

Abstract
Background:
Many patients with glioblastoma multiforme (GBM) develop deep venous thrombosis or pulmonary emboli. Cell-free circulating mitochondria increase after brain injury and are associated with coagulopathy.
Objectives:
This study evaluated whether mitochondria play a role in the GBM-induced hypercoagulable state.
Methods:
We examined the correlation between cell-free circulating mitochondria and venous thrombosis in patients with GBM and the impact of mitochondria on venous thrombosis in mice with inferior vena cava stenosis.
Results:
Using plasma samples of 82 patients with GBM, we found that patients with GBM had a higher number of mitochondria in their plasma (GBM with venous thromboembolism [VTE],: 2.8 × 107 mitochondria/mL; GBM without VTE, 1.9 × 107 mitochondria/mL) than that in healthy control subjects (n = 17) (0.3 × 107 mitochondria/mL). Interestingly, patients with GBM and VTE (n = 41) had a higher mitochondria concentration than patients with GBM without VTE (n = 41). In a murine model of inferior vena cava stenosis, intravenous delivery of mitochondria resulted in an increased rate of venous thrombosis compared with that in controls (70% and 28%, respectively). Mitochondria-induced venous thrombi were neutrophil-rich and contained more platelets than those in control thrombi. Furthermore, as mitochondria are the only source of cardiolipin in circulation, we compared the concentration of anticardiolipin immunoglobulin G in plasma samples of patients with GBM and found a higher concentration in patients with VTE (optical density, 0.69 ± 0.04) than in those without VTE (optical density, 0.51 ± 0.04).
Conclusion:
We concluded that mitochondria might play a role in the GBM-induced hypercoagulable state. We propose that quantifying circulating mitochondria or anticardiolipin antibody concentrations in patients with GBM might identify patients at increased risk of VTE.
Keywords: anticardiolipin antibody, glioblastoma, mitochondria, neutrophils, venous thrombosis
1 |. INTRODUCTION
Glioblastoma multiforme (GBM), a high-grade astrocytic brain tumor, is associated with high frequency of venous thromboembolism (VTE). Approximately 25% to 30% of patients with GBM develop deep venous thrombosis or pulmonary emboli, and a much higher percentage of them (90%) develop intratumor microvascular thrombosis [1–4]. Majority of VTEs occur within 3 to 6 months of diagnosis of GBM, cause significant complications, and are associated with a worse prognosis [5]. Despite the high incidence of VTEs, administration of prophylactic anticoagulants to all patients with GBM is associated with an unacceptable frequency of bleeding complications [6] and is not recommended beyond the postoperative period [7,8].
Considering recent data supporting a role for circulating cell-free mitochondria in thrombosis and coagulopathy after brain injury [9], we examined whether circulating mitochondria cause venous thrombosis in GBM. Cell injury or death releases intracellular molecules known as damage-associated molecular patterns (DAMPs) that activate innate immunity and promote inflammation [10,11]. The mitochondrion is a source of DAMPs. Mitochondrial DNA, peptides, and lipids have a role in various inflammatory conditions [12]. After traumatic brain injury, intact or fragmented mitochondria, either free or within brain-derived microvesicles, are detected in blood and cause activation of endothelial cells, platelets, and the coagulation cascade. Cardiolipin on the mitochondrial membrane is a procoagulant phospholipid, and reactive oxygen species released from mitochondria activate platelets, endothelial cells, and neutrophils [9,13,14]. We found that patients with GBM and venous thrombosis had a higher concentration of mitochondria in their plasma than that in the plasma of patients with GBM without venous thrombosis. Glioblastoma cell lines release mitochondria, and injection of GBM cell–derived mitochondria results in neutrophil-rich venous thrombi in mice. Mitochondria are the only source of cardiolipin; hence, we measured anticardiolipin antibodies in plasma samples of patients with GBM and found that patients with venous thrombi had a higher concentration of anticardiolipin antibodies than that in patients with GBM without venous thrombi. However, in our cohort, the number of circulating mitochondria did not correlate with the titer of anticardiolipin immunoglobulin G (IgG). We propose that mitochondria released from GBM cells promote venous thrombosis and that quantifying free circulating mitochondria or anticardiolipin antibodies may predict patients at a higher risk of venous thrombosis.
2 |. MATERIAL AND METHODS
All the experiments in the study were approved by the Intuitional Review Board and Institutional Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center (MDA CC).
2.1 |. Patient population
We used the MDACC VTE Repository to identify patients with GBM and VTE. The VTE repository is a long-term project aimed at identifying and tracking every venous thromboembolic event diagnosed by radiologic imaging since 2010 at MDACC. In the VTE repository, patients are categorized into definitive, probable, possible, or no VTE groups. In addition to the radiologic diagnosis, all the clinical, laboratory, therapeutic, and pathologic (including genetic) information about each patient is captured. Currently, this repository contains >300 million data endpoints on 60 383 patients. To identify patients with VTE from radiologic studies, complex parsing rules and dictionaries of VTE concept, body site, document section header, and negation word were developed and used in IBM Watson Content Analytics 3.5. VTE findings were combined with an existing cancer diagnosis, electronic medical rector system clinical and pharmacy data, clinical laboratory data, and genetic data for a unique and powerful cross-reference capability. Automated natural language processing to identify VTEs from diagnostic imaging reports was validated.
Among 476 patients with GBM, 139 had venous thrombosis (pulmonary emboli or deep venous thrombosis). We could identify plasma samples of 41 of 139 patients with GBM and VTE and 41 patients with GBM without VTE in the Brain Tumor Center. We confirmed the presence or absence of VTE in the medical records of these patients by reviewing the individual medical charts. Plasma samples from these patients were collected at the time of their initial visit to MDACC and before any therapeutic intervention or VTE, except for one patient who was diagnosed with VTE at the time of presentation.
2.2 |. Mitochondria concentration in plasma
Plasma samples (200 μl) were centrifuged at 1600 × g for 10 minutes at 4 °C to collect cell-free plasma, from which genomic DNA was extracted using the QIAprep Spin Miniprep Kit (27104; Qiagen). Mitochondrial-specific DNA was amplified as we have previously described [13,15]. Briefly, mitochondria DNA was amplified using amfiSure qGreen Q-PCR Master Mix (Q5602; GenDEPOT) with the following primers: forward 5′-TCTCCAGTGGCCAACAGTGTT-3′ and reverse 5′-GCCCTCTTGTTCCCATCAAC-3′. Amplification and data acquisition were performed using the CFX Connect Real-Time PCR Detection System and software (Bio-Rad). The number of mitochondria was calculated by dividing mitochondrial DNA in nanograms per milliliter divided by the DNA content of a single mitochondrion (≈ 5 × 10−8 ng) [16–18].
2.3 |. Cell culture and extraction of extracellular vesicles
The human GBM cell line U87G was cultured in Dulbecco’s modified eagle medium supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin-streptomycin. Cells were kept at 37 °C in a humidified incubator with 20% O2 and 5% CO2. Extracellular vesicle (EV) extraction from the U87G cell line was performed as described [19]. Briefly, cancer cells were cultured in optimal conditions until reaching 80% of confluence. Cells were then washed with phosphate-buffered saline (PBS) to remove the medium remnant and supplemented with a Dulbecco’s modified eagle medium containing 2% exosome-depleted fetal bovine serum (A2720803; Gibco). After 48 hours, media was collected and spun down for 5 minutes at 200 × g. The supernatant was collected and centrifuged for 10 min at 1500 × g. Finally, the supernatant was centrifuged for 3 hours at 100 000 relative centrifugal force at 4 °C to pellet EVs and organelles.
2.4 |. Transmission electron microscopy and flow cytometry on GBM EVs
For transmission electron microscopy, EVs were fixed with a solution containing 3% glutaraldehyde plus 2% paraformaldehyde in 0.1-M cacodylate buffer at pH 7.3. Fixed EVs were washed in 0.1-M sodium cacodylate buffer and treated with 0.1% Millipore-filtered cacodylate buffered tannic acid (MilliporeSigma), postfixed with 1% buffered osmium tetroxide, and stained with 1% uranyl acetate. The samples were dehydrated in increasing concentrations of ethanol, infiltrated, and embedded in the LX-112 medium. The samples were polymerized in an oven at 60 °C for approximately 3 days. Ultrathin sections were cut in a Leica Ultracut microtome (Leica), stained with uranyl acetate and lead citrate, and examined in a JEM 1010 transmission electron microscope (JEOL) at an accelerating voltage of 80 kV. Digital images were obtained using the AMT Imaging System (Advanced Microscopy Techniques Corp).
