

Abstract
Schinzel-Giedion syndrome is a rare condition characterized by dysmorphic features, neurologic features, urogenital abnormalities, and radiographic changes. The etiology has been traced to mutations in the SETBP1 gene. We report a Filipino patient with features suggestive of Schinzel-Giedion Syndrome and the first to be confirmed through molecular testing.
Keywords: Schinzel-Giedion, SETBP1, coarse facies, midface retraction
INTRODUCTION
Schinzel and Giedion (1978) first described a pair of siblings who presented with severe midface retractions, multiple skull anomalies, clubfeet, cardiac, and renal malformations.1 Prior to describing the etiology, the diagnosis of Shinzel-Giedieon Syndrome (SGS) was based on clinical characteristics proposed by the group of Lehman (2008)2 based on the presence of developmental delay and typical facial features (prominent forehead, midface retractions, and short upturned nose) associated with hydronephrosis or two of the skeletal malformations (sclerotic skull base, wide occipital synchondrosis, increased cortical density or thickness, and broad ribs). It was only in 2010 that the mutations in the SET binding protein 1 (SETBP1) have been implicated in SGS.3 There have been about 80 cases described until 2018.4 We report a Filipino child whose features were suggestive of SGS and the diagnosis was confirmed through molecular testing.
CASE
The patient is the only child of a non-consanguineous couple of Filipino descent. She was born pre-term (32 weeks age of gestation) to a 25-year-old healthy primigravid via spontaneous vaginal delivery. At birth, she had a poor cry and respiratory distress. In addition, she was described to be hirsute and with facial edema (Figure 1). She was admitted to the NICU for oxygen support and treatment of pneumonia. Her neonatal course was further complicated by probably necrotizing enterocolitis and hyperbilirubinemia. She was sent home well after 10 days.
Figure 1.
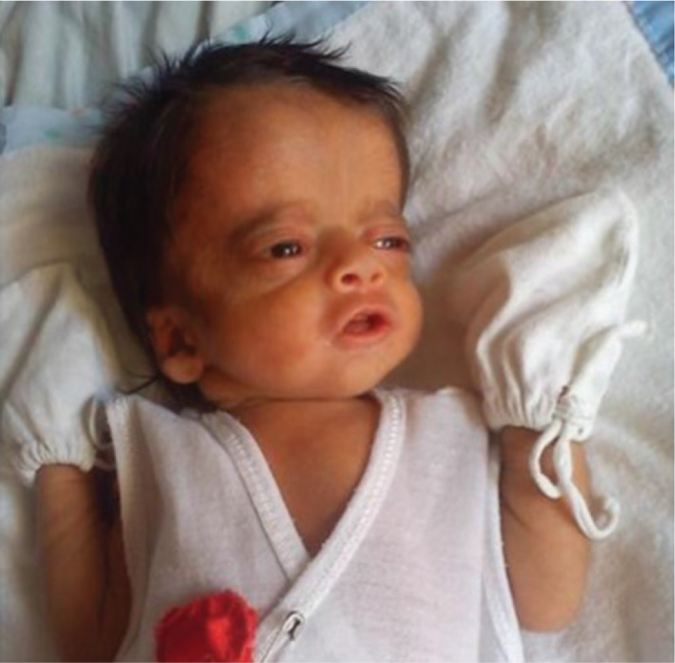
The patient at two weeks of age showed a wide prominent forehead, midface retraction, slightly upturned nose, low set ears, and mild coarse features.
At five months of age, she was unable to track objects, had a head lag, and could not rollover. Her developmental delays became more apparent with time. A cranial computer tomography (CT) Scan done at one year of age showed ventriculomegaly with associated volume loss and a thin corpus callosum. She had occasional episodes where she would have stiffening of the extremities and become cyanotic. There was also difficulty with swallowing and feeding. The family was advised to seek consult from various subspecialists.
