

Abstract
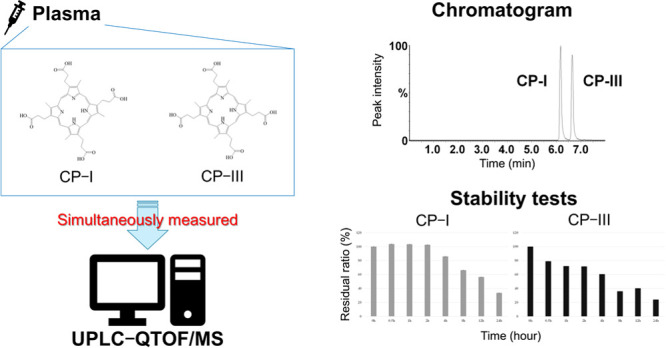
Organic anion transporting polypeptides (OATP) 1B1 and 1B3 are expressed in liver cells and are involved in drug uptake in the liver. OATP1B activity varies due to polymorphisms and is decreased by OATP1B inhibitors. Variability of OATP1B activity impacts the pharmacokinetics of OATP1B substrate drugs through drug–drug interactions. Lately, coproporphyrin-I (CP-I) and -III (CP-III) have been featured as quantitative endogenous biomarkers for evaluating the activity of OATP1B. CP-III has been reported to be transported not only by OATP1B but also by OATP2B1 in vitro. We have established a highly sensitive assay for the simultaneous measurement of CP-I and CP-III using a small volume of human plasma. The sample was pretreated by solid-phase extraction and analyzed by ultra-performance liquid chromatography coupled to quadrupole time-of-flight mass spectrometry (UPLC-QTOF/MS). This method that uses 100 μL of plasma met the acceptance criteria of the US Food and Drug Administration guidance for bioanalytical method validation, and the lower limit of quantification was 0.01 ng/mL for both coproporphyrins. The clinical application of the method was evaluated by measuring plasma CP-I and CP-III concentrations in healthy volunteers and rheumatoid arthritis (RA) patients. The measured concentrations were within the calibration range (0.01–50 ng/mL). Using this novel method to measure plasma concentrations of CP-I and CP-III may contribute to the evaluation of the activities of OATP1B1, OATP1B3, and OATB2B1 in healthy individuals and patients with various clinical conditions including RA.
Introduction
Some drug transporters have been reported to affect the pharmacokinetics of drugs and contribute to drug–drug interactions in various clinical conditions.1 Organic anion transporting polypeptides (OATPs) are a family of membrane transporters expressed in a variety of tissues including absorptive/excretory cells of the liver, kidney, and intestine and transport mainly organic anions across membranes.2 OATP1B1 and 1B3 (encoded by SLCO1B1 and 1B3, respectively) are expressed in hepatocytes and contribute to the hepatic uptake of substrate drugs such as hydroxymethylglutaryl (HMG)-CoA reductase inhibitors, repaglinide, fexofenadine, and some antihepatitis C virus drugs3−6 Rifampicin and cyclosporin A are OATP inhibitors that increase the plasma concentrations of OATP1B substrates by drug–drug interactions through inhibiting OATP1B uptake function.7,8 As genetic factors, there are two major single-nucleotide polymorphisms in SLCO1B1: A388G and T521C. OATP1B1*5 (c.388A-c.521C) and OATP1B1*15 (c.388G-c.521C) have been reported to decrease the transporting activities of OATP1B1.3,9−12 Thus, in vivo OATP1B activity varies widely among patients, and phenotyping of the activity of OATP1B may serve as a valuable approach for personalizing the dosage of OATP1B substrate drugs.
Recently, some probe drugs and potential biomarkers for phenotyping in vivo drug transporter activities are utilized in drug–drug interaction studies and pharmacokinetic approaches in patients.13−15 Especially, endogenous biomarkers seem to be very useful for phenotyping in vivo drug transporter activities because the administration of probe drugs is not needed. Plasma concentrations of two types of porphyrins, coproporphyrin-I (CP-I) and -III (CP-III), have been reported to be highly sensitive and specific for evaluating in vivo OATP1B activity as endogenous biomarkers.16−19 Additionally, an in vitro study has shown that CP-III is transported not only by OATP1B but also by OATP2B1.20 Plasma concentrations of CP-I and CP-III in healthy volunteers are relatively low in several reports (approximately 0.15–1.5 and 0.025–0.15 ng/mL, respectively).9,21,22 Three methods for the simultaneous quantification of CP-I and CP-III in human plasma were established and fully validated.21,23,24 The method developed by King-Ahmad et al.24 required a relatively large volume (200 μL) of the human plasma sample and had a lower limit of quantification (LLOQ) of 0.1 ng/mL for CP-I and CP-III. The method reported by Kandoussi et al.23 used 100 μL of the human plasma sample with an LLOQ of 0.05 ng/mL for CP-I and CP-III. The method of Njumbe Ediage et al.21 was the most sensitive, with an LLOQ of 0.02 ng/mL for CP-I and CP-III but required 200 μL of the human plasma sample. A method using a smaller sample volume is preferred for wider application to diverse individuals including infants and older patients. Furthermore, a better LLOQ is preferred for evaluation in various settings, especially for measuring CP-III that has lower blood levels than CP-I.9,21,22 Using ultra-performance liquid chromatography coupled to quadrupole time-of-flight mass spectrometry (UPLC-QTOF/MS), we developed an assay for simultaneous CP-I and CP-III measurements, which is more sensitive than previous methods and uses a smaller volume of human plasma (100 μL).
Rheumatoid arthritis (RA) is an autoimmune inflammatory disease characterized by high autoantibody production and cytokine release, especially IL-6 and TNF-α. RA associated with elevated levels of IL-6 and TNF-α has been reported to downregulate OATP1B.25 Therefore, monitoring of the activity of OATP1B is important in RA patients with changes in inflammatory state or medications, and a sensitive and precise assay will benefit these patients. In this study, we applied our novel method to measure the plasma concentrations of CP-I and CP-III in healthy volunteers and RA patients.