For flow cytometry, EVs were resuspended in 100-μl PBS. MitoTracker Green FM (M7514, 1:250; Invitrogen) was added to resuspend EVs, incubated for 10 min at dark, and further diluted with 400 ml of PBS, centrifuged for 70 minutes at 4 °C at 21,000 × g. The pellet was resuspended in 200 μl of PBS and analyzed using the Cytek Aurora flow cytometry system (Cytek Biosciences).
2.5 |. Isolation and quantification of mitochondria from U87G cells
Mitochondria were extracted and purified from the U87G cells using a Qproteome Mitochondria Isolation Kit (37612; Qiagen). Flow cytometry was used to quantify purified mitochondria. Briefly, samples were incubated with MitoTracker Green FM (M7514, 1:250; Invitrogen) for 20 minutes at room temperature in the dark. Submicron 0.5- and 1-μm particles (F13839; Invitrogen) were used as size references in flow cytometry. Samples were serially diluted until linearity between mitochondria counts vs dilution factor was observed. A linear regression model was applied to calculate the sample’s mitochondrial concentration. Cytek Aurora flow cytometer and FlowJo 10.8 software (BD Biosciences) were used to run and analyze the samples, respectively.
2.6 |. Murine model of inferior vena cava stenosis
A murine model of deep venous thrombosis (inferior vena cava [IVC] stenosis) was generated in B67BL/6 mice as described previously [19]. After isoflurane anesthesia and ethanol/iodine skin sterilization, a ventral midline abdominal incision was made. The intestines were moved to the side using cotton swabs covered with saline-soaked sterile gauze. The right IVC and its side branches were identified. The infrarenal IVC was then isolated from the aorta. A surgical silk suture of size 4–0 was placed securely around the IVC, just caudal to the lowest renal vein. A 27-gauge needle was temporarily placed over the IVC as a “spacer” while the ligature was tied to provide a small lumen and prevent complete IVC ligation. The internal organs were returned to the abdominal cavity, and the muscle and skin were subsequently closed with running sutures and clips, respectively. The mice were administered 1.25 × 107 mitochondria or saline through tail vein injection. Twenty-four hours later, digital subtraction angiography was performed to evaluate the formation of an IVC thrombus. Under anesthesia, iodinated contrast (Visipaque, GE Healthcare) was injected via tail vein catheterization. Digital subtraction angiography was performed using a commercial angiography system (Artis-Q Siemens) at a frame rate of 7.5 frames/s during contrast injection. Thrombus length measurements were obtained from the uncompressed, diagnostic-quality images using a standard PACS software platform (IntelliSpace PACS).
2.7 |. Immunostaining of thrombi
Formalin-fixed paraffin-embedded blood clots were sectioned at 4-μm thickness. Slides were deparaffinized with xylene and decreasing ethanol concentrations and then rehydrated with PBS. Diva decloaker (DV2004LX; Biocare Medical) was used as a heat-induced antigen retriever, and nonspecific binding was blocked using 1% bovine serum albumin in PBS. SYTOX Green Nucleic Acid Stain (S7020; 1:100; Invitrogen) dye, antiplatelet CD42b (SC-7070; 1:100; Santa Cruz Biotechnology), antineutrophil Ly6G (551459; 1:200; BD Biosciences), and antimitochondrial COX-IV (4844s; 1:100; Cell signaling) primary antibodies were diluted in blocking serum and incubated overnight at 4 °C in a humidified chamber. Slides were incubated with a Cy3 conjugated secondary donkey anti-goat IgG (705-165-147), anti-mouse (712-165-153), or anti-rabbit (711-165-152) (1:1000; Jackson ImmunoResearch) and 4′,6′-diamidino-2-phenylindole DAPI (D9542; 1:1000; Millipore Sigma). If needed, red blood cells’ autofluorescence signal was quenched (SP-8400–15; Vector Laboratories) for 3 minutes. Images were obtained using an Olympus FV1000 confocal microscope (Olympus). Each clot was imaged using a 10× (platelets) and 20× (neutrophils) objective lens. Composite images were generated using the Image J Stitching plugin [20]. Platelet or neutrophil area from clots was quantified by measuring the Cy3 fluorescence signal using ImageJ software.
2.8 |. Enzyme-linked immunoassay
Antibodies directed against cardiolipin were quantified in plasma samples using anti-human cardiolipin IgG ELISA (DCM113; Eagle Biosciences).
2.9 |. Statistical analysis
All statistical analyses were performed using the GraphPad Prism 8 software. A 1-way analysis of variance, Holm-Sidak test, Pearson’s or nonparametric correlation, or a 2-tailed unpaired t-test with Welch’s correction were used to determine the statistical significance of comparisons. Logistic regression analysis was used to adjust the effect of mitochondrial levels on the VTE risk for demographic and clinical covariates. Log-rank test was used to compare survival in the groups of patients with GBM with and without VTE. P values ≤0.05 were considered statistically significant.
3 |. RESULTS
3.1 |. Patients with GBM and VTE had a higher number of circulating mitochondria than GBM patients without VTE
All of our patients were diagnosed with GBM. Most of our patients (93%) were diagnosed before 2016. Among 467 patients with GBM, we identified 139 patients with VTE (deep venous thrombosis or pulmonary emboli) throughout their follow-up period at MDACC. We had access to the plasma samples of 41 patients through the Brain Tissue Bank in our institution. We chose an equal number of patients with GBM and without VTE with available plasma samples registered in the Brain Tissue Bank. The demographic and clinical characteristics of the patients are summarized in the Table. Among 41 patients with VTE, 13 (32%) patients had pulmonary embolism only, 23 (56%) patients had deep vein thrombosis only, and 5 (12%) patients had both deep vein thrombosis and pulmonary embolism.
TABLE.
The patients’ characteristics.
| Characteristic | VTE (N = 41) | No VTE (N = 41) | P value |
|---|---|---|---|
| Age (y), mean (SD) | 58.40 (9.18) | 58.03 (10.88) | .870 |
| Male sex, n (%) | 26 (60.5) | 22 (59.5) | .927 |
| Chemotherapy, yes; n (%) | 24 (53.3) | 31 (68.9) | .130 |
| Surgery, n (%) | 29 (64.4) | 32 (71.1) | .499 |
| XRT, n (%) | 27 (60) | 32 (71.1) | .276 |
| Bevacizumab, n (%) | 8 (17.8) | 13 (28.9) | .213 |
| No. of circulating mitochondria per milliliter, mean (SD) | 2.824 × 107 (2.427 × 107) | 1.911 × 107 (1.433 × 107) | .042 |
| Anticardiolipin antibody level (OD),a mean (SD) | 0.680 (0.253) | 0.476 (0.276) | .002 |
| Pulmonary thromboembolism, n (%) | 18 (42.9) | ||
| Deep vein thrombosis, n (%) | 28 (66.7) | ||
| Time from diagnosis to death (mo), median (95% CI) | 17.00 (14.9–19.09) | 21.00 (8.62–33.38) | .633b |
OD, optical density; VTE, venous thromboembolism.
From 34 patients without VTE and 35 patients with VTE.
Log-rank test of equality of survival distributions.
Within the group of patients who had experienced a VTE event, the median time to event was 4 months (95% CI, 1.3–6.6 months). We compared the concentration of mitochondria particles in cell-free plasma samples of patients with GBM (n = 82) and normal control subjects (n = 17). Patients with GBM and VTE had a significantly higher concentration of mitochondria (2.8 ± 0.28 × 107/mL) than that in patients with GBM without VTE (1.9 ± 0.22 × 107/mL) (p = .042). Level of mitochondria remained significantly associated with VTE status (p = .036) even after adjustment for age, sex, and clinical variables. Additionally, both groups of patients with GBM had a higher concentration of mitochondria than the control subjects (0.3 ± 0.1 × 107/mL) (p ≤ .001 and p = .01 for patients with and without VTE compared with controls, respectively) (Figure 1). The number of circulating free mitochondria detected in plasma samples of our healthy control subject is very similar to the previously reported numbers [18].
FIGURE 1.
