

Abstract
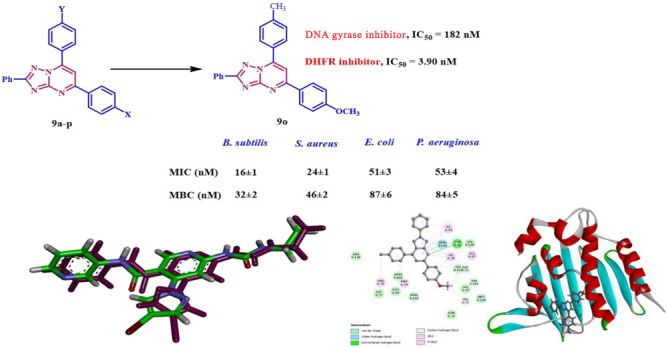
A series of 1,2,4-triazolo[1,5-a]pyrimidine-based derivatives were developed and prepared by reacting chalcones 8a–p with 3-phenyl-1,2,4-triazole-5-amine (5). The novel compounds were analyzed using several spectroscopic techniques, and their antimicrobial efficacies against six pathogens (Gram-negative, Gram-positive, and fungi) were tested. Most of the tested compounds exhibited significant antimicrobial activity compared to ciprofloxacin and fluconazole. Four compounds (9d, 9n, 9o, and 9p) showed promising results. Their minimal inhibitory concentration (MIC) values were between 16 and 102 μM, similar to ciprofloxacin’s 10–90 μM values. MIC values against the tested fungal species were between 15.50 and 26.30 μM, higher than fluconazole’s 11.50–17.50 μM values. Compounds 9n and 9o, in particular, showed excellent bactericidal activity. Compounds 9n and 9o, the most effective antibacterial agents, were further evaluated for their inhibitory effects on bacterial DNA gyrase and DHFR enzymes as possible molecular targets. The results indicated that 9n and 9o demonstrated a similar level of activity against DNA gyrase and DHFR when compared to the reference drugs ciprofloxacin and trimethoprim. We conducted molecular docking to investigate the binding mechanism and evaluate the reactivity of the intriguing compounds. Compounds 9n and 9o demonstrated favorable binding interactions with the essential amino acids necessary for the inhibition of E. coli DNA gyrase and DHFR enzymes.
1. Introduction
The global challenge of dealing with new bacterial diseases has gotten increasingly difficult due to the rising prevalence of multidrug resistance (MDR) microorganisms.1,2 This highlights the imperative necessity to develop novel, highly effective antibacterial agents. Multiple mechanisms contribute to mutations in microbial genomes, resulting in the development of resistance to established antibiotics. For example, abuse of antibiotics can lead to the emergence of resistant genotypes.3,4 Researchers are under pressure to find novel antimicrobial compounds as the incidence of infectious diseases and multidrug-resistant bacterial strains rises.5 The ultimate goal of medicinal chemistry is to develop new therapeutic compounds with superior pharmacological properties, drug tolerance, and reduced side effects.6
DNA gyrase has long been recognized as a promising target for antibacterial medicines.7 DNA gyrase belongs to the type II family of topoisomerases, which regulate the topological state of DNA in cells.8 DNA gyrase is a tetrameric enzyme comprising two subunits, GyrA and GyrB. The GyrB subunit breaks down ATP and causes DNA to supercoil negatively. This is necessary to maintain DNA structure during the replication process. DNA gyrase is a vital enzyme found in all types of bacteria. Blocking hinders the process of DNA production, ultimately leading to the death of the bacterial cells.9 In a therapeutic setting, two categories of antibiotics have proven gyrase to target DNA gyrase effectively. These categories include fluoroquinolones and aminocoumarins. Nevertheless, the increasing occurrence of antibiotic-resistant bacterial strains reduces the effectiveness of fluoroquinolone medications.10 Unfavorable pharmacokinetics and safety concerns have hindered the widespread use of the coumarin class of antibiotics.11,12
On the other hand, dihydrofolate reductase (DHFR) is a crucial component of the folate metabolic process. DHFR is essential for the biological production of RNA, DNA, and cellular proteins in different species.13,14 The enzyme is a promising focus for medicinal chemistry, and its inhibitors have been employed in treating severe ailments such as cancer, bacterial infections, malaria, tuberculosis, and others. Almost all cells contain DHFR. It plays a crucial role in biochemically sustaining the active state of the folate requirement.15,16 The dihydrofolate reductase enzyme converts 7,8-dihydrofolate to 5,6,7,8-tetrahydrofolate with the help of NADPH. This is the first step in making cofactors needed to make amino acids, thymidylate, and nucleotides like purines and pyrimidines.17,18 Thus, blocking the DHFR enzyme inhibits the DNA synthesis pathway. This inhibition is useful in various clinical settings because it causes rapidly growing cells to die.19
Bulow and Haas initially reported the 1,2,4-triazolo[1,5-a]pyrimidine heterocycle (TP, Figure 1).20 This scaffold can be found in natural products, like tobramycin (I, Figure 1), a TP derivative that was isolated from the broth of marine Streptomyces sp.21 However, most biologically active TP compounds are not in nature. Over time, the TP scaffold has found several applications in medicinal chemistry. Many studies have looked at TP derivatives as possible isosteric replacements for purines because they have structures similar to the purine ring. However, the TP heterocycle is a useful scaffold that can be used for more than just isosteric replacement techniques.22 It is often used to find biologically active molecules with good ADME-PK properties. Researchers have discovered numerous TP derivatives in recent years that show significant promise in a variety of therapeutic domains, including cancer treatment,23,24 neurological illnesses,25 and infectious diseases.26,27 Additionally, numerous TP derivatives have demonstrated potential as agrochemicals.28
Figure 1.

Structure of 1,2,4-triazolo[1,5-a]pyrimidine (TP) and compounds I–IV.
Essramycin (I, Figure 1), the first 1,2,4-triazolo[1,5-a]pyrimidine antibiotic was found in the culture broth of marine Streptomyces species, shows significant activity against Gram-positive and Gram-negative bacteria, with MIC ranging from 2.0 to 8.0 μg/mL.21 At a 6.25 μg/mL dose, triazolopyrimidine-6-carboxylic acid (II, Figure 1) inhibited M. tuberculosis H37RV growth by 92%. It also had antibacterial action against B. cereus and was nontoxic to mammalian cells. Additionally, compounds with a hydrazide group in the sixth position, IIIa and IIIb (Figure 1), showed strong antibacterial activity against Gram-positive and Gram-negative bacterial strains, with MIC values as low as 2 μg/mL. Also, IIIa and IIIb had IC50 values of 61 μg/mL against Gram-positive strains and 40 μg/mL against Gram-negative strains in the DNA displacement experiment.29
A recent study30 explored the design, synthesis, and screening of a series of 1,2,4-triazolo[1,5-a]pyrimidine derivatives. The compounds’ efficiency against bacterial and fungal infections and their safety profile were evaluated. Many novel compounds have been developed that are highly effective at killing Gram-positive and Gram-negative bacteria, with minimum inhibitory concentrations (MICs) ranging from 0.25 to 2.0 μg/mL. Compound IV (Figure 1) outperformed ciprofloxacin in inhibiting DNA gyrase (IC50 = 0.68 μM vs 0.85 μM). Molecular docking at the active site of DNA gyrase revealed a binding mechanism and docking scores comparable to those of ciprofloxacin.
Based on the information previously provided and our ongoing interest in developing new antimicrobials that target DNA gyrase and DHFR,31−35 we present the design, synthesis, and biological evaluation of a new group of 1,2,4-triazolo[1,5-a]pyrimidine-based derivatives 9a–p (Figure 2) that inhibit bacterial DNA gyrase and DHFR enzymes. Compounds 9a–p have been tested for antibacterial activity against two Gram-positive bacteria (S. aureus and B. subtilis), two Gram-negative bacteria (E. coli and P. aeruginosa), and two fungal strains (A. flavus and C. albicans), using ciprofloxacin and fluconazole as reference compounds. The most active compounds will have their minimum inhibitory concentration (MIC), and minimum bactericidal concentration (MBC) determined. We will additionally evaluate the most potent compounds as inhibitors of bacterial DNA gyrase and DHFR. Finally, molecular docking study will be conducted to assess their binding affinity to the target receptors.
Figure 2.

Structures of compounds 9a–p.
2. Results and Discussion
2.1. Chemistry
Scheme 1 outlines the synthesis of target compounds (9a–p). The key intermediate 3-phenyl-1,2,4-triazole-5-amine (5) was synthesized by two different pathways. Ethanol and sulfuric acid first converted aromatic acid 1 into ethyl ester 2. We then refluxed ethyl ester 2 with hydrazine hydrate to produce aromatic acid hydrazide 3. Acid hydrazide 3 interacted with aqueous S-methylisourea sulfate in the presence of sodium hydroxide to form aroylaminoguanidine 4, which underwent cyclization by fusion at 250 °C to yield 3-phenyl-1,2,4-triazole-5-amine 5.36 We used an alternate approach of thermal condensation of aromatic acid 1 with aminoguanidine sulfate at 210 °C under solvent-free conditions to produce 3-phenyl-1,2,4-triazole-5-amine 5.37
Scheme 1. Synthesis of the Key Intermediate 3-Phenyl-1,2,4-triazole-5-amine 5.

