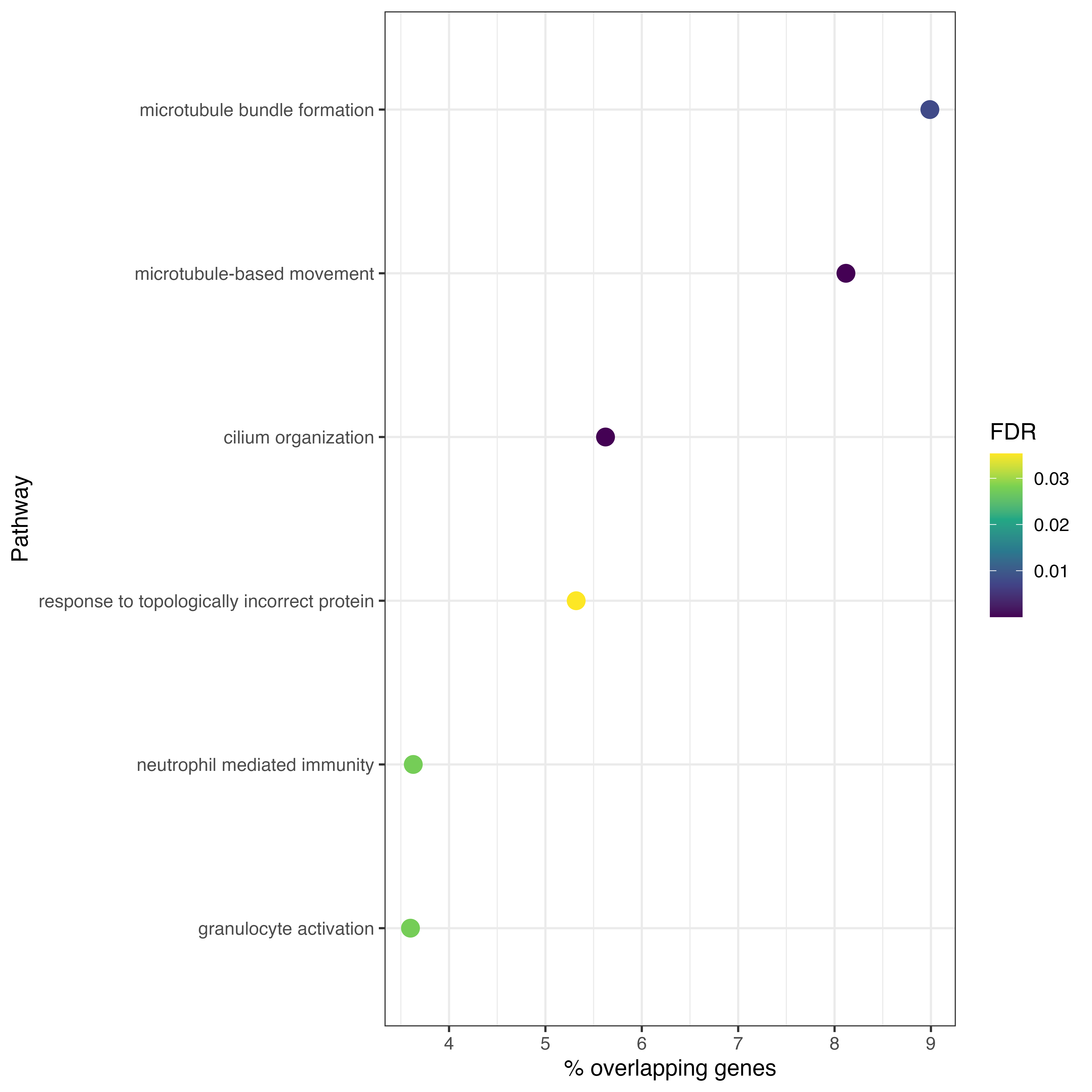{kind=link}
Supplementary figure S6c. GO pathway showing top 50 pathways in myeloid cells including leukocyte migration. ERJ-01947-2023.Figure_S6c (1.3MB, png)
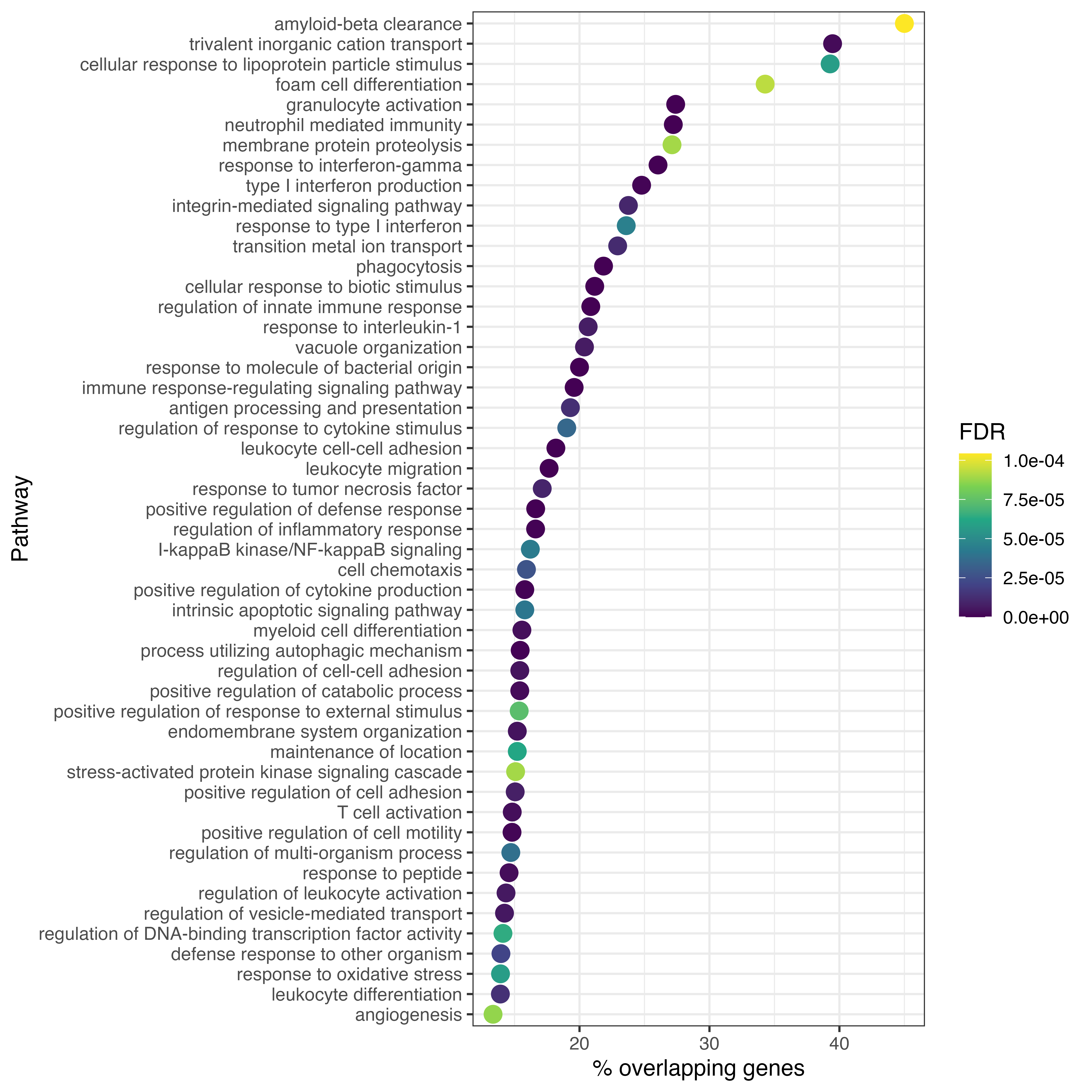{kind=link}
Shareable PDF
Acknowledgements
We would like to acknowledge the support of Yvonne Chung, Stephen Yu and Bernie Zhao (Biomedical Research Centre, The University of British Columbia, Vancouver, BC, Canada) and Andrew Warkman and Adriana Suarez-Gonzalez (10X Genomics, Pleasanton, CA, USA).
Footnotes
Ethics statement: Participants provided informed consent (University of British Columbia/Providence Health Care research ethics board approval H21-02149 and H19-02222).