She was first seen by a clinical geneticist at 2 years and 1 month of age. At the time of examination, she occasionally cooed and had a social smile. Her weight was 8 kg, length was 70 cm, and head circumference was 42.5 cm, all less than the 3rd percentile. She had mild hirsutism. She had an open and flat anterior fontanelle (1.5 x 1.5 cm), a wide prominent forehead, and a prominent metopic ridge. She had mild coarsening of her features, bushy eyebrows, and an infraorbital crease. She had midface retraction and retrognathia (Figure 2). Her breath sounds were clear, and no murmur was appreciated. She did not have any hepatosplenomegaly but her abdomen was globular. Her hands remained close-fisted, and she had bilateral club feet. She had an absent vaginal opening and hypoplastic labia. She was spastic and hyperreflexic.
Figure 2.

Patient at (A) 2 years and 1 month of age, (B) 4 years and 2 months, and (C) 6 years old.
She was evaluated by a pediatric neurologist at 2 years and 10 months of age. At the time, the patient continued to have episodes of generalized stiffening associated with cyanosis occurring three times per week. These episodes were diagnosed as tonic seizures and she was started on levetiracetam at a dose of 20 mg/kg/day, which decreased the frequency of seizures. An electroencephalogram (EEG) showed continuous slowing of the background activity with frequent epileptiform discharges in the left and right frontotemporal regions. At four years old, the patient had more frequent seizures and the levetiracetam dose was increased. Valproate was added to her seizure regimen. She developed intractable epilepsy exhibiting multiple seizure types, including axial tonic seizures evolving to bilateral tonic seizures, focal impaired awareness behavior arrest seizures, focal tonic seizures, focal clonic seizures, myoclonic seizures, and generalized tonic seizures.
The results of other diagnostic tests are listed in Table 1.
Table 1.
Summary of diagnostic examination results
| Test | Results |
|---|---|
| Abdominal ultrasound | Unremarkable right hepatic lobe, gallbladder, spleen, and kidneys; non-visualized left lobe of the liver and pancreas |
| Chromosomal analysis | Normal female (46, XX) |
| 2D Echocardiogram | No structural heart problems |
| Otoacoustic emissions test (OAE) | Hearing loss, bilateral |
| Skeletal survey at 6 years of age (Figures 3 and 4) | Sclerosis in the base of the skull Sclerotic changes in the C1 and C2 vertebral bodies Thoracolumbar dextroscoliosis 13 pairs of ribs Cortical thinning and shortening of the proximal end of the bilateral fibula Cystic lucencies at the base of the metacarpals |
Figure 3.

The patient’s skull radiograph showed sclerotic changes.
Figure 4.
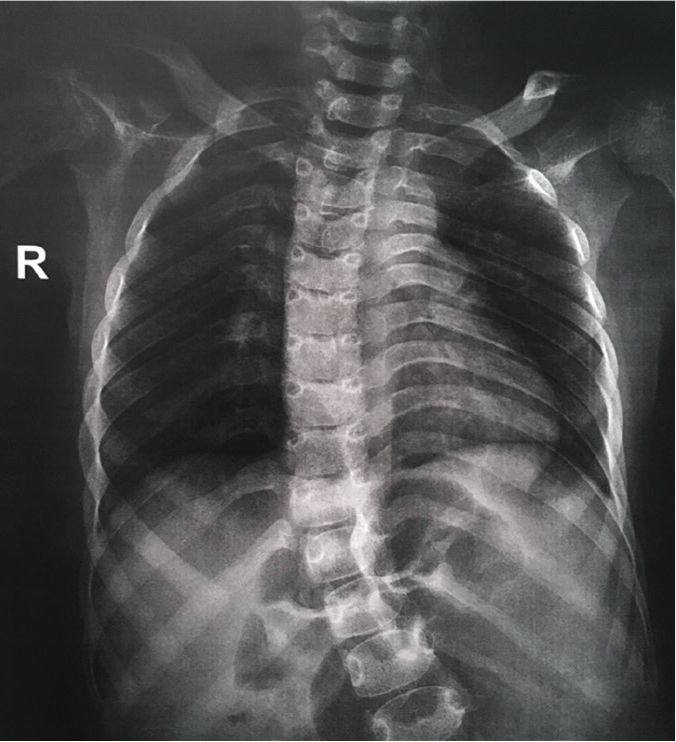
The patient’s chest radiograph showed 13 pairs of ribs.