The synthesis of compounds 8a–p and new compounds 9a–p is outlined in Scheme 2. P-substituted acetophenones 6a–d were reacted with aryl aldehydes 7a–d in methanolic sodium hydroxide to afford chalcones 8a–p in good yields.38 The new compounds 9a–p were synthesized by cyclocondensation of amino triazole 5 with chalcone derivatives 8a–p. The reaction unfolded in three stages. Initially, the aminotriazole’s nucleophilic amino group reacts with the chalcone carbonyl. Second, when triazole NH2 is introduced into the chalcone double bond, a cyclization reaction takes place. The procedure ends with the elimination of two hydrogens atoms, followed by aromatization.
Scheme 2. Synthesis of New Compounds 9a–p.

The structures of the newly synthesized compounds 9a–p were verified by 1H NMR, 13C NMR, LC-MS, and elemental analysis. Compound 9k is a good example of this group. Its 1H NMR spectrum showed two singlet signals of three protons at δ 2.44 and 2.38 ppm, which are two methyl groups. The 13C NMR spectrum confirmed this, revealing two singlet signals at approximately δ 21.70 and 21.53 ppm. Furthermore, the spectrum revealed the distinctive signals of aromatic protons. Additionally, the LC-MS spectra revealed a signal at m/z 377.00, corresponding to [M + H]+.
2.2. Biology
2.2.1. In Vitro Antimicrobial Assay
The antimicrobial properties of the target compounds (9a–p) were evaluated against two Gram-positive bacteria (S. aureus and B. subtilis), two Gram-negative bacteria (E. coli and P. aeruginosa), and two fungal strains (A. flavus and C. albicans). The modified disk diffusion method39,40 was used to determine the inhibition zones (IZ, mm/ml) and the minimal inhibitory concentration (MIC, nM). Ciprofloxacin and fluconazole were the positive controls used in the experiment.
The target compounds 9a–p demonstrated significant antibacterial activity against both Gram-positive and Gram-negative bacteria. The inhibition zones (IZ) of all tested pathogens ranged from 9 to 45 mm, comparable to ciprofloxacin’s IZ of 40 mm. Notably, compounds 9d, 9n, 9o, and 9p exhibited larger or similar inhibition zones to those of the reference ciprofloxacin against the examined bacterial strains, as shown in Table 1. These findings not only shed light on the potential of these compounds as effective antibacterial agents but also inspire hope for the future of antibacterial research.
Table 1. Inhibition Zone Diameter of Compounds 9a–p.
|
Inhibition
Zone (IZ) Diameter (mm/mL) |
||||||
|---|---|---|---|---|---|---|
|
Bacterial
species |
|
|||||
|
(G+) |
(G–) |
Fungi |
||||
| Sample | B. subtilis | S. aureus | E. coli | P. aeruginosa | A. flavus | C. albicans |
| 9a | 12 | 14 | 14 | 12 | 7 | 10 |
| 9b | 21 | 23 | 23 | 24 | 11 | 15 |
| 9c | 11 | 15 | 13 | 13 | 7 | 10 |
| 9d | 35 | 35 | 33 | 34 | 27 | 30 |
| 9e | 10 | 12 | 12 | 11 | 5 | 8 |
| 9f | 14 | 15 | 15 | 18 | 8 | 12 |
| 9g | 19 | 20 | 20 | 18 | 9.0 | 11 |
| 9h | 9 | 11 | 11 | 12 | 5 | 7 |
| 9i | 28 | 29 | 30 | 29 | 17 | 20 |
| 9j | 20 | 21 | 20 | 20 | 10 | 13 |
| 9k | 32 | 32 | 30 | 30 | 22 | 25 |
| 9l | 23 | 26 | 23 | 22 | 15 | 17 |
| 9m | 31 | 32 | 30 | 30 | 20 | 23 |
| 9n | 40 | 41 | 40 | 40 | 32 | 36 |
| 9o | 43 | 45 | 43 | 42 | 35 | 38 |
| 9p | 38 | 39 | 37 | 38 | 30 | 31 |
| Ciprofloxacin | 40 | 40 | 40 | 40 | NA | NA |
| Fluconazole | NA | NA | NA | NA | 40 | 40 |
The compound 9o (X = OMe, Y = Me) was identified as the most effective antibacterial agent in our study. It exhibited IZ values of 43 and 45 mm against Gram-positive pathogens B. subtilis and S. aureus and 43 and 42 mm against Gram-negative strains E. coli and P. aeruginosa, surpassing the reference ciprofloxacin (IZ = 40 mm/ml) against all species tested. Our findings not only suggest that the substitution pattern of the phenyl groups at positions 5 and 7 may indeed play a crucial role in the antibacterial action but also provide a deeper understanding of the role of the phenyl group in the antibacterial action. Compounds 9c (X = H, Y = Me), 9g (X = Cl, Y = Me), and 9k (X = Me, Y = Me) are structurally similar to 9o, but they have H, Cl, and Me groups at the para-position of the phenyl group at position 5 of the 1,2,4-triazolopyrimidine scaffold. These compounds exhibited weaker antibacterial activity than 9o, with IZ values ranging from 11 to 15 mm for 9c, 18–20 mm for 9g, and 30–32 mm for 9k (9o; IZ = 42–45 mm). This reiterates the importance of the phenyl group at the fifth position’s para-position in tolerating the methoxy group for antibacterial action and the order of reactivity as X = OMe > Me > Cl > H.
Also, the substitution pattern of the para-position of the phenyl group at position 7 may influence the antibacterial effect of 9a–p. Compound 9n (X = OMe, Y = Cl), which substitutes a chlorine atom for the methyl group in 9o, demonstrated the second-highest activity against all tested species, with an IZ diameter of approximately 40 mm. Its activity was comparable to that of ciprofloxacin. Another example is compound 9p (X = OMe, Y = OCH3), which is the methoxy derivative of 9o. With IZ diameters of approximately 38 mm against the four species under examination, it exhibited nearly identical activity as 9n. These results demonstrate that the antibacterial action tolerated methyl and methoxy groups, along with a chlorine atom at the para-position of the phenyl group at position 7, in the activity order Y = Me > Cl > OMe. The unsubstituted derivative at the para-position of the phenyl group at position 7, compound 9m (X = OMe, Y = H), was the least active of the methoxy derivatives (9m–p). The IZ diameter of 9m, of approximately 30 mm, versus the four studied species indicates that the H atom is not favored for antibacterial activity.
In addition, the newly synthesized compounds 9a–p showed IZ diameter values ranging from 5 to 38 mm against A. flavus and C. albicans, whereas the reference fluconazole had a value of 40 mm. Once again, compounds 9d, 9n, 9o, and 9p exhibited the highest activity level against the fungal species tested. The diameter of the inhibition zones ranged from 27 to 38 mm. These findings from Table 1 indicate that the identical principles governing the antibacterial effect can also be applied to the antifungal effect. We experimented to determine the minimal inhibitory concentrations (MIC, nM) of the most potent derivatives 9d, 9n, 9o, and 9p. Tables 2 and 3 present the results.
Table 2. MIC and MBC (nM) Values of Compounds 9d, 9n, 9o, and 9p.
| Bacterial
species |
||||||||
|---|---|---|---|---|---|---|---|---|
|
(G+) |
(G–) |
|||||||
| B. subtilis |
S. aureus |
E. coli |
P. aeruginosa |
|||||
| Compound | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC |
| 9d | 38 ± 2 | 64 ± 3 | 39 ± 2 | 71 ± 4 | 73 ± 5 | 102 ± 7 | 75 ± 5 | 109 ± 7 |
| 9n | 29 ± 2 | 49 ± 2 | 33 ± 2 | 65 ± 2 | 65 ± 4 | 93 ± 6 | 64 ± 5 | 94 ± 6 |
| 9o | 16 ± 1 | 32 ± 2 | 24 ± 1 | 46 ± 2 | 51 ± 3 | 87 ± 6 | 53 ± 4 | 84 ± 5 |
| 9p | 35 ± 2 | 52 ± 3 | 37 ± 2 | 69 ± 3 | 69 ± 4 | 97 ± 6 | 68 ± 5 | 98 ± 6 |
| Ciprofloxacin | 10 ± 1 | 19 ± 1 | 30 ± 2 | 45 ± 2 | 60 ± 4 | 90 ± 6 | 60 ± 5 | 90 ± 6 |
Table 3. MIC Values of Compounds 9d, 9n, 9o, and 9p against A. flavus and C. albicans.
| Fungi
(μM) |
||
|---|---|---|
| A. flavus | C. albicans | |
| Compound | MIC | MIC |
| 9d | 21.50 ± 1 | 26.30 ± 1 |
| 9n | 18.50 ± 1 | 21.90 ± 1 |
| 9o | 15.50 ± 1 | 19.50 ± 1 |
| 9p | 19.90 ± 1 | 23.70 ± 1 |
| Fluconazole | 11.50 ± 1 | 17.50 ± 1 |
2.2.2. Minimum Inhibitory Concentration (MIC) Assay
We assessed the antibacterial activity of the most potent components, 9d, 9n, 9o, and 9p, using a 2-fold serial dilution approach on a 96-well microtiter plate.40Table 2 presented these compounds’ MICs against the examined organisms, with ciprofloxacin as the reference medication. The outcomes of this in vitro assay test align with the results of the antimicrobial sensitivity test.