Author contributions: F.V. Gerayeli, X. Li and C.X. Yang ran the analysis and co-wrote the method section. J. Tuong, S.M. Vahedi and G.J. Koelwyn analysed the IPA analysis and contributed to the method section. F.V. Gerayeli, H.Y. Park, S. Milne, X. Li, C.X. Yang, C.Y. Cheung, J. Tuong, G.J. Koelwyn and D.D. Sin were involved in interpretation of the results. F.V. Gerayeli, S. Milne, C.Y. Cheung, E. Guinto, R.L. Eddy, C. Gilchrist, J.S.W. Yang, T. Stach, D. Yehia, T. Shaipanich and J.M. Leung contributed to the experiment and data acquisition. F.V. Gerayeli, H.Y. Park and D.D. Sin prepared the first draft of the manuscript. All authors contributed to revision of the draft manuscript and approved the final draft of the manuscript.
Conflict of interest: F.V. Geraveli reports grants from MiTACS Accelerate. S. Milne reports payment or honoraria for lectures, presentations, manuscript writing or educational events from Chiesi Australia, The Limbic Australia and Research Review Australia, support for attending meetings from Sanofi Australia, Chiesi Australia and AstraZeneca, and a leadership role with Thoracic Society of Australia and New Zealand (NSW/ACT). R.L. Eddy reports grants from Michael Smith Health Research BC, Canadian Respiratory Research Network and Natural Sciences and Engineering Research Council Canada, consultancy fees from VIDA Diagnostics Inc., payment or honoraria for lectures, presentations, manuscript writing or educational events from Thorasys Thoracic Medical Systems Inc., and support for attending meetings from Canadian Institutes of Health Research – Institute of Circulatory and Respiratory Health. C. Gilchrist reports grants from Canadian institutes of Health Research. J. Leipsic reports consultancy fees from Heartflow, and stock (or stock options) with Heartflow. J.M. Leung reports support for the present study from Canadian Institutes of Health Research, grants from Canadian Institutes of Health Research and BC Lung Foundation, payment or honoraria for lectures, presentations, manuscript writing or educational events from BC Lung Foundation and University of British Columbia, participation on a data safety monitoring board with Enhance Quality Safety and Patient experience in Chronic Obstructive Pulmonary Disorder (EQuiP COPD), and leadership roles with Canadian Respiratory Research Network and the CanCOLD Study. D.D. Sin reports payment or honoraria for lectures, presentations, manuscript writing or educational events from GSK, AstraZeneca and Boehringer Ingelheim, and participation on a data safety monitoring board or advisory board with NHLBI. The remaining authors have no potential conflicts of interest to disclose.
Support statement: This study was supported by Canadian Institutes of Health Research (CIHR), Genome British Columbia, and Mitacs. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.World Health Organization (WHO) . WHO Coronavirus (COVID-19) Dashboard. https://covid19.who.int/. Date last accessed: 1 November 2023.
- 2.Greenhalgh T, Knight M, A'Court C, et al. Management of post-acute covid-19 in primary care. BMJ 2020; 370: m3026. doi: 10.1136/bmj.m3026 [DOI] [PubMed] [Google Scholar]
- 3.Proal AD, VanElzakker MB. Long COVID or post-acute sequelae of COVID-19 (PASC): an overview of biological factors that may contribute to persistent symptoms. Front Microbiol 2021; 12: 698169. doi: 10.3389/fmicb.2021.698169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis HE, McCorkell L, Vogel JM, et al. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol 2023; 21: 133–146. doi: 10.1038/s41579-022-00846-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis HE, Assaf GS, McCorkell L, et al. Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. EClinicalMedicine 2021; 38: 101019. doi: 10.1016/j.eclinm.2021.101019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gentilotti E, Górska A, Tami A, et al. Clinical phenotypes and quality of life to define post-COVID-19 syndrome: a cluster analysis of the multinational, prospective ORCHESTRA cohort. EClinicalMedicine 2023; 62: 102107. doi: 10.1016/j.eclinm.2023.102107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woodruff MC, Bonham KS, Anam FA, et al. Chronic inflammation, neutrophil activity, and autoreactivity splits long COVID. Nat Commun 2023; 14: 4201. doi: 10.1038/s41467-023-40012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Government of Canada . COVID-19: Longer-Term Symptoms Among Canadian Adults – Highlights. https://health-infobase.canada.ca/covid-19/post-covid-condition/. Date last accessed: 1 November 2023.