Results
Chromatographic and Mass Spectrometric Analysis
Electrospray ionization detected intense [M + H]+ signals for CP-I and CP-III. Figure 1 shows the product ion scan data of the precursor and product ions of CP-I and CP-III. The limits of detection and LLOQ were 0.004 and 0.01 ng/mL, respectively, for both CP-I and CP-III. Figure 2 illustrates the UPLC-QTOF/MS chromatograms of the analytes, CP-I and CP-III, and their internal standards (CP-I–15N4 and CP-III–15N4) in various samples comprising blank matrix only (2% human serum albumin; HSA), LLOQ sample prepared in 2% HSA, high quality control (QC) sample prepared in pooled blank human plasma, representative plasma sample of a healthy volunteer, and representative plasma sample of an RA patient. The isolated peaks of the analytes and internal standards were observed at approximately 6.2 min for CP-I and CP-I–15N4 and at approximately 6.7 min for CP-III and CP-III–15N4. The resolution of the two peaks between CP-I and CP-III, and between their internal standards, was 2.2.
Figure 1.

Product ion scan data for CP-I and CP-III. CP-I, coproporphyrin-I; CP-III, coproporphyrin-III; m/z, mass-to-charge ratio.
Figure 2.

Chromatograms of CP-I, CP-III, CP-I–15N4, and CP-III–15N4 in samples with matrix (2% human serum albumin solution or human plasma) using ultra-performance liquid chromatography coupled to quadrupole time-of-flight mass spectrometry (UPLC-QTOF/MS). The samples as shown from top to down are blank matrix (2% human serum albumin solution), LLOQ sample prepared in blank matrix (0.01 ng/mL), high QC sample prepared in pooled blank human plasma (40 ng/mL), a representative plasma sample of a healthy volunteer (calculated concentrations of CP-I and CP-III at 0.43 and 0.07 ng/mL, respectively), and a representative plasma sample of an RA patient (calculated concentrations of CP-I and CP-III at 0.65 and 0.07 ng/mL, respectively). CP-I, coproporphyrin-I; CP-III, coproporphyrin-III; LLOQ, lower limit of quantitation; QC, quality control; and RA, rheumatoid arthritis.
Validation Results
For CP-I, the calibration curve had a coefficient of determination (r2) of 0.993 or greater for the calibration range of 0.01–50 ng/mL. Table 1 indicates the within-run and run-to-run accuracy and precision. Within-run accuracy and precision at LLOQ ranged from 94.0 to 103.0% and 2.06 to 2.33% coefficient of variation (CV), respectively. Run-to-run accuracy and precision at LLOQ were 99.0 and 3.99%, respectively. Within-run accuracy and precision for the three QCs (low QC, mid QC, and high QC) were within the ranges of 91.7–113.3 and 1.60–5.04% CV, respectively. Run-to-run accuracy and precision for the three QCs were within the ranges of 97.3–102.0 and 5.43–5.67% CV, respectively. For blank plasma, the within-run precision for CP-I varied from 2.28 to 4.01% CV, while the run-to-run precision was 3.84% CV.
Table 1. Validation Results of Accuracy and Precision for CP-I Concentrations in Human Plasmaa.
| nominal CP-I concentrations (ng/mL) | ||||||
|---|---|---|---|---|---|---|
| endogenous | LLOQ sample | low QC sample | mid QC sample | high QC sample | ||
| 0.01 + endo | 0.03 + endo | 15 + endo | 40 + endo | |||
| within-run | ||||||
| 1 | mean (ng/mL) | 0.471 | 0.471 | 0.485 | 15.7 | 38.9 |
| accuracy (%) | 97.9 [95.7–101.5] | 96.9 [92.7–101.5] | 101.8 [97.0–105.2] | 96.1 [91.8–101.4] | ||
| precision (%CV) | 2.28 | 2.06 | 2.72 | 2.87 | 3.03 | |
| 2 | mean (ng/mL) | 0.489 | 0.493 | 0.485 | 15.0 | 39.2 |
| accuracy (%) | 98.7 [94.0–101.4] | 93.4 [91.7–96.1] | 97.1 [94.5–99.4] | 96.9 [91.9–103.6] | ||
| precision (%CV) | 4.01 | 2.33 | 1.60 | 1.71 | 3.97 | |
| 3 | mean (ng/mL) | 0.499 | 0.510 | 0.537 | 16.6 | 43.2 |
| accuracy (%) | 100.3 [96.4–103.0] | 101.6 [98.8–106.0] | 107.0 [99.0–113.3] | 104.6 [97.6–111.6] | ||
| precision (%CV) | 2.41 | 2.30 | 2.54 | 5.04 | 5.04 | |
| run-to-run | ||||||
| accuracy (%) | 99.0 | 97.3 | 102.0 | 99.0 | ||
| precision (%CV) | 3.84 | 3.99 | 5.43 | 5.47 | 5.67 | |
CP-I, coproporphyrin-I; LLOQ, lower limit of quantitation; QC, quality control; Endo, endogenous level; CV, coefficient of variation.
For CP-III, the calibration curve had r2 of 0.993 or greater for the calibration range of 0.01–50 ng/mL. As shown in Table 2, the within-run accuracy and precision at LLOQ were within the ranges of 81.3–116.9 and 3.90–9.22% CV, respectively. Run-to-run accuracy and precision at the LLOQ were 95.5 and 11.0% CV, respectively. Overall within-run accuracy and precision for the three QCs were within the ranges of 85.9–108.4 and 1.90–8.57% CV, respectively. Overall run-to-run accuracy and precision for the three QCs were within the ranges of 92.1–94.2 and 5.49–8.21% CV, respectively. For blank plasma, the within-run precision for CP-III varied from 8.08 to 14.8% CV, while the run-to-run precision was 17.4% CV.
Table 2. Validation Results of Accuracy and Precision for CP-III Concentrations in Human Plasmaa.
| nominal CP-III concentrations (ng/mL) | ||||||
|---|---|---|---|---|---|---|
| endogenous | LLOQ sample | low QC sample | mid QC sample | high QC sample | ||
| 0.01 + endo | 0.03 + endo | 15 + endo | 40 + endo | |||
| within-run | ||||||
| 1 | mean (ng/mL) | 0.0355 | 0.0438 | 0.0606 | 14.2 | 35.9 |
| accuracy (%) | 96.3 [81.3–103.3] | 92.5 [87.0–97.7] | 94.5 [89.5–101.0] | 90.0 [87.6–92.8] | ||
| precision (%CV) | 14.8 | 8.05 | 3.99 | 4.44 | 2.06 | |
| 2 | mean (ng/mL) | 0.0422 | 0.0527 | 0.0678 | 13.7 | 37.1 |
| accuracy (%) | 101.0 [88.2–116.9] | 94.0 [85.9–103.9] | 91.3 [89.5–94.8] | 92.7 [87.4–98.9] | ||
| precision (%CV) | 9.43 | 9.22 | 6.22 | 1.90 | 4.32 | |
| 3 | mean (ng/mL) | 0.0500 | 0.0522 | 0.0695 | 14.6 | 38.4 |
| accuracy (%) | 89.3 [81.7–91.7] | 106.8 [87.5–96.3] | 96.9 [90.0–107.0] | 95.8 [86.5–108.4] | ||
| precision (%CV) | 8.08 | 3.90 | 4.03 | 6.69 | 8.57 | |
| run-to-run | ||||||
| accuracy (%) | 95.5 | 92.1 | 94.2 | 92.9 | ||
| precision (%CV) | 17.4 | 11.0 | 8.21 | 5.49 | 6.64 | |
CP-III, coproporphyrin-III; LLOQ, lower limit of quantitation; QC, quality control; Endo, endogenous level; CV, coefficient of variation.