The compound 9o (X = OMe, Y = CH3) was the most effective against S. aureus, E. coli, and P. aeruginosa, with MIC values of 24, 51, and 53 nM. It was more potent than ciprofloxacin against the tested species, although its MIC value against B. subtilis was 16 nM, 1.6 times less effective than ciprofloxacin (MIC = 10 nM). Compound 9n (X = OMe, Y = Cl) had the second-highest activity level. As shown in Table 2, the tested compound had higher MIC values than ciprofloxacin, which means it was less effective against S. aureus, E. coli, and P. aeruginosa. Nevertheless, It exhibited 3 times lower efficacy than ciprofloxacin against B. subtilis. Among the tested species, compound 9p (X = OMe, Y = OMe) was most effective against S. aureus and P. aeruginosa, with MICs of 69 and 68 nM, respectively. The data were similar to ciprofloxacin’s, with a MIC of 60 nM. Finally, compound 9d (X = H, Y = OCH3) demonstrated the lowest efficacy. It has a lower antibacterial efficacy than ciprofloxacin against all pathogens examined.
2.2.3. Minimum Bactericidal Concentration (MBC Assay)
The MBC differs from the MIC. The MIC test determines the minimum inhibitory concentration of an antimicrobial agent that effectively inhibits microorganism growth. In contrast, the MBC test reveals the minimum bactericidal concentration that leads to the death of these organisms. Unlike the MBC, which leads to mortality, the MIC hinders bacterial growth without causing death.41 The common expression for MBC is MBC50, signifying the antibiotic concentration that eliminates 50% of the initial bacterial population.42
Components 9d, 9n, 9o, and 9p exhibited potent bactericidal action overall. For Gram-positive bacteria, the MBC ranged from 32 to 71 nM. For ciprofloxacin, the MBC ranged from 19 to 45 nM, as shown in Table 2. Compounds 9o (X = OMe, Y = CH3) and 9n (X = OMe, Y = Cl) are the most effective antibacterial agents, demonstrating bactericidal activity of 32 and 49 nM, respectively, against B. subtilis, compared to ciprofloxacin’s MBC value of 19 nM, being 1.7- and 2.6-folds less potent than ciprofloxacin as a bactericidal agent. Compound 9o exhibited an MBC50 value of 46 nM against S. aureus, indicating that it was almost as efficient as ciprofloxacin (MBC50 = 46 nM) in killing the organism tested.
In the case of Gram-negative bacteria, compound 9o had the lowest MBC values among the compounds tested against E. coli and P. aeruginosa. This compound has MBC values of 87 and 84 nM, respectively, which makes it more efficient than ciprofloxacin (MBC = 90 nM) against E. coli and P. aeruginosa species. Compounds 9n (X = OMe, Y = Cl) and 9p (X = OMe, Y = OMe) demonstrated significant bactericidal action. Their MBC values were 93 and 97 nM for E. coli and 94 and 98 nM for P. aeruginosa, respectively, similar to ciprofloxacin’s value of 90 nM. This means that 9n and 9p are also effective at killing bacteria. Compound 9d (X = H, Y = OCH3) was the least effective bactericidal agent. It has poorer efficacy than ciprofloxacin against all tested pathogens.
2.2.4. Antifungal Assay
We evaluated the antifungal activity of compounds 9d, 9n, 9o, and 9p using a 2-fold serial dilution approach.43Table 3 shows these compounds’ MICs (μM) against A. flavus and C. albicans fungi, with fluconazole as the reference drug.
Compared to fluconazole, the studied compounds demonstrated moderate to good antifungal efficacy against the selected fungus species. Compounds 9d, 9n, 9o, and 9p reported MIC values of 15.50 to 21.50 μM against A. flavus, while fluconazole had a MIC of 11.50 μM. The studied compounds have MIC values ranging from 19.50 to 26.50 μM against C. albicans, with the reference fluconazole having a MIC of 17.50 μM. Compound 9o (R = OMe, X = CH3) exhibited high potency as an antibacterial and antifungal agent. It possessed MIC values of 15.50 μM against A. flavus and 19.50 μM against C. albicans, similar to the MIC values of the reference fluconazole (11.50 and 17.50 μM, respectively). The relatively low antifungal efficacy of the investigated compounds did not motivate us to evaluate their MBC values.
2.2.5. DNA Gyrase and DHFR Inhibitory Assay
The inhibitory efficacy of the most potent antibacterial agents, derivatives 9d, 9n, 9o, and 9p, was assessed against E. coli DNA gyrase and DHFR.44Table 4 presents the results as IC50 values for the tested compounds and the reference medicines (Novobiocin and Trimethoprim).
Table 4. IC50 Values of Compounds 9d, 9n, 9o, and 9p against DNA Gyrase and DHFR Enzymes.
| IC50 (μM) |
||
|---|---|---|
| Compound | E. coli DNA gyrase | DHFR E. coli |
| 9d | 217 ± 13 | 5.90 ± 0.20 |
| 9n | 190 ± 11 | 4.60 ± 0.20 |
| 9o | 182 ± 10 | 3.90 ± 0.10 |
| 9p | 205 ± 13 | 5.40 ± 0.20 |
| Novobiocin | 170 ± 10 | -- |
| Trimethoprim | -- | 5.20 ± 0.20 |
The results of this assay were consistent with the antibacterial activity findings. Compound 9n (X = OMe, Y = Cl) and 9o (X = OMe, Y = Me), the most potent antibacterial agents, demonstrated inhibitory effects on E. coli DNA gyrase, with IC50 values of 197 and 185 nM, respectively. These values were compared to the reference compound novobiocin, which had an IC50 value of 170 nM. However, compounds 9d (X = H, Y = OMe) and 9p (X = OMe, Y = OMe) had a noteworthy inhibitory impact on DNA gyrase, with IC50 values of 217 and 205, respectively. These values indicate that they are less effective than novobiocin.
Using Trimethoprim as a reference medication, we further tested Compounds 9d, 9n, 9o, and 9p for their inhibitory efficacy against the DHFR enzyme. The DHFR enzyme was strongly blocked by compounds 9n and 9o, with IC50 values of 4.60 and 3.90 μM, respectively, lower than the IC50 value of 5.20 for trimethoprim. Compounds 9n and 9o were more powerful than the reference trimethoprim and had significant antibacterial action. Compounds 9d and 9p showed IC50 values of 5.90 and 5.40 μM, respectively. These values were comparable to the reference trimethoprim (IC50 = 5.20 μM). These findings demonstrate the potential antibacterial activity of compounds 9n and 9o as dual inhibitors of DNA gyrase and DHFR enzymes, which may necessitate structural modifications to produce more potent derivatives with improved pharmacologic and pharmacokinetic properties.
2.3. Molecular Docking Study
2.3.1. E. coli DNA Gyrase B
Docking analysis was used to evaluate the potential binding interactions of the newly synthesized compounds within the E. coli DNA gyrase B active site, predicting their binding pattern and determining their capacity to satisfy the necessary structural properties for binding interactions. Autodock 4.245 was used to dock compounds 9b and 9o into the crystal structure of the active site of E. coli DNA gyrase B complexed with the pyridine-3-carboxamide ligand inhibitor (PDB ID: 6F86).46 We first verified the docking setup by self-docking the cocrystallized thiazole inhibitor in the active site of E. coli DNA gyrase B. The self-docking validation recreated the cocrystallized ligand inhibitor, proving that the docking method was right for the planned research, Figure 3. The minimal RMSD of 1.38 between the experimental cocrystallized inhibitor pose and the docked pose, along with the docking pose’s ability to replicate all essential interactions made by the cocrystallized ligands at the active site, demonstrate this.
Figure 3.

Validation of the accuracy and performance of docking process for E. coli DNA gyrase. The docked ligand inhibitor (purple) and native ligand inhibitor (green) demonstrated an RMSD of 1.38 Å.
