

Abstract
The combination of decreasing food intake and increasing energy expenditure represents a powerful strategy for counteracting cardiometabolic diseases such as obesity and type 2 diabetes1. Yet current pharmacological approaches require conjugation of multiple receptor agonists to achieve both effects2–4, and so far, no safe energy-expending option has reached the clinic. Here we show that activation of neurokinin 2 receptor (NK2R) is sufficient to suppress appetite centrally and increase energy expenditure peripherally. We focused on NK2R after revealing its genetic links to obesity and glucose control. However, therapeutically exploiting NK2R signalling has previously been unattainable because its endogenous ligand, neurokinin A, is short-lived and lacks receptor specificity5,6. Therefore, we developed selective, long-acting NK2R agonists with potential for once-weekly administration in humans. In mice, these agonists elicit weight loss by inducing energy expenditure and non-aversive appetite suppression that circumvents canonical leptin signalling. Additionally, a hyperinsulinaemic–euglycaemic clamp reveals that NK2R agonism acutely enhances insulin sensitization. In diabetic, obese macaques, NK2R activation significantly decreases body weight, blood glucose, triglycerides and cholesterol, and ameliorates insulin resistance. These findings identify a single receptor target that leverages both energy-expending and appetite-suppressing programmes to improve energy homeostasis and reverse cardiometabolic dysfunction across species.
Subject terms: Receptor pharmacology, Metabolic disorders, Homeostasis, Type 2 diabetes, Obesity
In mouse and nonhuman primate models, treatment with selective, long-acting neurokinin 2 receptor agonists aids weight loss by suppressing appetite and increasing energy expenditure, as well as by increasing insulin sensitivity.
Main
The development of long-acting pharmacotherapies based on the incretin hormones—glucagon-like peptide-1 (GLP-1), glucose-dependent insulinotropic peptide (GIP) and glucagon (GCG)—has been transformative for the treatment of cardiometabolic diseases such as obesity and type 2 diabetes (T2D)2,7–11. GLP-1 receptor agonists and GLP-1R/GIPR dual agonists are able to help an appreciable number of individuals living with obesity to achieve sustained weight loss similar to the levels obtained through the current ‘gold standard’, bariatric surgery12–14. Yet for people living with both obesity and T2D, a group of more than 380 million people globally, the weight-lowering efficacy of GLP-1-derived pharmacotherapies is significantly reduced compared with those individuals with obesity but without T2D10,12,15. The mechanistic underpinnings of this blunted weight-lowering efficacy of GLP-1 receptor agonists remain unknown but represent a major unmet need for the population with T2D. Additionally, although the current approved options and next generation of treatments (for example, amylin receptor agonists16) have made major strides in producing lasting and more tolerable appetite suppression, a key gap is the lack of a means to increase energy expenditure. The importance of targeting energy expenditure is particularly relevant given the steady decline in basal metabolic rate in the population over the past 40 years17.
In the 1930s, the mitochondrial uncoupler dinitrophenol demonstrated the powerful therapeutic potential of energy dissipation on weight loss18. However, dinitrophenol and more recent attempts to leverage energy expenditure, such as peripheral beta-adrenergic activation, have been burdened with narrow safety windows, cardiovascular concerns and translational hurdles across species1. Most recently, glucagon receptor (GCGR) agonism has proved to be an especially promising candidate for boosting catabolic metabolism3,4,19, although increases in heart rate and hepatic glucose production and questions surrounding loss of lean mass20,21 could potentially limit its applicability, particularly in the context of T2D. However, polypharmaceutical approaches that conjugate GCGR agonists to insulinotropic drivers such as GLP-1, with or without GIP (that is, triple or dual agonists, respectively) may help mitigate these undesirable effects and enable GCGR agonism to meaningfully contribute to treating cardiometabolic indications22.
From a T2D standpoint, despite insulin-sensitizing thiazolidinediones, GLP-1-based therapies and the sodium–glucose cotransporter 2 inhibitor (sGLT2i) class of glucose-lowering agents, some patients still progress to a regimen of increasing insulin doses to combat insulin resistance, which is likely to serve to further exacerbate weight gain. To this end, new mechanisms of glucose and lipid clearance have become attractive targets. Nevertheless, the landmark clinical breakthroughs ushered in by the incretin family solidly underscore the unprecedented therapeutic potential of G-protein-coupled receptors (GPCRs) across cardiometabolic indications. GPCRs are already the most druggable proteins throughout pharmacology owing, in large part, to their cell-type selectivity and surface accessibility23. Therefore, we initially set out to identify other GPCRs that might address remaining critical unmet needs for people living with both obesity and T2D. We identified a conserved GPCR pathway capable of not only peripherally promoting energy expenditure and insulin sensitization but also centrally reducing appetite.
Genetics of NK2R and metabolic health
To first enrich for candidate receptors that might be especially effective in a diabetic context, we leveraged the publicly available database, HugeAMP Type 2 Diabetes Knowledge Portal (T2D-KP)24, which contains human genetic associations assembled from over 350 studies. We compiled and ranked the loci of more than 380 non-odorant GPCRs according to their significance of association with haemoglobin A1c (HbA1c) levels, a primary clinical indicator of glucose control and diabetes progression25. We found that the most significant association was with the region containing the neurokinin 2 receptor (NK2R) gene (also known as tachykinin receptor 2 (TACR2)) (Fig. 1a). NK2R is a Gq-coupled receptor that is classically studied for its role in the gastrointestinal tract and the central nervous system5 (CNS) but—to our knowledge—has not previously been linked to glucose homeostasis or cardiometabolic health. However, the presence of two closely adjacent genes, tetraspanin 15 (TSPAN15) and hexokinase 1 (HK1), complicates the investigation of HbA1c and other trait associations to NK2R variants, particularly since HK1 is already significantly linked to HbA1c levels through effects on erythrocyte metabolism and turnover26,27.
Fig. 1. NK2R agonism is genetically and functionally linked to cardiometabolic protection.

a, Ranked P values for HbA1c associations of 381 non-odorant GPCR loci (±50 kb) from T2D-KP. b, Gq signalling and HbA1c associations of the NK2R missense variants I23T and R323H (n = 2 per variant). c, TWAS of HK1, NK2R and TSPAN15 and HbA1c levels, with and without adjustment for BMI. d,e, Obesity-related anthropometric associations of the NK2R non-coding variant rs139900276 in the Greenlandic cohort (d) and NK2R expression stratified by rs139900276 genotype (e). WHR, waist–hip ratio. f, Pharmacokinetics of NKA (n = 3 mice). g–k, In vivo effects of a twice daily subcutaneous (s.c.) injection of vehicle or 1 mg kg−1 NKA (g) on oxygen consumption (n = 6 mice per group) (h), food intake (i) and body weight (j) and white adipose tissue weight (k) at study conclusion (n = 6 (vehicle), n = 7 mice (NKA)). l, Insulin tolerance test of DIO mice treated with vehicle (n = 6) or NKA (n = 7) twice daily for 12 days. m, Pharmacokinetics of EB0014 (n = 3 mice). n,o, In vivo effects of daily subcutaneous injections of vehicle or 1 mg kg−1 EB0014 (n) on oxygen consumption (o; n = 6 mice per group). p–r, Dose-dependent changes in food intake (p), weight loss (q) and body composition (r) after 12 days of daily injections (n = 10 (vehicle, 0.1 mg kg−1), n = 9 (0.3 mg kg−1), n = 8 mice (1 mg kg−1)). Arrowheads indicate time of injection of vehicle or agonist (h,o). Data are mean ± s.e.m. (b,f,h–m,o–r); box plots present median and Tukey’s whiskers (e). *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001. Detailed statistics are in the Supplementary Information and Source Data.
Given this strong link between HbA1c and HK1, we initially focused on coding variants in NK2R from an exome sequencing dataset28. Functional investigation revealed that four missense variants, I23T (rs5030920; minor allele frequency (MAF) = 23.6%), R323H (rs61732393; MAF = 0.10%), V54I (rs151093941; MAF = 0.025%) and A161T (rs148031991; MAF = 0.102%), reduced NK2R signalling capacity of which two, I23T and R323H, were further associated with increased HbA1c levels in the UK Biobank (Fig. 1b and Extended Data Fig. 1a). We additionally found modest associations between these variants and fasting glucose, cholesterol, fat distribution, liver enzymes and total bilirubin (Extended Data Table 1). However, gene-based association tests for HbA1c, which were dominated by HK1 (Extended Data Table 2), suggested that missense variants alone would not provide a meaningful resolution of the specific effects attributable to NK2R.
Extended Data Fig. 1. Effect of NK2R genetic variants on receptor signaling.

a, Gq signalling and HbA1c associations of NK2R missense variants, V54I, A161T, T363A, T346M and H395R, (n = 2 per variant). b, Haplotype plot of HbA1c associations in the HKDC1-HK1-NK2R-TSPAN15 locus. Data are represented as mean ± s.e.m. Two-sided association tests without multiple corrections, a.
Extended Data Table 1.
Associations of NK2R variants R3232H (rs61732393-T), I23T (rs5030920-G), V54I (rs151093941-T) and A161T (rs148031991-T) with cardiometabolic traits and minor allele frequencies (MAF) in five different ancestries
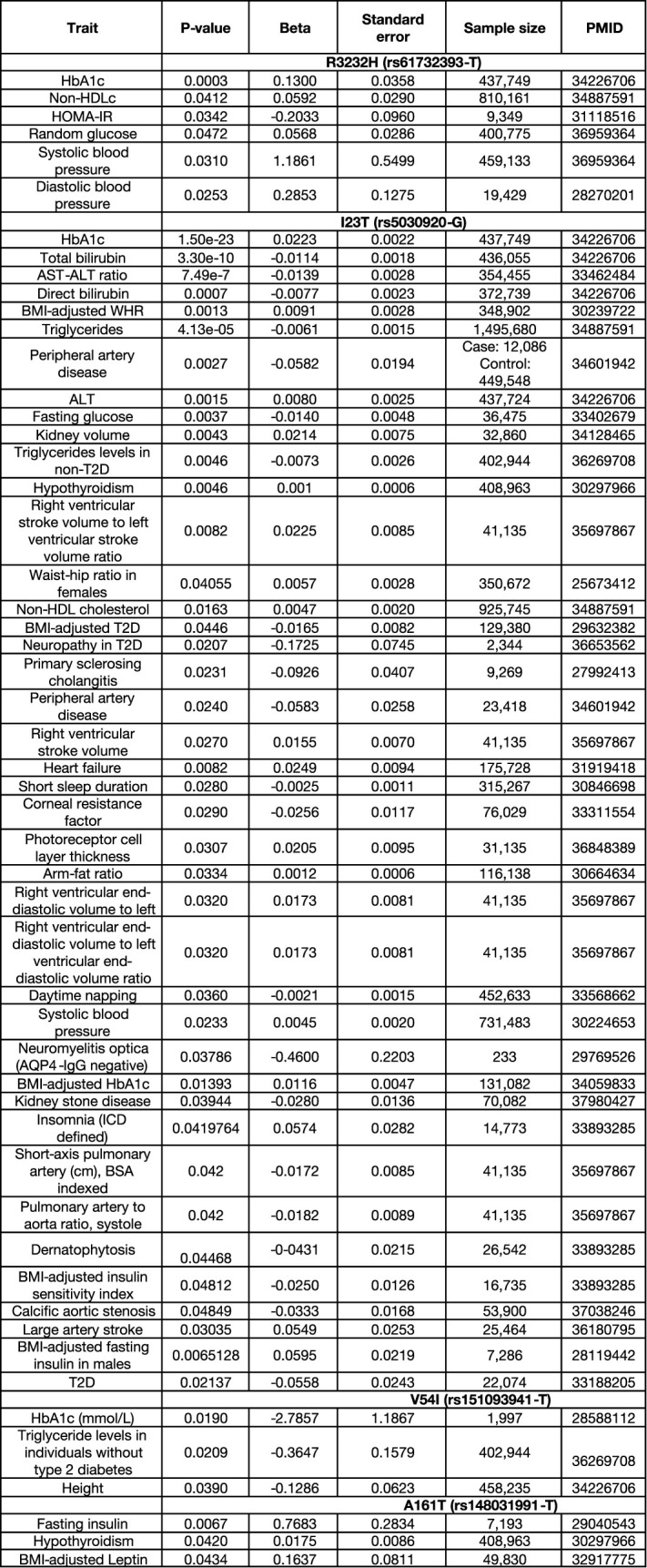
African American (AFR), Admixed American (AMR), East Asian (EAS), European (EUR) and South Asian (SAS). The beta (effect size), standard error, p-value and sample size are from the largest published European study for each trait, referenced with their PubMed ID (PMID). The association tests were two-sided without multiple corrections. HbA1c: Glycated hemoglobin; Non-HDLc: Non High-Density Lipoprotein Cholesterol; HOMA-IR: homeostatic model assessment – insulin resistance; AST-ALT ratio: Aspartate transferase - alanine transaminase ratio; BMI: body mass index; WHR: Waist-hip ratio; ALT: Alanine transaminase.
Extended Data Table 2.
Burden tests results for HK1, NK2R and TSPAN15 from Genebass