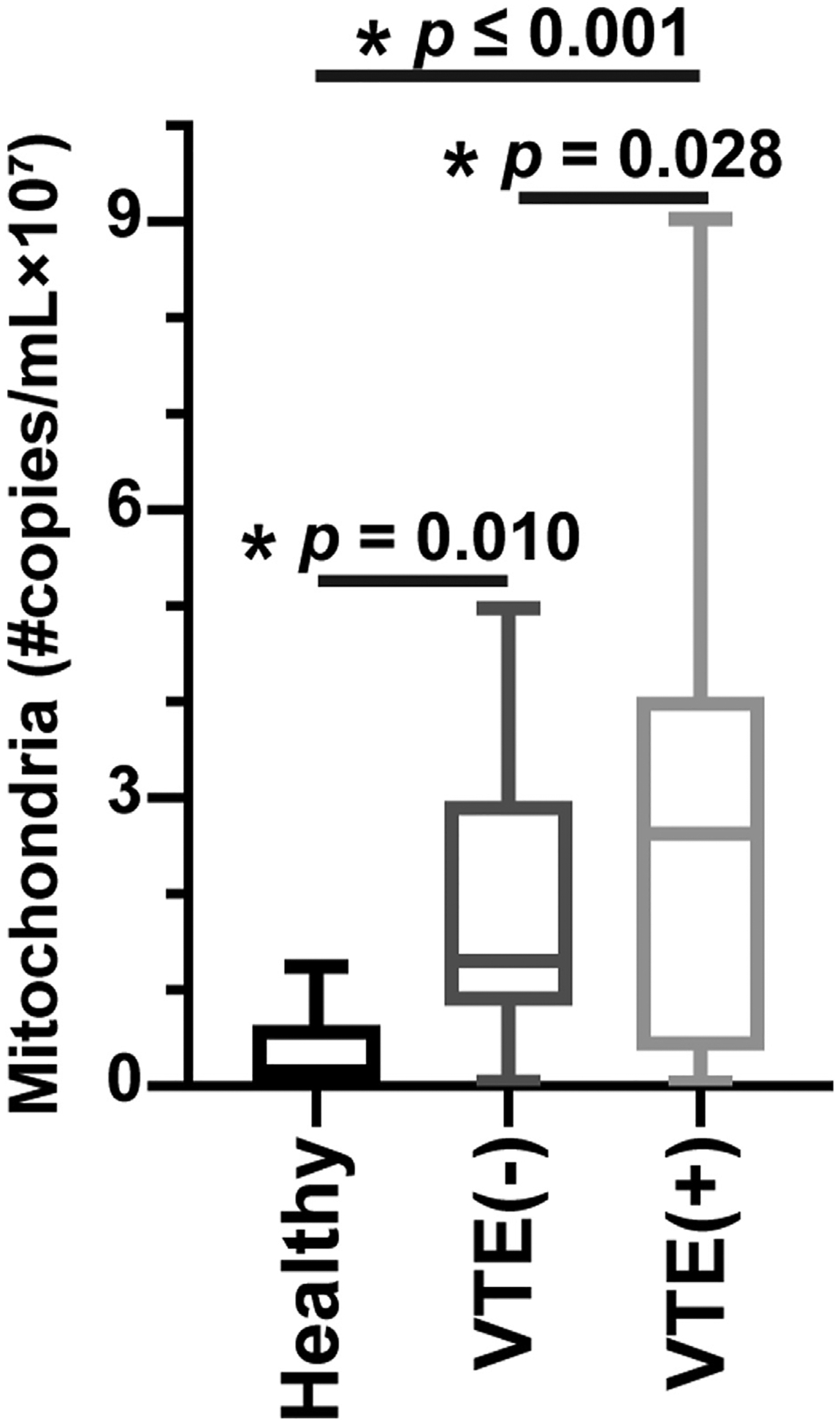
Mitochondria in blood and venous thrombosis in glioblastoma multiforme (GBM). The number of free circulating mitochondria in plasma samples of 41 patients with GBM and venous thromboembolism (VTE), 41 patients with GBM without VTE, and 17 healthy control subjects were shown.
3.2 |. GBM cancer cells release mitochondria in vitro
We examined the presence of mitochondria in tissue culture media incubated for 24 hours with U87 GBM cells. After removing cells and large cell fragments by centrifuging collected media at 1600 × g, mitochondria were detected by flow cytometry using MitoTraker green. Mitochondria (or mitochondria fragments) made up approximately 9% of EVs in the supernatant of U87 cells (EVs not stained with mitotracker served as negative control) (Figure 2A–C). Electron microscopy on collected media confirmed the presence of mitochondria released from U87 cells (Figure 2D–F).
FIGURE 2.
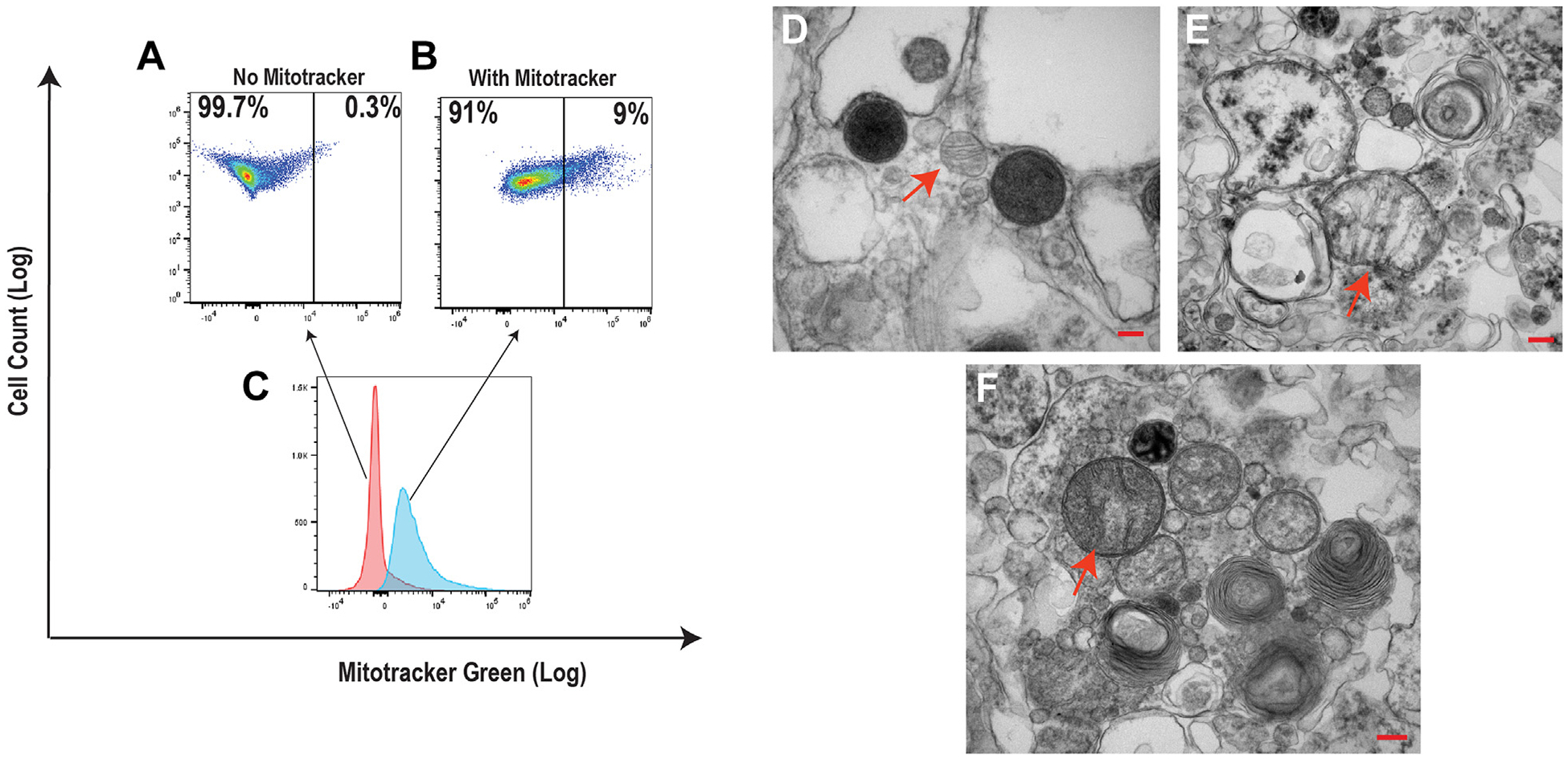
Extracellular mitochondria in glioblastoma multiforme cell culture media. Extracellular vesicles in the supernatant of U87 glioblastoma multiforme cancer cells were analyzed by flow cytometry (gated on particles 100–1000 nm in size), using MitoTracker green to detect mitochondria. Dot plots of (A) nonstained particles and (B) particles stained with mitotracker green. (C) Overlay histograms of A and B. Transmission electron microscopy showing mitochondria (red arrows) in the supernatant of media of cultured U87 cells after 48 hours. (D, E) Free extracellular mitochondria and (F) mitochondria embedded in the extracellular vesicles.