The calculated docking energy score was S = −11.76 kcal/mol. Asn46 forms hydrogen bonds with the oxygen function of urea, while Asp73 interacts with the two nitrogen atoms of urea via hydrogen bonding. The pyridine ring of N-pyridine carboxamide interacts with Arg136 as a π-cation, and the pyridine ring of pyridine carboxamide interacts with Glu50 as a π-anion. The remaining amino acids in the pocket, including Arg76, Pro79, Ile94, Ile78, Ala47, Val71, Asp73, Gly77, Gln72, Met166, Val43, Thr165, Val120, and Val167, interact with the methoxy and substituted triazolopyrimidine functional groups through π-alkyl, alkyl, or van der Waals interactions, see Figure 4 and Table 5.
Figure 4.

2D diagram representation of (left side) and 3D diagram representation (right side) of thiazole inhibitor docked into E. coli DNA gyrase B active site showing his binding interactions with the amino acids binding site.
Table 5. Binding Scores, Amino Acid Interactions, and Bond Lengths of the Selected Compounds within the Active Site of E. coli DNA Gyrase B.
| Interacting
residues |
||||
|---|---|---|---|---|
| Hydrophobic
interaction |
||||
| Compound | Binding score (S value) kcal/mol | H-bonding | π-cation, π-anion, π-alkyl interactions | van der Waals interactions |
| 9n | –11.42 | Asn46 | Arg76, Pro79, Ile94, Ile78, Ala47, Val71, Asp73 | Gly77, Glu50, Gln72, Met166, Val43, Thr165, Val120, Val167 |
| 9o | –11.28 | Asn46 | Arg76, Pro79, Ile94, Ile78, Ala47, Val71, Asp73 | Arg136, Gly77, Glu50, Gln72, Met166, Val43, Thr165, Val120, Val167, |
| Ligand inhibitor | –11.76 | GLY77, Asn46, ASP73 | Glu50, Arg136, Ala47, Pro79, Ile94, Ile78 | Met166, Gln72, Val71, Val167, Val43, Thr165, Arg76 |
The molecular docking analysis revealed that compounds 9n and 9o exhibited a favorable fit within the binding site of E. coli DNA gyrase B. The docking energy scores for these compounds were −11.42 and −11.28 kcal/mol, respectively. The two most active molecules, 9n and 9o, engage with Asn46 through hydrogen bonding via triazole-pyrimidine N4. The substituted triazolopyrimidine functionals form interactions with amino acids lining the pocket through π-alkyl, alkyl, or van der Waals interactions. These interactions are illustrated in Figures 5 and 6 and summarized in Table 5.
Figure 5.

2D diagram representation (left side) and 3D diagram representation (right side) of compound 9n docked into E. coli DNA gyrase B active site showing his binding interactions with the amino acids binding site.
Figure 6.

2D diagram representation (left side) and 3D diagram representation (right side) of compound 9o docked into E. coli DNA gyrase B active site showing his binding interactions with the amino acids binding site.
2.3.2. E. coli DNA Dihydrofolate Reductase (ecDHFR)
Docking of ecDHFR setup was first validated by performing self-docking of the cocrystallized ligand, TMP, in the active site of ecDHFR (PDB ID: 6R7G).47 The self-docking validation replicated the cocrystallized thiazole, demonstrating that the docking methodology is appropriate for the targeted docking investigation. The small RMSD of 0.91 Å demonstrates this between the experimental cocrystallized inhibitor pose and the docked pose and the docking pose’s ability to mimic all essential interactions achieved by the cocrystallized ligands in the active site, Figure 7.
Figure 7.
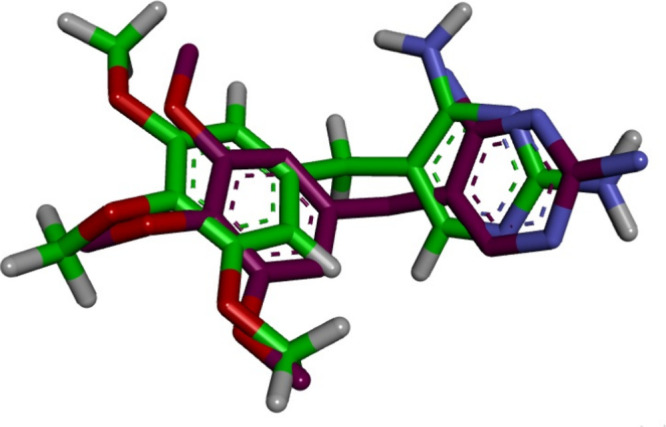
Validation of the accuracy of docking process for ecDHFR. The docked ligand inhibitor (purple) and native ligand inhibitor (green) demonstrated an RMSD of 0.91 Å.
The docking energy score (S) was −12.07 kcal/mol. Glu28, Met6, Ser97, and Tyr103 form hydrogen bonds with the amino groups of the 2,4-diaminopyrimidine function. Glu28 interacts with the pyrimidine ring via the μ-anion interaction. The remaining amino acids (Ala7, Ala8, Phe32, Leu64, Gly52, Met51, Val43, Met6) interact with the methoxyphenyl or pyrimidine rings through μ–μ or van der Waals interactions, Figure 8 and Table 6.
Figure 8.

2D diagram representation (left side) and 3D diagram representation (right side) of Trimethoprim docked into ecDHFR active site showing his binding interactions with the amino acids binding site.
Table 6. Binding Scores, Amino Acid Interactions, and Bond Lengths of the Selected Compounds within the Active Site of ecDHFR.
| Interacting
residues |
||||
|---|---|---|---|---|
| Hydrophobic
interaction |
||||
| Compound | Binding score (S value) kcal/mol | H-bonding | π–π, π–σ, π–alkyl interactions | van der Waals interactions |
| 9n | –12.37 | Tyr59, | Ala7, Ala8, Leu54, Phe32, Met51, Phe48, Ile15, Tyr103, | Ala53, Gly52, Glyn29, Glu28, Ly26, Ala25, Ser24, Trp23, Ile21, Ser97, Val43, Thr47, Gly99, Gly98, |
| 9o | –12.37 | Tyr59 | Ala7, Ala8, Leu54, Phe32, Met51, Phe48, Ile15, Tyr103, | Ala53, Gly52, Glyn29, Glu28, Ly26, Ala25, Ser24, Trp23, Ile21, Met6, Ser97, Val43, Thr47 |
| TMP | –12.07 | Glu28, Met6, Ser97, Tyr103 | Ala8, Leu54, Val43, Phe32, Met51 | Ala7, Leu64, Gly52, Met51, Met6, Glu28, Tyr59, Phe48, Thr47, Thr116, Ile21, Lys33, Gln29, |
The molecular docking studies revealed that compounds 9n and 9o orient and fit into the ecDHFR binding site similarly, with docking energy scores of −12.37 kcal/mol for both compounds. Compounds 9n and 9o form hydrogen bonds with Tyr59 via methoxy oxygen. The substituted triazolopyrimidine functionals interact with the key amino acids lining the pocket, including Ala7, Ala8, Leu54, Phe32, Met51, Phe48, Ile15, Tyr103, Ala53, Gly52, Glyn29, Glu28, Ly26, Ala25, Ser24, Trp23, Ile21, Met6, Ser97, Val43, Thr47. Interact using π–π, π–σ, and π-alkyl interactions. Phe48, Phe32, Lys9, Ile15, Tyr103, Ala7, Al8, Ala25, Lys26, Met51, Gly52, Ala53, Leu54, Tyr59 interact with the different phenyl or triazolopyrimidine rings by μ–μ, π–σ, π-alkyl, or van der Waals interactions, Figures 9 and 10.
Figure 9.

2D diagram representation (left side) and 3D diagram representation (right side) of 9n docked into ecDHFR active site showing his binding interactions with the amino acids binding site.
Figure 10.

2D diagram representation (left side) and 3D diagram representation (right side) of 9o docked into ecDHFR active site showing his binding interactions with the amino acids binding site.
2.4. Structure–Activity Relationship (SAR) Analysis
-
1.
The 1,2,4-triazolo[1,5-a]pyrimidine framework is crucial for efficacy. The N4 of the triazolo-pyrimidine moiety forms a hydrogen bond with the Asn46 amino acid residue, enhancing the binding to the receptor site of bacterial DNA gyrase. The triazolopyrimidine ring engages with several amino acids lining the pocket site of the DHFR enzyme by μ–μ, π–σ, π-alkyl, or van der Waals interactions.
-
2.
The para-position of the phenyl group at position 5 of the 1,2,4-triazolopyrimidine scaffold (X) is crucial for antibacterial efficacy and activity against bacterial DNA gyrase and DHFR, with the reactivity rank as X = OMe > Me > Cl > H. The oxygen atom in the methoxy group participates in hydrogen bonding with Tyr59 in the DHFR receptor site. The methoxy group can engage in interactions with the amino acids that delineate the pocket via π-alkyl, alkyl, or van der Waals forces in bacterial DNA gyrase.
-
3.