Three types of burden tests (Burden, SKAT and SKAT-O) for three types of exome variants (pLOF, missense and synonymous) were retrieved. Summary statistics of 9 NK2R synonymous variants are reported. The burden tests were two-sided without multiple corrections. A1: effect allele; A0: other allele; A1 frequency: allele frequency of the effect allele; P: p-value.
Therefore, to more comprehensively investigate NK2R genetic contributions, we assessed summary statistics on HbA1c associations in the greater NK2R-containing locus (including the adjacent genes hexokinase domain-containing 1 (HKDC1), HK1 and TSPAN15) from a European-ancestry meta-analysis comprising 438,069 individuals29. In this region, we identified 978 variants with genome-wide significant associations for HbA1c (P < 5 × 10−8) of which 16 were independent lead variants30 (r2 < 0.01 within ±1 Mb) (Table 1). Fine mapping of HbA1c associations using CARMA31 revealed 9 candidate causal variants with a posterior inclusion probability of less than 0.1 (Table 2). These variants represent four distinct signals within HKDC1 (three variants), HK1 (four variants), NK2R (one variant) and TSPAN15 (one variant) (Extended Data Fig. 1b). The NK2R variant rs791147 (MAF = 26.2–48.4%) is intronic and significantly associated with NK2R expression in several tissues, including the brain, adipose tissue, macrophages and skeletal muscle (Table 3), suggesting that it may regulate NK2R expression and HbA1c levels. For a better resolution of tissue-specific expression regulation and HbA1c associations, we performed gene-based transcriptome-wide association studies (TWASs) using PrediXcan. We found that increased NK2R expression in the nucleus accumbens (ACB) of the brain was associated with decreased HbA1c levels, with or without adjustment for body mass index (BMI), whereas expression of HK1 and TSPAN15 were not (Fig. 1c and Table 4).
Table 1.
Results for linkage disequilibrium (LD) clumping in the HKDC1–HK1–NK2R–TSPAN15 region (10:70929740–71367422)
| Variant | Base-pair location | A1 | A0 | AFR MAF | AMR MAF | EAS MAF | EUR MAF | SAS MAF | Beta | s.e. | P | Closest gene (consequence) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs143849423 | 10:70969723 | G | T | 0.0008 | 0.0140 | 0.0000 | 0.0159 | 0.0050 | 0.0363 | 0.0064 | 1.74 × 10−8 | HKDC1 (downstream gene variant) |
| rs72814215 | 10:70998703 | G | A | 0.0008 | 0.0060 | 0.0000 | 0.0139 | 0.0070 | 0.0580 | 0.0090 | 1.21 × 10−10 | HKDC1 (intron variant) |
| 10:71002040 | 10:71002040 | T | TG | NA | NA | NA | 0.4105 | NA | 0.0653 | 0.0019 | 7.52 × 10−257 | HKDC1 (intron variant) |
| 10:71069872 | 10:71069872 | T | TA | NA | NA | NA | 0.1777 | NA | 0.0204 | 0.0027 | 8.62 × 10−14 | HK1 (NA) |
| rs5030918 | 10:71078526 | A | C | 0.000 | 0.0010 | 0.0000 | 0.0149 | 0.0020 | 0.2872 | 0.0063 | 0 | HK1 (intron variant) |
| rs2015803 | 10:71081399 | T | C | 0.4675 | 0.4860 | 0.2480 | 0.2664 | 0.2700 | 0.0455 | 0.0021 | 6.32 × 10−101 | HK1 (intron variant) |
| 10:71118821 | 10:71118821 | AAG | A | NA | NA | NA | 0.0401 | NA | 0.3077 | 0.0049 | 0 | HK1 (NA) |
| rs9299503 | 10:71136050 | A | G | 0.3109 | 0.4510 | 0.1935 | 0.4930 | 0.1935 | 0.0533 | 0.0020 | 1.87 × 10−157 | HK1 (intron variant) |
| rs4745984 | 10:71143488 | C | A | 0.0144 | 0.3520 | 0.3254 | 0.0984 | 0.0700 | 0.0472 | 0.0031 | 1.79 × 10−53 | HK1 (intron variant) |
| 10:71144995 | 10:71144995 | C | CTT | NA | NA | NA | 0.0635 | NA | 0.0418 | 0.0039 | 1.20 × 10−26 | HK1 (NA) |
| rs117056999 | 10:71154564 | T | C | 0.0030 | 0.0100 | 0.0000 | 0.0288 | 0.0030 | 0.0400 | 0.0054 | 1.22 × 10−13 | HK1 (intron variant) |
| rs1236903 | 10:71198994 | C | G | 0.0030 | 0.0320 | 0.0000 | 0.0795 | 0.0150 | 0.0269 | 0.0040 | 1.77 × 10−11 | TSPAN15 (intergenic) |
| rs77356330 | 10:71205544 | C | T | 0.0030 | 0.0320 | 0.000 | 0.0378 | 0.0060 | 0.0308 | 0.0042 | 2.48 × 10−13 | TSPAN15 (regulatory region variant) |
| rs72811732 | 10:71227549 | A | T | 0.0008 | 0.0190 | 0.0000 | 0.0368 | 0.0030 | 0.0444 | 0.0053 | 7.96 × 10−17 | TSPAN15 (intron variant) |
| rs11598811 | 10:71288098 | A | G | 0.0023 | 0.0760 | 0.0000 | 0.0716 | 0.0150 | 0.0307 | 0.0037 | 1.46 × 10−16 | NEUROG3 (upstream gene variant) |
Results for the 16 independent variants (r2 < 0.01 in 1,000 kb) in the region with minor allele frequencies from 5 genetic ancestries: African (AFR), admixed American (AMR), East Asian (EAS), European (EUR) and South Asian (SAS). The variant ID is from dbSNP, the base-pair locations are in build 37 and the annotations are from Variant Effect Predictor. The association tests were two-sided without multiple corrections. A1, effect allele; A0, other allele; s.e., standard error; NA, variant not found in the reference panel.
Table 2.
HbA1c associations of the nine genome-wide significant and causal variants in the overlapping the HKDC1–HK1–NK2R–TSPAN15 region (10:70929740–71367422)
| Variant | Base-pair location | A1 | A0 | AFR MAF | AMR MAF | EAS MAF | EUR MAF | SAS MAF | Beta | s.e. | P | Consequence | PIP | Lead (r2) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs72814215 | 10:70998703 | G | A | 0.0008 | 0.0060 | 0.0000 | 0.0139 | 0.0070 | 0.0580 | 0.0090 | 1.21 × 10−10 | HKDC1 intron | 1.0000 | rs72814215 (r2 = 1.00) |
| rs112258245 | 10:71001310 | A | G | 0.0219 | 0.1100 | 0.0000 | 0.1670 | 0.0450 | 0.0334 | 0.0026 | 5.92 × 10−37 | HKDC1 intron | 0.6636 | rs5785903 (r2 = 0.26) |
| rs72814226 | 10:71001449 | G | C | 0.0219 | 0.110 | 0.0000 | 0.1670 | 0.0450 | 0.0333 | 0.0026 | 7.91 × 10−37 | HKDC1 intron | 0.3364 | rs5785903 (r2 = 0.26) |
| rs10823338 | 10:71065977 | G | T | 0.4145 | 0.5060 | 0.2649 | 0.2515 | 0.2700 | 0.0386 | 0.0022 | 1.03 × 10−67 | HK1 intron | 1.0000 | rs2015803 (r2 = 0.79) |
| rs16926246 | 10:71093392 | C | T | 0.1747 | 0.0810 | 0.0010 | 0.1412 | 0.0540 | 0.2642 | 0.0027 | 0 | HK1 intron | 1.0000 | rs5030918 (r2 = 0.07) |
| rs17476364 | 10:71094504 | T | C | 0.0015 | 0.0610 | 0.0010 | 0.0994 | 0.0170 | 0.3189 | 0.0030 | 0 | HK1 intron | 1.0000 | rs5030918 (r2 = 0.10) |
| rs6480402 | 10:71095378 | A | C | 0.3767 | 0.2590 | 0.1319 | 0.2942 | 0.4350 | 0.1301 | 0.0021 | 0 | HK1 intron | 1.0000 | rs5030918 (r2 = 0.02) |
| rs7911347 | 10:71170287 | A | C | 0.2912 | 0.4310 | 0.2619 | 0.4841 | 0.3190 | 0.0105 | 0.0019 | 2.99 × 10−8 | NK2R intron | 0.9965 | rs200572185 (r2 = 0.02) |
| rs142394825 | 10:71236395 | G | C | 0 | 0.0100 | 0 | 0.0119 | 0.0010 | 0.0888 | 0.0083 | 1.25 × 10−26 | TSPAN15 intron | 1.0000 | rs4745984 (r2 = 0.09) |
Results for the associations of 9 candidate causal (posterior inclusion probability (PIP) > 0.1) HbA1c variants in the 10:70929740–71367422 region. Fine mapping was computed with CARMA software utilizing HbA1c European summary statistics from ref. 29. All variants are located in introns, three in HKDC1, four in HK1, one in NK2R and one in TSPAN15. Additionally, the minor allele frequencies for five different genetic ancestries were added: African, admixed American, East Asian, European and South Asian. Finally, for each candidate causal variant the lead variant with the highest correlation measured in r2 was added. The variant ID is from dbSNP, the base-pair locations are in build 37 and the annotations from Variant Effect Predictor. The association tests were two-sided without multiple corrections.
Table 3.
eQTL associations for r7911347-A available in Open Target Genetics
| rs7911347-A | Adipose (Fusion) | Adipose (TwinsUK) | Brain DLPFC (ROSEMAP) | Muscle (FUSION) | Macrophage (ref. 61) | Monocyte influenza 6 h (ref. 62) |
|---|---|---|---|---|---|---|
| HK1 | NA | NA | NA | NA | NA | NA |
| NK2R | 0.397, P = 3.2 × 10−33 | 0.370, P = 1.2 × 10−19 | 0.165, P = 2.7 × 10−10 | 1.11, P = 4.0 × 10−63 | 0.294, P = 8.3 × 10−13 | NA |
| TSPAN15 | NA | NA | 0.106, P = 3.5 × 10−5 | NA | NA | 0.549, P = 1.9 × 10−13 |
Results are included for the tissues with reported expression quantitative trait loci (eQTL) associations for HK1, NK2R and TSPAN15. No eQTL associations were found for HK1. The association tests were two-sided without multiple corrections. NA, not applicable.
Table 4.
TWAS and sensitivity analysis of the association between NK2R expression and HbA1c levels in the ACB
| Gene | TWAS | Sensitivity analysis | Trait | ||
|---|---|---|---|---|---|
| Effect size | P value | Effect size | P value | ||
| NK2R | −0.147312885 | 5.57322 × 10−48 | −0.13674113 | 1.75 × 10−7 | HbA1c |
| NK2R | −0.047902365 | 1.60712 × 10−14 | −0.05624646 | 2.29 × 10−2 | HbA1c (BMI-adjusted) |
| HK1 | −0.005043655 | 0.647995 | 0.02991872 | 5.08 × 10−1 | HbA1c |
| HK1 | 0.000288765 | 0.9628904 | −0.01008011 | 8.27 × 10−1 | HbA1c (BMI-adjusted) |
| TSPAN15 | −0.032080736 | 0.004094338 | −0.05479775 | 4.60 × 10−1 | HbA1c |
| TSPAN15 | 0.003103694 | 0.5906543 | −0.01864292 | 9.05 × 10−1 | HbA1c (BMI-adjusted) |
P values were calculated using a two-sided z-test. Since only three genes were considered, nominal P values without applying a genome-wide false-discovery rate correction are reported.
We next complemented the large-scale analyses of Western Europeans with an assessment of NK2R variant associations in the more isolated Greenlandic population, which has a unique genetic architecture due to its geographical location and population history. The Greenlandic population has similar rates of cardiometabolic disease as Europeans and has previously revealed valuable genetic insights that are masked or absent in larger European cohorts32. Associations with traits related to glucose homeostasis were not found in the Greenlandic cohort; however, there were modest associations with obesity-related phenotypes (Extended Data Table 3). Specifically, the strongest associations were observed for the non-coding variant rs139900276 in the 5′ untranslated region of NK2R. This variant was significantly associated with reduced BMI, body weight, waist–hip ratio, waist–height ratio and fat percentage (Fig. 1d). Furthermore, carriers of rs139900276 in the Greenlandic population had significantly higher expression of NK2R (by transcriptional profiling of whole blood) (Fig. 1e), suggesting that NK2R expression was inversely related to obesity parameters. Collectively, the glycaemic and obesity genetic associations from different populations suggest that NK2R signalling may have a role in energy homeostasis in humans, and therefore represents an interesting target for metabolic investigation.
Extended Data Table 3.
Summary of association analyses with metabolic traits for rs139900276

Effect sizes reported in standard deviations for the rs139900276 CG-allele from association analyses run with an additive model. The association tests were two-sided without multiple corrections.
To evaluate the effect of pharmacological activation of NK2R signalling on mammalian metabolic homeostasis in vivo, the endogenous NK2R ligand, neurokinin A (NKA), was administered subcutaneously to diet-induced obese (DIO) mice. Mice were dosed twice daily, given that native NKA has a markedly short half-life of minutes (Fig. 1f). This regimen of NKA administration (Fig. 1g) was well tolerated and acutely increased oxygen consumption (Fig. 1h). During 9 days of repeated injections in DIO mice, food intake (Fig. 1i), body weight (Fig. 1j) and inguinal (iWAT) and epididymal (eWAT) white adipose tissue (Fig. 1k) were reduced in NKA-treated mice and insulin tolerance was improved relative to vehicle controls (Fig. 1l). Yet despite the beneficial effects of NK2R agonism, the rapid clearance of NKA precluded an accurate assessment of the bona fide therapeutic potential of NK2R. Therefore, we generated a longer-acting peptide (hereby referred to as EB0014) in which a 16-carbon fatty acid, called C16-gammaGlu (C16GG, the side chain used in the GLP-1R agonist liraglutide33), was covalently attached to Lys2 of native NKA to increase retention in the blood from minutes to hours, probably via albumin binding (Fig. 1m). Daily injections of EB0014 (Fig. 1n) robustly induced oxygen consumption (Fig. 1o), decreased food intake during the initial days of treatment (Fig. 1p) and dose-dependently reduced body weight (Fig. 1q), which was driven by loss of fat mass (Fig. 1r). Transient loose stools were initially noted in the mice at the highest dose but the compound was otherwise well tolerated. These functional data uncover a paradigm of homeostatic control whereby NK2R agonism is capable of simultaneously inducing energy expenditure and decreasing food intake. The genetic variants provide further support that this signalling pathway affects HbA1c in humans and pharmacological activation of NK2R might be a new route through which to counteract cardiometabolic diseases.
Development of long-acting NK2R agonists
Although NKA and long-acting EB0014 mediated several beneficial cardiometabolic effects, it remained unknown whether these effects were mediated by NKR2. NKA is a naturally unselective peptide within the tachykinin family, and signals through both NK1R and NK3R with nearly equal potency to its cognate receptor, NK2R6 (Fig. 2a). This lack of selectivity is a particular liability with respect to NK1R signalling, which is linked to CNS disorders, inflammation, cardiopulmonary disruption, bronchoconstriction and nociception (pain sensing)5. Thus, despite the encouraging efficacy of the protracted NKA molecule, EB0014, NK2R selectivity is necessary to pharmacologically leverage this signalling pathway for therapeutic gain. As a starting point for developing highly NK2R-specific long-acting agonists, we took advantage of a previously described truncated NKA analogue, neurokinin A (4–10)-S5K/L9mL/M10Nle (where mL is methylleucine and Nle is norleucine), which has been shown to exhibit some degree of selectivity34.
Fig. 2. Development and characterization of first-in-class selective, long-acting NK2R agonists.

a, Signalling schematic for the tachykinin receptor family along with respective endogenous ligands, substance P (SP), NKA and neurokinin B (NKB), adapted from ref. 6. b, Sequences of NKA and protracted, selective NK2R agonists. c, Ligand-induced Gq signalling of human tachykinin receptors in vitro (n = 2 per ligand). d, Pharmacokinetic profile of NK2R agonists (n = 3 mice per group). e–k, In vivo effects of a single injection of EB1002 (e; inset shows the implanted body temperature monitor) on oxygen consumption (f), fatty acid oxidation (g), body temperature (h), food intake (i), RER (j) and weight loss (k) in DIO mice. In f–j, arrowheads indicate time of injection of vehicle or EB1002. Plot colours in f–k match key in f. n = 6 (vehicle), n = 5 (EB1002) (f–i); n = 8 (vehicle), n = 9 (EB1002) (k). l–r, Evaluation of in vivo selectivity of EB1002 with or without pre-administration of the NK2R antagonist saredutant in DIO mice (l) on oxygen consumption (m), fatty acid oxidation (n), body temperature (o), food intake (p), RER (q) and weight loss (r). In m–q, arrowheads indicate 0.5 h pretreatment with vehicle or saredutant followed by EB1002. Plot colours in m–r match key below l. n = 6 per group (m,n,p–r); n = 5 per group (o). s,t, Glucose tolerance (s) and insulin level (t) of DIO mice (n = 8 (vehicle), n = 15 (325 nmol kg−1 EB1002), n = 16 (pair-fed with the EB1002-treated group)). u, Setup of the hyperinsulinaemic–euglycaemic clamp study. v,w, Glucose infusion rate (GIR) (v) and glucose uptake into iWAT and eWAT depots (w), for a hyperinsulinaemic–euglycaemic clamp of lean mice 16 h after a single injection of vehicle (n = 7) or EB1002 (n = 8). Data are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001. Detailed statistics are in the Supplementary Information and Source Data.
A single replacement of Phe at position 6 with a structurally similar Tyr conferred complete NK2R selectivity and generated the analogue neurokinin A (4–10)-S5K/F6Y/L9mL/M10Nle (called EB1001 here) (Fig. 2b). In the EB1000 compound series, we also changed the protraction strategy, substituting C16GG with a 2×(8-amino-3,6-dioxaoctanoic acid)-γGlu-C18-diacid moiety (C18DOGG; the side chain used in the GLP-1R agonist semaglutide) owing to its superior albumin affinity and water solubility compared with C16GG. We further optimized EB1001 by replacing the Nle at position 10 with a methoxinine (Mox) residue, to preserve hydrogen-bonding capacity, in an analogue, neurokinin A (4–10)-S5K/F6Y/L9mL/M10Mox (called EB1002 here) (Fig. 2b). Like EB1001, EB1002 is highly selective against NK1R on both mouse tachykinin receptors (Extended Data Fig. 2a,b) and human tachykinin receptors (Fig. 2c and Extended Data Fig. 2c) and has a markedly longer half-life in mice (10.3 h for EB1002 compared with 5.5 h for EB1001) (Fig. 2d).
Extended Data Fig. 2. Development and acute testing of NK2R selective agonists.

a, Mouse tachykinin receptor signaling with endogenous ligands and EB1001, b, mouse tachykinin receptor signaling with endogenous ligands and EB1002, and, c, human tachykinin receptor signaling with endogenous ligands and EB1001, (n = 2 per variant for a-c). d, Cumulative locomotor activity of DIO mice following a single injection of vehicle (n = 6 mice) or 325 nmol/kg (n = 5 mice). e, Serum liver enzymes (n = 4 (ALT EB1002), n = 5 mice for remaining data) and f, representative microscopy images of various tissues of CD-1 mice after injections with vehicle or increasing concentrations of EB1002 (see methods, 7500 nmol/kg EB1002 prior to euthanasia), scale bars 100 µm. g, Oxygen consumption, h, fatty acid oxidation, i, body temperature, j, food intake, k, RER, and, l, weight loss of DIO Nk2r KO mice following a single injection of vehicle or 325 nmol/kg EB1002 (n = 6 mice per group for e, f, h-j, l, n = 5 mice per group for g), downward triangles signify time of injection. m, clamped blood glucose, n, steady state mouse and human insulin levels and o, glucose uptake into metabolically active tissues for a hyperinsulinemic-euglycemic clamp of lean mice following a single injection of vehicle (n = 7 mice) or 325 nmol/kg EB1002 (n = 8 mice). p, in vitro glucose uptake (n = 6 each condition) and q, oxygen consumption of primary white adipocytes (WA) in response to various concentrations of EB1002 (n = 6 (vehicle), n = 8 (each EB1002 concentration)). r, ex vivo lipolysis of mature inguinal WA in response to various concentrations of EB1002 and 50 nM isoproterenol (n = 3 each condition). Representative western blot images and quantification of s, liver, t, gastrocnemius, u, BAT and, v, iWAT of DIO mice treated with vehicle or 325 nmol/kg EB1002 18 h prior to injections with a mock solution or insulin (n = 5 (vehicle/mock), n = 6 (EB1002/mock, vehicle/insulin) n = 7 mice (EB1002/insulin)). Data are represented as mean ± s.e.m. For all: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001. Unpaired two-tailed t-test of AUC for 48 h after injection, d, g-k; unpaired two-tailed t-test, e, l, n, o; repeated measures two-way ANOVA with Geisser-Greenhouse correction, significance indicates treatment effect, m; ordinary one-way ANOVA with Tukey’s multiple comparisons test, significance shows post hoc test for treatment effect, p, r; ordinary one-way ANOVA of AUC for baseline, compound (cpd), NE and FCCP increments, q; and ordinary two-way ANOVA with Sidak’s multiple comparison, significance indicates post hoc test between vehicle and EB1002 treated mice, s-v. Uncropped blots are presented in Supplementary Fig. 1.
To assess the mode of action of NK2R agonism, DIO mice were implanted with telemetric body temperature monitors and given a single subcutaneous injection of EB1002 (Fig. 2e). EB1002 administration led to a significant and sustained increase in oxygen consumption (Fig. 2f) and fatty acid oxidation (Fig. 2g) that was also reflected in a modest increase in core temperature (Fig. 2h). Concurrent with enhancing energy expenditure, a single administration of EB1002 also strongly decreased food intake (Fig. 2i) and respiratory exchange ratio (RER) (Fig. 2j) for more than 24 h. Collectively, these acute metabolic effects reduced body weight (Fig. 2k) without any overt signs of discomfort or changes in physical activity (Extended Data Fig. 2d). To further evaluate safety and toxicity, CD-1 mice were subjected to a dose escalation up to 7,500 nmol kg−1. This dose amounted to more than 20 times the dose that we used in our mouse efficacy models. We did not observe any changes in serum liver enzymes (Extended Data Fig. 2e) or pathohistological signs (Extended Data Fig. 2f and Extended Data Table 4). Consistent with previous knowledge of neurokinin biology5, transient appearance of loose stools was observed at the highest doses and thus served to inform tolerance limits for EB1002 dosing. Notably, the efficacy of EB1002 was abolished both in genetic Nk2r knockout (Extended Data Fig. 2g–l) and after acute pharmacological NK2R antagonism (Fig. 2l–r), confirming robust in vivo selectivity.
Extended Data Table 4.
Overview of gross changes in tissue architecture and cellar appearances following pharmacological pre-toxicology study