Based on her clinical features, the three differential diagnoses were: (1) Schinzel-Giedion syndrome, based on hypertrichosis, a prominent forehead, and retracted midface; (2) Hajdu Cheney syndrome, due to the hirsutism and the somewhat coarse facial features; however, in this syndrome joint laxity is prominent, and (3) Mowat-Wilson syndrome, based on the high forehead and frontal bossing; however, our patient did not present with a prominent nasal tip, ear abnormalities, or Hirschsprung disease, which are common features.
The diagnosis of Schinzel-Giedion was confirmed through molecular testing that showed a c.2612T>C change in the SETBP1 gene resulting in a p.I871T missense mutation.
She was last seen at eight years of age with very minimal developmental progress. She had already developed flexion contractures in the elbows and knees. She succumbed to a respiratory infection a few months after.
DISCUSSION
Similar to the cases reported in the literature, the patient presented with features suggestive of SGS including developmental delay and typical facies (midface retraction and prominent forehead). She did not present with hydro-nephrosis which occurs in 88% of patients and is part of the diagnostic criteria proposed by Lehman and colleagues, but she did have genitourinary abnormalities which are reported in 76% of cases.4
In addition to midfacial hypoplasia, patients may have a coarse appearance and deep grooves under the eyes as clues to diagnosis.5 The coarse features of this syndrome are reminiscent of a storage disorder.6 Verloes et al. (1993)7 described the face as a figure “8” shape and they observed changes in features with time such as vanishing hirsutism, less prominent retraction of the midface, and more obvious bitemporal narrowing. These changes were also seen in our patient (Figures 1 and 2).
The skeletal survey of the patient was done in childhood rather than infancy but still presented with a sclerotic skull base and rib anomalies, which are commonly reported in SGS. Our patient had thinning of the cortices rather than the usual description of a dense, thickened, and untertubulated cortex, which is part of the proposed diagnostic criteria.2 However, the relative frequencies of skeletal features among reported cases have not been established and reports vary.8 Further, the absence of certain features cannot rule out the diagnosis of SGS.9 We also report that our patient has a thoracolumbar dextroscoliosis, which is a feature not commonly seen in SGS. Sharma and Gonzales (2009)10 reported a case of a 10-year-old child with SGS who had a spinal deformity and they surmise that the rare association of scoliosis with SGS is because most patients die young and they usually do not have time to develop scoliosis.
Our patient’s cranial imaging showed ventriculomegaly, which is seen in 31% of cases.4 Watanabe et al. (2012) reports that patients with SGS show cortical and white matter atrophy, enlarged ventricles, and brainstem atrophy on MRI.11 Thinning of the corpus callosum was also reported, much like in our patient.12 These changes may account for the developmental delay and neurologic symptoms that are seen in all patients with SGS. Early signs of neurologic compromise are characterized by feeding problems that were also apparent in our patient.9 The natural history of SGS is characterized by an early onset of neurologic signs, progressive spasticity, seizures, and poor motor and developmental progress.8 Epilepsy is a common feature in patients with SGS with up to 73% reported to have seizures.13 Multiple seizure types have been reported in SGS including focal motor seizures, tonic seizures, tonic-clonic seizures, myoclonic seizures, and infantile spasms.12,13 Epilepsy in SGS is frequently refractory to treatment, similar to our patient. There have also been reports of SGS presenting with epileptic encephalopathy.14
It has been postulated that SGS predisposes to an increased risk of embryonic tumors.6 An increased prevalence of malignant tumors has been reported including sacrococcygeal teratoma, lumbosacral primitive neuroectodermal tumor, hepatoblastoma, kidney tumor, extradural ependymal tumor, and malignant germ cell tumors in 11 SGS patients.4 At present, there are no guidelines regarding the surveillance of patients with SGS.