The substitution pattern of the para-position of the phenyl group at position 7 (Y) may influence the activity of 9a–p, where the order of reactivity as Y = Me > Cl > OMe. This substitution may improve binding to receptor sites by interactions with amino acids that lining the pocket through π-alkyl, alkyl, or van der Waals forces.
3. Conclusions
We evaluated the antimicrobial efficacy of 16 novel compounds based on the 1,2,4-triazolo[1,5-a]pyrimidine scaffold against four bacterial strains and two fungus species. Compounds 9d, 9n, 9o, and 9p show notable antibacterial efficacy. Compounds 9n and 9o demonstrated significant bactericidal activity. Furthermore, compounds 9n and 9d have shown significant inhibitory action against bacterial DNA gyrase and DHFR as potential molecular targets. Moreover, the molecular docking analysis revealed that compounds 9n and 9o exhibited a favorable fit within the binding site of E. coli DNA gyrase and DHFR. Structure modifications are under process by adding more substitutions to the phenyl groups at positions 5 and 7 or replacing the phenyl group at position 2 with electron-donating or electron-withdrawing groups. This is undertaken to develop more potent lead compounds for in vitro, in vivo, and stability research.
4. Experimental Section
4.1. Chemistry
General Details: See Appendix A.
Synthesis and analytical data of compounds 5(36) and 8a–p35 were reported.
4.1.1. Synthesis of 2-(5-Cyano-6-oxo-4-phenyl-1,6-dihydro-pyrimidin-2-ylsulfanyl)-N-(4-phenyl-thiazol-2-yl)acetamide Derivatives (9a-p)
A mixture of 3-phenyl-1,2,4-triazole-5-amine (5) (0.30 g, 1.2 mmol) and benzylidene-acetophenone derivatives (8a-p) (1.2 mmol) was heated at 210 °C in a solvent-free condition for 1 h. The reaction mixture was cooled and crystallized from ethanol 95%.
4.1.1.1. 2,5,7-Triphenyl-1,2,4-triazolo[1,5-a]pyrimidine (9a)
Yield, 66%; m.p., 210–212 °C; 1H NMR (400 MHz, CDCl3) δ 8.33–8.23 (m, 2H, Ar–H), 8.18–8.08 (m, 4H, Ar–H), 7.58–7.50 (m, 2H, Ar–H), 7.46 (s, 1H, Ar–H), 7.44–7.32 (m, 5H, Ar–H), 6.95 (d, J = 8.5 Hz, 2H, Ar–H).13C NMR (101 MHz, CDCl3) δ 165.78, 162.44, 160.68, 157.25 (C = N carbons), 146.05, 137.94, 130.78, 130.59, 130.48, 129.37, 129.22, 128.77, 128.69, 128.66, 127.52, 114.44 (Ar carbons), 105.44 (triazolopyrimidine C6). LC-MS (m/z) calcd for C23H16N4; 348.14, Found: 349.20 [M + H]+. Anal. Calcd for C23H16N4: C, 79.29; H, 4.63; N, 16.08. Found: C, 79.13; H, 4.78; N, 16.11
4.1.1.2. 7-(4-Chlorophenyl)-2,5-diphenyl-1,2,4-triazolo[1,5-a]pyrimidine (9b)
Yield, 69%; m.p., 214–216 °C; 1H NMR (400 MHz, CDCl3) δ 8.35–8.28 (m, 2H, Ar–H), 8.16 (td, J = 7.9, 7.1, 2.8 Hz, 4H, Ar–H), 7.59–7.54 (m, 2H, Ar–H), 7.50 (s, 1H, Ar–H), 7.41 (dd, J = 5.2, 2.0 Hz, 4H, Ar–H), 6.98–6.93 (m, 2H, Ar–H). LC-MS (m/z) calcd for C23H15ClN4; 382.10, Found: 382.87 [M + H]+. Anal. Calcd for C23H15ClN4: C, 72.16; H, 3.95; N, 14.63. Found: C, 72.25; H, 3.82; N, 14.70
4.1.1.3. 2,5-Diphenyl-7-p-tolyl-1,2,4-triazolo[1,5-a]pyrimidine (9c)
Yield, 68%; m.p., 218–220 °C; 1H NMR (400 MHz, CDCl3) δ 8.48–8.37 (m, 2H, Ar–H), 8.31–8.16 (m, 2H, Ar–H), 8.11 (d, J = 7.9 Hz, 2H, Ar–H), 7.64–7.43 (m, 7H, Ar–H), 7.37 (d, J = 7.9 Hz, 2H, Ar–H), 2.46 (s, 3H, CH3). LC-MS (m/z) calcd for C24H18N4; 362.15, Found: 363.10 [M + H]+. Anal. Calcd for C24H18N4: C, 79.54; H, 5.01; N, 15.46. Found: C, 79.65; H, 4.89; N, 15.35.
4.1.1.4. 7-(4-Methoxyphenyl)-2,5-diphenyl-1,2,4-triazolo[1,5-a]pyrimidine (9d)
Yield, 72%; m.p., 228–230 °C; 1H NMR (400 MHz, CDCl3) δ 8.24 (dd, J = 6.7, 3.0 Hz, 2H), 8.12 (d, J = 8.6 Hz, 2H), 8.02 (d, J = 8.3 Hz, 2H), 7.38–7.29 (m, 7H), 6.97 (d, J = 8.6 Hz, 2H), 3.80 (s, 3H). LC-MS (m/z) calcd for C24H18N4O; 378.15, Found: 379.00 [M + H]+. Anal. Calcd for C24H18N4O: C, 76.17; H, 4.79; N, 14.81. Found: C, 76.07; H, 4.86; N, 14.95.
4.1.1.5. 5-(4-Chlorophenyl)-2,7-diphenyl-1,2,4-triazolo[1,5-a]pyrimidine (9e)
Yield, 76%; m.p., 260–262 °C; 1H NMR (400 MHz, CDCl3) δ 8.30–8.24 (m, 2H), 8.16–8.06 (m, 4H), 7.55 (d, J = 5.7 Hz, 2H), 7.47 (s, 1H), 7.39 (d, J = 6.8 Hz, 6H). LC-MS (m/z) calcd for C23H15ClN4; 382.10, Found: 383.00 [M + H]+. Anal. Calcd for C23H15ClN4: C, 72.16; H, 3.95; N, 14.63. Found: C, 72.25; H, 4.08; N, 14.70.
4.1.1.6. 5,7-Bis(4-Chlorophenyl)-2-phenyl-1,2,4-triazolo[1,5-a]pyrimidine (9f)
Yield, 70%; m.p., 202–204 °C; 1H NMR (400 MHz, CDCl3) δ 8.30 (dd, J = 6.7, 3.0 Hz, 2H, Ar–H), 8.14 (d, J = 8.2 Hz, 4H, Ar–H), 7.56 (d, J = 8.1 Hz, 2H, Ar–H), 7.50 (s, 1H, Ar–H), 7.47–7.37 (m, 5H, Ar–H). LC-MS (m/z) calcd for C23H14Cl2N4; 416.06, Found: 417.20 [M + H]+. Anal. Calcd for C23H14Cl2N4: C, 66.20; H, 3.38; N, 13.43. Found: C, 66.32; H, 3.45; N, 13.35.
4.1.1.7. 5-(4-Chlorophenyl)-2-phenyl-7-p-tolyl-1,2,4-triazolo[1,5-a]pyrimidine (9g)
Yield, 74%; m.p., 248–250 °C; 1H NMR (400 MHz, CDCl3) δ 8.26 (dd, J = 6.7, 3.0 Hz, 2H, Ar–H), 8.04 (dd, J = 15.4, 8.1 Hz, 4H, Ar–H), 7.35 (td, J = 20.0, 18.8, 10.2 Hz, 8H, Ar–H), 2.39 (s, 3H). LC-MS (m/z) calcd for C24H17ClN4; 396.11, Found: 397.10 [M + H]+. Anal. Calcd for C24H17ClN4: C, 72.63; H, 4.32; N, 14.12. Found: C, 72.75; H, 4.25; N, 14.20.
4.1.1.8. 5-(4-Chlorophenyl)-7-(4-methoxyphenyl)-2-phenyl-1,2,4-triazolo[1,5-a]pyrimidine (9h)
Yield, 73%; m.p., 236–238 °C; 1H NMR (400 MHz, DMSO) δ 8.50–8.40 (m, 4H, Ar–H), 8.29–8.22 (m, 2H, Ar–H), 8.11 (s, 2H, Ar–H), 7.67–7.64 (m, 2H, Ar–H), 7.57 (dq, J = 4.8, 3.4, 2.4 Hz, 2H, Ar–H), 7.24 (d, J = 9.0 Hz, 2H, Ar–H), 3.92 (s, 3H, OCH3). LC-MS (m/z) calcd for C24H17ClN4O; 412.11, Found: 413.20 [M + H]+. Anal. Calcd for C24H17ClN4O: C, 69.82; H, 4.15; N, 13.57. Found: C, 69.95; H, 4.06; N, 13.65.