Mice were either injected with vehicle or EB1002 in a dose-up fashion up to 7500 nmol/kg. Tissues were taken for pathohistological evaluation and compared in a pair-wise fashion. Comp. to vehicle – compared to vehicle; ncd – no change detected. 1) slightly increased amount of fibrosis around individual large vessels; 2) no epithelium in the sample; 3) Modest amount of liver tissue; 4) cornea, iris and lens not present; 5) few small areas with infiltration; 6) a smaller focus, possibly pressure influence from isolation; 7) less infiltration towards the lumen.
We next investigated the effects of NK2R agonism on glucose metabolism. EB1002 significantly improved glucose tolerance in DIO mice 24 h after a single dose compared to mice that were either treated with vehicle only or treated with vehicle and pair-fed with EB1002-treated animals (Fig. 2s,t), showing that the glycaemic correction occurred independent of changes in insulin secretion or food intake reduction. To determine whether the improvement in glucose control was due to altered insulin sensitivity, we performed a hyperinsulinaemic–euglycaemic clamp study in chow-fed mice (Fig. 2u). Mice treated with a single dose of EB1002 required a 50% higher glucose infusion rate during steady state compared to vehicle controls (Fig. 2v and Extended Data Fig. 2m,n), revealing a profound increase in insulin sensitivity. EB1002 trended to enhance oxidative skeletal muscle glucose disposal (Extended Data Fig. 2o) and significantly increased glucose uptake into white adipose depots (Fig. 2w). However, NK2R-agonist-induced effects on white adipocyte glucose uptake or other metabolic functions, including oxygen consumption and lipolysis, were not cell autonomous (Extended Data Fig. 2p–r). We next sought to determine how NK2R agonism impacted tissue-specific insulin signalling in vivo. EB1002 acutely and significantly increased insulin-induced AKT phosphorylation in liver, gastrocnemius and brown adipose tissue (BAT) (Extended Data Fig. 2s–u). Despite the increased insulin-stimulated glucose uptake in WAT we observed during the clamp, there was no clear change in insulin-induced AKT phosphorylation in iWAT after EB1002 administration (Extended Data Fig. 2v). Together, these findings reveal that selective, long-acting NK2R agonism elicits dual regulation of energy expenditure and food intake and potently improves insulin sensitivity.
NK2R agonism improves metabolic health
Repeated daily administration of EB1002 significantly and sustainably reduced body weight (Fig. 3a and Extended Data Fig. 3a) at least in part owing to transient suppression of food intake (Fig. 3b) in DIO mice. Notably, injecting animals every other day with EB1002 was just as effective as daily administration at producing longer-term weight loss over the course of 21 days (Extended Data Fig. 3a). However, we observed that the body weights of NK2R-treated animals began to increase gradually over the 21 days of injections, raising potential concerns of desensitization (Extended Data Fig. 3a). Thus, we tracked the weight re-gain of the cohort after injections were ceased and then re-administered a single injection of EB1002 to all mice (Extended Data Fig. 3b). Regardless of previous compound exposure, all mice exhibited similar weight loss (Extended Data Fig. 3b,c), demonstrating that there was no lasting desensitization induced by systemic NK2R agonism. Unexpectedly, the single injection of EB1002 resulted in 10–15% weight loss over 7 days (Extended Data Fig. 3c), suggesting a far more sustained efficacy than predicted by blood occupancy of the peptide.
Fig. 3. Improvement of systemic energy homeostasis by long-acting NK2R agonism.

a,b, Weight loss (a) and food intake (b) of DIO mice injected daily with vehicle or EB1002 (n = 6 per group). WT, wild type. c, Weight loss of DIO mice that were transitioned to chow diet or continued on HFD 5 days before daily injections (n = 6 (HFD vehicle), n = 8 (chow vehicle, chow EB1002 and HFD EB1002)). Arrowhead indicates first injection. d–g, Plasma concentrations of GLP-1 (d), leptin (e), glucagon (f) and PYY (g) of DIO mice injected daily with vehicle (n = 7) or EB1002 (n = 8). h, Body composition of DIO mice injected daily with vehicle (n = 11) or EB1002 (n = 15). i–k, Weight loss (i), food intake (j) and body composition (k) of DIO Nk2r-knockout (KO) mice injected daily with vehicle (n = 8) or EB1002 (n = 9). l, Weight loss of DIO mice injected daily with vehicle (n = 11), EB1002 (n = 15) or vehicle and pair-fed with EB1002-treated mice (n = 12). m–t, Schematic of DIO Ucp1-knockout mice at thermoneutrality (m) used to assess weight loss (n), blood glucose concentration (o), RER (p), fatty acid oxidation (q) and oxygen consumption (r), and linear regression of oxygen consumption versus body weight (s) and body temperature (t) after a single injection of vehicle, EB1002 or vehicle and pair-fed with EB1002-treated mice. In p–r,t, arrowheads indicate time of injection, diamonds indicate replenishment of food for the pair-fed group. In n–t, n = 6 per group. u,v, Weight loss (u) and food intake (v) of DIO, thermoneutrally housed Ucp1-knockout mice injected daily with vehicle (n = 6) or EB1002 (n = 7). w–z, Setup of triple-chip study to investigate the anatomical resolution of thermogenic output (w) with interscapular temperature (x; n = 3), hindlimb temperature (y; n = 5) and abdominal temperature (z; n = 5) of DIO mice after a single injection of EB1002 (indicated by arrowheads (x–z)). Data are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001. Detailed statistics are in the Supplementary Information and Source Data.
Extended Data Fig. 3. In vivo effects of repeated NK2R agonist dosing.

a, Weight loss of DIO mice that have been injected s.c. daily with vehicle or 325 nmol/kg EB1002 (q.d. EB1002), or every other day with 325 nmol/kg EB1002 (q.o.d. EB1002) (n = 10 mice per group), and b, weight trajectory of the same mice including a washout period and a single re-injection with 325 nmol/kg EB1002, with c, weight loss after re-injection. d, Faecal lipid content and e, faecal cholesterol content of DIO mice that have been injected s.c. daily with vehicle or 325 nmol/kg EB1002 (n = 6 mice (d1 vehicle and d7 vehicle and EB1002), n = 12 mice (d1 EB1002)). f, Weight loss of DIO mice that were transitioned to chow diet or continued on HFD 5 days prior to daily s.c. injections with vehicle or 325 nmol/kg EB1002, data normalized to d0 (n = 6 mice (HFD vehicle), n = 8 mice (chow vehicle, chow EB1002 and HFD EB1002)). g, Adipose tissue weights of DIO mice that have been injected s.c. daily with vehicle (n = 11 mice) or 325 nmol/kg EB1002 (n = 15 mice) for 7 days. h, Weight loss, i, body composition, j, adipose depot weights, k, blood glucose, l, food intake, m, RER, n, oxygen consumption, o, fatty acid oxidation, and p, body temperature of female DIO mice that have been injected s.c. daily with vehicle or 325 nmol/kg EB1002 (n = 6 mice per group (f, I, j-m), n = 17 mice per group (g, h), n = 6 mice (vehicle) and n = 5 mice (EB1002) for n), downward triangles signify time of injection. q, Adipose depot weights of DIO Nk2r KO mice that have been injected s.c. daily with vehicle (n = 8 mice) or 325 nmol/kg EB1002 (n = 9 mice). r, Interscapular temperature (n = 3 mice), s, hindlimb temperature (n = 5 mice), t, abdominal temperature (n = 5 mice) of DIO mice after a single s.c. injection of vehicle, downward triangles signify time of vehicle injection. u, Schematic setup of triple chip study to interrogate anatomical resolution of thermogenic output with v, interscapular temperature, w, hindlimb temperature, and x, abdominal temperature of BAT denervated DIO mice after a single injection of 325 nmol/kg EB1002 and vehicle (n = 3 mice), downward triangles signify time of injection of the indicated compound. y, Representative western blot images and quantification of BAT from sham operated or BAT denervated mice (n = 6 (sham), n = 4 mice (DNV), each BAT lobe has been analyzed separately). Data are represented as mean ± s.e.m. For all: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001. Repeated measures two-way ANOVA with Geisser-Greenhouse correction and Tukey’s multiple comparison test, significance indicates post hoc test for treatment effect at last timepoint, a, f; repeated measures two-way ANOVA with Geisser-Greenhouse correction, significance indicates treatment effect, c, h; repeated measures two-way ANOVA with Geisser-Greenhouse correction and Sidak’s multiple comparison test, significance indicates post hoc test for treatment effects, d, e; unpaired two-tailed t-test, g, i, j, q, y; repeated measures two-way ANOVA and Sidak’s multiple comparison, significance indicates time effect, k; unpaired two-tailed t-test of AUC of 24 h increments, l-p.
In the initial phase of systemic agonism, we found that EB1002 acutely increased faecal triglyceride content and trended to increase faecal cholesterol (Extended Data Fig. 3d,e), suggesting a potential for reduced lipid absorption and/or an induced release of lipids into the intestine. Moreover, the efficacy of NK2R agonism was not dependent on ingestion of high-fat diet (HFD), as DIO mice that were transitioned from HFD to chow diet 5 days before EB1002 treatment to mimic a dietary intervention, displayed similar weight loss to mice that were maintained on HFD (Fig. 3c and Extended Data Fig. 3f). We next investigated whether acute or repeated administration of an NK2R agonist affected the blood levels of hormones known to modulate feeding and metabolism. A single injection of EB1002 did not change the concentrations of GLP-1, leptin, glucagon or peptide YY (PYY) (Fig. 3d–g). However, leptin and glucagon were significantly decreased and PYY was significantly increased following a week of daily injections (Fig. 3d–g), suggesting that NK2R-agonist-induced whole-body metabolic improvements engage other homeostatic-regulating hormone pathways.
Body composition was assessed after 7 days of daily injections to determine the cause of the weight reduction. We found that NK2R-agonist-induced weight loss was solely attributable to decreased adiposity (Fig. 3h and Extended Data Fig. 3g) whereas lean mass was spared (Fig. 3h). There were no sex-dependent differences in response to NK2R agonism as EB1002-treated female DIO mice displayed similar changes in body weight, body composition, adipose depot weights, acute blood glucose control, food intake, RER, oxygen consumption, fatty acid oxidation and body temperature (Extended Data Fig. 3h–p). The effects of repeated daily EB1002 injections were abolished in animals lacking Nk2r (Fig. 3i–k and Extended Data Fig. 3q), reinforcing the selectivity of the agonist. The dependence of NK2R-agonist-mediated weight loss on both food intake (Fig. 2i) and energy-expending mechanisms (Fig. 2f), was further evidenced when comparing EB1002-treated animals to pair-fed controls (Fig. 3l).
The translational potential of energy-expending modes-of-action can be confounded by the reliance of rodent physiology on BAT thermogenesis. Therefore, we evaluated the efficacy of NK2R agonism in thermoneutrally housed DIO mice lacking the canonical BAT effector, uncoupling protein 1 (UCP1) (Fig. 3m). To distinguish effects from reduced food intake versus increased energy expenditure, we included a pair-fed Ucp1-knockout group that was matched to EB1002-treated mice. In DIO Ucp1-knockout mice, acute EB1002 treatment significantly reduced body weight and blood glucose compared to vehicle-injected and pair-fed mice (Fig. 3n,o), demonstrating NK2R agonism elicited UCP1-independent mechanisms of weight loss and glucose control. A single EB1002 injection into mice lacking Ucp1 reduced RER more robustly than pair feeding over 2 days (Fig. 3p) and increased fatty acid oxidation (Fig. 3q), oxygen consumption (Fig. 3r,s) and body temperature (Fig. 3t). The observation that oxygen consumption and body temperature of pair-fed mice were lower than those of the the vehicle group suggested that EB1002 induced an even greater degree of energy expenditure than previously estimated. Repeated daily injections of EB1002 in DIO Ucp1-knockout mice at thermoneutrality reduced weight and food intake (Fig. 3u,v) to similar magnitudes as our earlier studies in wild-type mice.
We next sought to investigate the anatomical contributions and dynamics of NK2R-induced energy expenditure. Whereas our standardized in vivo model involves implanting temperature probes that are free-moving throughout the peritoneal cavity, we surgically anchored telemetric temperature probes to three regions of each mouse to achieve spatial resolution (Fig. 3w): (1) embedded in the interscapular region, between the brown adipose tissue and shoulder muscles; (2) positioned along the upper thigh, running parallel to the quadriceps muscle; and (3) situated against the abdominal wall of the peritoneal cavity. Interscapular BAT depots were surgically denervated in a subset of the mice. Compared with vehicle injection in the same mice, a single subcutaneous injection of EB1002 robustly increased interscapular temperature acutely and continuously over 48 h (Fig. 3x and Extended Data Fig. 3r). However, hindlimb temperature was initially blunted before progressively increasing between 2 and 5 days after injection (Fig. 3y and Extended Data Fig. 3s), revealing a multiphasic, temporal distribution of thermogenic output. Abdominal temperature was initially blunted before returning to the pre-injection pattern (Fig. 3z and Extended Data Fig. 3t). Similar patterns in all three anatomical regions were observed in the BAT-denervated mice (Extended Data Fig. 3u–y). Although it remains unclear how NK2R agonism orchestrates the observed anatomical and temporal dynamics, the ability of a single administration of EB1002 to impart effects over the course of a week is in line with the timeframe of earlier weight loss studies in which one injection led to progressive weight reduction over 7 days (Extended Data Fig. 3b,c). Collectively, these findings show that long-acting NK2R-induced improvements in metabolic outcomes are driven by both reduced food intake and increased energy expenditure, which occurs dynamically across multiple tissues and independently of the canonical BAT activity.
NK2R agonism in the CNS and periphery
We next assessed NK2R agonism in the context of genetic obesity and canonical hypothalamic appetite-regulating circuitry35,36. EB1002 was subcutaneously administered to DIO mice or hyperphagic models lacking leptin (ob/ob) or the melanocortin 4 receptor (Mc4r-knockout) (Extended Data Fig. 4a). Surprisingly, the same dose of EB1002 (325 nmol kg−1) that moderately lowered food intake in non-hyperphagic DIO mice robustly attenuated appetite in both male and female ob/ob mice (Extended Data Fig. 4b,), indicating that NK2R agonism not only regulates food intake independently of leptin signalling but also seems to be potentiated in the absence of leptin. However, this potentiated action of NK2R agonism was not observed in mice lacking Mc4r (Extended Data Fig. 4b), suggesting that NK2R food intake regulation was still, to some extent, dependent on canonical appetite-regulating signalling downstream of leptin. The markedly increased efficacy of NK2R agonism in leptin-deficient mice was also reflected in body weight (Extended Data Fig. 4c) and blood glucose (Extended Data Fig. 4d), which were sustained significantly lower even 48 h after injection despite food intake beginning to increase back towards baseline (Extended Data Fig. 4e).
Extended Data Fig. 4. Anatomical resolution of NK2R agonism.