- 9.Milne S, Li X, Yang CX, et al. Inhaled corticosteroids downregulate SARS-CoV-2-related genes in COPD: results from a randomised controlled trial. Eur Respir J 2021; 58: 2100130. doi: 10.1183/13993003.00130-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerayeli FV, Eddy R, Sin DD. A proposed approach to pulmonary long COVID: a viewpoint. Eur Respir J 2024; 64: 2302302. doi: 10.1183/13993003.02302-2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerayeli FV, Milne S, Yang CX, et al. Single-cell RNA sequencing of bronchoscopy specimens: development of a rapid minimal handling protocol. Biotechniques 2023; 75: 157–167. doi: 10.2144/btn-2023-0017 [DOI] [PubMed] [Google Scholar]
- 12.10X Genomics . Chromium Single Cell 3' Reagent Kits User Guide (v3.1 Chemistry). 2019. https://cdn.10xgenomics.com/image/upload/v1660261285/support-documents/CG000204_ChromiumNextGEMSingleCell3_v3.1_Rev_D.pdf.
- 13.Young MD, Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience 2020; 9: giaa151. doi: 10.1093/gigascience/giaa151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Traag VA, Waltman L, van Eck NJ. From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep 2019; 9: 5233. doi: 10.1038/s41598-019-41695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franzén O, Gan LM, Björkegren JLM. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019; 2019: baz046. doi: 10.1093/database/baz046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hewitt RJ, Lloyd CM. Regulation of immune responses by the airway epithelial cell landscape. Nat Rev Immunol 2021; 21: 347–362. doi: 10.1038/s41577-020-00477-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaragosi LE, Deprez M, Barbry P. Using single-cell RNA sequencing to unravel cell lineage relationships in the respiratory tract. Biochem Soc Trans 2020; 48: 327–336. doi: 10.1042/BST20191010 [DOI] [PubMed] [Google Scholar]
- 18.Okuda K, Chen G, Subramani D, et al. Localization of secretory mucins MUC5AC and MUC5B in normal/healthy human airways. Am J Respir Crit Care Med 2019; 199: 715–727. doi: 10.1164/rccm.201804-0734OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis JD, Wypych TP. Cellular and functional heterogeneity of the airway epithelium. Mucosal Immunol 2021; 14: 978–990. doi: 10.1038/s41385-020-00370-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He L, Davila-Velderrain J, Sumida TS, et al. NEBULA is a fast negative binomial mixed model for differential or co-expression analysis of large-scale multi-subject single-cell data. Commun Biol 2021; 4: 629. doi: 10.1038/s42003-021-02146-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen R, Sang L, Jiang M, et al. Longitudinal hematologic and immunologic variations associated with the progression of COVID-19 patients in China. J Allergy Clin Immunol 2020; 146: 89–100. doi: 10.1016/j.jaci.2020.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiang CC, Korinek M, Cheng WJ, et al. Targeting neutrophils to treat acute respiratory distress syndrome in coronavirus disease. Front Pharmacol 2020; 11: 572009. doi: 10.3389/fphar.2020.572009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilk AJ, Rustagi A, Zhao NQ, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med 2020; 26: 1070–1076. doi: 10.1038/s41591-020-0944-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schulte-Schrepping J, Reusch N, Paclik D, et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 2020; 182: 1419–1440. doi: 10.1016/j.cell.2020.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Zhang K, Zhang Y, et al. Neutrophils in COVID-19: recent insights and advances. Virol J 2023; 20: 169. doi: 10.1186/s12985-023-02116-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Z, Ren L, Zhang L, et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 2020; 27: 883–890. doi: 10.1016/j.chom.2020.