The molecular basis of SGS has only been reported in 2010. Gain-of-function or dominant-negative mutation in SETBP1 is thought to be a causative mechanism in SGS.3 This gene is located on chromosome 18q21.1, is ubiquitously expressed and its function is not yet fully elucidated.9 A study by Banfi et al. (2021) illustrated that mutations in SETBP1 lead to an accumulation of SETBP1 and SET proteins and the P53 inhibition of neural cells which in turn cause a cancer-like behavior in neural progenitors.15 As a result, there is widespread DNA damage without apoptosis that in turn stimulates Poly (ADP-ribose) polymerase 1 [PARP-1] that become Poly (ADP-ribose) [PAR] polymers, which are toxic to the cells. This is the basis for the neurological pathology seen in patients with SGS. However, a definite genotype-phenotype correlation was not identified.16
SGS was previously thought to be inherited in an autosomal recessive manner based on the initial report that siblings were affected.1 It is now widely accepted to be due to an autosomal dominant disorder whose precise incidence is unknown.9 The reported recurrence in siblings could be due to gonadal mosaicism.3
Our patient’s seizures were managed with anti-epileptic medications and she continued to undergo physical and occupational therapy. Having confirmed the diagnosis, noted the inheritance pattern, and looking at the family history, our patient’s case was likely sporadic. The family was counseled that based on the information available at the time of the consult, the recurrence risk was low. At the time of this writing, the couple had another child who did not have features suggestive of SGS.
The reported prognosis of SGS is poor with most cases dying before two years of age usually due to apneic episodes.8 However, Herenger et al. (2015), report two children ages 6 and 15 years who were alive but with a worsening neurological condition.17
CONCLUSION
SGS should be considered in children presenting with global developmental delay, hirsutism, and midface retraction. However, it must be kept in mind that just like other syndromes, SGS may occur in a spectrum. The dysmorphic features represent an important clue to the syndrome and typical features may not be present in mild cases. While reaching a diagnosis early may not improve the prognosis, establishing a diagnosis is important for genetic counseling and anticipatory guidance.
Consent
Parental consent was given for the publication of the case and the pictures. Figure 1 is courtesy of the patient’s family.
Acknowledgment
Thanks to Prof. Ivan Lo and the Clinical Genetics Service Department of Health, Hong Kong for doing the mutation analysis of this patient.
Statement of Authorship
Both authors contributed in the conceptualization of work, acquisition and analysis of data, drafting and revising and approved the final version submitted.
Author Disclosure
Both authors declared no conflicts of interest.
Funding Source
This study has no funding support.
