

Abstract
The double-stranded RNA (dsRNA)-activated protein kinase (PKR) provides a fundamental control step in the regulation of protein synthesis initiation through phosphorylation of the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2α), a process that prevents polypeptide chain initiation. In such a manner, activated PKR inhibits cell growth and induces apoptosis, whereas disruption of normal PKR signaling results in unregulated cell growth. Therefore, tight control of PKR activity is essential for regulated cell growth. PKR is activated by dsRNA binding to two conserved dsRNA binding domains within its amino terminus. We isolated a ribosomal protein L18 by interaction with PKR. L18 is a 22-kDa protein that is overexpressed in colorectal cancer tissue. L18 competed with dsRNA for binding to PKR, reversed dsRNA binding to PKR, and did not directly bind dsRNA. Mutation of K64E within the first dsRNA binding domain of PKR destroyed both dsRNA binding and L18 interaction, suggesting that the two interactive sites overlap. L18 inhibited both PKR autophosphorylation and PKR-mediated phosphorylation of eIF-2α in vitro. Overexpression of L18 by transient DNA transfection reduced eIF-2α phosphorylation and stimulated translation of a reporter gene in vivo. These results demonstrate that L18 is a novel regulator of PKR activity, and we propose that L18 prevents PKR activation by dsRNA while PKR is associated with the ribosome. Overexpression of L18 may promote protein synthesis and cell growth in certain cancerous tissue through inhibition of PKR activity.
Protein synthesis is an important regulatory step in gene expression. Most translational control occurs at the level of polypeptide chain initiation, the rate-limiting step in protein synthesis. Cells respond to rapid changes in their environment by reversible covalent modification of the translational apparatus. Many physiological conditions that inhibit initiation of protein synthesis result in a decrease in the activity of eukaryotic translation initiation factor 2 (eIF-2) through phosphorylation of its alpha subunit, eIF-2α (27). eIF-2 is a heterotrimer that is essential for transferring initiator methionyl tRNA in a ternary complex with GTP to the 40S ribosomal subunit in the first step of polypeptide chain initiation. Upon 60S ribosomal subunit joining, GTP is hydrolyzed and the GDP–eIF-2 complex requires GTP exchange mediated by eIF-2B in order to promote another round of initiation. When eIF-2 is phosphorylated on serine 51 of the alpha subunit, it cannot undergo GDP/GTP exchange and forms a nondissociable complex between eIF-2B and eIF-2–GDP. Since intracellular levels of eIF-2B are approximately 10-fold lower than eIF-2, eIF-2B becomes sequestered by small increases in the level of eIF-2α phosphorylation and prevents polypeptide chain initiation events (62).
Three protein kinases that specifically phosphorylate eIF-2α at serine 51 have been identified and cloned: the heme-regulated inhibitor from rabbit reticulocytes, the GCN2 kinase in yeast and higher vertebrates, and the double-stranded RNA (dsRNA)-activated kinase PKR (73). PKR is a serine-threonine protein kinase ubiquitously expressed in mammalian cells that is associated with the ribosome and can be released with a high salt concentration (46, 51, 74). Immunofluorescence studies demonstrated that PKR is localized to the endoplasmic reticulum and the nucleolus (32, 33, 76). PKR was first identified as a component of the host defense mechanism induced at the transcriptional level by type 1 interferons (alpha/beta interferon [IFN-α/β]) (28, 72). Evidence has accumulated that PKR plays a critical role in growth control (13, 41, 59), dsRNA-dependent transcriptional regulation (53, 82, 89, 92), regulation of differentiation (65, 66), suppression of cell transformation (41, 59), and induction of apoptosis (47, 78, 90).