3.3 |. Injection of mitochondria induces venous thrombosis in mice
We found that 69.2% of the mice with IVC stenosis injected with mitochondria from U87G GBM cells (9 of 13 mice) developed IVC thrombosis compared with 28.6% of mice injected with PBS (2 of 7 mice) (Figure 3A). Furthermore, IVC thrombi in mitochondria-injected mice were heavier than those in PBS-injected control mice (Figure 3B). The average length of blood clots in mitochondria-injected mice was 9.1 ± 0.8 mm compared with 4.9 ± 2.1 mm (p = .61) in PBS-injected mice. The average weight of blood clots in mitochondria- and PBS-injected mice were 22.9 ± 3.3 and 6.7 ± 2.1 mg, respectively (p = .004).
FIGURE 3.
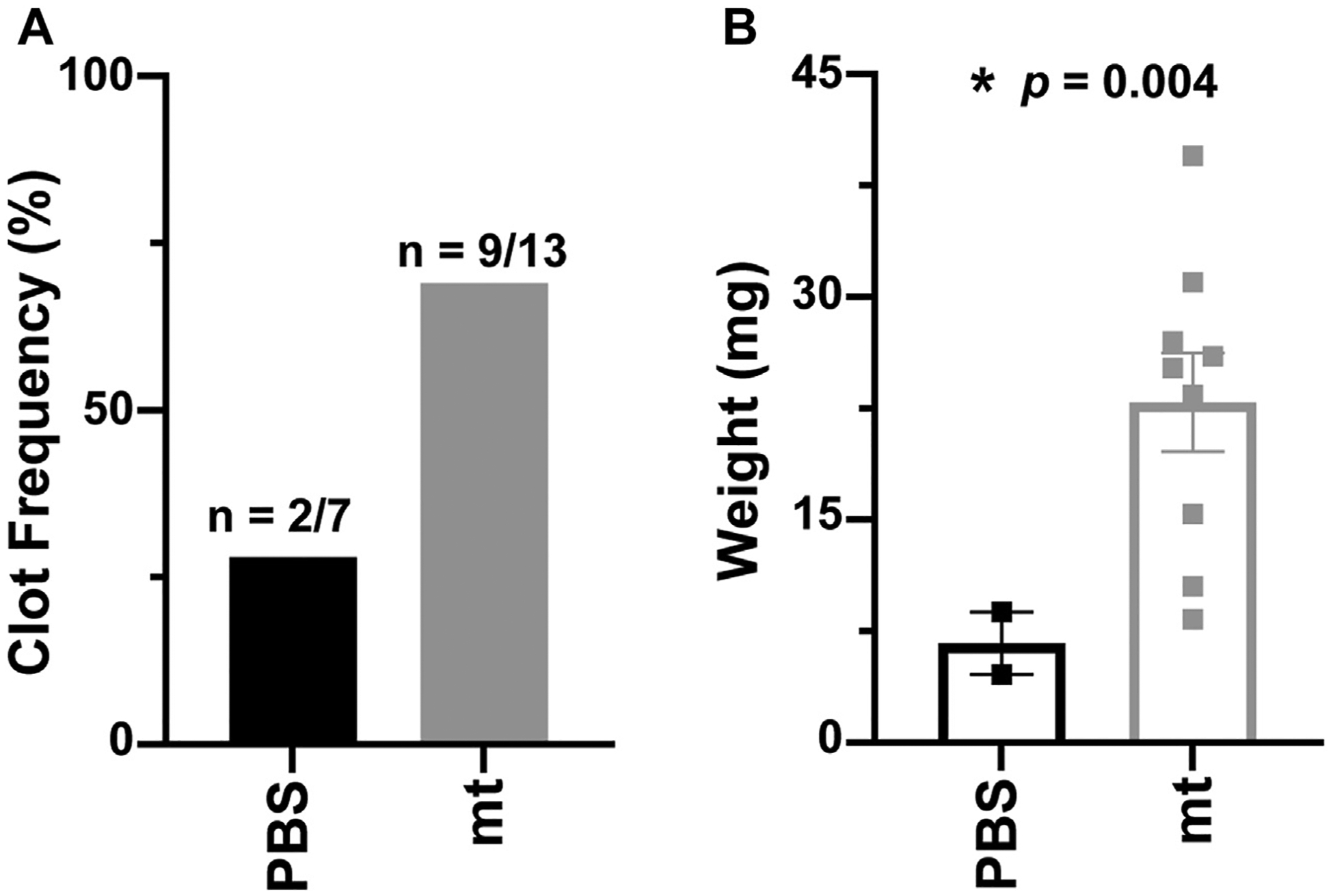
Mitochondria (mt)-induced venous thrombosis in mice with inferior vena cava (IVC) stenosis. (A) Frequency of IVC thrombosis and (B) weight of IVC thrombi resected from mice with IVC stenosis injected with mt or phosphate-buffered saline (PBS). A 2-tailed Student’s t-test with Welch correction was used to calculate p values.
3.4 |. Mitochondria-induced blood clots are rich in platelets and neutrophils
Immunostaining of thrombi demonstrated blood clots in mice with IVC stenosis injected with mitochondria from U87G GBM cells were rich in platelets (Figure 4A, B) and neutrophils (Figure 4C, D), compared to those injected with PBS. Likewise, mitochondria-induced venous thrombi showed more neutrophil extracellular traps (NETs) (Figure 5A). In addition, immunostaining of thrombi with COX-IV antimitochondrial antibody showed the involvement of mitochondria in blood clots induced after injection of mitochondria (Figure 5B).
FIGURE 4.
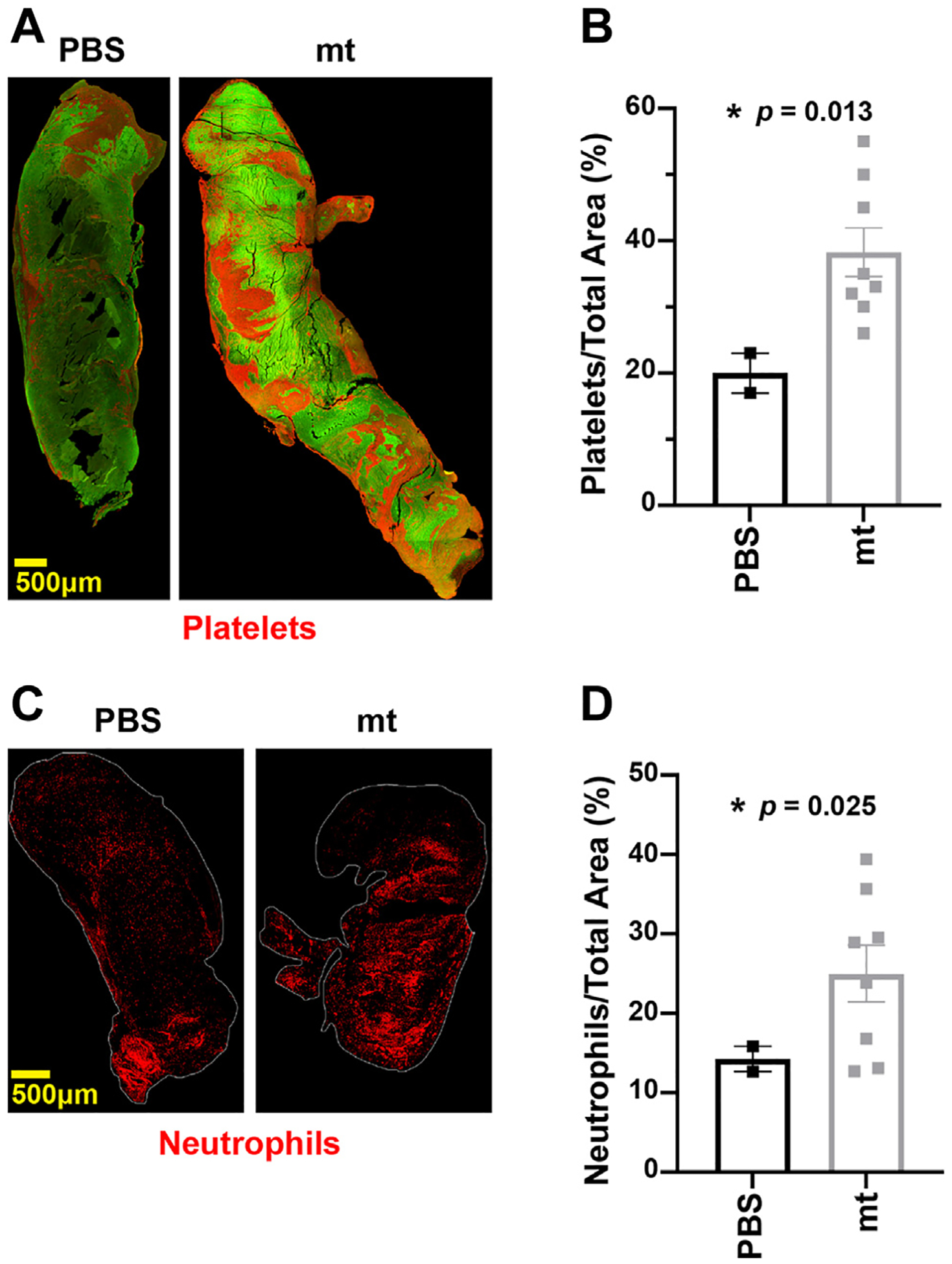
Mitochondria (mt)-induced venous thrombi are rich in platelets and neutrophils. (A) Representative immunofluorescence stain for platelets (CD42b) in PBS- and mt-induced venous thrombi. (B) Average surface area covered by platelets in PBS- and mt-induced venous thrombi. (C) Representative immunofluorescence stain for neutrophils (Lys6G) in phosphate-buffered saline (PBS)- and mt-induced venous thrombi. (D) Average surface area covered by neutrophils in PBS- and mt-induced venous thrombi.