4.1.1.9. 2,7-Diphenyl-5-p-tolyl-1,2,4-triazolo[1,5-a]pyrimidine (9i)
Yield, 70%; m.p., 238–240 °C1H NMR (400 MHz, DMSO) δ 8.36–8.30 (m, 2H, Ar–H), 8.22 (t, J = 7.4 Hz, 4H, Ar–H), 7.99 (s, 1H, Ar–H), 7.67 (d, J = 5.4 Hz, 3H, Ar–H), 7.53 (d, J = 5.9 Hz, 3H, Ar–H), 7.32 (d, J = 7.9 Hz, 2H, Ar–H), 2.35 (s, 3H, CH3). LC-MS (m/z) calcd for C24H18N4; 362.15, Found: 363.10 [M + H]+. Anal. Calcd for C24H18N4: C,79.54; H, 5.01; N, 15.46. Found: C,79.48; H, 5.11; N, 15.35.
4.1.1.10. 7-(4-Chlorophenyl)-2-phenyl-5-p-tolyl-1,2,4-triazolo[1,5-a]-pyrimidine (9j)
Yield, 72%; m.p., 240–242 °C. 1H NMR (400 MHz, CDCl3) δ 8.36–8.30 (m, 2H, Ar–H), 8.15 (d, J = 8.2 Hz, 2H, Ar–H), 8.06 (d, J = 7.8 Hz, 2H, Ar–H), 7.57 (d, J = 8.1 Hz, 2H, Ar–H), 7.47 (s, 4H, Ar–H), 7.23 (d, J = 7.8 Hz, 2H, Ar–H), 2.37 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 165.74, 160.77, 157.13 (C = N carbons), 145.86, 141.88, 137.86, 133.27, 132.12, 130.79, 130.56, 130.45, 129.67, 129.47, 129.08, 128.81, 128.62, 128.57, 128.31, 127.47, 127.44, 127.21(Ar carbons), 105.55 (triazolopyrimidine C6), 21.47 (CH3 carbons). LC-MS (m/z) calcd for C24H17ClN4; 396.11, Found: 397.10 [M + H]+. Anal. Calcd for C24H17ClN4: C,72.63; H, 4.32; N, 14.12. Found: C, 72.74; H, 4.25; N, 14.20.
4.1.1.11. 2-Phenyl-5,7-di-p-tolyl-1,2,4-triazolo[1,5-a]pyrimidine (9k)
Yield, 68%; m.p., 224–226 °C. 1H NMR (400 MHz, CDCl3) δ 8.39–8.30 (m, 2H, Ar–H), 8.18–8.06 (m, 4H, Ar–H), 7.56 (s, 1H, Ar–H), 7.48–7.37 (m, 4H, Ar–H), 7.37 (s, 1H, Ar–H), 7.28 (d, J = 8.0 Hz, 2H, Ar–H), 2.44 (s, 3H, CH3), 2.38 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 147.56, 142.44, 141.88 (C = N carbons), 133.72, 130.58, 129.80, 129.63, 129.43, 128.65, 127.70, 127.63, 127.44 (Ar carbons), 105.70 (triazolopyrimidine C6), 21.70, 21.53 (CH3 carbons). LC-MS (m/z) calcd for C25H20N4; 376.17, Found: 377.00 [M + H]+. Anal. Calcd for C25H20N4: C,79.76; H, 5.35; N, 14.88. Found: C, 79.68; H, 5.43; N, 14.75.
4.1.1.12. 7-(4-Methoxyphenyl)-2-phenyl-5-p-tolyl-1,2,4-triazolo[1,5-a]pyrimidine (9l)
Yield, 69%; m.p., 222–224 °C. 1H NMR (400 MHz, DMSO) δ 8.50–8.42 (m, 2H, Ar–H), 8.35–8.28 (m, 2H, Ar–H), 8.28–8.20 (m, 2H, Ar–H), 8.05 (s, 1H, Ar–H), 7.58 (s, 1H, Ar–H), 7.59–7.50 (m, 2H, Ar–H), 7.40 (d, J = 8.2 Hz, 2H, Ar–H), 7.31–7.20 (m, 2H, Ar–H), 3.92 (s, 3H, OCH3), 2.42 (s, 3H, CH3). LC-MS (m/z) calcd for C25H20N4O; 392.16, Found: 393.100 [M + H]+. Anal. Calcd for C25H20N4O: C,76.51; H, 5.14; N, 14.28. Found: C, 76.63; H, 5.20; N, 14.17.
4.1.1.13. 5-(4-Methoxyphenyl)-2,7-diphenyl-1,2,4-triazolo[1,5-a]pyrimidine (9m)
Yield, 70%; m.p., 222–224 °C. 1H NMR (400 MHz, DMSO) δ 8.45–8.35 (m, 4H, Ar–H), 8.27–8.20 (m, 3H, Ar–H), 8.11 (s, 2H, Ar–H), 7.81–7.76 (m, 2H, Ar–H), 7.60–7.55 (m, 2H, Ar–H), 7.19–7.12 (m, 2H, Ar–H), 3.87 (d, J = 6.0 Hz, 3H, OCH3). LC-MS (m/z) calcd for C24H18N4O; 378.15, Found: 379.130 [M + H]+. Anal. Calcd for C24H18N4O: C,76.17; H, 4.79; N, 14.81. Found: C, 76.30; H, 4.86; N, 14.75.
4.1.1.14. 7-(4-Chlorophenyl)-5-(4-methoxyphenyl)-2-phenyl-1,2,4-triazolo[1,5-a]pyrimidine (9n)
Yield, 70%; m.p., 226–228 °C. 1H NMR (400 MHz, DMSO) δ 8.41–8.35 (m, 2H, Ar–H), 8.35–8.29 (m, 2H, Ar–H), 8.23–8.11 (m, 2H, Ar–H), 8.01 (s, 1H, Ar–H), 7.77–7.67 (m, 2H, Ar–H), 7.60–7.44 (m, 3H, Ar–H), 7.12–7.04 (m, 2H, Ar–H), 3.84 (s, 3H, OCH3). LC-MS (m/z) calcd for C24H17ClN4O; 412.11, Found: 413.00 [M + H]+. Anal. Calcd for C24H17ClN4O: C, 69.82; H, 4.15; N, 13.57. Found: C, 69.94; H, 4.23; N, 13.68.
4.1.1.15. 5-(4-Methoxyphenyl)-2-phenyl-7-p-tolyl-1,2,4-triazolo[1,5-a]pyrimidine (9o)
Yield, 64%; m.p., 176–168 °C. 1H NMR (400 MHz, CDCl3) δ 8.33–8.23 (m, 2H, Ar–H), 8.09 (d, J = 8.4 Hz, 2H, Ar–H), 8.01 (d, J = 7.8 Hz, 2H, Ar–H), 7.43–7.33 (m, 4H, Ar–H), 7.30 (d, J = 7.9 Hz, 2H, Ar–H), 6.87 (d, J = 8.3 Hz, 2H, Ar–H), 3.74 (s, 3H, OCH3), 2.38 (s, 3H, CH3). LC-MS (m/z) calcd for C25H20N4O; 392.16, Found: 393.20 [M + H]+. Anal. Calcd for C25H20N4O: C,76.51; H, 5.14; N, 14.28. Found: C, 76.38; H, 5.21; N, 14.20.
4.1.1.16. 5,7-Bis(4-methoxyphenyl)-2-phenyl-1,2,4-triazolo[1,5-a]pyrimidine (9p)
Yield, 68%; m.p., 202–204 °C. 1H NMR (400 MHz, CDCl3) δ 8.35–8.26 (m, 2H, Ar–H), 8.24–8.14 (m, 2H, Ar–H), 8.18–8.05 (m, 2H, Ar–H), 7.48–7.33 (m, 4H, Ar–H), 7.11–7.01 (m, 2H, Ar–H), 6.98–6.87 (m, 2H, Ar–H), 3.86 (s, 3H, OCH3), 3.80 (s, 3H, OCH3). LC-MS (m/z) calcd for C25H20N4O2; 408.16, Found: 409.30 [M + H]+. Anal. Calcd for C25H20N4O2: C,73.51; H, 4.94; N, 13.72. Found: C, 73.63; H, 5.05; N, 13.57.
4.2. Biology
4.2.1. Antimicrobial Assay
The antimicrobial activity of the target compounds (9a-p) was tested against two Gram-positive bacteria (S. aureus and B. subtilis), two Gram-negative bacteria (E. coli and P. aeruginosa), and two fungal strains (A. flavus and C. albicans). The inhibition zones (IZ, mm/ml) and minimal inhibitory concentrations (MIC, nM) were determined using the modified disk diffusion method.39,40 The MIC values for selected compounds were determined using dose–response tests. The given values result from at least two studies, each with three replicates per concentration. Appendix A (Supplementary File) provides experimental details.