PKR contains two conserved dsRNA binding motifs in its amino terminus and a kinase domain in its carboxyl terminus. The dsRNA binding motifs are comprised of a stretch of approximately 65 amino acids that are present in at least 27 different proteins (39). PKR is synthesized in a latent form that requires activation by dsRNA (6, 12, 23, 55, 70, 75, 78, 81, 84, 85). The activation curve for dsRNA is bimodal: low concentrations of dsRNA activate and high concentrations of dsRNA actually inhibit PKR activation (29, 42). Although the amino-terminal dsRNA binding motif is more important than the carboxy-terminal dsRNA binding motif, the two dsRNA binding motifs together are required for high-affinity binding to dsRNA (6, 23, 55, 70). dsRNA viral genomes (e.g., reovirus) and viral mRNA transcripts with precise secondary stem loop structures resembling dsRNA are potent activators of PKR. Short RNA duplexes of 16 bp are capable of binding PKR; however, the binding efficiency and activation increases with length up to 85 bp (6, 52, 75). Further increases in size do not affect binding or activation. dsRNA binding to PKR induces dimerization with subsequent trans-autophosphorylation with activation of the eIF-2α kinase activity (57, 64, 81, 84, 85). eIF-2α is the best-characterized PKR substrate. In addition to eIF-2α phosphorylation, data support that activated PKR leads to activation of NFκB and IRF-1, transcription factors for proinflammatory and IFN-responsive genes, respectively (43, 44).
PKR provides a fundamental role in the IFN antiviral response. Although it has long been established that dsRNA is cytotoxic to cell cultures in vitro (50), it was only recently demonstrated that PKR is a negative regulator of cell growth and mediates cytotoxicity in response to dsRNA (18, 40, 47, 48, 79). Forced expression of wild-type human PKR suppresses growth in yeast (13) and induces apoptosis in mammalian cells (47, 78, 90). Further evidence to support a growth suppression activity for PKR is the observation that expression of a catalytically inactive mutant of PKR that acts as a trans-dominant negative to inhibit endogenous PKR produces a transformed phenotype in NIH 3T3 cells (3, 41) and deregulates growth in HeLa cells (59). Cells expressing the mutant PKR are oncogenic and form tumors when injected into nude mice. The mechanism by which PKR inhibits cell growth may require phosphorylation of eIF-2α, since expression of an S51A mutant eIF-2α, to inhibit PKR-mediated phosphorylation, prevents stress-induced apoptosis and also yields a transforming phenotype in NIH 3T3 cells (21, 78). In addition, expression of a S51D mutant eIF-2α is sufficient to induce apoptosis in mammalian cells (78). These studies provide compelling evidence for the antiproliferative effect of PKR activation and the pivotal role of eIF-2α phosphorylation in growth inhibition. As a consequence, cells and viruses have evolved numerous mechanisms to downregulate PKR activity. In this respect several cellular and viral inhibitors of PKR have been identified, although few of them are well characterized for their physiological importance in vivo (31, 35, 61, 68, 71).
Here we report the identification and characterization of a novel inhibitor of PKR, the 60S ribosomal protein L18. L18 is a 22-kDa protein that is overexpressed in human colorectal cancer tissue compared to normal colon tissue (4, 69). We demonstrate that L18 binds PKR to prevent its activation by dsRNA and thereby inhibits PKR-dependent eIF-2α phosphorylation in vitro. We also show that L18 inhibits phosphorylation of eIF-2α and stimulates translation initiation in vivo.
MATERIALS AND METHODS
Derivation of expression vectors.
Glutathione S-transferase (GST)-tagged human wild-type (GST-PKR) and K296P mutant (GST-PKR-K296P) PKR were constructed by PCR amplification with PKR-specific primers (5′-CGCGGATCCATGGCTGGTGATCTTTCAGC-3′ and 3′-CTTGCTGTGTGTACACTA-5′). Human L18 cDNA was isolated from a human HeLa cell cDNA library by PCR amplification with primers derived from the published sequence (5′-CGCCTGCAGGCCACCATGGGAGTGGACATCCGCCATAACAAG-3′ and 3′-CGGTCGGCTCCGATGTTTTTGATTCTTAAGCGC-5′). These primers introduce an ATG codon within a good context for initiation. The human L18 expression vector was constructed by cloning the L18 cDNA into the EcoRI and PstI restriction endonuclease sites of the expression vector pMTVA- (38). GST-L18 was made by cloning the L18 cDNA into the BamHI and EcoRI restriction endonuclease sites of pGEX2T (Pharmacia Biotech, Inc., Piscataway, N.J.). The expression vectors encoding adenosine deaminase (ADA; p9A) and eIF-2α (pD61eIF-2α) were described previously (37, 38). PKR expression vectors encoding wild-type, K296P mutant, K64E mutant, dsRNA binding domain (RBD; amino acid residues 1 to 243), first dsRNA binding domain (D1; amino acid residues 1 to 99), K64E mutant D1, and the second dsRNA binding domain (D2; amino acid residues 100 to 243) cloned into pETFVA- were as previously described (84).