FIGURE 5.
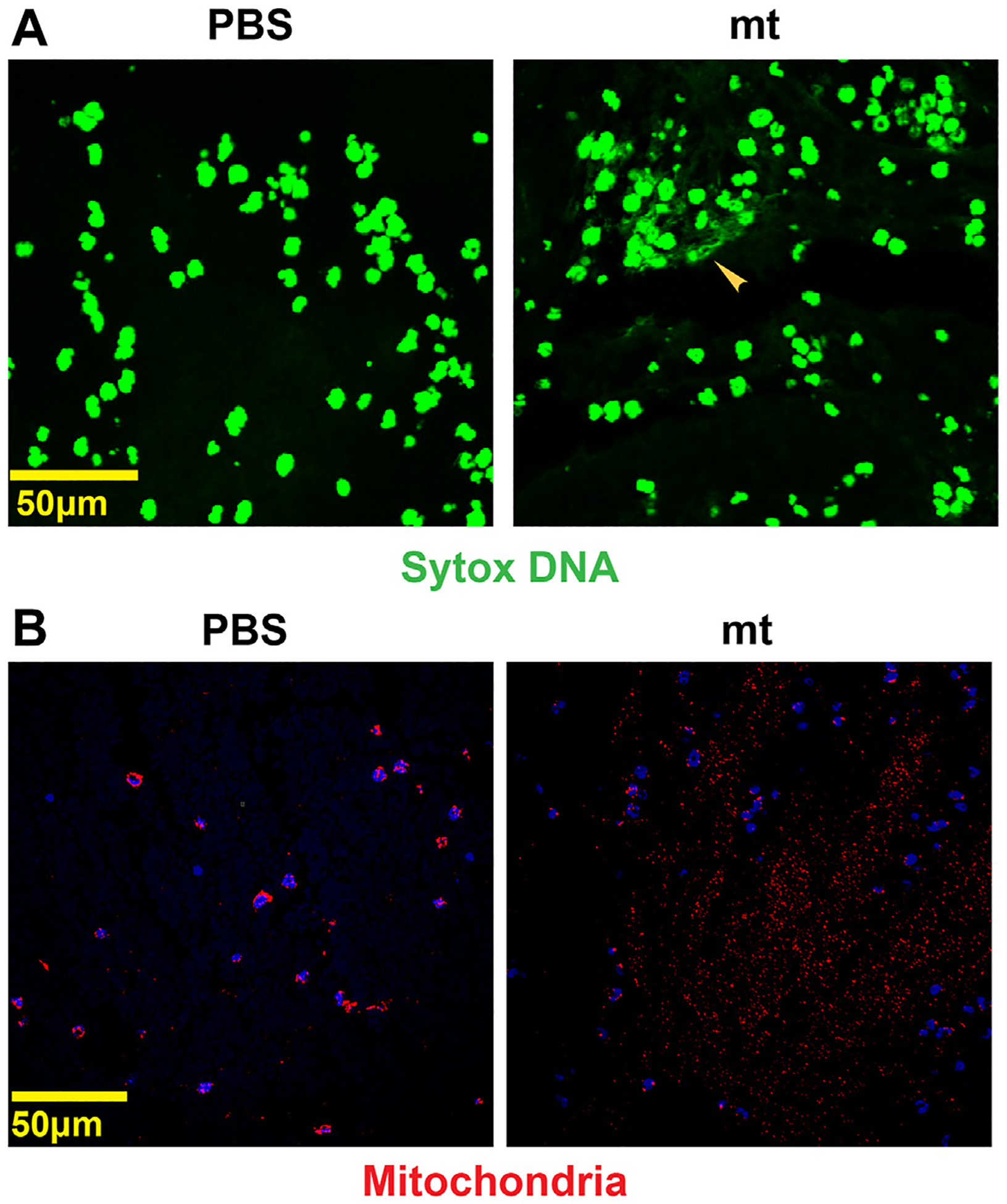
Mitochondria (mt)-induced venous thrombi have neutrophil extracellular traps. (A) Venous thrombi stained with Sytox-DNA (green) showing neutrophil extracellular traps in thrombus induced after mt injection compared to phosphate-buffered saline (PBS). (B) Staining with COX-IV antimitochondrial antibody demonstrated a large number of extracellular mt in thrombi induced after injection of mt compared to PBS.
3.5 |. Higher concentration of anticardiolipin antibodies in patients with GBM with venous thrombosis
Anticardiolipin antibodies are an important component of the antiphospholipid antibody repertoire and are associated with an increased risk of venous and arterial thrombosis. Mitochondria are the only source of cardiolipin, and we hypothesized that the presence of mitochondria in circulation might induce the generation of anticardiolipin antibodies. The association between cardiolipin antibody subtypes and thrombosis is more significant for IgG than immunoglobulin M [21]. Quantification of anticardiolipin IgG levels in the plasma samples of 38 patients with GBM and VTE, 37 patients with GBM without VTE, and 10 healthy control subjects demonstrated that patients with GBM and VTE had a significantly higher concentration of anticardiolipin IgG than that in patients without VTE (optical density values of 0.68 ± 0.043 and 0.48 ± 0.047, respectively; p ≤ .001) and that both groups had a higher concentration than controls (optical density, 0.27 ± 0.04) (p = .009 for patients with GBM and VTE vs controls, and p = .028 for patients with GBM without VTE vs controls) (Figure 6). After converting to U/mL, the anticardiolipin antibody titers were 30 ± 6 U/mL in controls, 50 ± 12 U/mL in patients with GBM and VTE, and 40 ± 14 U/mL in patients with GBM without VTE. We checked if the levels of mitochondria were correlated with the levels of anticardiolipin IgG, but it was not significant (p = .743). Both the concentration of mitochondria and the concentration of anticardiolipin IgG were significantly associated with VTE status (p = .021 and p = .007, respectively) when included simultaneously in the logistic regression model, adjusted for the clinical variables.
FIGURE 6.