The overall recovery rates of three QCs (low QC, mid QC, and high QC) ranged from 27.0 to 76.1% for CP-I and 23.0 to 364.8% for CP-III. Matrix effects ranged from 92.3 to 156.2% for CP-I and 58.4 to 203.2% for CP-III (Table 3). The overall recovery rates corrected by the internal standard ranged from 85.7 to 111.0% for CP-I and from 79.0 to 108.7% for CP-III. Matrix effects corrected by the internal standard ranged from 83.6 to 119.1% for CP-I and from 81.1 to 117.7% for CP-III (Table 3).
Table 3. Recovery Rates and Matrix Effects for CP-I and CP-III Concentrations in Human Plasmaa.
| CP-I | CP-III | |||||
|---|---|---|---|---|---|---|
| nominal concentrations (ng/mL) | low QC sample | mid QC sample | high QC sample | low QC sample | mid QC sample | high QC sample |
| 0.03 + endo | 15 + endo | 40 + endo | 0.03 + endo | 15 + endo | 40 + endo | |
| recovery rate | ||||||
| analyte recovery rate [%, mean (range)] | 44.6 [27.0–66.9] | 46.2 [32.4–76.1] | 41.4 [28.8–74.0] | 95.5 [26.5–364.8] | 79.4 [28.4–280.1] | 36.1 [23.0–67.8] |
| recovery rate corrected by internal standard [%, mean (range)] | 96.8 [87.1–111.0] | 93.9 [87.4–102.9] | 95.1 [85.7–101.3] | 93.7 [79.0–108.7] | 93.3 [84.5–106.7] | 92.0 [83.6–98.4] |
| matrix effect | ||||||
| analyte matrix effect [%, mean (range)] | 116.7 [92.3–156.2] | 110.6 [104.4–115.2] | 106.2 [92.4–116.4] | 120.0 [58.4–203.2] | 119.0 [106.4–134.8] | 113.7 [98.2–128.8] |
| matrix effect corrected by internal standard [%, mean (range)] | 101.3 [83.6–119.1] | 111.0 [99.5–117.3] | 102.9 [98.4–106.9] | 88.0 [81.1–92.4] | 110.2 [100.0–117.7] | 105.6 [96.5–110.1] |
CP-I, coproporphyrin-I; CP-III, coproporphyrin-III; QC, quality control; Endo, endogenous level.
Carryover was assessed by measuring three consecutive blank samples after the upper limit of the quantification sample. All blank samples were less than 20% of the LLOQ peak area, confirming no carryover.
Stability Tests of CP-I and CP-III
Freeze–thaw stability (three cycles) was tested using two QCs (low and high QC). No obvious concentration changes in the QC samples were observed, with accuracy ranging from 94.1 to 112.6% for CP-I and from 99.1 to 111.9% for CP-III. For autosampler stability (24 h), CP-I and CP-III remained stable, with accuracy ranging from 95.6 to 112.7% for CP-I and from 97.6 to 111.9% for CP-III. The photostability test under benchtop conditions was acceptable until 4 h for CP-I in human plasma, but photostability of CP-III was already unacceptable at 0.5 h (Figure 3A,B). Both CP-I and CP-III remained stable up to 180 days at various temperatures (room temperature, 4, −20, and −80 °C) (Figure 3C,D).
Figure 3.

Stability of CP-I and CP-III in human plasma under various conditions. Photostability of CP-I (A) and CP-III (B) in human plasma was investigated under lighted conditions at room temperature for 0.5, 1, 2, 4, 8, 12, and 24 h. The residual ratio was determined at each time point. Long-term stability of CP-I (C) and CP-III (D) in human plasma stored under dark conditions was investigated at room temperature, 4, −20, and −80 °C for 1, 8, 15, 30, 90, and 180 days. CP-I, coproporphyrin-I; CP-III, coproporphyrin-III.
CP-I and CP-III Concentrations in Healthy Volunteers and RA Patients
Clinical application of the validated UPLC-QTOF/MS method was tested by measuring plasma CP-I and CP-III concentrations in healthy volunteers and RA patients. Table 4 shows the clinical characteristics of the participants. There were significant differences in age and serum creatinine levels between the two groups.
Table 4. Clinical Characteristics of Healthy Volunteers and RA Patientsa,b.
| characteristic | healthy volunteers | RA patients | p-value |
|---|---|---|---|
| number of subjects | 8 | 36 | |
| males/females | 2/6 | 10/26 | >0.9999 |
| age (years) | 24.8 ± 2.77 [22–31] | 65.3 ± 14.0 [24–87] | <0.0001 |
| body weight (kg) | 53.1 ± 6.97 [44.0–65.0] | 53.9 ± 10.6 [28.5–77.0] | 0.803 |
| ALT (IU/L) | 13.3 ± 3.34 [9.0–19.0] | 19.7 ± 15.5 [8.1–90.3] | 0.223 |
| total bilirubin (mg/dL) | 0.600 ± 0.173 [0.40–1.0] | 0.647 ± 0.269 [0.25–1.50] | 0.566 |
| serum creatinine (mg/dL) | 0.596 ± 0.0840 [0.45–0.72] | 0.706 ± 0.138 [0.43–1.01] | 0.0135 |
| DAS28-CRP | 3.36 ± 0.711 [2.20–5.31] |
ALT, alanine aminotransaminase; DAS-28-CRP, disease activity score-28-C–reactive protein.
Data are expressed as numbers or mean ± standard deviation [range].
Plasma concentrations (median [range]) of CP-I were 0.413 [0.343–0.885] ng/mL in healthy subjects and 0.696 [0.383–1.41] ng/mL in RA patients, with a significant difference between the two groups based on the Mann–Whitney U test (p = 0.0011) (Figure 4A). Plasma concentrations of CP-III were 0.0525 [0.044–0.193] ng/mL in healthy volunteers and 0.0598 [0.035–0.132] ng/mL in RA patients, with no significant difference between the two groups (p = 0.852) (Figure 4B). The concentrations of CP-I and CP-III in all measured samples were within the calibration ranges of the validated method.