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.George PM, Reed A, Desai SR, et al. A persistent neutrophil-associated immune signature characterizes post-COVID-19 pulmonary sequelae. Sci Transl Med 2022; 14: eabo5795. doi: 10.1126/scitranslmed.abo5795 [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Zhou Z, Min W. Mitochondria, oxidative stress and innate immunity. Front Physiol 2018; 9: 1487. doi: 10.3389/fphys.2018.01487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dipankar P, Kumar P, Dash SP, et al. Functional and therapeutic relevance of Rho GTPases in innate immune cell migration and function during inflammation: an in silico perspective. Mediators Inflamm 2021; 2021: 6655412. doi: 10.1155/2021/6655412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeilhofer HU, Schorr W. Role of interleukin-8 in neutrophil signaling. Curr Opin Hematol 2000; 7: 178–182. doi: 10.1097/00062752-200005000-00009 [DOI] [PubMed] [Google Scholar]
- 31.Proost P, De Wolf-Peeters C, Conings R, et al. Identification of a novel granulocyte chemotactic protein (GCP-2) from human tumor cells. In vitro and in vivo comparison with natural forms of GRO, IP-10, and IL-8. J Immunol 1993; 150: 1000–1010. doi: 10.4049/jimmunol.150.3.1000 [DOI] [PubMed] [Google Scholar]
- 32.Nugteren S, Samsom JN. Secretory leukocyte protease inhibitor (SLPI) in mucosal tissues: protects against inflammation, but promotes cancer. Cytokine Growth Factor Rev 2021; 59: 22–35. doi: 10.1016/j.cytogfr.2021.01.005 [DOI] [PubMed] [Google Scholar]
- 33.Shao MX, Nadel JA. Neutrophil elastase induces MUC5AC mucin production in human airway epithelial cells via a cascade involving protein kinase C, reactive oxygen species, and TNF-α-converting enzyme. J Immunol 2005; 175: 4009–4016. doi: 10.4049/jimmunol.175.6.4009 [DOI] [PubMed] [Google Scholar]
- 34.Fischer BM, Voynow JA. Neutrophil elastase induces MUC5AC gene expression in airway epithelium via a pathway involving reactive oxygen species. Am J Respir Cell Mol Biol 2002; 26: 447–452. doi: 10.1165/ajrcmb.26.4.4473 [DOI] [PubMed] [Google Scholar]
- 35.Mehandru S, Merad M. Pathological sequelae of long-haul COVID. Nat Immunol 2022; 23: 194–202. doi: 10.1038/s41590-021-01104-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun J, Xiao J, Sun R, et al. Prolonged persistence of SARS-CoV-2 RNA in body fluids. Emerg Infect Dis 2020; 26: 1834–1838. doi: 10.3201/eid2608.201097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leiva-Juárez MM, Kolls JK, Evans SE. Lung epithelial cells: therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol 2018; 11: 21–34. doi: 10.1038/mi.2017.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carlier FM, de Fays C, Pilette C. Epithelial barrier dysfunction in chronic respiratory diseases. Front Physiol 2021; 12: 691227. doi: 10.3389/fphys.2021.691227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michalick L, Kuebler WM. TRPV4 – a missing link between mechanosensation and immunity. Front Immunol 2020; 11: 413. doi: 10.3389/fimmu.2020.00413 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary table S1. Curated gene list used for annotating the identified clusters from the Pegasus human lung, human immune legacy markers, and other resources including the Protein Atlas, PanglaoDB and the markers highlighted in literatures such as Hewitt et al. and Zaragosi et al. ERJ-01947-2023.Table_S1 (33.1KB, pdf)
Supplementary table S2. Results of the pre-ranked GSEA analysis of the differentially expressed genes of the pooled epithelial cells based on the negative binomial mixed model identified enriched pathways based on the reference Human Molecular Signatures Database (MSigDB), including gene ontology (GO), Reactom85, hallmark gene sets, and KEGG (KEGG MEDICUS, KEGG legacy gene sets). ERJ-01947-2023.Table_S2 (57.4KB, xlsx)
Supplementary table S3. The list of the genes correspondent to the predicted therapeutics based on the IPA database that could potentially reverse the transcriptomic signature across all epithelial cells. ERJ-01947-2023.Table_S3 (15.9KB, csv)