a, Genetic models of hyperphagic obesity used to interrogate NK2R agonist-dependent appetite suppression. b, Food intake following a single s.c. administration of vehicle or 325 nmol/kg EB1002 to DIO, male and female leptin deficient (ob/ob) and Mc4r KO mice (n = 6 mice (DIO, vehicle), n = 5 mice (DIO, EB1002), n = 8 mice (male ob/ob, vehicle; female ob/ob), n = 6 mice (male ob/ob, EB1002), n = 11 mice (Mc4r KO, vehicle), n = 8 mice (Mc4r KO, EB1002)). c, Weight loss, d, blood glucose and e, food intake of ob/ob mice housed at thermoneutrality following a single s.c. injection of vehicle or 325 nmol/kg EB1002 (n = 6 mice each group). f, Schematic of crossover study comparing in vivo effects of peripheral versus central delivery of EB1002 on g, RER, h, body temperature following s.c. or ICV injection (n = 6 (vehicle s.c injections and EB1002 ICV injections), n = 4 (EB1002 s.c. injections and vehicle ICV injections)), downward triangles signify time of injection (g, h). i, RER, j, oxygen consumption, k, food intake, l, weight loss and m, blood glucose of ob/ob mice following a single s.c. injection of vehicle or 40 nmol/kg EB1002 (n = 4 mice each group). Data are represented as mean ± s.e.m. For all: *P ≤ 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001. Unpaired two-tailed t-test of AUC b; Repeated measures two-way ANOVA with Geisser-Greenhouse correction, significance indicates treatment effect, c, e; Repeated measures two-way ANOVA with Sidak’s multiple comparison test, significance shows post hoc test for treatment effects, d; unpaired two-tailed t-test of AUC for 48 h after injection, g-k; unpaired two-tailed t-test, l; Repeated measures two-way ANOVA, m.
To distinguish between peripheral and central contributions of NK2R signalling, we performed a pharmacological study comparing subcutaneous and intracerebroventricular (ICV) administration of EB1002. HFD-fed ob/ob mice were housed in indirect calorimetric cages at thermoneutrality and metabolic readouts were assessed after a single subcutaneous injection of vehicle or EB1002. After wash-out, the mice received an ICV dose in a crossover design (Fig. 4a and Extended Data Fig. 4f). Central delivery of EB1002 robustly reduced food intake (Fig. 4b) and RER (Extended Data Fig. 4g) similarly to peripheral administration, indicating that appetite could be directly controlled by the CNS. However, ICV delivery of EB1002 significantly decreased oxygen consumption (Fig. 4c), consistent with an effect primarily on food intake without a simultaneous boosting or maintenance of energy expenditure as was observed following subcutaneous injection (Fig. 4c). In line with the oxygen consumption measurements, body temperature was more robustly increased by subcutaneous injection compared with ICV delivery, yet central administration was still able to elicit a distinct temperature increase during the light phase (Extended Data Fig. 4h). Thus, thermogenic contributions of NK2R signalling appear to emanate largely from peripherally accessible regions but direct contributions from the CNS cannot be entirely ruled out. Even without inducing energy expenditure, the EB1002 dose delivered centrally produced weight reductions similar to peripheral dosing (Fig. 4d), probably owing to the more marked suppression of food intake. Notably however, blood glucose was only significantly decreased by subcutaneous administration (Fig. 4e), suggesting that acute glycaemic control was weight-independent and required peripheral NK2R signalling. The effects of the ICV dose were CNS-specific as peripheral administration of the ICV concentration, equivalent to 40 nmol kg−1, elicited no effects on metabolic parameters (Extended Data Fig. 4i–m). These findings collectively suggest that NK2R activation acts via the periphery and CNS to simultaneously suppress appetite, increase energy expenditure and improve insulin sensitivity.
Fig. 4. Distinct central and peripheral actions of long-acting NK2R agonism.

a, Study design comparing effects of central and peripheral delivery of EB1002. b–e, Food intake (b), oxygen consumption (c), weight loss (d) and blood glucose concentration (e) following subcutaneous or ICV injection (all plot colours as in key in b). n = 6 (vehicle subcutaneous injections, EB1002 ICV injections), n = 4 (EB1002 subcutaneous injections, vehicle ICV injections) (b,c); n = 6 (vehicle subcutaneous injections), n = 10 (EB1002 subcutaneous injections), n = 8 (vehicle ICV injections), n = 13 (EB1002 ICV injections) (d,e). Arrowheads indicate injections (c). f,g, Representative FOS staining in the DVC (f; scale bar, 200 µm) and quantification of FOS-positive cells in the NTS, AP and DMV (g) of mice injected with vehicle (n = 6) or EB1002 (n = 8). h, Preference ratio for vehicle, semaglutide or EB1002 (n = 10 per group). i, Representative microscopy images and quantification of FOS colocalization with reporter neurons expressing GFP in the NTS of Leprcre L10 GFP (n = 3 (vehicle), n = 6 (EB1002)), Cckcre L10 GFP (n = 2 (vehicle), n = 3 (EB1002)) and Calcrcre Sun1 GFP mice (n = 6 (vehicle), n = 7 (EB1002)) injected with vehicle or EB1002. Scale bars, 50 µm. Plot colours as in key in b. j, Identity of FOS+ neurons in the NTS of EB1002-injected mice. k, Uniform manifold approximation and projection (UMAP) plot of expression data from 23,664 neurons coloured by populations according to ref. 39. l, Immediate early gene (IEG) expression in DVC snRNA-seq data from mice injected with vehicle or EB1002 (n = 6 samples with 5 mice per sample). m, Schematic of AAV injection into the DVC of Nk2r-floxed mice. n,o, Food intake (n) and weight loss (o) of Nk2rDVC-GFP and Nk2rDVC-cre mice injected with vehicle or EB1002 (n = 5 per group). p–r, Group average FOS heat map (p; scale bar, 500 µm), volcano plot of brain subregions (q) and heat map of activated brain regions involved in feeding, energy expenditure and reward (r) of wild-type DIO mice injected with vehicle or EB1002 (n = 8 per group). Data in j are mean, and data in all other graphs are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001. Detailed statistics are in the Supplementary Information and Source Data.
We next sought to gain insight into which brain regions might contribute to the NK2R-mediated suppression of feeding. We found that administration of EB1002 acutely and significantly reduced food intake during refeeding following an overnight fast (Extended Data Fig. 5a). Given that the activity of Agouti-related peptide-expressing neurons (AgRP neurons) mediates a significant portion of refeeding, this result suggests that EB1002 could, at least in part, overcome AgRP neuron-driven feeding. The activity of AgRP neurons can be blocked through the activation of neurons in the dorsomedial hypothalamus (DMH) or the dorsal vagal complex (DVC). To evaluate the potential activation of these sites (as well as other regions known to regulate food intake, including the paraventricular nucleus of the hypothalamus (PVH), arcuate nucleus (ARH) and the parabrachial nucleus (PBN)), we assessed FOS immunoreactivity (FOS-IR) 2 h after EB1002 administration in overnight-fasted mice. NK2R-agonist-induced changes in FOS-IR were detected only in the DVC (Extended Data Fig. 5b,c). Within this region, FOS-IR was primarily increased in neurons within the nucleus of the solitary tract (NTS) and to a lower extent in the area postrema (AP); we found no change in the dorsal motor nucleus of the vagus (DMV) (Fig. 4f,g). Whereas the AP and NTS each restrain food intake as part of the physiological response to a meal, some AP and NTS neurons also promote aversive signals37,38. Thus, we performed a saccharin-based conditioned taste avoidance test with EB1002 treatment, revealing no conditioned taste aversion formation by NK2R agonism (unlike the aversive GLP-1R agonist semaglutide) (Fig. 4h). We additionally assessed gastric emptying rate (also modulated by the DVC) but found no difference following acute administration of EB1002 when compared to vehicle (Extended Data Fig. 5d). Thus, although EB1002 activates neurons in the DVC, the appetite suppression by the compound appears to occur independently of aversive responses or alterations in gastric emptying.
Extended Data Fig. 5. Central effects of NK2R agonism.

a, Food intake in over-night fasted mice that have been s.c. injected with vehicle or 325 nmol/kg EB1002 at time of refeeding (n = 10 mice per group). b, Schematics of brain sections (adopted from The Allen Brain Atlas) and representative images of FOS staining (purple) with c, quantification of FOS positive cells in the paraventricular nucleus of the hypothalamus (PVH), the dorsomedial hypothalamic nucleus (DMH), arcuate nucleus (ARH), the parabrachial nucleus (PBN) and the dorsal vagal complex (DVC) of overnight fasted mice injected with vehicle or 325 nmol/kg EB1002 2 h prior to euthanasia (n = 4 mice per group), scale bar 200 µm. d, Liquid phase gastric emptying of mice s.c. injected with vehicle or 325 nmol/kg EB1002 (n = 10 mice each group). Representative microscopy images and quantification of FOS (purple) colocalization with reporter neurons (green) in the DVC of e, LeprCreL10 GFP (n = 3 (vehicle), n = 6 (EB1002)), f, CckCreL10 GFP (n = 2 (vehicle), n = 3 (EB1002)), and, g, CalcrCre Sun1 GFP mice (n = 6 (vehicle), n = 7 (EB1002)) injected with vehicle or EB1002, scale bar 200 µm. h, UMAP plot of 23,664 neurons colored by treatment, i, violin plot, and j, transcriptional case-control analysis of single nuclei RNA sequencing data of the DVC of mice s.c. injected with vehicle or 325 nmol/kg EB1002 2 h prior to euthanasia (n = 6 samples with 5 mice/sample). k, in situ hybridization of Nk2r (red), Calcr (green) and Glp1r (purple) in the DVC, scale bar: top panel 200 µm, bottom panel 50 µm. l, Representative microscopy images of the hit sites, scale bar 200 µm. m, Representative microscopy images of Fluoro-Jade C stain in the DVC of Nk2r floxed mice that received a bilateral injection of AAV-GFP (Nk2rDVC-GFP) or AAV-Cre (Nk2rDVC-Cre) into the DVC, scale bar 200 µm, (n = 3 mice per group). Data are represented as mean ± s.e.m. For all: *P ≤ 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001. Repeated measures two-way ANOVA with Geisser-Greenhouse correction, significance indicates treatment effect, a; unpaired two-tailed t-test, c, d, m; for j red color indicates *P < 0.05.
To map which neuronal populations in the DVC were activated by NK2R agonism, we used reporter mice that labelled Lepr-, Cck- or Calcr-expressing neurons. Whereas EB1002 activated a subset of neurons within these established hunger-regulating populations (Fig. 4i and Extended Data Fig. 5e–g), the majority of FOS-positive neurons were distinct from these canonical populations (Fig. 4j). To further characterize the cellular identity of DVC neurons activated by EB1002, we generated a single-nucleus RNA-sequencing (snRNA-seq) dataset of the DVC from vehicle- and EB1002-treated mice. Transcriptionally defined cell classes were identified with known neuronal and glial markers. The neuronal population was subclassified and labelled on the basis of a publicly available DVC atlas39, and 23,664 nuclei were categorized into 25 neuronal populations (Fig. 4k). Cells from both treatment groups were evenly distributed across all neuronal clusters (Extended Data Fig. 5h). Quantification of immediate early gene expression and transcriptional differences revealed Glu3 neurons39, a Gal- and Lepr-expressing population of glutamatergic NTS neurons (Extended Data Fig. 4i), to be the most responsive to EB1002 (Fig. 4l and Extended Data Fig. 5j).
Given that Nk2r is expressed in the NTS (Extended Data Fig. 5k) and ICV administration strongly regulated food intake (Fig. 4b), we assessed the importance of Nkr2 expression in the DVC for the response to EB1002. We directly injected GFP- or Cre-expressing adeno-associated virus (AAV) into the DVC of Nk2r-floxed mice (Fig. 4m and Extended Data Fig. 5l) (called Nk2rDVC-GFP or Nk2rDVC-cre, respectively, here) and found that EB1002 was still able to acutely suppress food intake and reduce body weight in Nk2rDVC-cre mice (Fig. 4n,o). Although we observed a trend towards increased cell death in the AAV-Cre-injected DVCs compared with AAV-GFP controls (Extended Data Fig. 5m), the preservation of EB1002 effects in these mice suggests that direct NK2R signalling in the DVC is not required for the efficacy of systemic NK2R agonism. These data do not preclude the possibility that NTS neurons, which would be downstream from a peripheral NK2R-induced signal, might mediate EB1002 action.
To comprehensively decode the CNS contributions of NK2R agonism in a global, unbiased manner, we next resolved whole-brain FOS activation using iDISCO (immunolabelling-enabled three-dimensional imaging of solvent-cleared organs) imaging after a peripheral administration of vehicle or EB1002, using DIO mice fasted for the 2 h dosing period (as opposed to the overnight-fasted chow-fed mice in Fig. 4f,g and Extended Data Fig. 5b,c). This analysis revealed the widespread activation of FOS by EB1002 compared with vehicle (Fig. 4p,q), suggesting potentially different responses to NK2R agonism for obese mice compared with lean mice. In DIO mice, EB1002 stimulated FOS activation in many brain regions involved in the control of food intake, reward and energy balance and/or targeted by clinically validated weight-lowering drugs40, including, but not limited to, the PVH, NTS, AP, DMV, PBN, ARH, DMH, ACB, lateral hypothalamic area (LHA) and parasubthalamic nucleus (PSTN) (Fig. 4r). The effect of EB1002 further extended to the ventral and lateral areas of the cortical plate (Fig. 4r), which are associated with the processing of olfactory and taste information, consistent with a potential role of NK2R agonism on sensory processing related to food intake. These complex networks of NK2R action in the CNS suggest that long-acting NK2R agonism exerts systemic control through the coordinated engagement of numerous homeostasis and behaviour-regulating neuronal populations.
Effects of NK2R agonism in primates
In light of translational hurdles between rodents and higher mammals, we next sought to explore NK2R agonism in a cohort of obese male and female rhesus macaques (nonhuman primates) with cardiometabolic disease. At the time when the macaque study was initiated, EB1001 was the most advanced NK2R agonist developed, and was therefore the compound selected. EB1001 was administered daily through subcutaneous injection in a dose-escalation study design over eight weeks to determine tolerability and explore potential efficacy (Fig. 5a). The dosing period was followed by a two-week wash-out period. To our knowledge, selective NK2R agonism has not previously been tested in nonhuman primates, and the neurokinin receptor family has been linked to several adverse side effects5. Therefore, a dedicated primate behavioural specialist monitored the animals throughout the study, and compound exposure was assessed at each dose (Extended Data Fig. 6a). In line with most obesity and T2D therapeutic agents, gastrointestinal events were the primary side effects. Although there were no incidents of nausea, emesis or diarrhoea at any dose, there was a peak in looser stools at the highest dose of 480 nmol kg−1 (Extended Data Fig. 6b), along with a concurrent spike in subdued, withdrawn behaviour (Extended Data Fig. 6c). Gastrointestinal effects abruptly ceased after a return to the preceding tolerated dose of 240 nmol kg−1, suggesting a profile driven by maximum serum concentration (Cmax), and withdrawn behaviour followed similarly. Importantly, there were no indications of anxiety or anxiety-like behaviours (for example, agitation, pacing or unrest) at any dose (Fig. 5b). Moreover, there was no adverse effect on cardiopulmonary (Fig. 5c and Extended Data Fig. 6d), hepatic (Fig. 5d and Extended Data Fig. 6e) or renal (Extended Data Fig. 6f) parameters. Thus, these dose-escalation findings establish a safety window for evaluating NK2R agonism in obese macaques.
Fig. 5. NK2R agonism safely counteracts cardiometabolic disease in diabetic, obese macaques.

a, Schematic of EB1001 dose-escalation study in rhesus macaques (nonhuman primates (NHPs)). b–d, Anxiety-like behaviour (b), heart rate (c) and blood alanine transaminase concentration over the course of the dose-escalation study (n = 10 macaques). e, Stratification of macaque groups on the basis of diabetic status. f–k, Changes in body weight (f), food intake (g), fasting blood glucose (h), insulin level (i), HOMA-IR (j) and insulin:C-peptide ratio (k) for normoglycaemic (n = 3) and diabetic (n = 7) macaques. All plot colours as in key in f. l, Changes in triglyceride concentrations for all macaques over the course of the study (n = 10 macaques). m–o, Stratification of macaque groups on the basis of baseline cholesterol levels (m) and changes in total cholesterol (n) and LDL cholesterol (o) over the course of the dose escalation. All plot colours as in key in n. n = 4 macaques (low baseline cholesterol), n = 6 macaques (high baseline cholesterol). Data are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001. Detailed statistics are in the Supplementary Information and Source Data.
Extended Data Fig. 6. Effects of NK2R agonism in nonhuman primates.