Figure 4.

Plasma concentrations of CP-I (A) and CP-III (B) in healthy volunteers and RA patients. The levels of CP-I and CP-III in healthy volunteers and RA patients did not show Gaussian distribution based on the Shapiro–Wilk test. Thus, comparison between the two groups was analyzed by the Mann–Whitney U test. The horizontal bars in each group indicate the median value. CP-I, coproporphyrin-I; CP-III, coproporphyrin-III; and RA, rheumatoid arthritis.
Discussion
Previous studies have reported the role of OATPs (including the roles of OATP1B1, OATP1B3 and OATP2B1) in the hepatic uptake of various drugs. OATP-mediated drug–drug interactions may pose problems when using inhibitors of OATPs or when analyzing phenotypes of OATPs.16,17,20,26,27 Therefore, it is crucial to evaluate the activities of OATPs in the clinical setting. Various studies have concurred that CP-I and CP-III are valuable endogenous biomarkers for assessing OATP activities.16,17,20,26,27 Some studies have reported that plasma CP-I concentrations in healthy volunteers range from approximately 0.15 to 1.5 ng/mL, while CP-III concentrations range from 0.025 to 0.15 ng/mL.9,21,22 Three liquid chromatographic assays have been developed for the simultaneous quantification of CP-I and CP-III in human plasma.21,23,24 The method reported by Njumbe Ediage et al.21 had an LLOQ of 0.02 ng/mL for both CP-I and CP-III, which is the lowest among the three methods. Considering the individual variations of plasma CP-I and CP-III concentrations, all three previously reported methods are inadequate for accurate measurement of low CP-III concentrations in human plasma. For this reason, it was necessary to establish a more sensitive quantification method having an LLOQ.
In the present research, we established and validated a novel highly sensitive simultaneous quantification method for CP-I and CP-III using UPLC-QTOF/MS, and we utilized the method to measure the two analytes in plasma samples of healthy volunteers and RA patients. The use of UPLC-QTOF/MS makes it possible to lower the LLOQ for CP-I and CP-III compared to previous studies because of the advantages of reduced solvents, sharper peaks, and shorter retention times. Additionally, the noise level was reduced, which improved the signal-to-noise ratio. QTOF/MS with higher resolution and specificity compared to the tandem mass spectrometry method also contributed to the achievement of an LLOQ. Therefore, our method shows promise for measuring plasma CP-I and CP-III concentrations in individuals with diverse backgrounds and clinical conditions. Moreover, the quantification method developed by Njumbe Ediage et al.,21 which had the lowest LLOQ among previously reported methods, required 200 μL of the plasma sample, whereas we successfully reduced the plasma volume to 100 μL by applying SPE for sample pretreatment. The Oasis MAX 96-well μElution plate has a mixed-mode polymeric sorbent that has anion-exchange and reversed adsorption phase and is suitable for extracting acidic compounds such as CP-I and CP-III. It removes matrix effects by effective washing and eluting processes, resulting in lower noise levels and more stable baselines. These advanced techniques make it easier to measure analytes in patients from whom only a minute volume of plasma can be collected, such as infants and older individuals.
CP-I and CP-III exhibit identical m/z values and the same precursor and product ions due to their isotopic relationship. Separating the CP-I and CP-III peaks proved challenging when attempting to simultaneously quantify CP-I and CP-III. In our previous study, we were able to separate the peaks of CP-I and CP-III using UHPLC-MS/MS, but the peak boundaries were unclear.22 In the current study, by optimizing the gradient conditions and columns as shown in the Materials and Methods section, the CP-I and CP-III peaks were clearly separated, and the peak boundaries were more defined (retention time: 6.2 min for CP-I, 6.7 min for CP-III) than in our previous method.
The validation results for CP-I and CP-III demonstrated accuracy and precision within the acceptable ranges provided in the guidance published by the Food and Drug Administration (FDA).28 Without correction by internal standards, matrix effects and recovery rates varied widely. The reason for these findings is not clear. Probably, residues of CP-I and CP-III remained in the solid phase. However, correction by internal standards improved the variability of matrix effects and recovery rates and confirmed reproducibility. Effective sample pretreatment that minimizes both ion suppression and ion enhancement may have contributed to favorable validation results. Additionally, we carried out various tests to assess the stability of CP-I and CP-III using our novel UPLC-QTOF/MS method. The photostability test (Figure 3A,B) reveals that CP-I in human plasma has good stability under lighted benchtop conditions until 4 h, whereas CP-III loses stability within 0.5 h. Also, the long-term stability test (Figure 3C,D) reveals that both analytes in human plasma are stable when stored in the dark at room temperature, 4, −20, and −80 °C for a maximum of 180 days. Considering the instability of CP-III under laboratory lighting conditions, light shielding using shading tubes should always be implemented during experiments that involve simultaneous measurement of CP-I and CP-III concentrations.
Clinical application of our novel UPLC-QTOF/MS quantification method was evaluated by analyzing plasma levels of CP-I and CP-III in a group of eight healthy volunteers and 36 RA patients. The concentrations of CP-I and CP-III in all of the plasma samples fell within the calibration range, verifying the applicability of our novel method for clinical use. Plasma CP-I and CP-III concentrations in healthy volunteers measured by our new method were comparable with past reports,9,21,22 and plasma concentrations of CP-I in RA patients were comparable with a previous study.29 Healthy volunteers were significantly younger compared to RA patients (p < 0.0001). The clinical significance of the age difference between healthy individuals and RA patients on plasma CP-I and CP-III concentrations has to be examined in future clinical studies. As presented in Figure 4A,B, when comparing plasma concentrations of CP-I and CP-III between healthy volunteers and RA patients, the CP-I concentration was significantly higher in RA patients than in healthy volunteers (p = 0.0011), while no significant difference in the CP-III concentration was observed (p = 0.852). Considering that CP-III is transported not only by OATP1B but also by OATP2B1,20 it may not reflect OATP1B activity accurately. Taking into account these findings and the characteristics of CP-I and CP-III as biomarkers reported previously,16,17,20,26,27 OATP1B activity decreases in RA patients. Hence, CP-I may be a more specific biomarker than CP-III for the assessment of OATP1B activity. However, because CP-III is transported by both the enzymes of OATP1B and OATP2B1, CP-III has the potential to be a biomarker for the evaluation of OATP2B1 activity, and further studies are expected. To the best of our knowledge, this is the first study to analyze plasma CP-III concentrations in RA patients and compare the plasma concentrations of CP-I and CP-III between healthy volunteers and RA patients.