Supplementary figure S1. The comparison of composition distribution in the clusters between the PLC and control groups. Wilcoxon rank-sum test of the composition of the clusters between the PLC and control groups revealed the neutrophil cluster to be of statistical significance (p<0.05). a) Epithelial cells. ERJ-01947-2023.Figure_S1a (202.6KB, png)
Supplementary figure S1. b) Immune cells. ERJ-01947-2023.Figure_S1b (208.5KB, png)
Supplementary figure S2. Cytospin slides in PLC and control samples. Haematoxylin and eosin staining, original magnification ×35. a) Image representative of a PLC sample showing neutrophil presence. Neutrophils have been highlighted using yellow arrow. ERJ-01947-2023.Figure_S2a (1.4MB, tif)
Supplementary figure S2. b) Image representative of a control sample, showing lack of neutrophil presence. ERJ-01947-2023.Figure_S2b (1.1MB, tif)
Supplementary figure S3. RNA velocity and pseudotime analysis show majority of cells being in quiescent state. a) The RNA velocity dynamical map showing the full transcriptional dynamics of splicing kinetic estimating the parameters of reaction rates and latent cell-specific variables across all the identified clusters. b) The RNA velocity pseudotime based on the distance measures on the velocity graph, as well as expression of MKI67 proliferation marker across all the identified clusters in both PLC and control. Majority of the cells appears to be non-proliferating and at the quiescent state. c) Velocity confidence, calculated based on the rate of differentiation on the basis of the length of the velocity vector. It is providing a measure of confidence, as it showcases if the velocity direction is indeterminate. ERJ-01947-2023.Figure_S3 (515.3KB, tif)
Supplementary figure S4a. Figure S4 shows the top 10-13 IPA canonical pathways across all identified clusters that had >2% presence in the analysis. Excluding cells in transition. ERJ-01947-2023.Figure_S4a (1MB, jpg)
Supplementary figure S4b. ERJ-01947-2023.Figure_S4b (1MB, jpg)
Supplementary figure S4c. ERJ-01947-2023.Figure_S4c (1.1MB, jpg)
Supplementary figure S4d. ERJ-01947-2023.Figure_S4d (1.2MB, jpg)
Supplementary figure S4e. ERJ-01947-2023.Figure_S4e (909.9KB, jpg)
Supplementary figure S4f. ERJ-01947-2023.Figure_S4f (964.3KB, jpg)
Supplementary figure S4g. ERJ-01947-2023.Figure_S4g (1,002.9KB, jpg)
Supplementary figure S4h. ERJ-01947-2023.Figure_S4h (1.1MB, jpg)
Supplementary figure S4i. ERJ-01947-2023.Figure_S4i (1.1MB, jpg)
Supplementary figure S4j. ERJ-01947-2023.Figure_S4j (914.9KB, jpg)
Supplementary figure S4k. ERJ-01947-2023.Figure_S4k (1MB, jpg)
Supplementary figure S4l. ERJ-01947-2023.Figure_S4l (1.1MB, jpg)
Supplementary figure S4m. ERJ-01947-2023.Figure_S4m (994.8KB, jpg)
Supplementary figure S4n. ERJ-01947-2023.Figure_S4n (1,019.6KB, jpg)
Supplementary figure S4o. ERJ-01947-2023.Figure_S4o (965.2KB, jpg)
Supplementary figure S4p. ERJ-01947-2023.Figure_S4p (1.1MB, jpg)
Supplementary figure S4q. ERJ-01947-2023.Figure_S4q (1MB, jpg)
Supplementary figure S4r. ERJ-01947-2023.Figure_S4r (1.1MB, jpg)
Supplementary figure S4s. ERJ-01947-2023.Figure_S4s (1.1MB, jpg)
Supplementary figure S5. Enrichment of the epithelial cells from the differentially expressed genes based on the mixed effect model. ERJ-01947-2023.Figure_S5 (387.6KB, png)
Supplementary figure S6a. Venn diagram of the most abundant cell types upregulated genes. After applying cut off threshold of <0.05 and log2FC >0.5 we identified 10 overlapping upregulated genes between them. ERJ-01947-2023.Figure_S6a (2.3MB, png)
Supplementary figure S6b. GO pathway showing top 50 enriched pathways in the secretory cells including neutrophil mediated immunity. ERJ-01947-2023.Figure_S6b (327.3KB, png)
Supplementary figure S6c. GO pathway showing top 50 pathways in myeloid cells including leukocyte migration. ERJ-01947-2023.Figure_S6c (1.3MB, png)
This PDF extract can be shared freely online.
Shareable PDF ERJ-01947-2023.Shareable (1.3MB, pdf)