4.2.2. DNA Gyrase and DHFR Inhibitory Assays
The inhibitory efficacy of derivatives 9d, 9n, 9o, and 9p, the most potent antibacterial agents, was assessed against E. coli DNA gyrase and DHFR.44 Inspiralis assay kits measured inhibitory activity against DNA gyrase and DHFR in E. coli. IC50 values were determined using seven different inhibitor concentrations and then calculated using the GraphPad Prism 6.0 software. We established IC50 values for the most important inhibitors using three independent measurements and provided the final results as mean values. Refer to Appendix A for more details.
4.3. Docking Experiment
Two enzymes examined were E. coli DNA gyrase (PDB ID 6F86) and E. coli dihydrofolate reductase B (PDB ID: 7r6g). The cocrystallized structures of the two enzymes were downloaded from the Protein Data Bank,43,44 Chem Office tools (ChemDraw ultra 8.0 2D and 3D) were used to prepare the energy-minimized chemical structures of the ligands; Open Babel GUI was used to convert the images of the ligands into their different file formats. The two enzymes were prepared by removing unnecessary cocrystallized ligands and water and adding polar hydrogen and charges using Autodock 4.2 and Discovery Studio 2024. The active sites of the enzymes were taken from the attributes of SPD Sphere of the corresponding enzyme, which were x,y,z = 61.680259, 28.330852, 64.290148, radius= 10.105432 for 6F86; x,y,z = 10.093469, −53.275716, −7.239296, radius = 10.093469, – 53.275716, – 7.239296 for 7RG6.
Acknowledgments
The authors acknowledge the support by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2023R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c08365.
1H NMR, 13C NMR, LCMS spectra, and elemental analysis of compounds 9a–p (DOCX)
The authors declare no competing financial interest.
Supplementary Material
References
- Morris S.; Cerceo E. Trends, epidemiology, and management of multi-drug resistant gram-negative bacterial infections in the hospitalized setting. Antibiotics 2020, 9 (4), 196. 10.3390/antibiotics9040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alara J. A.; Alara O. R. An Overview of the Global Alarming Increase of Multiple Drug Resistant: A Major Challenge in Clinical Diagnosis. Infect. Disord. Drug Targets 2024, 24 (3), 26–42. 10.2174/1871526523666230725103902. [DOI] [PubMed] [Google Scholar]
- Muteeb G.; Rehman M. T.; Shahwan M.; Aatif M. Origin of antibiotics and antibiotic resistance, and their impacts on drug development: A narrative review. Pharmaceuticals 2023, 16 (11), 1615. 10.3390/ph16111615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romandini A.; Pani A.; Schenardi P. A.; Pattarino G. A. C.; De Giacomo C.; Scaglione F. Antibiotic resistance in pediatric infections: global emerging threats, predicting the near future. Antibiotics 2021, 10 (4), 393. 10.3390/antibiotics10040393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terreni M.; Taccani M.; Pregnolato M. New antibiotics for multidrug-resistant bacterial strains: latest research developments and future perspectives. Molecules 2021, 26 (9), 2671. 10.3390/molecules26092671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y.; Frutos-Beltrán E.; Kang D.; Pannecouque C.; De Clercq E.; Menéndez-Arias L.; Liu X.; Zhan P. Medicinal chemistry strategies for discovering antivirals effective against drug-resistant viruses. Chem. Soc. Rev. 2021, 50 (7), 4514–4540. 10.1039/D0CS01084G. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziz S. A.; Cirnski K.; Herrmann J.; Abdel-Aal M. A.; Youssif B. G.; Salem O. I. Novel fluoroquinolone hybrids as dual DNA gyrase and urease inhibitors with potential antibacterial activity: Design, synthesis, and biological evaluation. J. Mol. Struct. 2023, 1271, 134049 10.1016/j.molstruc.2022.134049. [DOI] [Google Scholar]
- Al-Wahaibi L. H.; Amer A. A.; Marzouk A. A.; Gomaa H. A.; Youssif B. G.; Abdelhamid A. A. Design, synthesis, and antibacterial screening of some novel heteroaryl-based ciprofloxacin derivatives as DNA gyrase and topoisomerase IV inhibitors. Pharmaceuticals 2021, 14 (5), 399. 10.3390/ph14050399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T.; Sankhe K.; Suvarna V.; Sherje A.; Patel K.; Dravyakar B. DNA gyrase inhibitors: Progress and synthesis of potent compounds as antibacterial agents. Biomedicine & Pharmacotherapy 2018, 103, 923–938. 10.1016/j.biopha.2018.04.021. [DOI] [PubMed] [Google Scholar]
- Biedenbach D. J.; Farrell D. J.; Mendes R. E.; Ross J. E.; Jones R. N. Stability of linezolid activity in an era of mobile oxazolidinone resistance determinants: results from the 2009 Zyvox® Annual Appraisal of Potency and Spectrum program. Diagnostic microbiology and infectious disease 2010, 68 (4), 459–467. 10.1016/j.diagmicrobio.2010.09.018. [DOI] [PubMed] [Google Scholar]
- Maxwell A. The interaction between coumarin drugs and DNA gyrase. Molecular microbiology 1993, 9 (4), 681–686. 10.1111/j.1365-2958.1993.tb01728.x. [DOI] [PubMed] [Google Scholar]
- Mustafa Y. F. Emerging trends and future opportunities for coumarin-heterocycle conjugates as antibacterial agents. Results in Chemistry 2023, 6, 101151 10.1016/j.rechem.2023.101151. [DOI] [Google Scholar]
- Abali E. E.; Skacel N. E.; Celikkaya H.; Hsieh Y. C. Regulation of human dihydrofolate reductase activity and expression. Vitamins & Hormones 2008, 79, 267–292. 10.1016/S0083-6729(08)00409-3. [DOI] [PubMed] [Google Scholar]
- Sehrawat R.; Rathee P.; Khatkar S.; Akkol E.; Khayatkashani M.; Nabavi S. M.; Khatkar A. Dihydrofolate reductase (DHFR) inhibitors: A comprehensive review. Curr. Med. Chem. 2024, 31 (7), 799–824. 10.2174/0929867330666230310091510. [DOI] [PubMed] [Google Scholar]
- Raimondi M. V.; Randazzo O.; La Franca M.; Barone G.; Vignoni E.; Rossi D.; Collina S. DHFR inhibitors: reading the past for discovering novel anticancer agents. Molecules 2019, 24 (6), 1140. 10.3390/molecules24061140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M.; Chauhan P. M. Dihydrofolate reductase as a therapeutic target for infectious diseases: opportunities and challenges. Future medicinal chemistry 2012, 4 (10), 1335–1365. 10.4155/fmc.12.68. [DOI] [PubMed] [Google Scholar]
- Schweitzer B. I.; Dicker A. P.; Bertino J. R. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990, 4 (8), 2441–2452. 10.1096/fasebj.4.8.2185970. [DOI] [PubMed] [Google Scholar]
- Bertacine Dias M. V.; Santos J. C.; Libreros-Zuniga G. A.; Ribeiro J. A.; Chavez-Pacheco S. M. Folate biosynthesis pathway: mechanisms and insights into drug design for infectious diseases. Future medicinal chemistry 2018, 10 (8), 935–959. 10.4155/fmc-2017-0168. [DOI] [PubMed] [Google Scholar]
- Hassan A. S.; Askar A. A.; Naglah A. M.; Almehizia A. A.; Ragab A. Discovery of new Schiff bases tethered pyrazole moiety: Design, synthesis, biological evaluation, and molecular docking study as dual targeting DHFR/DNA gyrase inhibitors with immunomodulatory activity. Molecules 2020, 25 (11), 2593. 10.3390/molecules25112593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bülow C.; Haas K. Synthetische Versuche zur Darstellung von Derivaten des heterokondensierten, heterocyclischen 1.3-Triazo-7.0′-pyrimidins. Berichte der deutschen chemischen Gesellschaft 1909, 42 (4), 4638–4644. 10.1002/cber.19090420468. [DOI] [Google Scholar]
- El-Gendy M.; Shaaban M.; Shaaban K. A.; El-Bondkly A. M.; Laatsch H. Essramycin: a first triazolopyrimidine antibiotic isolated from nature. Journal of antibiotics 2008, 61 (3), 149–157. 10.1038/ja.2008.124. [DOI] [PubMed] [Google Scholar]