Recently, a novel method for simultaneous quantification of CP-I and CP-III in monkey plasma was reported.19 This method can be measured down to an LLOQ of 0.01 ng/mL using 50 μL of the plasma sample. Further studies are required to examine the applicability of this method to human samples and various clinical settings. Although requiring 100 μL of the plasma sample, our method has some advantages comparing with the recent study,19 such as simpler pretreatment, shorter run-time, and directly applicable to humans.
We have developed and validated a novel method for simultaneous quantification of CP-I and CP-III using UPLC-QTOF/MS. This method is expected to enhance the understanding of intra- and interindividual variations of OATP1B1, OATP1B3, and OATP2B1 activities. Although we applied the novel method to analyze the plasma concentrations of CP-I and CP-III in healthy volunteers and RA patients, the numbers of subjects studied were small. It is imperative to conduct larger-scale clinical studies to further clarify the activities of the OATP family (including OATP1B1, OATP1B3, and OATP2B1) in patients with various clinical conditions by measuring plasma CP-I and CP-III as biomarkers.
In conclusion, a novel method was developed and validated for simultaneously measuring CP-I and CP-III levels in human plasma using UPLC-QTOF/MS. The clinical applicability of this validated method was demonstrated by measuring plasma CP-I and CP-III levels in both healthy volunteers and RA patients. This new quantification method has the potential to enhance the research on in vivo activities of OATP1B1, OATP1B3, and OATB2B1 in healthy individuals and patients with various clinical conditions including RA.
Materials and Methods
Chemicals
The standards of CP-I and CP-III were purchased from Frontier Specialty Chemicals, Inc. (Logan, Utah, USA). The internal standards that are stable isotope-labeled CP-I (CP-I–15N4) and CP-III (CP-III–15N4) were purchased from Cosmo Bio Co., Ltd. (Koto-ku, Tokyo, Japan). HSA used to prepare calibration samples was purchased from Sigma-Aldrich (St. Louis, Missouri, USA). All other reagents (formic acid, 28% ammonia solution, phosphoric acid, dimethyl sulfoxide, methanol, and acetonitrile) were of high-purity analytical grade and were purchased from FUJIFILM Wako Pure Chemical Corporation (Chuo-Ku, Osaka, Japan).
Stock and Working Solutions
CP-I and CP-III stock solutions used for calibration and QC were prepared separately, as described below. CP-I and CP-III were weighed and dissolved in dimethyl sulfoxide in volumetric flasks (25 mg/100 and 10 mg/50 mL, respectively). The internal standards CP-I–15N4 and CP-III–15N4 were also weighed and dissolved in dimethyl sulfoxide in volumetric flasks (0.50 mg/250 mL and 0.25 mg/50 mL, respectively).
Working solutions for calibration were prepared by diluting the stock solutions for calibration with dimethyl sulfoxide. Each solution contained mixtures of CP-I and CP-III at the same concentration. The limits of detection and LLOQ concentration were set at signal-to-noise ratios of 3 and 10, respectively. Nine calibration working solutions were prepared, each containing 0.1, 0.2, 0.5, 2, 5, 20, 50, 200, or 500 ng/mL (final concentration: 0.01, 0.02, 0.05, 0.2, 0.5, 2, 5, 20, or 50 ng/mL, respectively) of CP-I and CP-III. These were used for performing validation and analyzing the concentrations of CP-I and CP-III in subject’s plasma.
Working solutions for QC were prepared by diluting the stock solutions for QC with dimethyl sulfoxide. Each solution contained a mixture of CP-I and CP-III at the same concentration. Four QC working solutions comprising LLOQ, low QC, mid QC, and high QC were prepared, containing 0.1, 0.3, 150, and 400 ng/mL (final concentrations: 0.01, 0.03, 15, and 40 ng/mL), respectively, of CP-I and CP-III.
Healthy Volunteers
Blank plasma samples were obtained from healthy volunteers (n = 6) who had not taken any medication within 2 months of blood collection. These plasma samples were used for the analysis of QC samples in the validation process and for comparison with those of RA patients. Healthy volunteers abstained from food or drink for at least 8 h prior to blood collection. Demographic and clinical data were investigated, including age, sex, body weight, serum creatinine, serum total bilirubin, and serum alanine aminotransferase (ALT). Blood samples collected in tubes containing ethylenediaminetetraacetate (EDTA) anticoagulant were centrifuged at 3000 rpm for 5 min at 4 °C, and the supernatant was separated (plasma samples). Plasma samples were stored at –80 °C until measurement. This study was reviewed and approved by the Ethics Committee of Meiji Pharmaceutical University (approval number: 202128). All volunteers were given an appropriate explanation of the purpose of this study and signed informed consent before participation.
RA Patients
Blood samples were collected from 36 RA patients aged over 18 years who attended the outpatient clinic of the Oita University Hospital. These patients had not taken the OATP1B inhibitors represented by rifampicin and cyclosporin A within at least 3 months before blood collection. Blood samples were collected in tubes containing EDTA-3K anticoagulant and centrifuged at 3500 rpm for 5 min at 4 °C. Plasma samples were separated into shaded tubes and stored in a deep freezer (−80 °C) until measurement. All participants were given appropriate explanations about the purpose of the study and signed an informed consent form before participation. Demographic and clinical data were investigated, including age, sex, body weight, serum creatinine, serum total bilirubin, serum ALT, and Disease Activity Score 28–C-reactive protein (DAS28–CRP, an index of disease activity in RA patients). All 36 participants did not meet the exclusion criteria of body mass index >30 kg/m2, total bilirubin >1.5 mg/dL, and ALT > 100 IU/L. This study was reviewed and approved by the Ethics Committees of Meiji Pharmaceutical University (approval number: 202221) and Oita University Hospital (approval number: 2195).