a, Exposure to EB1001 after s.c. injections of different doses. b, stool composition, c, changes in withdrawn behaviour, d, blood oxygenation, e, blood aspartate transaminase (AST), f, creatinine and blood urea nitrogen levels, g, body weight, and h, food intake during the dose escalation study (n = 10 nonhuman primates (NHPs) for a-h). i, Changes in triglycerides (raw-left side and %-right side), normoglycemic (n = 3 NHPs) and diabetic NHPs (n = 7 NHPs). j, Summary comparison of effects of NK2R agonism between mice and NHPs. Data are represented as mean ± s.e.m. For all: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001. Repeated measures one-way ANOVA with Geisser-Greenhouse correction and Dunnett’s multiple comparison test, significance indicates post-test between vehicle and treatments, b; repeated measures one-way ANOVA with Geisser-Greenhouse correction, c-h; Repeated measures two-way ANOVA with Geisser-Greenhouse correction, i.
Even using the early generation tool peptide EB1001, we were able to gain insights into the potential efficacy of NK2R agonism in macaques. Notably, although all macaques in the cohort were obese, their glucose control varied from normoglycaemic to a range across the diabetes spectrum, from mild to severe insulin resistance (stratified by criteria adapted from ref. 41) (Fig. 5e). Over the course of the eight-week dose-up, EB1001 led to a modest but significant reduction in body weight across the whole cohort (Extended Data Fig. 6g), which was driven by non-plateauing weight loss in the diabetic group that was maintained throughout the two-week period following treatment cessation (Fig. 5f). After an initial trending difference in food intake between normoglycaemic and diabetic animals (Fig. 5g), all macaques uniformly exhibited an approximate 25% decrease in food intake (Extended Data Fig. 6h). Technical limitations prevented the measurement of energy expenditure in the macaques.
Nonetheless, despite not being able to determine the maximal weight-lowering potential of NK2R agonism by the conclusion of the study, there were several improvements in cardiometabolic parameters. Before reaching the full extent of weight loss, EB1001 markedly reduced fasting blood glucose concentrations (Fig. 5h) and insulin levels (Fig. 5i), leading to a uniform correction of insulin resistance (as assessed by homeostatic model assessment–insulin resistance (HOMA-IR)) (Fig. 5j) across the heterogenous diabetic group of macaques. These findings on glucose control underscore the genetic association that we uncovered between NK2R variants and HbA1c levels in humans. Notably, the improvements in glucose control were still present two weeks after the final compound dosing. Despite the marked improvement in glucose control, NK2R agonism did not produce any incidents of hypoglycaemia in the macaques at any point during the dose escalation. In addition, the insulin:C-peptide ratio, a non-invasive indicator of hepatic steatosis42, was also significantly decreased in diabetic macaques following EB1001 administration (Fig. 5k), suggesting an NK2R-agonist-induced reduction in liver fat. In line with these results, EB1001 rapidly and robustly lowered plasma triglycerides in all macaques (Fig. 5l), irrespective of diabetic status (Extended Data Fig. 6i). Furthermore, NK2R agonism significantly reduced both total and low-density lipoprotein (LDL) cholesterol in animals with high starting cholesterol (Fig. 5m–o) (as stratified by ref. 43). Together, in obese, diabetic macaques, NK2R agonism decreased body weight and food intake without causing emesis, and efficiently improved insulin sensitivity and lipid control. The weight loss and glucose control appeared to be even more pronounced in the diabetic macaques. Notably, the efficacy parameters and side-effect profile of NK2R agonism that were assessed in mice and macaques were fully translatable (Extended Data Fig. 6j).
Discussion
We set out to identify GPCR pathways that regulate energy homeostasis and could be leveraged to improve cardiometabolic health. In our search, we uncovered a novel drug target whereby agonism of NK2R elicited increased peripheral energy expenditure and insulin sensitization, as well as central control of appetite. Our motivation to design selective NK2R agonists was inspired by the revelation of genetic associations to obesity and HbA1c. The association of HbA1c with NK2R variants appears to have been previously overshadowed by the neighbouring gene HK1. Given the strength of the association between HbA1c and HK1 variants26 and the close genomic proximity of HK1 and NK2R, fully disentangling variant associations between these two genes is difficult. Nevertheless, our fine-mapping analysis and TWASs revealed associations between HbA1c and NK2R that were independent of HK1 variants. Notably, the HbA1c associations in the HK1–NK2R–TSPAN15 region have not been ascribed to glycaemic control, but are instead linked to haemoglobin biology by affecting the turnover or metabolism of erythrocytes44. To that end, the anaemic phenotype of mice that are deficient in Hk1 (ref. 45) is fully consistent with this nonglycaemic influence on HbA1c. However, NK2R agonism significantly counteracted hyperglycaemia and improved insulin sensitivity acutely in mice and chronically in macaques, suggesting that NK2R signalling acts on glucose control and not on erythrocyte viability. Further supporting the erythrocyte independence of NK2R action are the findings that blood oxygenation was significantly improved and blood urea nitrogen remained stable in macaques after prolonged NK2R-agonist administration. This genomic region has also been previously linked to cholesterol27, which is less likely to be attributed to the erythrocyte-based, proanaemic outcome elicited by HK1 deficiency. NK2R agonism lowered triglycerides in all macaques and significantly decreased LDL cholesterol in macaques with higher baseline cholesterol, hinting that NK2R may potentially be the gene responsible for this association to this region.
Key questions raised by the genetic findings include what the mechanistic underpinnings are of the obesity association with NK2R variants found in the Greenlanders, and why this pattern is not observed in the Western European population. Similarly, why is the HbA1c association found in Western Europeans and not in Greenlanders? It is, of course, tempting to speculate that the pronounced differences in diet and environment may be contributing factors. Moreover, on the basis of the extensive understanding of the genetics of human obesity46,47, the effect sizes of the reported NK2R variant imply that disruption of NK2R signalling does not have a causal pathophysiological role in the development of obesity. However, the generation of long-acting, potent NK2R agonists raises the possibility that sustained pharmacological activation may ultimately contribute meaningfully to weight management in humans, regardless of the physiological role of this receptor. Future investigation into the mode of action of NK2R and comparison between pharmacological and physiological contributions to energy homeostasis should provide clarifying insights.
Owing to the high bar set by current and late-stage pipeline strategies for the treatment of obesity2, for which more than 20% weight loss in humans is now achievable13,48, the modest weight loss and the gastrointestinal-related tolerability ceiling observed in preclinical studies of these nonoptimized NK2R agonists would probably limit the standalone targeting of this receptor for cardiometabolic indications. This prospect is made even more likely in light of the profound recent advances and an overall shift to leveraging multiple modes of action in unimolecular polyagonists2, exemplified by clinically validated compounds such as tirzepatide (acting on GLP-1 and GIP13, with average weight loss of up to 20.9%), survodutide (acting on GLP-1 and GCG49, with average weight loss of up to 14.9%) and retatrutide (acting on GLP-1, GIP and GCG48, with average weight loss of up to 22.1%). In the context of polyagonism, the energy-expending feature of NK2R agonism could provide an attractive alternative to glucagon, given that in preclinical models, NK2R agonism increased oxygen consumption but did not affect heart rate and spared lean mass. Moreover, the non-aversive appetite suppression, weight-lowering efficacy and insulin sensitization in diabetic obese animals would represent additional strengths over the current pharmaceutical toolkit. Thus, polyagonism strategies that incorporate NK2R activation may provide the greatest opportunity to exploit this new biology for next-generation biopharmaceuticals.
This work reveals the beneficial effects of pharmacological NK2R activation, but the physiological roles of NK2R signalling in metabolism are likely to be more nuanced and complex. Native NKA has a half-life on the order of minutes5 and is cleared with high efficiency that prevents appreciable circulation. Thus, endogenous NK2R signalling is largely dictated by local paracrine actions of NKA released from resident secretory cells. Conversely, EB1001 and EB1002 are present in circulation for hours and will theoretically activate all accessible NK2R receptors throughout the body. To that end, the collective effects of EB1001 or EB1002-induced central anorexigenic control and peripheral increase in energy expenditure and insulin sensitivity may be physiologically elicited separately by NKA–NK2R signalling axes in different organ systems and biological contexts. Specifically, determining whether peripheral energy expenditure and central anorectic control are coordinated together and functionally linked will be a critical point to resolve how endogenous NK2R signalling affects systemic energy homeostasis. From a therapeutic standpoint, it will be further advantageous to mechanistically delineate how the beneficial cardiometabolic and gastrointestinal effects are regulated. The dose-escalation study in macaques supports a pharmacological model whereby cardiometabolic efficacy parameters are largely dictated by compound exposure, whereas effects on the gastrointestinal are driven by Cmax. Notably, although leveraging exposure-driven efficacy and minimizing gastrointestinal motility is essential for the cardiometabolic therapeutic window, efforts to capitalize on the acute, high-dose effects are being targeted as a potential way to help restore gut motility in individuals who have undergone spinal cord trauma and suffer from diminished neuronal control of gastrointestinal function. Therefore, resolving the complex cell, tissue and interorgan contributions that collectively shape the whole-body responses to physiological and pharmacological NK2R activation could enable us to clinically harness the full potential of NK2R biology across indications.
Another key, outstanding question surrounding the mode of action of NK2R is which tissue or tissues are responsible for the energy expenditure. NK2R is expressed throughout the body, however, the current understanding of NKA action is centred largely around its role as a local, non-circulating messenger in the gastrointestinal tract or as a neurotransmitter in the CNS. Given that we find white adipose depots to be the organs with the most significant NK2R-agonist-induced glucose disposal, and previous work in Caenorhabditis elegans has implicated NK2R signalling in adipose catabolism50, these tissues represent intriguing candidates. Palamiuc et al. also found that C. elegans tachykinin signalling functionally interacted with central serotonergic signalling50, which is known to influence catabolic pathways and holds considerable potential for the treatment of cardiometabolic disease51. The lack of direct effect of NK2R agonists on isolated adipocytes suggests the requirement for other cell types or interactions (for example, innervation) in the in vivo tissue environment. Moreover, the anatomical and temporal resolution of NK2R-agonist-induced effects on heat production implicate contributions from multiple organs at different times following compound administration. However, our findings from BAT-denervated animals and thermoneutrally housed Ucp1-knockout mice probably preclude a role of canonical brown adipocytes.
The ability of EB1002-mediated NK2R agonism to suppress food intake and body weight in ob/ob mice, which are obese owing to CNS alterations in appetite control, together with the ability of ICV-delivered EB1002 to reduce feeding and body weight, reveal the importance of NK2R-regulated CNS pathways for the salient actions of EB1002. The important task of defining the neural systems by which NK2R signalling acts to control metabolism and food intake will require substantial additional work, however. Although neurons in the DVC (especially the NTS) were most strongly activated by EB1002 under multiple conditions and we identified several NTS cell types that were activated by EB1002, ablating Nk2r in the DVC did not affect EB1002-induced appetite suppression and weight loss. Consistently, iDISCO imaging of whole brains from EB1002-treated DIO mice revealed that NK2R agonism elicited FOS activation in many brain regions (including in many areas linked to appetite, energy expenditure, body weight regulation, reward and the processing of olfactory and taste information). Although one or more of these brain regions presumably mediate the effects of EB1002 on energy balance, the lack of strong detection and spatial resolution of Nk2r expression data in the CNS from single-nuclei RNA-sequencing brain atlases render it unclear how much of the NK2R-dependent FOS activation is due to afferent signals conveyed from the periphery, direct action on neurons in a particular brain region, or signalling between interconnected CNS regions. Furthermore, our studies were all performed using a 2 h timepoint, when EB1002 is maximally high in the blood. Thus, future investigations to map Nk2r and FOS co-expression and incorporating multiple timepoints may better decode the cell types and complex trajectory of neuronal signalling that mediate NK2R control of energy homeostasis.
Given the sexual dimorphism present in cardiovascular metabolism linked to body weight and energy homeostasis52 and the translational blind spots in exclusively testing new therapeutic mechanisms in male preclinical models53, we evaluated key features of NK2R agonism in female DIO mice. Effects on body weight, blood glucose, energy expenditure, fatty acid oxidation, body temperature, food intake and RER were all preserved between sexes. We similarly observed a potentiated response on food intake in female ob/ob mice as in males, indicating that the interaction between leptin and NK2R signalling pathways was independent of sex. We also evaluated NK2R agonism in obese female macaques. The assigned females were ovariectomized in earlier studies unrelated to this research. Although this allowed the maintenance of male–female social housing pairs without the possibility of breeding, the females are more in line with a postmenopausal model. Thus, even though the female macaques responded similarly to males, we could not determine whether there were any interactions between oestrogen and NK2R signalling or how female steroid hormones might affect efficacy in primates.
These findings on dual appetite and energy-expending control by NK2R are particularly surprising given that the tachykinin receptors, NK1R, NK2R and NK3R, are among the oldest known neuropeptide receptor families, with the NK1R ligand substance P even being called the ‘pioneering peptide’54. Substance P and NK1R have been implicated in metabolic homeostasis through driving food aversion55, which is in line with the emetic role of NK1R56. This is in contrast to our findings that NK2R agonism is non-aversive in mice and that there were no emetic events following any dosing of an NK2R agonist in macaques. NK1R antagonists have even been posited for obesity therapy57, yet no changes in weight or cardiometabolic parameters were reported after several clinical trials58. Exploring the efficacy of NK2R activation has long been impeded by the well-established cross-reactivity of its native ligand NKA with the other tachykinin receptors and the difficulty in generating NK2R-selective molecules that would avoid potential NK1R-linked adverse effects, such as gastrointestinal inflammation, anxiety, bronchoconstriction and pain nociception5, or NK3R-linked adverse effects, such as hot flushes59. Yet decades-old studies in humans comparing infusion of NKA and substance P already hinted at the potential of NK2R agonism to increase energy dissipation without changes in heart rate60, but these findings were never expounded upon.
One of the ubiquitous and most pronounced hurdles faced by rodent-based biomedical research and development is translation to humans. We were fortunate to have the opportunity to test the early generation, prototypic NK2R agonist EB1001 in a cohort of older macaques. Every parameter that we were able to measure translated from our observations in rodents to the diabetic, obese macaques, with the added benefit of reduced cholesterol and triglycerides. The recent advances in bioharmaceutical therapies for T2D and obesity have set incredibly high innovation bars12,13. However, we believe that features of NK2R agonism such as energy expenditure with no signals of increased cardiovascular risk, lean mass sparing, lack of nausea and, crucially, insulin sensitization could complement the current drug repertoire, particularly in the context of individuals living with both obesity and T2D. Nonetheless, given the translational uncertainty that exists even from nonhuman primate to human pharmacology, the true potential of NK2R agonism and its position and utilization among current cardiometabolic pharmacotherapies will only be realized in clinical trials.
Methods
Mice