REFERENCES
- 1.Schinzel A, Giedion A. A syndrome of severe midface retraction, multiple skull anomalies, clubfeet, and cardiac and renal malformation in sibs. Am J Med Genet. 1978; 1(4):361-75. [DOI] [PubMed] [Google Scholar]
- 2.Lehman AM, McFadden D, Pugash D, Sangha K, Gibson WT, Patel MS. Schinzel-Giedion Syndrome: Report of splenopancreatic fusion and proposed diagnostic criteria. Am J Med Genet A. 2008; 146A(1):1299-306. [DOI] [PubMed] [Google Scholar]
- 3.Hoischen A, van Bon BW Gilissen C, Arts P, van Lier B, Steehouwer Met al. De novo mutations of SETBP1 cause Schinzel-Giedion Syndrome. Nat Genet. 2010; 42(6):483-5. [DOI] [PubMed] [Google Scholar]
- 4.Liu WL, He ZX, Li F, Ai R, Ma H. Schinzel-Giedion syndrome: a novel case, review and revised diagnostic criteria. J of Genet 2018; 97(1):35-46. [PubMed] [Google Scholar]
- 5.Okamoto N, Takeuci M, Kitajima H, Hosokawa S. A patient with Schinzel-Giedion syndrome and a review of 20 patients. Jpn J Hum Genet. 1995; 40(2):189-93. [DOI] [PubMed] [Google Scholar]
- 6.Elliott AM, Meagher-Villemure K, Oudjhane K and Der Kaloustian VM. Schinzel-Giedion syndrome: further delineation of the phenotype. Clin Dysmoprh 1996; 5(2):135-42. [DOI] [PubMed] [Google Scholar]
- 7.Verloes A, Moes D, Palumbo L, Elmer C, Francois A, Bricteux G. Schinzel-Giedion syndrome. Eur J Pediatr. 1993; 152(5):421-3. [DOI] [PubMed] [Google Scholar]
- 8.AL-Mudaffer M, Oley C, Price S, Hayes I, Stewart A, Hall CMet al. Clinical and Radiological Findings in Schinzel-Giedion Syndrome. Eur J Pediatr. 2008; 167(12):1399-407. [DOI] [PubMed] [Google Scholar]
- 9.Carvalho E, Honjo R, Magalhaes M, Yamamoto G, Rocha K, Naslavsky Met al. Schinzel-Giedion Syndrome in two Brazilian patients: report of a novel mutation in STEBP1 and literature review of clinical features. Am J Med Genet Part A. 2015; 167A(5):1039-46. [DOI] [PubMed] [Google Scholar]
- 10.Sharma AK, Gonzales JA. Scoliosis in a case of Schinzel-Giedion syndrome. HESJ. 2009; 5(2):120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watanabe S, Murayama A, Haginoya K, Tanaka S, Togashi N, Abukawa Det al. Schinzel-Giedion syndrome: a further cause of early myoclonic encephalopathy and vacuolating myelinopathy. Brain Dev. 2012; 34(2):151-5. [DOI] [PubMed] [Google Scholar]
- 12.Ko JM, Lim BC, Kim KJ, Hwang YS, Ryu HW, Lee JHet al. Distinct neurological features in a patient with Schinzel-Giedion syndrome caused by a recurrent SETBP1 mutation. Child Nerv Syst. 2013; 29(4):525-9. [DOI] [PubMed] [Google Scholar]
- 13.Grosso S, Pagano C, Cioni M, Di Bartolo RM, Morgese G, Balestri P. Schinzel-Giedion syndrome: a further cause of West Syndrome. Brain Dev. 2003; 25(4):294-8. [DOI] [PubMed] [Google Scholar]
- 14.Leonardi E, Bettella E, Pelizza MF, Aspromote MC, Pollu R, Boniver Cet al. Identification of SETBP1 mutations by gene panel sequencing in individuals with intellectual disability and epileptic encephalopathy. Front Neurol. 2020; 11:593446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banfi F, Rubio A, Zaghi M, Massimino L, Fagnocchi G, Bellini Eet al. SETBP1 Accumulation induces P53 inhibition and genotoxic stress in neural progenitors underlying neurodegeneration in Schinzel-Giedion syndrome. Nat Commun. 2021. Jun 30;12(1):4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyake F, Kuroda Y, Naruto T, Ohashi I, Takano K, Kurosawa K. West syndrome in a patient with Schinzel-Giedion syndrome. J of Child Neurol. 2015; 30(7):932-6. [DOI] [PubMed] [Google Scholar]
- 17.Herenger Y, Stoetzel C, Scahefer E, Scheidecker S, Maniere M, Pelletier Vet al. Long term follow up of two independent patients with Schinzel-Giedion carrying SETBP1 Mutations. Eur J Med Genet. 2015; 58(9):479-87. [DOI] [PubMed] [Google Scholar]