Sample Preparation
Plasma samples were pretreated by solid-phase extraction (SPE) for the measurement of CP-I and CP-III concentrations. An Oasis MAX 96-well μElution plate, 2 mg sorbent per well, 30 μm (Waters, Milford, Massachusetts, USA) was used for SPE. In each 1.5 mL shaded safe-lock tube, 100 μL of subject’s plasma sample, 430 μL 4% phosphoric acid, 10 μL of dimethyl sulfoxide, and 10 μL of CP-I and CP-III internal standard solution (200 ng/mL each of CP-I–15N4 and CP-III–15N4) were added. The tubes were vortex-mixed and centrifuged (5 min at 12,000 × g, 10 °C). Five hundred μL of the mixture was applied to the Oasis MAX μElution plate pretreated with 200 μL of methanol and water. Then, the wells were washed with 200 μL of 1.25% ammonia aqueous solution and 200 μL of methanol. After 35 μL of 2% formic acid in methanol/acetonitrile (1/1, v/v) was added to each well, the analytes were eluted into a 96-well collection plate (Waters, Milford, Massachusetts, USA). Each extracted sample was diluted with 35 μL of a 2% formic acid aqueous solution. The collection plate was closed with a sealed film (Waters, Milford, Massachusetts, USA) and transferred to the sample manager for direct injection into the UPLC-QTOF/MS system. All SPE procedures were performed using a 96-well positive pressure unit (Waters, Milford, Massachusetts, USA).
Calibration samples were prepared by adding 100 μL of HSA solution (blank matrix) to 10 μL of a calibration working solution containing both CP-I and CP-III. QC samples were prepared by adding 100 μL of pooled blank human plasma (blank sample) to 10 μL of QC working solution containing both CP-I and CP-III. Each calibrator or QC sample was then mixed with 430 μL 4% phosphoric acid and 10 μL of CP-I and CP-III internal standard solution (200 ng/mL each of CP-I–15N4 and CP-III–15N4). The mixtures were vortexed and processed as for subjects’ plasma samples.
Instrumental Analysis Parameters
The UPLC-QTOF/MS system (Waters, Milford, Massachusetts, USA) consisted of a Waters Acquity Premier system and a Waters Xevo G2-XS QTOF mass spectrometer. A Waters ACQUITY UPLC HSS T3 Column (1.8 μm, 2.1 × 150 mm) was used with a Waters ACQUITY UPLC HSS T3 VanGuard precolumn (1.8 μm, 2.1 × 5 mm) at 40 °C for chromatographic separation. The eluent consisted of 0.1% aqueous formic acid and 5% acetonitrile with 2 mM ammonium formate (solution A) and acetonitrile with 0.1% formic acid and 5% water containing 2 mM ammonium formate (solution B). The gradient was started at 60% A for 0.5 min; the ratio was changed linearly to 40% A within the next 7.5 min, and the ratio was immediately changed to 5% A and maintained for another 2 min; then, the ratio was returned to 60% A and maintained for another 2.5 min. The flow rate was 0.25 mL/min, and the injection volume was 10 μL. The ionization parameters were as follows: spray voltage 3.0 kV, cone voltage 30 V, source temperature 150 °C, cone gas flow 100 L/h, desolvation gas flow 800 L/h, and desolvation temperature 450 °C.
TOF MS/MS analysis was carried out with electrospray ionization (ESI) positive mode using argon as a collision-induced dissociation gas. The MS/MS transitions monitored in the positive ion mode were m/z 655.2759 → m/z 596.2655 for CP-I and CP-III and m/z 659.2618 → m/z 600.2504 for the internal standards. The TOF cone voltage and collision energy were set at 30 and 48 V, respectively. and the scan time was set at 0.5 s for all analytes.
Validation of the Analytical Method
The validation method was based on the guidance of the FDA.28 Within-run and run-to-run accuracy and precision were evaluated by analyzing blank and QC samples [a total of 30 samples comprising pooled blank plasma sample (plasma from healthy volunteers plus internal standard), LLOQ, low QC, mid QC, and high QC in plasma, six replicates each]. The acceptance criterion for accuracy was ±15% of nominal concentrations, with the exception of ±20% for LLOQ. The acceptance criterion for precision was ±15% CV, with the exception of ±20% CV for LLOQ. Recovery rates from blank plasma were determined using low QC, mid QC, and high QC samples (same samples as used in the within-run and run-to-run accuracy and precision tests). The peak area of the extracted QC sample at each concentration was compared with the peak area of the extracted blank plasma sample spiked at that QC concentration postextraction (representing 100% recovery rate). The matrix effects were accessed using low QC, mid QC, and high QC samples. The peak area of the extracted blank plasma sample spiked at each QC concentration was compared with the peak area of the extracted matrix-free sample (using water instead of blank plasma) containing standards and internal standards (representing 100% recovery rate). The recovery rate and matrix effect were calculated by the following equations: recovery rate (%) = peak area of the analyte in the extracted sample/peak area of the analyte in extracted blank plasma spiked at postextraction × 100; matrix effect (%) = peak area of the analyte in the extracted blank plasma spiked at the postextraction/peak area of the analyte in aqueous solution × 100. When the assessment of recovery rates and matrix effects was implemented, six repetitive runs were performed using the blank plasma samples of six different healthy volunteers. Stability in the autosampler was evaluated by repeated measurements of low QC and high QC samples after leaving in the autosampler at 10 °C for 24 h. Freeze–thaw stability was measured using low QC and high QC samples subjected to three freeze–thaw cycles and then calculating QC accuracy.
Long-Term Stability Tests of CP-I and CP-III under Various Conditions
Stability of the CP-I and CP-III concentrations in human plasma stored at different temperatures for various durations was evaluated. Pooled blank plasma in a 1.5 mL shaded safe-lock tube was spiked with 10 μL of CP-I and CP-III (1 ng/mL each). These samples were stored at room temperature, 4, −20, and −80 °C, and measured after storage for 1, 8, 15, 30, 90, and 180 days. Triplicate samples were evaluated under each condition. Concentrations of CP-I and CP-III were determined by the developed quantification method using UPLC-QTOF/MS. The measured concentrations of CP-I and CP-III for each condition were compared with the control (pooled blank plasma not spiked with CP-I or CP-III measured under the same temperature and time conditions) and assessed by the rate of variation as a percentage. CP-I and CP-III were considered to be stable when the variation of each sample was within 15%.
Photostability Test of CP-I and CP-III under Benchtop Conditions
Stability of CP-I and CP-III in human plasma under the light conditions that were expected during the assay was tested as follows. Pooled blank plasma in a 1.5 mL transparent tube was spiked with 10 μL of CP-I and CP-III (0.1 ng/mL each), and triplicate samples were tested. These samples were left in a lighted room (100–110 lx) at room temperature, and the residual ratio was evaluated over time (time course: 0, 0.5, 1, 2, 4, 8, 12, and 24 h). Concentrations of CP-I and CP-III were determined by the developed quantification method using UPLC-QTOF/MS. The residual ratio was calculated by comparing the mean concentration of the test samples with the mean concentration of a sample immediately after spiking (0 h). CP-I and CP-III were considered to be stable when the residual ratio of each analyte was more than 85%.