Mouse studies performed at the University of Copenhagen were approved by The Danish Animal Experiments Inspectorate (permit number: 2018-15-0201-01441 and 2023-15-0201-01386) and the University of Copenhagen. Animal experiments performed at the University of Michigan were approved by the University of Michigan Committee on the Use and Care of Animals (protocol no. 00011066) and in accordance with Association for the Assessment and Approval of Laboratory Animal Care and National Institutes of Health guidelines. Studies in Fig. 1p–r using EB0014 were conducted by Gubra. Mice were housed in solid bottom cages with environmental enrichment at 22 °C (±2 °C) in 55% (±10%) humidity and a 12-h light:dark cycle (light from 06:00 to 18:00). Mice had ad libitum access to water and standard chow diet (SAFE D30, Safe Diets) or 60% HFD (Rodent Diet with 60 kcal% fat, D12492i, Research Diets) to induce obesity as indicated in the figure captions and results. For studies with DIO mice, mice were challenged with HFD for a minimum of 12 weeks starting from 8 weeks of age and were used in pharmacology studies once the mean cohort size was at least 35 g. Groups were randomized for vehicle and compound treatment. Pharmacological DIO studies in wild-type male and female mice were performed on animals with a C57Bl/6 NRj genetic background (Janvier Labs). The description of backgrounds for the genetic models used are as follows: B6.V-Lepob/JRj (ob/ob) mice and RjOrl:SWISS (CD-1) mice were purchased from Janvier Labs. B6.129S4-Mc4rtm1Lowl/J (Mc4r-knockout), Ccktm1.1(cre)Zjh/J, B6.129-Leprtm3(cre)Mgmj/J, Calcrtm1.1(cre)Mgmj/J, B6;129-Gt(ROSA)26Sortm5(CAG-Sun1/sfGFP)Nat/J and Gt(ROSA)26Sortm1(CAG-EGFP/Rpl10)Dolsn mice were purchased from the Jackson Laboratory. The Ucp1-knockout line was provided by K. Kristiansen. B6N-Tacr2tm1Zpg (Nk2r-floxed) mice were generated by GenOway on a C57Bl/6N background by inserting loxP sites around exon 1, which encodes 130 out of the 384 amino acids in the full-length receptor. Ucp1-knockout, Nk2r-knockout and Nk2r-floxed lines were bred in-house at 22 °C ( ± 2 °C). Ccktm1.1(cre)Zjh/J and B6.129-Leprtm3(cre)Mgmj/J mice were crossed with Gt(ROSA)26Sortm1(CAG-EGFP/Rpl10)Dolsn mice to create CckCreL10 GFP and LeprCre L10 GFP mice respectively. Calcrtm1.1(cre)Mgmj/J mice were crossed with B6;129-Gt(ROSA)26Sortm5(CAG-Sun1/sfGFP)Nat/J to create CalcrCre Sun1 GFP mice. Studies were generally performed at 22 °C (±2 °C). Studies in Ucp1-knockout mice and the ob/ob one time injection and subcutaneous/ICV crossover study were performed in thermoneutrally (29 °C (±2 °C)) housed mice.
Mice were trained with mock injections for all injection studies for at least 5 days. NKA (1 mg kg−1; Almac) dissolved in Gelofusine (B. Braun) was administered subcutaneously twice daily for 9 consecutive days. EB0014 (synthesized by Almac) was dissolved in DMSO to a 17 mg ml−1 solution and further in 0.9% saline with 3% BSA. EB0014 was subcutaneously administered once daily at 1 mg kg−1 for indirect calorimetry measurements and at 0.01 mg kg−1, 0.1 mg kg−1, 0.3 mg kg−1 and 1 mg kg−1 for the dose-dependent weight loss study.
EB1002 (synthesized by Novo Nordisk or PolyPeptide) was dissolved in 8 mM phosphate and 240 mM propylene glycol (pH 8.2) and administered once, daily for 7 or 21 consecutive days or every other day for 21 days via subcutaneous injections at 40 nmol kg−1 or 325 nmol kg−1 between 15:00 and 16:00. The NK2R antagonist, saredutant (SR 48968, MedChemExpress) was injected into the peritoneum at 5 mg kg−1 in 0.9% saline with 1% Tween80 30 min prior to EB1002 injections. Body weight, blood glucose and food weight were taken immediately before injections and 24 h after injections. The amount of food for pair-fed groups was determined on the basis of at least two previous studies. To mimic a diet intervention, DIO mice were switched to chow diet 5 days prior to first injection and compared to DIO mice maintained on HFD. The effect of EB1002 on food intake in Extended Data Fig. 5a was assessed in overnight-fasted mice that were refed at the time of injection.
The insulin tolerance test was performed in DIO mice after the 9-day injection regimen, 12 h after the final injection with 1 mg kg−1 NKA. Insulin tolerance test was carried out on 4-h fasted mice by intraperitoneal injection of 0.75 U kg−1 insulin. Twenty-four hours prior to the glucose tolerance test, DIO mice were subcutaneously injected with vehicle or 325 nmol kg−1 EB1002. Vehicle-treated mice had either ad libitum access to HFD or were pair-fed to EB1002-treated mice. Glucose tolerance test was performed the following day on 4-h fasted mice by intraperitoneal injection of 1 g kg−1 glucose. Insulin levels were measured using the Mouse/Rat Insulin Kit (mesoscale) according to the manufacturer’s protocol.
GLP-1, leptin, glucagon and PYY were measured in plasma samples using the U-PLEX Gut Hormone Combo 1 (ms) SECTOR Kit (MSD, K15307K-1) according to the manufacturer’s protocol.
To evaluate the aversive potential in mice, a saccharin-conditioned taste avoidance was performed as described63. On the conditioning day, mice were exposed to a new flavour (0.15% saccharin) followed by a subcutaneous injection of vehicle, 10 nmol kg−1 semaglutide or 325 nmol kg−1 EB1002.
Liquid phase gastric emptying was performed in 4-h fasted mice. Mice received an injection of vehicle or EB1002 30 min prior to an oral gavage of 4 mg paracetamol in 0.9% NaCl. Blood samples were collected via the tail vein 15 min after gavage and paracetamol levels were measured using Acetaminophen L3K assay kit (Sekisui Diagnostics, 506-30) according to the manufacturer’s protocol.
Tissue-specific insulin signalling was assessed in mice 18 h after injection with vehicle or EB1002. 2 h fasted mice were sedated with 75 mg kg−1 pentobarbital (intraperitoneal injection) and received a retro orbital injection with 1.5 U kg−1 insulin. Tissues were dissected 10 min after insulin administration, snap-frozen and analysed as described in ‘Immunoblotting’.
Total lean and fat mass were measured by magnetic resonance imaging using the minispec LF90 II body composition analyser (Bruker).
Metabolic characterization
Oxygen consumption, carbon dioxide production and food intake were recorded using the Promethion core system (Sable Systems International) with data processing using OneClickMacroV2.52.1 and Macrointerpretersetup_v2_47 or the Phenomaster Home Cage System (TSE Systems) with PhenoMaster software v.8.2.9. Generally, mice were transferred to training cages 3–7 days prior to the measurement and were allowed to acclimate in the measurement chambers for at least 24 h or until a stable baseline was observed. Fatty acid oxidation was calculated using the formula64: energy expenditure (kcal/h) × (1-RER)/0.3.
Core body temperature and gross motor activity measurements
Continuous body temperature and motor activity were measured with G2 E-Mitter telemetric devices (Starr Life Sciences) or with radio frequency identification (RFID) temperature transponders (UCT-2112 microchips, Unified Information Devices). Probes were implanted into the peritoneal cavity under sterile conditions. In brief, mice were anaesthetized with isoflurane and received 5 mg kg−1 Rimadyl (ScanVet, 027693) and 8 mg kg−1 Lidocaine (AstraZeneca). The sterile probe was inserted into the peritoneum via a midline incision. After closure, mice were allowed to recover on a heated surface and received 5 mg kg−1 Rimadyl for 3 consecutive days. Mice were allowed to recover for at least 7 days before study start. E-mitter data was recorded by placing ER4000 Receivers under the cages within the TSE cabinet. Temperature data was integrated in the Phenomaster software. RFID temperature transponder data was recorded by using UID Mouse Matrix system (Unified Information Devices) in combination with the Sable system.
Triple-chip study
RFID temperature transponders (UCT-2112 microchips, Unified Information Devices) were surgically implanted and anchored in female mice for continuous and simultaneous measurement of temperature in three distinct anatomical locations. Surgical and post-operative procedures were performed as described above. Ventromedial abdominal chip (abdominal temperature): following a midline incision, the chip was inserted in the abdominal cavity. Distolateral femoral chip (hindlimb temperature): following a transverse left gluteal incision, a subcutaneous lateral femoral pocket was prepared, and the chip was inserted. Dorsomedial intrascapular chip (interscapular temperature) and bilateral BAT denervation: following a midline dorsal incision, the interscapular BAT was detached from the underlying muscle layer. Bilateral denervation was performed as described65. The chip was inserted with the tip containing the temperature probe placed between the BAT and the dorsal muscle. The three temperatures were recorded using the UID Mouse Matrix system (Unified Information Devices).
Brain cannulation and ICV injections
For administration of compounds directly into the brain of awake mice, a cannula was placed into the cerebral ventricle. B6.V-Lepob/JRj (ob/ob) mice were anaesthetized with isoflurane and received 5 mg kg−1 Rimadyl (ScanVet, 027693) and 8 mg kg−1 Lidocaine (AstraZeneca). Mice were fixated in a stereotaxic frame (Kopf Instruments) and the scalp was opened to expose the skull. Coordinates were zeroed on bregma and moved to −0.3 mm in the anteroposterior axis and −1.0 mm in the mediolateral axis. A small hole was drilled into the skull and the guide cannula (C315GS-4/Spc 2 mm, PlasticsOne) was inserted and fixated using a G-bond layer (GC America) and G-ænial universal flow (GC America). Finally, a dummy (C315DCS-4/Spc 2.5 mm, PlasticsOne) was screwed onto the guide cannula and mice were allowed to recover on a heated surface and received 5 mg kg−1 Rimadyl for 2 consecutive days following the surgery. To test the correct placement of the cannula, mice received an infusion of 1.5 μl of 24 μM angiotensin II (Sigma) in artificial cerebrospinal fluid (CSF) (Harvard Apparatus) via an injector (C315IS-4/Spc 2.5 mm, PlasticsOne) and an infusion pump (Harvard Apparatus) at a rate of 2 μl min−1 5 days after surgery. After the infusion, the injector was left in place for an additional 45 s to minimize backflow. Then, the injector was removed, and the dummy was placed on the cannula. Mice were monitored for angiotensin II-induced water intake and responsive mice were subsequently used for ICV injection studies. One week prior to study start, mice were housed at thermoneutrality. Mice first received a single subcutaneous injection of vehicle or 325 nmol kg−1 EB1002 and after a wash-out period they were infused with 1.5 μl artificial CSF or 2 nmol EB1002 in 1.5 μl artificial CSF as described above in a crossover design.
AAV injections into the DVC of Nkr2-floxed mice
Nk2rfl/fl mice were anaesthetized using isoflurane. Mouse heads were oriented at a 90° angle and an incision was made at the caudal aspect of the skull to expose the brainstem. Using the obex of the skull as a guide, 50 nl of AAV-GFP or AAV-CMV-CRE-GFP were injected into each site of the DVC at a depth (z) of 0.350 mm. Virus was injected at a rate of 15–35 nl min−1. The pipette remained in the DVC for an additional 3 min to allow viral particles to disperse, and then the pipette was slowly removed. Mice received prophylactic analgesic carprofen (5 mg kg−1) before and for 24 h after surgery and were monitored for 10 days following the procedure to ensure recovery for surgical intervention. Mice were fed a 60% HFD starting 6 weeks after surgery for 5 weeks. To assess the acute food intake response to EB1002, mice were injected subcutaneously with either vehicle or 325 nmol kg−1 EB1002 in a crossover design. Food intake was measured at hours 0, 1, 2, 4 and 24 and body weights were recorded at 0 and 24 h post-injection. After termination, hit sites of all mice were confirmed using immunohistochemistry and mice with a confirmed bilateral hit site were included in the analysis.
Hyperinsulinaemic–euglycaemic clamp and peripheral glucose uptake
Hyperinsulinaemic–euglycaemic clamps were performed in conscious, unrestrained male mice at 24 weeks of age as previously described66. In short, catheters were implanted under aseptic conditions into the right jugular vein (C20PU-MJV1458; Instech) and the left common carotid artery (C10PU-MCA1459; Instech), exteriorized in the scapular region and secured using a dual-channel vascular access button (VABM2B/25R25; Instech) under general isoflurane anaesthesia. Mice were subcutaneously injected preoperatively with Carprofen (5 mg kg−1, Norodyl Vet, Scanvet) for analgesia and a mixture of Lidocaine (7 mg kg−1, Xylocain, AstraZeneca) and Bupivacaine (7 mg kg−1, Marcaine, Orifarm) at the incision sites for local anaesthesia. Mice were allowed to recover for 7–10 days. Mice were treated with either vehicle or 325 nmol kg−1 EB1002 24 h prior to clamp experiment. Clamp studies were performed in 4 h fasted mice. At 15 min and 5 min before the start of the clamp, blood samples were taken for determination of basal glucose and insulin levels. At 0 min, the clamp was initiated with continuous infusion of human insulin (4mU kg−1 min−1, Actrapid; Novo Nordisk, Denmark). Donor red blood cells were washed and used to compensate for the blood loss to experimental mice during the repeated sampling (5 μl min−1 of 50% RBC in 10 U ml−1 heparinized saline). Blood glucose samples were taken every 10 min (Contour XT, Bayer) and blood glucose was then adjusted using a variable infusion of 50% glucose. Both human and mouse insulin levels (Mercodia) were determined at 100 and 120 min. After 120 min, a 13 μCi bolus of 2-[1-14C]-deoxy-d-glucose was given and blood samples taken at 122, 125, 135, 145 and 155 min and specific activity for 2-[1-14C]-deoxy-d-glucose was determined in these samples. After euthanization, tissues for tissue-specific 2-[1-14C]-deoxy-d-glucose uptake were sampled and snap-frozen in liquid nitrogen. Samples from tissue-specific glucose uptake were processed as previously described66.
Pharmacological pre-toxicology evaluation
For the evaluation of potential toxicological effects CD-1 outbred mice were daily either injected with vehicle or with increasing concentrations of EB1002 (150 nmol kg−1 (2 consecutive days), 300 nmol kg−1 (2 consecutive days), 600 nmol kg−1 (2 consecutive days), 1,200 nmol kg−1 (2 consecutive days), 2,400 nmol kg−1 (2 consecutive days), 4,800 nmol kg−1 (2 consecutive days) and finally 7,500 nmol kg−1 (3 consecutive days)). Liver, spleen, heart, kidney, oesophagus, stomach, duodenum, colon, testis, cornea, bladder and thymus were dissected, fixed in 10% formalin and stained with haematoxylin and eosin for pathohistological analysis performed by the Histology department, HistoCore, at the University of Copenhagen. Slides were compared pairwise. Serum liver enzymes were analysed by the Veterinary Diagnostic Laboratory core at the University of Copenhagen.
Quantification of faecal cholesterol and triacylglycerides
Faecal samples were homogenized in cold methanol containing butylated hydroxytoluene (1 mg ml−1). After centrifugation (10 min at 10,000g, 4 °C), the supernatants were transferred into fresh vials and the methanol was evaporated by vacuum centrifugation. Pellets were re-dissolved in 0.1 M potassium phosphate, pH 7.4, 0.05 M NaCl, 5 mM cholic acid and 0.1% Triton X-100. Cholesterol and triacylglycerides were quantified using colorimetric kits from DiaSys (Cholesterol (113009910704) and triacylglycerides (157109910026)) according to the manufacturer’s instructions.
NKA and compound detection in the blood
Pharmacokinetic profiles were determined in lean mice. Mice were subcutaneously dosed with 1 mg kg−1 NKA, 666.3 nmol kg−1 EB0014, 325 nmol kg−1 EB1001 and 325 nmol kg−1 EB1002. Blood samples were taken starting at 5 min until 60 h as indicated in respective graphs. Plasma levels of NKA were determined using a Human Neurokinin A kit Elisa Kit (Ray Biotech). Plasma levels of EB0014, EB1001 and EB1002 were determined using LC-MS after precipitation with acetonitrile containing 0.1% formic acid.
In situ hybridization
Wild-type mice were anaesthetized with 75 mg kg−1 pentobarbital and transcardially perfused with 4% paraformaldehyde (PFA) in PBS. Whole brains were dissected, further fixated in 4% PFA in PBS for 48 h at 4 °C and subsequently dehydrated using the automated Excelsior AS tissue processor (Thermo Scientific) and embedded in paraffin. Paraffin-embedded brains were cut into 4 μm sections using the Microm Ergostar HM 200 (Marshall Scientific) and mounted onto glass slides. After deparaffination and rehydration, RNA molecules were detected using the RNAscope Multiplex Fluorescent V2 Assay kit (ACDbio) according to manufacturer’s protocol. Nk2r, Calcr and Glp1r were detected with the RNAscope probes Mm-Tacr2 (441311), Mm-Calcr-C2 (494071-C2) and Mm-Glp1r-C3 (418851-C3), and 3-plex Negative probe (320871) and 3-plex-mouse Positive probe (320881, all ACDbio) were used as negative and positive control respectively. Signals were visualized with fluorophores at 570 nm (OP-001003) for channel 1, 520 nm (OP-001006) for channel 2 and 690 for channel 3 (OP-001001, all 1:1000, Akoya Bioscience). Sections were mounted with ProLong Gold antifade reagent with DAPI (P36935, Invitrogen). For analysis, 4 mice were used and 2–3 sections per animal were imaged.
Immunohistochemistry
Mice were fasted overnight and injected subcutaneously with vehicle or 325 nmol kg−1 2 h prior to euthanasia. Mice anaesthetized with isoflurane and transcardially perfused with PBS followed by 4% PFA in PBS. Whole brains were dissected, further fixated in 4% PFA in PBS for 4 h at 22 °C. After 48 h in a 30% sucrose solution, brains were cut into 30 µm sections using a sliding microtome SM2010R (Leica) with a freezing stage and temperature controller (BFS40-MPA, Physiotemp). Free floating sections were blocked in 3% donkey serum with 0.1% Triton X-100 in PBS and incubated overnight at room temperature using the following antibodies, against FOS (Cell Signalling, 2250, 1:1,000) and GFP (Aves Laboratories, 1020, 1:1,000). Sections were washed, incubated with a secondary antibody conjugated to Alexa Fluor 488 and 568 (Invitrogen, A-11039, A-11011, 1:250) and mounted.
To assess neuronal degradation in the DVC of Nk2rDVC-GFP and Nk2rDVC-cre mice, the Fluoro-Jade C (FJC), RTD Ready-to-Dilute Staining Kit for identifying Degenerating Neurons (VWR) was applied according to manufacturer’s instructions. Fluoro-Jade C positive neurons were counted for quantification of neuronal degradation.
Microscopy
Fluorescence microscopy was performed using a Zeiss Axio Observer microscope with Axiocam 702 mono camera or with an Olympus BX53 microscope.
Whole-brain FOS imaging
Two-hour fasted obese wild-type mice received a single subcutaneous dose of vehicle or 325 nmol kg−1 EB1002 before being terminated and perfused with 4% PFA 2 h later. The brains were subsequently isolated, postfixed, and transferred to Gubra. At Gubra, the samples were cleared, stained for FOS, and imaged at single-cell resolution using a light sheet microscope as previously described40. The data from individual brains was mapped into an average mouse brain atlas template, and the number of FOS labelled cells was quantified in more than 800 brain regions. For the calculation of fold changes, brain regions without FOS signal (n = 25) were excluded.
Immunoblotting
Protein was isolated and western blots were run as described previously67. Proteins were detected using the following antibodies, AKT (Cell Signaling, 9272, 1:1,000), pAKT T308 (Cell Signalling, 9275, 1:1,000), Tyrosine hydroxylase (Abcam, ab137869, 1:1,000), UCP1 (Abcam, ab10983, 1:7,500) and peroxidase-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L) (Jackson Immuno Research, 111-035-144, 1:5,000). Images were acquired using an Odyssey Fc Imager (LI-COR). Uncropped images can be found in the source data.