- Badrey M. G.; Gomha S. M.; Zaki M. E.; Farag B.; El-Reedy A. A. Cyanauric Chloride as a key precursor and a core component for Three-Armed Triazolopyrimidines: Recent finding about SARs and docking analyses. Results in Chemistry 2024, 7, 101337 10.1016/j.rechem.2024.101337. [DOI] [Google Scholar]
- Singleton J. D.Synthesis and Biological Evaluation of Pyrazolo [1, 5-a] pyrimidines and (4-Hydroxy-6-trifluoromethylpyrimidin-2-yl) guanidines; Brigham Young University: 2021. [Google Scholar]
- Gomha S. M.; El-Sayed A. A. A.; Zaki M. E. A.; Alrehaily A.; Elbadawy H. M.; Al-Shahri A. B. A.; Alsenani S. R.; Abouzied A. S. Synthesis, In vitro and In silico Studies of Novel bis-triazolopyridopyrimidines from Curcumin Analogues as Potential Aromatase Agents. Chem. Biodiversity 2024, 21, e202400701 10.1002/cbdv.202400701. [DOI] [PubMed] [Google Scholar]
- Gregory J. A.; Hickey C. M.; Chavez J.; Cacace A. M. New therapies on the horizon: Targeted protein degradation in neuroscience. Cell Chemical Biology 2024, 31 (9), 1688–1698. 10.1016/j.chembiol.2024.08.010. [DOI] [PubMed] [Google Scholar]
- Felicetti T.; Pismataro M. C.; Cecchetti V.; Tabarrini O.; Massari S. Triazolopyrimidine nuclei: Privileged scaffolds for developing antiviral agents with a proper pharmacokinetic profile. Curr. Med. Chem. 2022, 29 (8), 1379–1407. 10.2174/0929867328666210526120534. [DOI] [PubMed] [Google Scholar]
- Ouf S. A.; Gomha S. M.; Farag B.; Zaki M. E.; Ewies M. M.; Sharawy I. A.; Khalil F. O.; Mahmoud H. K. Synthesis of novel Bis-1, 2, 4-Triazolo [3, 4-b][1, 3, 4] Thiadiazines from natural camphoric acid as potential anti-candidal agents. Results in Chemistry 2024, 7, 101406 10.1016/j.rechem.2024.101406. [DOI] [Google Scholar]
- Abdelkhalek A. S.; Attia M. S.; Kamal M. A. Triazolopyrimidine derivatives: an updated review on recent advances in synthesis, biological activities and drug delivery aspects. Curr. Med. Chem. 2024, 31 (14), 1896–1919. 10.2174/0929867330666230228120416. [DOI] [PubMed] [Google Scholar]
- Ghaly M. A.; El-Bendary E. R.; Shehata I. A.; Bayomi S. M.; Habib E. Synthesis, antimicrobial activity, DNA-binding affinity and molecular docking of certain 1, 2, 4-triazolo [1, 5-a] pyrimidines as nalidixic acid isosteres. J. Am. Sci. 2012, 8, 617–628. [Google Scholar]
- Abd El-Aleam R. H.; George R. F.; Hassan G. S.; Abdel-Rahman H. M. Synthesis of 1, 2, 4-triazolo [1, 5-a] pyrimidine derivatives: Antimicrobial activity, DNA Gyrase inhibition and molecular docking. Bioorganic Chemistry 2020, 94, 103411 10.1016/j.bioorg.2019.103411. [DOI] [PubMed] [Google Scholar]
- Al-Wahaibi L. H.; Mahmoud M. A.; Alzahrani H. A.; Abou-Zied H. A.; Gomaa H. A. M.; Youssif B. G. M.; Bräse S.; Rabea S. M. Discovery of new Schiff bases of the disalicylic acid scaffold as DNA gyrase and topoisomerase IV inhibitors endowed with antibacterial properties. Front. Chem. 2024, 12, 1419242 10.3389/fchem.2024.1419242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbastawesy M. A.; Mohamed F. A.; Zaki I.; Alahmdi M. I.; Alzahrani S. S.; Alzahrani H. A.; Gomaa H. A.; Youssif B. G. Design, synthesis and antimicrobial activity of novel quinoline-2-one hybrids as promising DNA gyrase and topoisomerase IV inhibitors. J. Mol. Struct. 2023, 1278, 134902 10.1016/j.molstruc.2023.134902. [DOI] [Google Scholar]
- Frejat F. O. A.; Cao Y.; Wang L.; Zhai H.; Abdelazeem A. H.; Gomaa H. A. M.; Youssif B. G. M.; Wu C. New 1, 2, 4-oxadiazole/pyrrolidine hybrids as topoisomerase IV and DNA gyrase inhibitors with promising antibacterial activity. Arch. Pharm. 2022, 355 (7), 2100516 10.1002/ardp.202100516. [DOI] [PubMed] [Google Scholar]
- Frejat F. O. A.; Cao Y.; Zhai H.; Abdel-Aziz S. A.; Gomaa H. A.; Youssif B. G.; Wu C. Novel 1, 2, 4-oxadiazole/pyrrolidine hybrids as DNA gyrase and topoisomerase IV inhibitors with potential antibacterial activity. Arabian Journal of Chemistry 2022, 15 (1), 103538 10.1016/j.arabjc.2021.103538. [DOI] [Google Scholar]
- Hofny H. A.; Mohamed M. F.; Gomaa H. A.; Abdel-Aziz S. A.; Youssif B. G.; El-Koussi N. A.; Aboraia A. S. Design, synthesis, and antibacterial evaluation of new quinoline-1, 3, 4-oxadiazole and quinoline-1, 2, 4-triazole hybrids as potential inhibitors of DNA gyrase and topoisomerase IV. Bioorganic Chemistry 2021, 112, 104920 10.1016/j.bioorg.2021.104920. [DOI] [PubMed] [Google Scholar]
- Hassan A. Y.; Sarg M. T.; Bayoumi A. H.; El-Deebb M. A. Synthesis and Anticancer Evaluation of Some Novel 5-Amino[1,2,4]Triazole Derivatives. J. Heterocycl. Chem. 2018, 55, 1450. 10.1002/jhet.3184. [DOI] [Google Scholar]
- Al-Wahaibi L. H.; Abdel-Rahman M. H.; El-Adl K.; Youssif B. G. M.; Bräse S.; Abdel-Aziz S. A. New Diaryl-1,2,4-triazolo[3,4-a]pyrimidine Hybrids as Selective COX-2/sEH Dual Inhibitors with Potent Analgesic/Anti-inflammatory and Cardioprotective Properties. ACS Omega 2024, 9 (31), 33494–33509. 10.1021/acsomega.4c00870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghraby M. T. E.; Salem O. I. A.; Youssif B. G. M.; Sheha M. M. Design, synthesis, and modelling study of new 1,2,3-triazole/chalcone hybrids with antiproliferative action as epidermal growth factor receptor inhibitors. Chem. Biol. Drug Des 2023, 101 (3), 749–759. 10.1111/cbdd.14178. [DOI] [PubMed] [Google Scholar]
- Alkhaldi A. A.; Abdelgawad M. A.; Youssif B. G. Synthesis, antimicrobial evaluation and docking studies of new pyrazolone derivatives. Tropical Journal of Pharmaceutical Research 2019, 17 (11), 2235–2241. 10.4314/tjpr.v17i11.18. [DOI] [Google Scholar]
- Manso T.; Lores M.; de Miguel T. Antimicrobial activity of polyphenols and natural polyphenolic extracts on clinical isolates. Antibiotics 2022, 11 (1), 46. 10.3390/antibiotics11010046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdullahi Z. U.; Musa S. S.; Abu-Odah H.; Ahmed A.; Lawan A. A.; Bello U. M. Bactericidal effects of snake venom phospholipases A2: a systematic review and analysis of minimum inhibitory concentration. Physiologia 2023, 3 (1), 30–42. 10.3390/physiologia3010003. [DOI] [Google Scholar]
- Sun N.; Li M.; Cai S.; Li Y.; Chen C.; Zheng Y.; Li X.; Fang Z.; Lv H.; Lu Y.-J. Antibacterial evaluation and mode of action study of BIMQ, a novel bacterial cell division inhibitor. Biochemical and biophysical research communications 2019, 514 (4), 1224–1230. 10.1016/j.bbrc.2019.05.086. [DOI] [PubMed] [Google Scholar]
- Wiegand I.; Hilpert K.; Hancock R. E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nature protocols 2008, 3 (2), 163–175. 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- Durcik M.; Tammela P.; Barančoková M.; Tomašič T.; Ilaš J.; Kikelj D.; Zidar N. Synthesis and evaluation of N-phenylpyrrolamides as DNA gyrase B Inhibitors. ChemMedChem. 2018, 13 (2), 186–198. 10.1002/cmdc.201700549. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of computational chemistry 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narramore S.; Stevenson C. E.; Maxwell A.; Lawson D. M.; Fishwick C. W. New insights into the binding mode of pyridine-3-carboxamide inhibitors of E. coli DNA gyrase. Bioorganic & medicinal chemistry 2019, 27 (16), 3546–3550. 10.1016/j.bmc.2019.06.015. [DOI] [PubMed] [Google Scholar]
- Krucinska J.; Lombardo M. N.; Erlandsen H.; Estrada A.; Si D.; Viswanathan K.; Wright D. L. Structure-guided functional studies of plasmid-encoded dihydrofolate reductases reveal a common mechanism of trimethoprim resistance in Gram-negative pathogens. Commun. Biol. 2022, 5 (1), 459. 10.1038/s42003-022-03384-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.