Data Analysis and Statistics
Plasma concentrations of CP-I and CP-III were analyzed by using analyte-specific TOF MS/MS quantifier transitions. The calibration curve of each analyte was obtained by calculating the peak area ratio (peak area of the calibrator divided by the peak area of the internal standard), and weighted linear regression (1/x) was implemented using Target Lynks XS (Waters, Milford, Massachusetts, USA). Data obtained in this study are expressed as the mean ± standard deviation [range] for parametric data or median [range] for nonparametric data. Data normality was evaluated using the Shapiro–Wilk test. A comparison of clinical background data between healthy volunteers and RA patients was performed using the χ2 test or Welch’s t-test for parametric data, or Mann–Whitney U test for nonparametric data. Differences in the plasma concentrations of CP-I and CP-III between healthy volunteers and RA patients were analyzed by the Mann–Whitney U test. A p-value less than 0.05 was considered to indicate significant statistical differences between groups. All statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, CA, USA).
Acknowledgments
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The authors declare no competing financial interest.
References
- Zamek-Gliszczynski M. J.; Taub M. E.; Chothe P. P.; Chu X.; Giacomini K. M.; Kim R. B.; Ray A. S.; Stocker S. L.; Unadkat J. D.; Wittwer M. B.; et al. Transporters in Drug Development: 2018 ITC Recommendations for Transporters of Emerging Clinical Importance. Clin Pharmacol Ther 2018, 104 (5), 890–899. 10.1002/cpt.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetter A.; Kullak-Ublick G. A. Drugs and hepatic transporters: A review. Pharmacol. Res. 2020, 154, 104234. 10.1016/j.phrs.2019.04.018. [DOI] [PubMed] [Google Scholar]
- Mykkanen A. J. H.; Taskinen S.; Neuvonen M.; Paile-Hyvarinen M.; Tarkiainen E. K.; Lilius T.; Tapaninen T.; Backman J. T.; Tornio A.; Niemi M. Genomewide Association Study of Simvastatin Pharmacokinetics. Clin Pharmacol Ther 2022, 112 (3), 676–686. 10.1002/cpt.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Q.; Liu J. Y.; Yin J. Y.; Yang G. P.; Liu S. K.; Zheng Y.; Xie P.; Guo C. X.; Luo M.; Zhou H. H.; et al. Repaglinide-irbesartan drug interaction: effects of SLCO1B1 polymorphism on repaglinide pharmacokinetics and pharmacodynamics in Chinese population. Eur. J. Clin Pharmacol 2018, 74 (8), 1021–1028. 10.1007/s00228-018-2477-6. [DOI] [PubMed] [Google Scholar]
- Niemi M.; Kivisto K. T.; Hofmann U.; Schwab M.; Eichelbaum M.; Fromm M. F. Fexofenadine pharmacokinetics are associated with a polymorphism of the SLCO1B1 gene (encoding OATP1B1). Br. J. Clin. Pharmacol. 2005, 59 (5), 602–604. 10.1111/j.1365-2125.2005.02354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furihata T.; Matsumoto S.; Fu Z.; Tsubota A.; Sun Y.; Matsumoto S.; Kobayashi K.; Chiba K. Different interaction profiles of direct-acting anti-hepatitis C virus agents with human organic anion transporting polypeptides. Antimicrob. Agents Chemother. 2014, 58 (8), 4555–4564. 10.1128/AAC.02724-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K.; Ikeda Y.; Fujita T.; Yoshida K.; Azuma Y.; Haruyama Y.; Yamane N.; Kumagai Y.; Sugiyama Y. Identification of the rate-determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin. Pharmacol. Ther. 2011, 90 (4), 575–581. 10.1038/clpt.2011.142. [DOI] [PubMed] [Google Scholar]
- Watanabe T.; Tanaka R.; Suzuki Y.; Sato H.; Negami J.; Yoshijima C.; Oda A.; Ono H.; Tatsuta R.; Ohno K.; et al. Positive correlation between organic anion transporter 1B function indicated by plasma concentration of coproporphyrin-I and blood concentration of cyclosporin A in real-world patients. Br. J. Clin. Pharmacol. 2023, 89 (5), 1672–1681. 10.1111/bcp.15640. [DOI] [PubMed] [Google Scholar]
- Suzuki Y.; Sasamoto Y.; Koyama T.; Yoshijima C.; Nakatochi M.; Kubo M.; Momozawa Y.; Uehara R.; Ohno K. Substantially Increased Plasma Coproporphyrin-I Concentrations Associated With OATP1B1*15 Allele in Japanese General Population. Clin Transl Sci. 2021, 14 (1), 382–388. 10.1111/cts.12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercep I.; Radman I.; Trkulja V.; Bozina T.; Simicevic L.; Budimir E.; Ganoci L.; Bozina N. Loss of function polymorphisms in SLCO1B1 (c.521T > C, rs4149056) and ABCG2 (c.421C > A, rs2231142) genes are associated with adverse events of rosuvastatin: a case-control study. Eur. J. Clin Pharmacol 2022, 78 (2), 227–236. 10.1007/s00228-021-03233-7. [DOI] [PubMed] [Google Scholar]
- Neuvonen M.; Hirvensalo P.; Tornio A.; Rago B.; West M.; Lazzaro S.; Mathialagan S.; Varma M.; Cerny M. A.; Costales C.; et al. Identification of Glycochenodeoxycholate 3-O-Glucuronide and Glycodeoxycholate 3-O-Glucuronide as Highly Sensitive and Specific OATP1B1 Biomarkers. Clin Pharmacol Ther 2021, 109 (3), 646–657. 10.1002/cpt.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J. M.; Jang E. J.; Yee J.; Song T. J.; Kim D. H.; Park J.; Gwak H. S. Association between SLCO1B1 genetic polymorphisms and bleeding risk in patients treated with edoxaban. Sci. Rep 2023, 13 (1), 15967. 10.1038/s41598-023-43179-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodieux F.; Gotta V.; Pfister M.; van den Anker J. N. Causes and Consequences of Variability in Drug Transporter Activity in Pediatric Drug Therapy. J. Clin. Pharmacol. 2016, 56 (Suppl 7), S173–S192. 10.1002/jcph.721. [DOI] [PubMed] [Google Scholar]