snRNA-seq
Mice were injected subcutaneously with vehicle or 325 nmol kg−1 EB1002 2 h prior to euthanasia. After decapitation, the brain was removed, and the DVCs were isolated from the brainstem under a surgical microscope. A 1 mm coronal section of the hindbrain from bregma −7.0 to −8.0 mm was cut with a razor blade. From the coronal section, a 1.4 mm square containing the DVC was snap-frozen. Samples from the same experimental condition were pooled (n = 5 mice per sample). Nuclei were extracted as previously described68. Sorting of 2n nuclei was performed by flow cytometry using a BD FACSAria IIIu Influx cell sorter (BD Biosciences). The gating was set according to size and granularity using FSC and SSC to capture singlets and remove debris. To detect DraQ5-positive nuclei fluorescence was set at 647 nm and 670 nm. Each sample was sorted into separate tubes, each with a total of 20,000 nuclei per 40 µl. Sequencing libraries were generated using 10x Genomics Chromium Single-Cell 3′ Reagent kit according to the standardized protocol. Paired-end sequencing was performed using an Illumina NovaSeq 6000.
snRNA-seq analysis
Raw sequencing data were demultiplexed, aligned to the mouse reference genome GRCm38 (mm10) and counted using cellranger (version 7.0.0; 10x Genomics). Ambient RNA molecules were removed from cellranger raw count matrix files with cellbender:remove-background (v0.3)69. Filtered count matrices were analysed in R with Seurat (v4.3.0)70. Nuclei with at least 1000 detectable genes were retained. Genes expressed in at least 10 nuclei were retained. ScDblFinder (v1.15.4)71 with standard parameters was run on individual 10x lanes to remove doublets. The count data were normalized with NormalizeData and scaled with ScaleData function. Genes were then defined as variable using FindVariableFeatures and used as input into principal components analysis with RunPCA. The top 30 principal components were retained and used for further dimensionality reduction using RunUMAP and clustered using a resolution of 0.8 with FindClusters. To perform cell-type-specific quality control, the nuclei were split into two broad categories, neuronal and nonneuronal, using CellAnnotatorR(v), on the basis of the expression of neuronal marker gene Rbfox3 or Snap25 and the absence of nonneuronal markers. Neuronal data was further filtered by removing nuclei with unique molecular identifiers (UMIs) in the first and 99th percentile. Library complexity was calculated by dividing the log of the total genes detected per nuclei by the log of the total UMIs per nuclei. Nuclei in the first percentile of this metric were removed. Add_Mito_Ribo_Seurat was used to identify and remove nuclei with >1% mitochondria and ribosomal genes. We finally removed outliers by isOutlier with nmads = 5 run on individual 10x lanes from the package SingleCellExperiment72. After all quality control, a total of 23,664 neurons remained. Neuronal cell types were labelled by projecting labels from a previously published dataset39 (GSE166649) using Seurat FindtransferAnchors and TransferData function.
Immediate early gene analysis
To calculate an IEG score, rapid primary response genes73 (Fosb, Npas4, Fos, Junb, Nr4a1, Arc, Egr2, Egr1, Maff, Ier2, Klf4, Dusp1, Gadd45g, Dusp5, Btg2, Ppp1r15a and Amigo3) were used to score each nuclei with the AddModuleScore function from Seurat. For each cluster-sample combination we calculated the average activity score which we then modelled by the interaction of cell type and treatment. Estimated marginal means were compared between treatments within each cluster. P values were corrected for multiple testing using the Benjamini–Hochberg method (adjusting for the number of cell populations).
Perturbed cell-type analysis
We used the scDist package to calculate the transcriptional distance between treatment and controls while also controlling for individual-to-individual variability74.
Primary cell studies
Primary white adipocytes were isolated for wild-type mice and cultures as described75. For in vitro oxygen consumption measurements, cells were replated in Seahorse XF96 Cell Culture Microplates (Agilent Technologies) on day 3 and oxygen consumption in response to vehicle or EB1002 (10 nM, 1 µM and 10 µM) was measured using a Seahorse XFe96 Extracellular Flux Analyzer (Agilent Technologies) on day 7 as described75. For in vitro glucose uptake, differentiated cells were starved for 2 h before treatment with Krebs-Ringer buffer containing 5 mM glucose, and vehicle or EB1002 (10 nM, 1 µM and 10 µM). After 2 h, cell media was incubated with a reaction mix mix (200 mM Tris-HCl, 500 mM MgCl2, 5.2 mM ATP, 2.8 mM NADP, and 6 μg ml−1 hexokinase and glucose-6-phosphate dehydrogenase mixture (10737275001, Roche Diagnostics)) for 15 min and glucose content was measured spectrophotometrically at 340 nm (Hidex sense, Hidex). Glucose uptake was calculated on the basis of disappearance of glucose from the media. Cells have been tested negative for mycoplasma contamination.
Ex vivo lipolysis
To assess the effects of EB1002 on the lipolytic function of adipocytes, mature adipocytes were isolated from iWAT of wild-type chow-fed mice. In brief, adipose depots were minced and digested in Krebs-Ringer buffer containing 2% BSA and 0.2% collagenase type I (Worthington Biochemical) at 37 °C. Cells were passed through a 100-µm cell strainer, centrifuged (10 min, 10g, 4 °C) and the floating mature adipocyte fraction was washed three times. Cells were incubated with vehicle, EB1002 (10 nM, 1 µM and 10 µM) or 50 nM isoproterenol for 2 h at 37 °C. Finally, non-esterified free fatty acids were measured in the medium using the Fujifilm NEFA HR R2 kit according to manufacturer’s protocol.
Macaque studies
The macaque studies were conducted in compliance with all federal regulations, including the US Animal Welfare Act. Studies were reviewed and approved by the OHSU/ONPRC Institutional Animal Care and Use Committee. The ONPRC is accredited by AAALAC International. For these studies, 10 rhesus macaques (5 males and 5 ovariectomized females,12–23 years of age with a body weight ranging from 7–24 kg) were pair-housed (1 male and 1 female) in custom designed cages (Carter2 Systems) in shared rooms under fixed photoperiodic conditions (lights on from 07:00 to 19:00). The cages meet the minimum European Union accepted standards for housing nonhuman primates (2.0 m2 enclosure size, 3.6 m3 enclosure volume, and 1.8 m enclosure height). Shelves, verandas, solid flooring and changeable plastic toys were available for the monkeys. The commercially available HFD (5L0P, Lab/Test Diets) was provided ad libitum twice every day. Food intake was measured daily, and animals were separated for 1–2 h periods when individual food intake was measured. Paired food intake was measured for the remaining feeding times when animals were socially pair-housed.
EB1001 was dissolved in 8 mM phosphate and 240 mM propylene glycol (pH 8.2) and administered for 8 consecutive weeks via subcutaneous injections between 08:00 and 09:00 prior to the morning meal. All animals were started on the 30 nmol kg−1 dose with every other day dosing (q48h) for 1 week, followed by daily dosing of the subsequent higher doses. Dose escalations were as follows (nmol kg−1): 60 (1 week), 90 (1 week), 120 (1 week), 240 (2 weeks), 480 (4 days) and 240 (10 days). Body weight was measured on a weekly basis using the same, calibrated digital scale. Heart rate and oxygen saturation were recorded in sedated animals using a pulse oximeter (Model 7500, Nonin Medical).
Blood samples were collected in conscious animals prior to morning meal (overnight fast) and daily dose administration. Blood glucose was measured on a Biosen clinical analyser (EKF Diagnostics) and C-peptide and insulin were measured on a Cobas e411 analyser (Roche Diagnostics). Remaining chemistry parameters alanine aminotransferase (ALT), aspartate transaminase (AST), total cholesterol (Chol), creatinine (CREA), glucose triglyceride (TG), blood urea nitrogen (BUN) and LDL cholesterol were analysed using the Pentra C400 (Horiba Medical).
Behaviour analysis was performed by members of the ONPRC Behavioural Sciences Unit blinded to the experimental design. Observations were taken directly on a mobile device and average behaviour scores were calculated on the basis of events such as anxiety, stereotypy, eye poking or withdrawn behaviour. Additional cage side observations included signs of nausea (gaping and hunched posture), emesis and stool consistency.
Association of missense variants with cardiometabolic traits
To assess whether the 4 missense NK2R variants (R3232H, I23T, V54I and A161T) are associated with cardiometabolic traits, their associations in T2D Knowledge Portal (HbA1c phenotype page, accessed 21 May 2024; https://t2d.hugeamp.org/phenotype.html?phenotype=HBA1C; RRID:SCR_003743)24 were queried. For each variant, only the associations that presented a P value < 0.05 for the latest and largest European genome-wide association study (GWAS) per trait were included. Additionally, the minor allele frequency in five ancestries are reported: AFR, AMR, EAS, EUR and SAS retrieved from 1000 Genomes reference panel data from dbSNP30,76.
SNP-level associations in the HK1–NK2R–TSPAN15 region
Utilizing the latest and largest European GWAS of HbA1c available in T2D Knowledge Portal (query on 10/05/2024) (HbA1c phenotype page, accessed 21 May 2024; https://t2d.hugeamp.org/phenotype.html?phenotype=HBA1C (RRID:SCR_003743))24, summary statistics from Jurgens et al.29 were retrieved and 1,944 common variants (minor allele frequency > 1%) located 100 kilobases (kb) upstream from the HK1 transcription start site (TSS) and 100 kb downstream from the TSPAN15 TSS (10:70929740–71367422) were investigated further.
The number of lead, independent, genome-wide significant variants were assessed by performing LD clumping with with plink1.9 (ref. 77) and utilizing the European 1000 Genomes reference panel phase 3 version 5 (ref. 30). An r2 threshold of 0.01 and distance threshold of 1,000 kilobases (kb) were used.
Fine mapping of HbA1c associations in 10:70929740–71367422 locus with CARMA31, a software designed to correct for differences in LD between summary statistics and LD reference panels were performed. CARMA with the default settings, utilizing European 1000 Genomes reference panel phase 3 version 5 with annotations30 were performed, and those variants that presented a posterior inclusion probability of >0.1 were investigated further. In dbSNP76, minor allele frequencies in five genetic ancestries were retrieved: AFR, AMR, EAS, EUR and SAS.
Finally, Open Target Genetics (OTG)78 was utilized to query the variant-to-gene (V2G) scores used for gene prioritization and the associations of causal variants with eQTLs.
Linkage disequilibrium analysis
All analyses were performed in Rstudio (2022.07.2 + 576) with R (4.1.3). Data were loaded and manipulated using data.table (1.14.2) and tidyverse (1.3.1). LD operations were performed using plink1.9 (ref. 77), ggLD (https://github.com/mmkim1210/ggLD), and LDLink79. All results can be reproduced by following the code available at https://github.com/MarioGuCBMR/nk2r_hk1_genetics.
TWAS of NK2R and HbA1c
Elastic Net model of SPrediXcan methods were used to predict the association between gene and traits80–82. The Elastic Net-based GTEx v8 eQTL models were downloaded from https://predictdb.org/post/2021/07/21/gtex-v8-models-on-eqtl-and-sqtl/. The summary statistics of GWAS for BMI83, HbA1c levels (total sample based)83, and BMI-adjusted HbA1c levels (total sample based)84 were used to conduct the association analysis. The SNPlocs.Hsapiens.dbSNP144.GRCh37 Bioconductor package85 was used to convert the genomic coordinates of SNPs to rsID for the summary statistics of HbA1c and BMI-adjusted HbA1c. Sensitivity analysis was performed using the CAVIAR fine-mapped variants86 for NK2R in nucleus accumbens of the brain tissue.
Genetic association studies in Greenlanders
Blood samples from Greenlanders collected in three population surveys (data are available at ref. 87 and https://www.sdu.dk/da/sif/rapporter/2011/inuit_health_in_transition and https://www.sdu.dk/da/sif/rapporter/2019/befolkningsundersoegelsen_i_groenland) were genotyped using the Multi-Ethnic Global Array (Illumina). After quality control up to 5,758 individuals were available for association analyses. Metabolic phenotypes were measured as previously described88, and association analyses were performed with a linear mixed model, to account for relatedness and admixture, assuming an additive genetic model and adjusting for age, sex, cohort using GEMMA89. We performed association tests for all 132 variants within the coding region of NK2R and the 3′ and 5′ untranslated regions ±1,000 bp. The strongest associations across traits related to body composition were observed for the non-coding rs139900276 variant. RNA was extracted from peripheral blood for a subset of 499 individuals. The procedure for RNA extraction, sequencing, quality control, and quantification has previously been described88. NK2R expression according to rs139900276 genotype was tested with a linear mixed model adjusted for sex, age, and top ten principal components from a principal components analysis of the normalized expression matrix.
Mutation of human wild-type NK2R
SNPs identified by GWAS were introduced to human wild-type NK2R coding sequence (NP_001048.2) in a custom pCDNA3.1(+) vector (Genscript) using PCR-based QuickChange Site-Directed Mutagenesis (Agilent) according to manufacturer’s protocol. Mutated vector was introduced to Escherichia coli to amplify DNA and indicated mutations were confirmed by forward and reverse strand Sanger sequencing (Eurofins). PCR primers used to introduce mutations are as follows (forward, reverse): I23T (CAACACCACGGGCACGACAGCCTTCTCCA, TGGAGAAGGCTGTCGTGCCCGTGGTGTTG), V54I (TGACGGGTAATGCCATCATCATCTGGATCATCCTG, CAGGATGATCCAGATGATGGCATTACCCGTCA), A161T (CTGGTGGCTCTCACCCTGGCCTCCC, GGGAGGCCAGGGTGAGAGCCACCAG), R323H (CCGGCTTGCCTTCCATTGCTGCCCATGG, CCCATGGGCAGCAATGGAAGGCAAGCCGG), T346M (CGACCTCCCTCTCCATGAGAGTCAACAGGTG, CACCTGTTGACTCTCATGGAGAGGGAGGTCG), T363A (TGGCTGGGGACGCAGCCCCCTCC, GGAGGGGGCTGCGTCCCCAGCCA), H395R (TTGCCCCCACCAAAACTCGTGTTGAAATTTGAGGATC, GATCCTCAAATTTCAACACGAGTTTTGGTGGGGGCAA).
Receptor activity assays
NKA-induced activation of mutated human NK2R and substance P-, NKA-, NKB-, EB1001- and EB1002-induced activation of mouse and human wild-type NK1R, NK2R and NK3R were measured by inositol-1,4,5-trisphosphate [3H] Radioreceptor assay (IP3 assay). IP3 assays were carried out using COS-7 cells (ATCC, CRL-1651) transiently transfected by calcium phosphate transfection with one of the mutated NK2R variants or the wild-type receptors as previously described75. The IP3 assay was performed the day after transfection. In brief, cells were washed and pre-incubated in assay buffer (HBSS, 10 mM LiCl, 0.2% w/v ovalbumin) for 30 min followed by 120 min incubation with substance P, NKA, NKB, EB1001 or EB1002 at 37 °C as indicated in result section and figure legends. After incubation, plates were immediately placed on ice, and cells were lysed (10 mM formic acid). After 30 min incubation 1 mg per well SPA YSI beads were pipetted into a solid white 96 wells plate and 35 μl of the lysis solution was transferred to the plate. Plates were mixed, briefly centrifuged and left at room temperature for 8 h before counting in a MicroBeta plate counter (Perkin Elmer). Cells have been tested negative for mycoplasma contamination.
Sample size determination and blinding
No statistical methods were applied to predetermine sample size for in vivo pharmacology experiments. Sample sizes were determined on the basis of previous experience with related experimental setups75. Studies were not blinded.
Statistical analysis
Statistical analyses were performed using GraphPad Prism v.9.5.1 or SPSS v.29.0.2.0 (IBM). Sample numbers and statistical analysis methods are provided in the figure legends. Data are presented as mean ± s.e.m. unless otherwise specified.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-024-08207-0.
Supplementary information
This file contains extended legends for Figs. 1–5 (statistical information) and Supplementary Fig. 1 (uncropped western blots for Extended Data Figs. 2s–v and 3y).
Source data
Source Data Figs. 1–5 and Source Data Extended Data Figs. 2–6
Acknowledgements
The authors thank the members of the Gerhart-Hines laboratory for discussions; L. Thisted and G. Hansen for in vivo pharmacological studies; U. Roostalu and T. Topilko for iDISCO whole-brain imaging; L. Steuernagel, C. A. Bauder and J. C. Brüning for assistance with snRNA-seq of the DVC; K. Kristiansen for providing the Ucp1-knockout mice; C. Kukat and M. Germer for fluorescence-activated cell sorting of nuclei for single-nuclei sequencing experiments; and the Cologne Center for Genomics for library preparation and sequencing. We acknowledge the Rodent Metabolic Phenotyping Platform at the Novo Nordisk Foundation Center for Basic Metabolic Research (CBMR) for technical and computational expertise and support. M.M.B. and M.S. received support from the American Lebanese Syrian Associated Charities. This project was supported by the Novo Nordisk Foundation (NNF21SA0072102 and NNF22OC0074128 to TOK; International Postdoctoral Fellowship NNF20OC0060969 to L.D.; NNF18OC0033444 to the Center for Adipocyte Signaling for F.S., M.S., M.M.B. and Z.G.-H.). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (Starting Grant aCROBAT agreement no. 639382 to Z.G.-H. and Proof-of-concept Grant GET-UP BAT agreement no. 768783 to Z.G.-H., Consolidator Grant HEAT-UP agreement no. 101088636 to Z.G.-H.). A.W. was supported by the Mühlbauer-foundation, and A.W. and J.H. received support from the Deutsche Forschungsgesellschaft (450149205-TRR333/1 and 335447717-SFB1328). R.J.S. was supported by the National Institutes of Health/National Institutes of Diabetes and Digestive and Kidney Diseases (P30DK089503, R01DK133140). The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent research centre at the University of Copenhagen, partially funded by an unrestricted donation from the Novo Nordisk Foundation (NNF18CC0034900 and NNF23SA0084103). Figs. 1b,g,n, 2a,e,l,u, 3m,w, 4a,m and 5a and Extended Data Figs. 1a, 3u and 4f were created with BioRender.com.
Extended data figures and tables
Author contributions