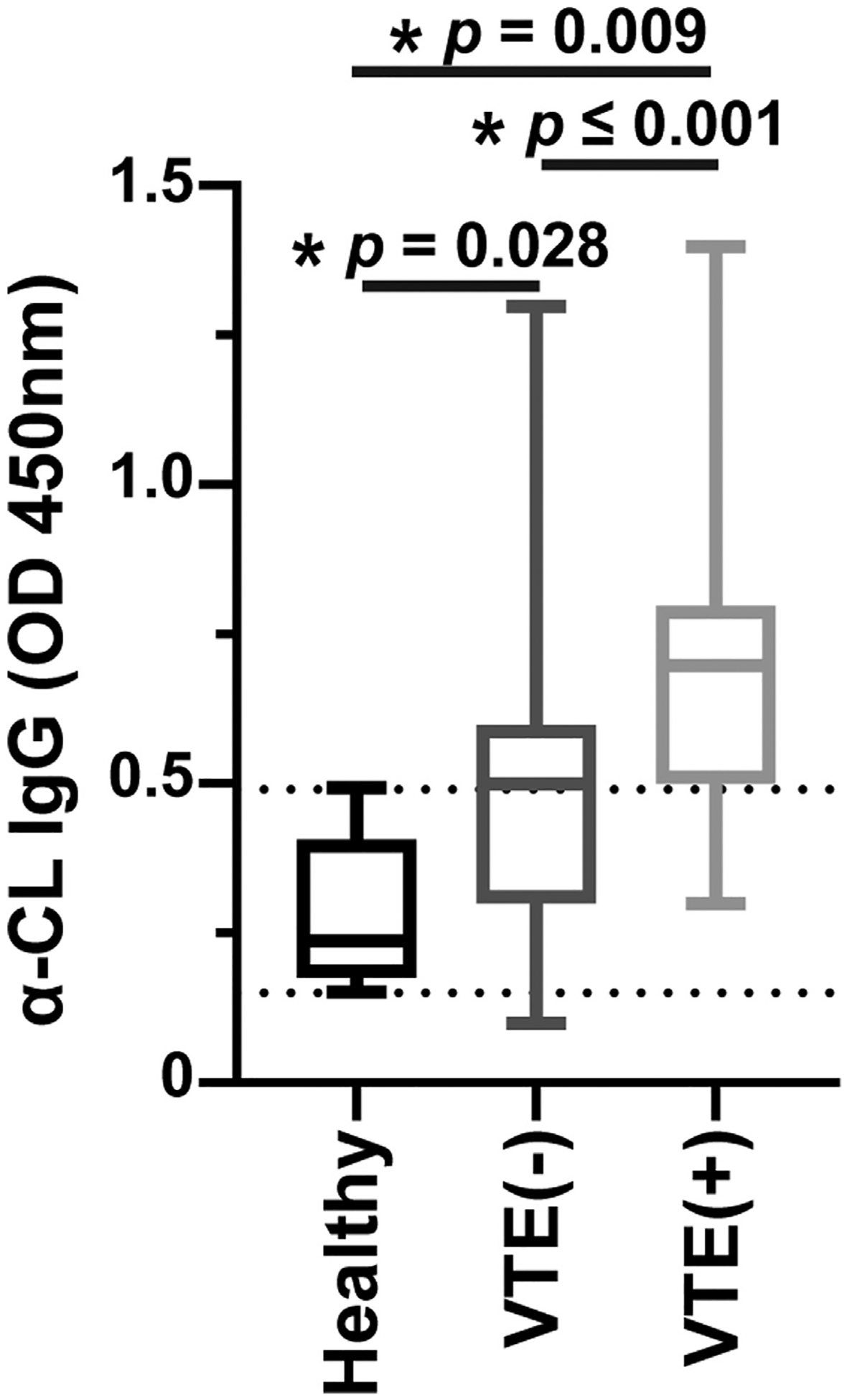
Anticardiolipin immunoglobulin G (IgG) in plasma samples of patients with GBM and control subjects: the concentration of anticardiolipin IgG in plasma samples of 38 patients with GBM and venous thromboembolism (VTE), 37 patients with GBM without VTE, and 10 healthy control subjects were measured using enzyme-linked immunoassay (ELISA) and showed as optical density (OD) units. A 2-tailed Student’s t-test with Welch correction was used to calculate p values. α-CL, anticardiolipin.
4 |. DISCUSSION
Many patients with cancer develop venous thromboembolic events, but prophylactic anticoagulation in the outpatient setting is controversial and recommended only in high-risk settings [22,23]. However, what factors comprise the high-risk setting is not clear. Advanced age, medical comorbidities (eg, diabetes mellitus and obesity) [24], tumor histology, cancer stage (higher stages have higher risk), anemia, thrombocytosis, and leukocytosis [25,26] have been reported as predictive risk factors for cancer-associated VTE. A combination of some of these clinical and laboratory factors is included in scoring models [27,28] to guide physicians in managing the risk of VTE in patients with GBM. However, because the molecular pathogenesis of cancer-associated VTE is poorly understood, available predictive models do not utilize cancer-specific risk factors. Consequently, current scoring systems are not universally accepted and have not provided enough evidence to establish a standard of care for outpatient thromboprophylaxis in patients with cancer.
Several risk factors for VTE in GBM have been described, including tumor size, surgical interventions (biopsy or subtotal surgical resection), immobilization, and antivascular endothelial growth factor therapy. More recently, tumor-related risk factors, such as isocitrate dehydrogenase 1 wild-type status [29], increased podoplanin expression [2,30,31], and intratumor thrombosis [32,33], were correlated to a higher risk of venous thrombosis. Procoagulant EVs expressing tissue factor (TF) released from GBM cells may contribute to local or systemic hypercoagulopathy [34]. Ectopic expression of TF by cancer cells, its dissemination by cancer cell–derived EVs, and activation of the coagulation cascade is a reasonable hypothesis explaining the higher rate of VTE in cancer [35,36]. We have also shown a role for TF-expressing EVs released by cancer cells in venous thrombosis in ovarian cancer [37]. However, despite the expression of TF by various types of cancer, a clear link among the expression of TF in brain tumors [38], the presence of TF-bearing EVs, and the frequency of VTE has not been established in cancer-induced thrombosis [39].
Cell injury or death releases intracellular molecules known as DAMPs that activate innate immunity and promote inflammation. The mitochondrion is a source of DAMPs, and mitochondria-derived DNA, peptides, and lipids might have a role in various inflammatory conditions [12]. We have found that not only mitochondrial content but also intact or partially damaged mitochondria are released from injured cells and cancer cells into extracellular fluid and blood (either as free or EV-embedded mitochondria). We found that these extracellular circulating mitochondria promote thrombosis. Normal cells at baseline and under stress, apoptotic or necrotic cells, and cancer cells release intact mitochondria [10,11,40]. Mitochondria are detected in plasma samples of healthy individuals and patients with cancer [18,41,42]. In healthy subjects, mitochondria in circulation are originated from endothelial cells, platelets, and neutrophils. In this study, we showed that GBM cells also release mitochondria. Numerous studies have shown cell-free mitochondrial DNA as a stress marker and an inducer of inflammation [11,14,43]. However, recent studies demonstrated that most cell-free mitochondrial DNA (90% of it) is, in fact, within circulating mitochondria [18,41]. Cell-free circulating mitochondria are respiratory competent and have active metabolism [18]. We speculate that most consequences attributed to cell-free mitochondria DNA are the results of free mitochondria in blood. The role of free-circulating mitochondria in VTE in cancer is comparable to their role in coagulopathy after traumatic brain injury. Traumatic brain injury is associated with systemic coagulopathy induced by the release of brain-derived procoagulant EVs. A critical component of these EVs are mitochondria originating from damaged brain tissue and circulating in the blood as intact or fragmented mitochondria surrounded by a lipid bilayer membrane or as free mitochondria [9,13]. Extracellular mitochondria are metabolically active, produce reactive oxygen species activating platelets and endothelial cells, and propagate systemic inflammation. We investigated whether a similar mechanism is involved in venous thrombosis in GBM. Extracellular mitochondria promote coagulation and thrombosis through various pathways. Extracellular mitochondria activate endothelial cells and platelets in the traumatic brain injury model in mice [13,44]. Additionally, cardiolipin, an anionic phospholipid transported to the outer membrane from the inner membrane in circulating mitochondria, is released from injured cells [13,44]. Cardiolipin acts like phosphatidylserine (another procoagulant anionic phospholipid) and forms a platform for the assembly of the coagulation cascade [13].
We found that intact or partially damaged mitochondria are released from cancer cells into media. We compared the concentration of mitochondria in plasma samples of patients with GBM and found that these patients had a higher number of mitochondria in their plasma than healthy subjects. Furthermore, mitochondria were significantly higher in patients with GBM and VTE than in those without VTE. Although this difference was not large, a high number of circulating cell-free mitochondria in patients with cancer may be an additional risk factor to consider in clinical management. We could not determine if the circulating mitochondria are intact or fragmented. Our goal was to identify a practical biomarker that can be used to determine VTE risk in GBM. Using mitochondrial DNA is a relatively easy way to estimate the number of mitochondria. A limitation of our study was that we did not have serial blood collections from patients and could not monitor the change in the number of circulating mitochondria over time. We examined the impact of free mitochondria on venous thrombosis in a murine model of IVC stenosis [19]. We monitored the development of IVC thrombosis in mice by real-time angiography and avoided the inaccuracy of postmortem examination of IVC to detect blood clots. Mitochondria injection into tail veins induced thrombosis in IVC more frequently, larger in size, and heavier than the control PBS buffer. Our studies did not identify the primary source of cell-free mitochondria in blood and the dominant mechanism responsible for mitochondria-induced venous thrombosis. Mitochondria activate platelets, neutrophils, and endothelial cells. Histologic examination of thrombi showed that mitochondria-induced clots had more platelet and neutrophils. Brain-derived extracellular mitochondria bind platelets and activate them in an oxidant-dependent manner [9]. Circulating mitochondria are metabolically active and continue oxidative phosphorylation, producing reactive oxygen species that activate platelets, endothelial cells, and neutrophils [14,45–48]. Proteins synthesized in mitochondria are tagged with formyl methionine. Formyl methionine is chemotactic for neutrophils, and reactive oxygen species promote the formation of NETs [42,49]. Interestingly, in our mouse model of venous thrombosis, mitochondria-induced blood clots were rich in neutrophils and NETs. The contribution of NETs to cancer thrombosis was suggested [50,51]; however, the mechanism of the generation of NETs in cancer remains unknown.