- Chu X.; Liao M.; Shen H.; Yoshida K.; Zur A. A.; Arya V.; Galetin A.; Giacomini K. M.; Hanna I.; Kusuhara H.; et al. Clinical Probes and Endogenous Biomarkers as Substrates for Transporter Drug-Drug Interaction Evaluation: Perspectives From the International Transporter Consortium. Clin Pharmacol Ther 2018, 104 (5), 836–864. 10.1002/cpt.1216. [DOI] [PubMed] [Google Scholar]
- Maeda K. Recent progress in in vivo phenotyping technologies for better prediction of transporter-mediated drug-drug interactions. Drug Metab Pharmacokinet 2020, 35 (1), 76–88. 10.1016/j.dmpk.2019.12.004. [DOI] [PubMed] [Google Scholar]
- Barnett S.; Ogungbenro K.; Menochet K.; Shen H.; Lai Y.; Humphreys W. G.; Galetin A. Gaining Mechanistic Insight Into Coproporphyrin I as Endogenous Biomarker for OATP1B-Mediated Drug-Drug Interactions Using Population Pharmacokinetic Modeling and Simulation. Clin Pharmacol Ther 2018, 104 (3), 564–574. 10.1002/cpt.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Cheeti S.; Yoshida K.; Choo E.; Chen E.; Chen B.; Gates M.; Singel S.; Morley R.; Ware J.; et al. Effect of OATP1B1/1B3 Inhibitor GDC-0810 on the Pharmacokinetics of Pravastatin and Coproporphyrin I/III in Healthy Female Subjects. J. Clin Pharmacol 2018, 58 (11), 1427–1435. 10.1002/jcph.1261. [DOI] [PubMed] [Google Scholar]
- Neuvonen M.; Tornio A.; Hirvensalo P.; Backman J. T.; Niemi M. Performance of Plasma Coproporphyrin I and III as OATP1B1 Biomarkers in Humans. Clin Pharmacol Ther 2021, 110 (6), 1622–1632. 10.1002/cpt.2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T.; Mano Y. Significant increases in detection capability for assessment of organic anion transporting polypeptide-mediated drug-drug interaction in cynomolgus monkeys by modifying pretreatment of Coproporphyrin I and III, and glycochenodeoxycholate-3-sulfate in plasma. Anal. Chim. Acta 2024, 1322, 343056 10.1016/j.aca.2024.343056. [DOI] [PubMed] [Google Scholar]
- Bednarczyk D.; Boiselle C. Organic anion transporting polypeptide (OATP)-mediated transport of coproporphyrins I and III. Xenobiotica 2016, 46 (5), 457–466. 10.3109/00498254.2015.1085111. [DOI] [PubMed] [Google Scholar]
- Njumbe Ediage E.; Dillen L.; Vroman A.; Diels L.; Kunze A.; Snoeys J.; Verhaeghe T. Development of an LC-MS method to quantify coproporphyrin I and III as endogenous biomarkers for drug transporter-mediated drug-drug interactions. J. Chromatogr B Analyt Technol. Biomed Life Sci. 2018, 1073, 80–89. 10.1016/j.jchromb.2017.12.008. [DOI] [PubMed] [Google Scholar]
- Suzuki Y.; Sasamoto Y.; Yoshijima C.; Tanaka R.; Ono H.; Ando T.; Shin T.; Mimata H.; Itoh H.; Ohno K. Simultaneous quantification of coproporphyrin-I and 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid in human plasma using ultra-high performance liquid chromatography coupled to tandem mass spectrometry. J. Pharm. Biomed Anal 2020, 184, 113202 10.1016/j.jpba.2020.113202. [DOI] [PubMed] [Google Scholar]
- Kandoussi H.; Zeng J.; Shah K.; Paterson P.; Santockyte R.; Kadiyala P.; Shen H.; Shipkova P.; Langish R.; Burrrell R.; et al. UHPLC-MS/MS bioanalysis of human plasma coproporphyrins as potential biomarkers for organic anion-transporting polypeptide-mediated drug interactions. Bioanalysis 2018, 10 (9), 633–644. 10.4155/bio-2017-0246. [DOI] [PubMed] [Google Scholar]
- King-Ahmad A.; Clemens S.; Ramanathan R.; Zhang Y.; Raha N.; Zhang Y.; Holliman C.; Rodrigues A. D.; Li F. A fully automated and validated human plasma LC-MS/MS assay for endogenous OATP biomarkers coproporphyrin-I and coproporphyrin-III. Bioanalysis 2018, 10 (9), 691–701. 10.4155/bio-2017-0270. [DOI] [PubMed] [Google Scholar]
- Caris J. A.; Benzi J. R. L.; de Souza F. F. L.; de Oliveira R. D. R.; Donadi E. A.; Lanchote V. L. Rheumatoid arthritis downregulates the drug transporter OATP1B1: Fluvastatin as a probe. Eur. J. Pharm. Sci. 2020, 146, 105264 10.1016/j.ejps.2020.105264. [DOI] [PubMed] [Google Scholar]
- Lai Y.; Mandlekar S.; Shen H.; Holenarsipur V. K.; Langish R.; Rajanna P.; Murugesan S.; Gaud N.; Selvam S.; Date O.; et al. Coproporphyrins in Plasma and Urine Can. Be Appropriate Clinical Biomarkers to Recapitulate Drug-Drug Interactions Mediated by Organic Anion Transporting Polypeptide Inhibition. J. Pharmacol Exp Ther 2016, 358 (3), 397–404. 10.1124/jpet.116.234914. [DOI] [PubMed] [Google Scholar]
- Shen H.; Dai J.; Liu T.; Cheng Y.; Chen W.; Freeden C.; Zhang Y.; Humphreys W. G.; Marathe P.; Lai Y. Coproporphyrins I and III as Functional Markers of OATP1B Activity: In Vitro and In Vivo Evaluation in Preclinical Species. J. Pharmacol Exp Ther 2016, 357 (2), 382–393. 10.1124/jpet.116.232066. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration. Bioanalytical Method Validation Guidance for Industry. 2018. https://www.fda.gov/media/70858/download (accessed 2023 June 30).
- Ono H.; Tanaka R.; Suzuki Y.; Oda A.; Ozaki T.; Tatsuta R.; Maeshima K.; Ishii K.; Ohno K.; Shibata H.; et al. Factors Influencing Plasma Coproporphyrin-I Concentration as Biomarker of OATP1B Activity in Patients With Rheumatoid Arthritis. Clin Pharmacol Ther 2021, 110 (4), 1096–1105. 10.1002/cpt.2375. [DOI] [PubMed] [Google Scholar]