Conceptualization was done by F.S., T.M., J.B.H., T.W.S. and Z.G.-H. The peptide chemistry design campaign was led by J.B.H., M.T.-C. and M.B.F.G. The macaque study was conducted by L.B., M.K. and P.K. In vivo methodology was developed by K.A.S. and T.S.N. Human genetics analysis was carried out by M.K.A., M.S., Y.W., M.G.U., M.E.J., I.M., A.A., N.G., T.H., M.M.B. and T.O.K. In vivo mouse phenotyping studies were carried out by F.S., T.M., J.H.E., H.B.B., T.S.N., J.M.B., L.D., W.T.Y., C.K.K., A.J.T., S. Kernodle, K.A.S., C.T.V., W.I.h.P., A.L.B., M.G.-M., D.M.R., T.H.P., P.C.N.R., R.J.S., A.W., J.H., S. Kooijman and J.B.H. In situ hybridization was conducted by O.D. Pharmacological cell signalling studies were carried out by D.P.C., J.E.P., T.W.S. and A.P. Graphical visualization was done by F.S. The original draft was written by F.S., T.M., J.B.H., T.W.S. and Z.G.-H. The manuscript was edited and revised by all co-authors.
Peer review
Peer review information
Nature thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Data availability
snRNA-seq data have been deposited to the NCBI Gene Expression Omnibus under accession number GSE276735. Source data are provided with this paper.
Code availability
The source code used to analyse the snRNA-seq data and produce figures is available at https://github.com/perslab/Sass-2024/.
Competing interests
T.M., J.H.E., J.B.H., D.P.C., M.B.F.G., M.T.-C., T.W.S. and Z.G.-H. work or have worked in some capacity for Embark Laboratories ApS, a company developing therapeutics for the treatment of T2D and obesity. P.K. is a consultant for Embark Laboratories ApS. The use of and chemical composition for NK2R agonists are patented by the University of Copenhagen and Embark Laboratories ApS, respectively. R.J.S. has received research support from Novo Nordisk, Fractyl, AstraZeneca, Congruence Therapeutics, Eli Lilly, Bullfrog AI, Glycsend Therapeutics and Amgen. R.J.S. has served as a paid consultant for Novo Nordisk, Eli Lilly, CinRx, Fractyl, Structure Therapeutics, Crinetics and Congruence Therapeutics. R.J.S. has equity in Calibrate, Rewind and Levator Therapeutics. The other authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Frederike Sass, Tao Ma
Contributor Information
Jakob B. Hansen, Email: jbh@embarklaboratories.com
Zachary Gerhart-Hines, Email: zpg@sund.ku.dk.
Extended data
is available for this paper at 10.1038/s41586-024-08207-0.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-024-08207-0.
References
- 1.Christoffersen, B. Ø. et al. Beyond appetite regulation: targeting energy expenditure, fat oxidation, and lean mass preservation for sustainable weight loss. Obesity30, 841–857 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Müller, T. D., Blüher, M., Tschöp, M. H. & DiMarchi, R. D. Anti-obesity drug discovery: advances and challenges. Nat. Rev. Drug Discov.21, 201–223 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coskun, T. et al. LY3437943, a novel triple glucagon, GIP, and GLP-1 receptor agonist for glycemic control and weight loss: from discovery to clinical proof of concept. Cell Metab.34, 1234–1247.e9 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Finan, B. et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med.21, 27–36 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Steinhoff, M. S., von Mentzer, B., Geppetti, P., Pothoulakis, C. & Bunnett, N. W. Tachykinins and their receptors: contributions to physiological control and the mechanisms of disease. Physiol. Rev.94, 265–301 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maggi, C. A. & Schwartz, T. W. The dual nature of the tachykinin NK1 receptor. Trends Pharmacol. Sci.18, 351–355 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Drucker, D. J. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab.27, 740–756 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Perdomo, C. M., Cohen, R. V., Sumithran, P., Clément, K. & Frühbeck, G. Contemporary medical, device, and surgical therapies for obesity in adults. Lancet401, 1116–1130 (2023). [DOI] [PubMed] [Google Scholar]
- 9.Müller, T. D. et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab.30, 72–130 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergmann, N. C., Davies, M. J., Lingvay, I. & Knop, F. K. Semaglutide for the treatment of overweight and obesity: a review. Diabetes Obes. Metab.25, 18–35 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lau, D. C. W., Batterham, R. L. & le Roux, C. W. Pharmacological profile of once-weekly injectable semaglutide for chronic weight management. Expert Rev. Clin. Pharmacol.15, 251–267 (2022). [DOI] [PubMed] [Google Scholar]
- 12.Wilding, J. P. H. et al. Once-weekly semaglutide in adults with overweight or obesity. N. Engl. J. Med.384, 989–1002 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Jastreboff, A. M. et al. Tirzepatide once weekly for the treatment of obesity. N. Engl. J. Med.387, 205–216 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Garvey, W. T. et al. Two-year effects of semaglutide in adults with overweight or obesity: the STEP 5 trial. Nat. Med.28, 2083–2091 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies, M. et al. Semaglutide 2.4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): a randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet397, 971–984 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Lau, D. C. W. et al. Once-weekly cagrilintide for weight management in people with overweight and obesity: a multicentre, randomised, double-blind, placebo-controlled and active-controlled, dose-finding phase 2 trial. Lancet398, 2160–2172 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Speakman, J. R. et al. Total daily energy expenditure has declined over the past three decades due to declining basal expenditure, not reduced activity expenditure. Nat. Metab.5, 579–588 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cutting, W. C., Mehrtens, H. G. & Tainter, M. L. Actions and uses of dinitrophenol: promising metabolic applications. J. Am. Med. Assoc.101, 193 (1933). [Google Scholar]
- 19.Capozzi, M. E., D’Alessio, D. A. & Campbell, J. E. The past, present, and future physiology and pharmacology of glucagon. Cell Metab.34, 1654–1674 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knerr, P. J. et al. Next generation GLP-1/GIP/glucagon triple agonists normalize body weight in obese mice. Mol. Metab.63, 101533 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedrichsen, M. H. et al. Results from three phase 1 trials of NNC9204-1177, a glucagon/GLP-1 receptor co-agonist: Effects on weight loss and safety in adults with overweight or obesity. Mol. Metab.78, 101801 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tschöp, M. H. et al. Unimolecular polypharmacy for treatment of diabetes and obesity. Cell Metab.24, 51–62 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schiöth, H. B. & Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov.16, 829–842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Costanzo, M. C. et al. The Type 2 Diabetes Knowledge Portal: An open access genetic resource dedicated to type 2 diabetes and related traits. Cell Metab.35, 695–710.e6 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherwani, S. I., Khan, H. A., Ekhzaimy, A., Masood, A. & Sakharkar, M. K. Significance of HbA1c test in diagnosis and prognosis of diabetic patients. Biomark. Insights11, 95–104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonnefond, A. et al. Genetic variant in HK1 is associated with a proanemic state and A1C but not other glycemic control-related traits. Diabetes58, 2687–2697 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gjesing, A. P. et al. Studies of a genetic variant in HK1 in relation to quantitative metabolic traits and to the prevalence of type 2 diabetes. BMC Med. Genet.12, 99 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albrechtsen, A. et al. Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia56, 298–310 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jurgens, S. J. et al. Analysis of rare genetic variation underlying cardiometabolic diseases and traits among 200,000 individuals in the UK Biobank. Nat. Genet.54, 240–250 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.1000 Genomes Project Consortium. A global reference for human genetic variation. Nature526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang, Z. et al. CARMA is a new Bayesian model for fine-mapping in genome-wide association meta-analyses. Nat. Genet.55, 1057–1065 (2023). [DOI] [PubMed] [Google Scholar]
- 32.Moltke, I. et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature512, 190–193 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Madsen, K. et al. Structure−activity and protraction relationship of long-acting glucagon-like peptide-1 derivatives: importance of fatty acid length, polarity, and bulkiness. J. Med. Chem.50, 6126–6132 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Chassaing, G. et al. Selective agonists of NK-2 binding sites highly active on rat portal vein (NK-3 bioassay). Neuropeptides19, 91–95 (1991). [DOI] [PubMed] [Google Scholar]
- 35.Yeo, G. S. H. et al. The melanocortin pathway and energy homeostasis: from discovery to obesity therapy. Mol. Metab.48, 101206 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman, J. M. Leptin and the endocrine control of energy balance. Nat. Metab.1, 754–764 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Grill, H. J. & Hayes, M. R. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab.16, 296–309 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng, W. et al. Hindbrain circuits in the control of eating behaviour and energy balance. Nat. Metab.4, 826–835 (2022). [DOI] [PubMed] [Google Scholar]
- 39.Ludwig, M. Q. et al. A genetic map of the mouse dorsal vagal complex and its role in obesity. Nat. Metab.3, 530–545 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Hansen, H. H. et al. Whole-brain activation signatures of weight-lowering drugs. Mol. Metab.47, 101171 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bingdi Wang, X. W. et al. Dysglycemia and dyslipidemia models in nonhuman primates: part i. model of naturally occurring diabetes. J. Diabetes Metab.10.4172/2155-6156.S13-010 (2015).
- 42.Inokuchi, T., Watanabe, K., Kameyama, H. & Orita, M. Altered basal C-peptide/insulin molar ratios in obese patients with fatty liver. Jpn. J. Med.27, 272–276 (1988). [DOI] [PubMed] [Google Scholar]
- 43.Yin, W. et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J. Lipid Res.53, 51–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soranzo, N. et al. Common variants at 10 genomic loci influence hemoglobin A1C levels via glycemic and nonglycemic pathways. Diabetes59, 3229–3239 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peters, L. L. et al. Downeast anemia (dea), a new mouse model of severe nonspherocytic hemolytic anemia caused by hexokinase (HK1) deficiency. Blood Cells. Mol. Dis.27, 850–860 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Loos, R. J. F. & Yeo, G. S. H. The genetics of obesity: from discovery to biology. Nat. Rev. Genet.23, 120–133 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farooqi, S. & O’Rahilly, S. Genetics of obesity in humans. Endocr. Rev.27, 710–718 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Jastreboff, A. M. et al. Triple-hormone-receptor agonist retatrutide for obesity—a phase 2 trial. N. Engl. J. Med.389, 514–526 (2023). [DOI] [PubMed] [Google Scholar]
- 49.Le Roux, C. W. et al. Glucagon and GLP-1 receptor dual agonist survodutide for obesity: a randomised, double-blind, placebo-controlled, dose-finding phase 2 trial. Lancet Diabetes Endocrinol.12, 162–173 (2024). [DOI] [PubMed] [Google Scholar]
- 50.Palamiuc, L. et al. A tachykinin-like neuroendocrine signalling axis couples central serotonin action and nutrient sensing with peripheral lipid metabolism. Nat. Commun.8, 14237 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garfield, A. S. & Heisler, L. K. Pharmacological targeting of the serotonergic system for the treatment of obesity. J. Physiol.587, 49–60 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trouwborst, I., Goossens, G. H., Astrup, A., Saris, W. H. M. & Blaak, E. E. Sexual dimorphism in body weight loss, improvements in cardiometabolic risk factors and maintenance of beneficial effects 6 months after a low-calorie diet: results from the randomized controlled DiOGenes trial. Nutrients13, 1588 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allegra, S., Chiara, F., Di Grazia, D., Gaspari, M. & De Francia, S. Evaluation of sex differences in preclinical pharmacology research: how far is left to go? Pharmaceuticals16, 786 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hökfelt, T., Pernow, B. & Wahren, J. Substance P: a pioneer amongst neuropeptides. J. Intern. Med.249, 27–40 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Bai, L. et al. Enteroendocrine cell types that drive food reward and aversion. eLife11, e74964 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saito, R., Takano, Y. & Kamiya, H.-O. Roles of substance P and NK(1) receptor in the brainstem in the development of emesis. J. Pharmacol. Sci.91, 87–94 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Karagiannides, I. et al. Substance P as a novel anti-obesity target. Gastroenterology134, 747–755 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quartara, L., Altamura, M., Evangelista, S. & Maggi, C. A. Tachykinin receptor antagonists in clinical trials. Expert Opin. Investig. Drugs18, 1843–1864 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Mullard, A. FDA approves first-in-class NK3 receptor antagonist for hot flushes. Nat. Rev. Drug Discov.22, 526 (2023). [DOI] [PubMed] [Google Scholar]
- 60.Evans, T., Dixon, C., Clarke, B., Conradson, T. & Barnes, P. Comparison of neurokinin A and substance P on cardiovascular and airway function in man. Br. J. Clin. Pharmacol.25, 273–275 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nédélec, Y. et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell167, 657–669.e21 (2016). [DOI] [PubMed]
- 62.Quach, H. et al. Genetic adaptation and Neandertal admixture shaped the immune system of human population. Cell167, 643–656.e17 (2016). [DOI] [PMC free article] [PubMed]
- 63.Cheng, W. et al. Calcitonin receptor neurons in the mouse nucleus tractus solitarius control energy balance via the non-aversive suppression of feeding. Cell Metab.31, 301–312.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang, Q. et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab.33, 833–844.e5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fischer, A. W., Schlein, C., Cannon, B., Heeren, J. & Nedergaard, J. Intact innervation is essential for diet-induced recruitment of brown adipose tissue. Am. J. Physiol.316, E487–E503 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi, H. et al. Skeletal muscle O-GlcNAc transferase is important for muscle energy homeostasis and whole-body insulin sensitivity. Mol. Metab.11, 160–177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Negoita, F. et al. CaMKK2 is not involved in contraction-stimulated AMPK activation and glucose uptake in skeletal muscle. Mol. Metab.75, 101761 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dowsett, G. K. C. et al. A survey of the mouse hindbrain in the fed and fasted states using single-nucleus RNA sequencing. Mol. Metab.53, 101240 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fleming, S. J. et al. Unsupervised removal of systematic background noise from droplet-based single-cell experiments using CellBender. Nat. Methods20, 1323–1335 (2023). [DOI] [PubMed] [Google Scholar]
- 70.Stuart, T. et al. Comprehensive integration of single-cell data. Cell177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Germain, P.-L., Lun, A., Garcia Meixide, C., Macnair, W. & Robinson, M. D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Research10, 979 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Amezquita, R. A. et al. Orchestrating single-cell analysis with Bioconductor. Nat. Methods17, 137–145 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tyssowski, K. M. et al. Different neuronal activity patterns induce different gene expression programs. Neuron98, 530–546.e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nicol, P. B. et al. Robust Identification of Perturbed Cell Types in Single-Cell RNA-Seq Data. Preprint at bioRxiv10.1101/2023.05.06.539326 (2023). [DOI] [PMC free article] [PubMed]
- 75.Sveidahl Johansen, O. et al. Lipolysis drives expression of the constitutively active receptor GPR3 to induce adipose thermogenesis. Cell184, 3502–3518.e33 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sherry, S. T., Ward, M. & Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res.9, 677–679 (1999). [PubMed] [Google Scholar]
- 77.Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet.81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ghoussaini, M. et al. Open targets genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res.49, D1311–D1320 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Machiela, M. J. & Chanock, S. J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics31, 3555–3557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun.9, 1825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barbeira, A. N. et al. Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol.22, 49 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet.47, 1091–1098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jurgens, S. J. et al. Adjusting for common variant polygenic scores improves yield in rare variant association analyses. Nat. Genet.55, 544–548 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen, J. et al. The trans-ancestral genomic architecture of glycemic traits. Nat. Genet.53, 840–860 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pagès, H. SNPlocs.Hsapiens.dbSNP144.GRCh37: SNP locations for Homo sapiens (dbSNP Build 144). R package version 0.99.20 10.18129/B9.bioc.SNPlocs.Hsapiens.dbSNP144.GRCh37 (2017).
- 86.Hormozdiari, F., Kostem, E., Kang, E. Y., Pasaniuc, B. & Eskin, E. Identifying causal variants at loci with multiple signals of association. Genetics198, 497–508 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bjerregaard, P. et al. Inuit health in Greenland: a population survey of life style and disease in Greenland and among Inuit living in Denmark. Int. J. Circumpolar Health62, 3–79 (2003). [DOI] [PubMed] [Google Scholar]
- 88.Andersen, M. K. et al. The derived allele of a novel intergenic variant at chromosome 11 associates with lower body mass index and a favorable metabolic phenotype in Greenlanders. PLoS Genet.16, e1008544 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou, X. & Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet.44, 821–824 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains extended legends for Figs. 1–5 (statistical information) and Supplementary Fig. 1 (uncropped western blots for Extended Data Figs. 2s–v and 3y).
Source Data Figs. 1–5 and Source Data Extended Data Figs. 2–6
Data Availability Statement
snRNA-seq data have been deposited to the NCBI Gene Expression Omnibus under accession number GSE276735. Source data are provided with this paper.
The source code used to analyse the snRNA-seq data and produce figures is available at https://github.com/perslab/Sass-2024/.