The source of circulating free mitochondria in the blood of patients with cancer can be blood cells, endothelial cells, cancer cells, or other cellular components of the tumor microenvironment. Extracellular mitochondria originate from glial cells and can be detected in blood, brain, and cerebrospinal fluid [44,52,53]. We could not identify the primary source of cell-free mitochondria in the blood of patients with cancer (cancer cells, stroma, endothelium, or cells remote from the tumor). In the future, we plan to use mutations in the mitochondrial genome of cancer cells to trace circulating mitochondria back to cancer cells.
Because mitochondria exclusively express cardiolipin, we hypothesized that free-circulating mitochondria might induce anticardiolipin antibodies. Anticardiolipin antibodies, particularly the IgG variant, are associated with an increased risk of venous and arterial thrombosis in antiphospholipid antibody syndrome [21,54]. We found that patients with GBM and venous thrombosis had a higher average titer of anticardiolipin IgG in blood plasma than patients with GBM and without venous thrombi. The presence of antiphospholipid antibodies in patients with cancer and the associated risk of venous thrombosis have been reported [55]. Future studies should examine whether this association has a predictive value; in other words, whether patients with GBM with high titers of anticardiolipin antibodies are at an increased risk of venous thrombosis. In our study, the number of circulating mitochondria and anticardiolipin IgG levels were independently associated with VTE. We examined the correlation between the number of mitochondria and the titer anticardiolipin IgG in our cohort and did not find a correlation and concluded that anticardiolipin antibodies could not be used as a surrogate marker for circulating cell-free mitochondria. In addition to the limited number of samples, other possible explanations for the lack of correlation could be that either mitochondrial cardiolipin has no role in generating anticardiolipin antibodies or other systemic factors (such as inflammation or inflammation-associated immune suppression) determine antibody production to cardiolipin. Larger studies are needed to examine the association between circulating mitochondria and anticardiolipin antibodies in venous thrombosis and other disorders.
Essentials.
Venous thrombosis is a common complication in patients with glioblastoma multiforme (GBM).
We examined the impact of cell-free mitochondria on venous thrombosis.
Cell-free circulating mitochondria were associated with venous thrombosis in patients with GBM and induced venous thrombosis in mice with inferior vena cava stenosis.
Anticardiolipin immunoglobulin G is elevated in the plasma of patients with GBM with venous thrombosis.
ACKNOWLEDGMENTS
This study was supported, in part, by the National Institutes of Health grant CA231141 to V.A.-K. and O.G., the National Institutes of Health grant CA177909 to V.A.-K., the Liz Tilberis Early Career Development Award from the Ovarian Cancer Research Alliance, Ovarian Cancer Research in honor of Liza Chance from the Foundation of Women Cancer, and the National Institutes of Health grant P50 CA217685 (to M.S.C.). We acknowledge the Brain Tumor Center and the Neuropathology Satellite Tumor Bank of the University of Texas MD Anderson Cancer Center for providing the plasma specimens. In addition, the authors gratefully acknowledge the assistance of Katherine Dixon, Malea Williams, and Crystal Dupuis for the in vivo inferior vena cava stenosis model generation and angiography experiments, Cancer Center Support Grant (CCSG) NIH P30CA016672, and High-Resolution Electron Microscopy Facility for transmission electron microscopy studies.
Footnotes
DECLARATION OF COMPETING INTERESTS
There are no competing interests to disclose.
REFERENCES
- [1].Riedl J, Ay C. Venous thromboembolism in brain tumors: risk factors, molecular mechanisms, and clinical challenges. Semin Thromb Hemost. 2019;45:334–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tawil N, Bassawon R, Meehan B, Nehme A, Montermini L, Gayden T, De Jay N, Spinelli C, Chennakrishnaiah S, Choi D, Adnani L, Zeinieh M, Jabado N, Kleinman CL, Witcher M, Riazalhosseini Y, Key NS, Schiff D, Grover SP, Mackman N, et al. Glioblastoma cell populations with distinct oncogenic programs release podoplanin as procoagulant extracellular vesicles. Blood Adv. 2021;5:1682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Muster V, Gary T. Incidence, therapy, and bleeding risk-cancer-associated thrombosis in patients with glioblastoma. Cancers (Basel). 2020;12:1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Spinelli C, Tawil N, Adnani L, Rak J, Choi D. Extracellular vesicle mediated vascular pathology in glioblastoma. Subcell Biochem. 2021;97:247–73. [DOI] [PubMed] [Google Scholar]
- [5].Semrad TJ, O’Donnell R, Wun T, Chew H, Harvey D, Zhou H, White RH. Epidemiology of venous thromboembolism in 9489 patients with malignant glioma. J Neurosurg. 2007;106:601–8. [DOI] [PubMed] [Google Scholar]
- [6].Perry JR, Julian JA, Laperriere NJ, Geerts W, Agnelli G, Rogers LR, Malkin MG, Sawaya R, Baker R, Falanga A, Parpia S, Finch T, Levine MN. PRODIGE: a randomized placebo-controlled trial of dalteparin low-molecular-weight heparin thromboprophylaxis in patients with newly diagnosed malignant glioma. J Thromb Haemost. 2010;8:1959–65. [DOI] [PubMed] [Google Scholar]
- [7].Jo JT, Schiff D, Perry JR. Thrombosis in brain tumors. Semin Thromb Hemost. 2014;40:325–31. [DOI] [PubMed] [Google Scholar]
- [8].Kaptein FHJ, Stals MAM, Kapteijn MY, Cannegieter SC, Dirven L, van Duinen SG, van Eijk R, Huisman MV, Klaase EE, Taphoorn MJB, Versteeg HH, Buijs JT, Koekkoek JAF, Klok FA. Incidence and determinants of thrombotic and bleeding complications in patients with glioblastoma. J Thromb Haemost. 2022;20:1665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhao Z, Zhou Y, Hilton T, Li F, Han C, Liu L, Yuan H, Li Y, Xu X, Wu X, Zhang F, Thiagarajan P, Cap A, Shi FD, Zhang J, Dong JF. Extracellular mitochondria released from traumatized brains induced platelet procoagulant activity. Haematologica. 2020;105:209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Maeda A, Fadeel B. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. 2014;5: e1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Trumpff C, Michelson J, Lagranha CJ, Taleon V, Karan KR, Sturm G, Lindqvist D, Fernström J, Moser D, Kaufman BA, Picard M. Stress and circulating cell-free mitochondrial DNA: a systematic review of human studies, physiological considerations, and technical recommendations. Mitochondrion. 2021;59:225–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, Vandenabeele P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011;32:157–64. [DOI] [PubMed] [Google Scholar]
- [13].Zhao Z, Wang M, Tian Y, Hilton T, Salsbery B, Zhou EZ, Wu X, Thiagarajan P, Boilard E, Li M, Zhang J, Dong JF. Cardiolipin-mediated procoagulant activity of mitochondria contributes to traumatic brain injury-associated coagulopathy in mice. Blood. 2016;127: 2763–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, Paré A, Rousseau M, Naika GS, Lévesque T, Laflamme C, Marcoux G, Lambeau G, Farndale RW, Pouliot M, Hamzeh-Cognasse H, Cognasse F, Garraud O, Nigrovic PA, Guderley H, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. 2014;124:2173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tian Y, Salsbery B, Wang M, Yuan H, Yang J, Zhao Z, Wu X, Zhang Y, Konkle BA, Thiagarajan P, Li M, Zhang J, Dong JF. Brain-derived microparticles induce systemic coagulation in a murine model of traumatic brain injury. Blood. 2015;125:2151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Miller FJ, Rosenfeldt FL, Zhang C, Linnane AW, Nagley P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copy number with age. Nucleic Acids Res. 2003;31:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sonenshein GE, Holt CE. Molecular weight of mitochondrial DNA in Physarum polycephalum. Biochem Biophys Res Commun. 1968;33: 361–7. [DOI] [PubMed] [Google Scholar]
- [18].Al Amir Dache Z, Otandault A, Tanos R, Pastor B, Meddeb R, Sanchez C, Arena G, Lasorsa L, Bennett A, Grange T, El Messaoudi S, Mazard T, Prevostel C, Thierry AR. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 2020;34: 3616–30. [DOI] [PubMed] [Google Scholar]
- [19].Sasano T, Gonzalez-Delgado R, Muñoz NM, Carlos-Alcade W, Cho MS, Sheth RA, Sood AK, Afshar-Kharghan V. Podoplanin promotes tumor growth, platelet aggregation, and venous thrombosis in murine models of ovarian cancer. J Thromb Haemost. 2022;20: 104–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25: 1463–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kelchtermans H, Pelkmans L, de Laat B, Devreese KM. IgG/IgM antiphospholipid antibodies present in the classification criteria for the antiphospholipid syndrome: a critical review of their association with thrombosis. J Thromb Haemost. 2016;14: 1530–48. [DOI] [PubMed] [Google Scholar]
- [22].Key NS, Khorana AA, Kuderer NM, Bohlke K, Lee AYY, Arcelus JI, Wong SL, Balaban EP, Flowers CR, Francis CW, Gates LE, Kakkar AK, Levine MN, Liebman HA, Tempero MA, Lyman GH, Falanga A. Venous thromboembolism prophylaxis and treatment in patients with cancer: ASCO clinical practice guideline update. J Clin Oncol. 2020;38:496–520. [DOI] [PubMed] [Google Scholar]
- [23].Lyman GH, Carrier M, Ay C, Di Nisio M, Hicks LK, Khorana AA, Leavitt AD, Lee AYY, Macbeth F, Morgan RL, Noble S, Sexton EA, Stenehjem D, Wiercioch W, Kahale LA, Alonso-Coello P. American Society of Hematology 2021 guidelines for management of venous thromboembolism: prevention and treatment in patients with cancer. Blood Adv. 2021;5:927–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer-associated venous thrombosis. Blood. 2013;122:1712–23. [DOI] [PubMed] [Google Scholar]
- [25].Khorana AA, Francis CW, Culakova E, Lyman GH. Risk factors for chemotherapy-associated venous thromboembolism in a prospective observational study. Cancer. 2005;104:2822–9. [DOI] [PubMed] [Google Scholar]
- [26].Connolly GC, Khorana AA, Kuderer NM, Culakova E, Francis CW, Lyman GH. Leukocytosis, thrombosis and early mortality in cancer patients initiating chemotherapy. Thromb Res. 2010;126:113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood. 2008;111:4902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Agnelli G, Gussoni G, Bianchini C, Verso M, Mandalà M, Cavanna L, Barni S, Labianca R, Buzzi F, Scambia G, Passalacqua R, Ricci S, Gasparini G, Lorusso V, Bonizzoni E, Tonato M. PROTECHT Investigators. Nadroparin for the prevention of thromboembolic events in ambulatory patients with metastatic or locally advanced solid cancer receiving chemotherapy: a randomised, placebo-controlled, double-blind study. Lancet Oncol. 2009;10:943–9. [DOI] [PubMed] [Google Scholar]
- [29].Unruh D, Schwarze SR, Khoury L, Thomas C, Wu M, Chen L, Chen R, Liu Y, Schwartz MA, Amidei C, Kumthekar P, Benjamin CG, Song K, Dawson C, Rispoli JM, Fatterpekar G, Golfinos JG, Kondziolka D, Karajannis M, Pacione D, et al. Mutant IDH1 and thrombosis in gliomas. Acta Neuropathol. 2016;132:917–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Riedl J, Preusser M, Nazari PM, Posch F, Panzer S, Marosi C, Birner P, Thaler J, Brostjan C, Lötsch D, Berger W, Hainfellner JA, Pabinger I, Ay C. Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood. 2017;129:1831–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tawil N, Chennakrishnaiah S, Bassawon R, Johnson R, D’Asti E, Rak J. Single cell coagulomes as constituents of the oncogene-driven coagulant phenotype in brain tumours. Thromb Res. 2018;164(Suppl 1):S136–42. [DOI] [PubMed] [Google Scholar]
- [32].Rodas RA, Fenstermaker RA, McKeever PE, Blaivas M, Dickinson LD, Papadopoulos SM, Hoff JT, Hopkins LN, Duffy-Fronckowiak M, Greenberg HS. Correlation of intraluminal thrombosis in brain tumor vessels with postoperative thrombotic complications: a preliminary report. J Neurosurg. 1998;89:200–5. [DOI] [PubMed] [Google Scholar]
- [33].Costa B, Eisemann T, Strelau J, Spaan I, Korshunov A, Liu HK, Bugert P, Angel P, Peterziel H. Intratumoral platelet aggregate formation in a murine preclinical glioma model depends on podoplanin expression on tumor cells. Blood Adv. 2019;3:1092–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Passamonti SM, Artoni A, Carrabba G, Merati G, Abbattista M, Capecchi M, Castellani M, Marenghi C, Trombetta E, Giammattei L, Caroli M, Bucciarelli P, Scalambrino E, Peyvandi F, Martinelli I. Plasma levels of extracellular vesicles and the risk of post-operative pulmonary embolism in patients with primary brain tumors: a prospective study. J Thromb Thrombolysis. 2021;52:224–31. [DOI] [PubMed] [Google Scholar]
- [35].Kasthuri RS, Taubman MB, Mackman N. Role of tissue factor in cancer. J Clin Oncol. 2009;27:4834–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ruf W Tissue factor and cancer. Thromb Res. 2012;130(Suppl 1): S84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sasano T, Cho MS, Rodriguez-Aguayo C, Bayraktar E, Taki M, Afshar-Kharghan V, Sood AK. Role of tissue-factor bearing extracellular vesicles released from ovarian cancer cells in platelet aggregation in vitro and venous thrombosis in mice. Thrombosis Update. 2021;2:100020. [Google Scholar]
- [38].Thaler J, Preusser M, Ay C, Kaider A, Marosi C, Zielinski C, Pabinger I, Hainfellner JA. Intratumoral tissue factor expression and risk of venous thromboembolism in brain tumor patients. Thromb Res. 2013;131:162–5. [DOI] [PubMed] [Google Scholar]
- [39].Thaler J, Ay C, Mackman N, Bertina RM, Kaider A, Marosi C, Key NS, Barcel DA, Scheithauer W, Kornek G, Zielinski C, Pabinger I. Microparticle-associated tissue factor activity, venous thromboembolism and mortality in pancreatic, gastric, colorectal and brain cancer patients. J Thromb Haemost. 2012;10:1363–70. [DOI] [PubMed] [Google Scholar]
- [40].Zhu M, Barbas AS, Lin L, Scheuermann U, Bishawi M, Brennan TV. Mitochondria released by apoptotic cell death initiate innate immune responses. Immunohorizons. 2018;2:384–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stephens OR, Grant D, Frimel M, Wanner N, Yin M, Willard B, Erzurum SC, Asosingh K. Characterization and origins of cell-free mitochondria in healthy murine and human blood. Mitochondrion. 2020;54:102–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16:1438–44. [DOI] [PubMed] [Google Scholar]
- [43].Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhao Z, Zhou Y, Li M, Zhang J, Dong JF. Extracellular mitochondria in traumatic brain injury induced coagulopathy. Semin Thromb Hemost. 2020;46:167–75. [DOI] [PubMed] [Google Scholar]
- [45].Arthur JF, Gardiner EE, Kenny D, Andrews RK, Berndt MC. Platelet receptor redox regulation. Platelets. 2008;19:1–8. [DOI] [PubMed] [Google Scholar]
- [46].Pearlstein DP, Ali MH, Mungai PT, Hynes KL, Gewertz BL, Schumacker PT. Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia. Arterioscler Thromb Vasc Biol. 2002;22:566–73. [DOI] [PubMed] [Google Scholar]
- [47].Li AE, Ito H, Rovira II, Kim KS, Takeda K, Yu ZY, Ferrans VJ, Finkel T. A role for reactive oxygen species in endothelial cell anoikis. Circ Res. 1999;85:304–10. [DOI] [PubMed] [Google Scholar]
- [48].Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, Washburn NR, Devera ME, Liang X, Tör M, Billiar T. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. [DOI] [PubMed] [Google Scholar]
- [49].Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. 2017;8:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Olsson AK, Cedervall J. NETosis in cancer-platelet-neutrophil crosstalk promotes tumor-associated pathology. Front Immunol. 2016;7:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mauracher LM, Posch F, Martinod K, Grilz E, Däullary T, Hell L, Brostjan C, Zielinski C, Ay C, Wagner DD, Pabinger I, Thaler J. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J Thromb Haemost. 2018;16:508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chou SH, Lan J, Esposito E, Ning M, Balaj L, Ji X, Lo EH, Hayakawa K. Extracellular mitochondria in cerebrospinal fluid and neurological recovery after subarachnoid hemorrhage. Stroke. 2017;48:2231–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schreiber K, Sciascia S, de Groot PG, Devreese K, Jacobsen S, Ruiz-Irastorza G, Salmon JE, Shoenfeld Y, Shovman O, Hunt BJ. Antiphospholipid syndrome. Nat Rev Dis Primers. 2018;4:18005. [DOI] [PubMed] [Google Scholar]
- [55].Reinstein E, Shoenfeld Y. Antiphospholipid syndrome and cancer. Clin Rev Allergy Immunol. 2007;32:184–7. [DOI] [PubMed] [Google Scholar]