Cell culture, DNA transfection, and preparation of cell extracts.
COS-1 cells were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum and 100 U of penicillin and 100 μg of streptomycin per ml. COS-1 cells were transfected by the calcium phosphate precipitation method as described previously (2). Where indicated, cells were cotransfected with pPUR (Clontech), where the amount of pPUR DNA represented 20% of the total DNA in the transfection cocktail and where cells were selected by propagation in 10 μg of puromycin per ml. COS-1 cell extracts were prepared in Nonidet P-40 [NP-40] lysis buffer (50 mM Tris-HCl [pH 7.4], 75 mM NaCl, 0.1% NP-40) with protease inhibitor cocktail mix (Boehringer Mannheim, Indianapolis, Ind.) as described elsewhere (37). Where indicated, COS-1 cells were labeled at 60 h posttransfection with [35S]methionine and [35S]cysteine (100 μCi/ml; 1,000 Ci/mmol; Amersham Corp., Arlington Heights, Ill.) in methionine- and cysteine-free medium for 20 min, and then cell extracts were prepared. Extracts were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and gels were fixed in 30% methanol and 10% acetic acid, treated with En3Hance (New England Nuclear Corp., Boston, Mass.), and prepared for autoradiography.
GST fusion proteins.
GST fusion proteins were made by using the protocol described earlier (24). GST fusion proteins were expressed in Escherichia coli DH5α and inoculated into Luria-Bertani broth containing 100 μg of ampicillin per ml. When the cultures reached an optical density at 600 nm of 0.6 to 0.9, isopropylβ-d-thiogalactopyranoside (IPTG) was added to a final concentration of 0.4 mM, and the cultures were incubated for 3 h at 30°C. Bacteria were then centrifuged and washed in phosphate-buffered saline. The pellet was finally resuspended in lysis buffer (20 mM Tris-HCl [pH 7.4], 1 M NaCl, 1 mM EDTA, 10% glycerol) containing protease inhibitor cocktail, sonicated at 4°C, and centrifuged for 30 min at 15,000 rpm. The supernatant was collected and incubated with washed glutathione-Sepharose 4B beads for 1 h at 4°C with gentle shaking. The beads were then washed three times with buffer A containing 1 M NaCl, followed by washing in buffer A containing 0.1 M NaCl. The protein-bound beads were finally resuspended in buffer A containing 0.1 M NaCl and 0.01% sodium azide and then stored at 4°C until further use. Protein concentrations were determined by a protein assay kit (Bio-Rad Laboratories, Hercules, Calif.).
Affinity chromatography, protein binding assays, and identification of novel proteins interacting with PKR.
Wild-type and mutant K296P PKR-GST fusion proteins bound to glutathione-Sepharose 4B beads or beads containing only GST were packed in a 10-ml Bio-Rad column and preequilibrated with affinity chromatography buffer (ACB; 10 mM HEPES [pH 7.9], 0.1 M NaCl, 0.1 mM EDTA, 10% glycerol, 0.05% NP-40, 1 mg of bovine serum albumin (BSA) per ml 1 mM phenylmethylsulfonyl fluoride, and 1 mM dithiothreitol). COS-1 cell extract was loaded onto the column. The beads were washed with 15 to 20 column volumes of ACB without BSA. The bound proteins were then eluted with ACB containing 1 M NaCl without NP-40 and BSA. The eluted proteins were subjected to SDS-PAGE, transferred onto polyvinylidene difluoride (PVDF) membranes, and stained with Coomassie blue. The band migrating at 22 kDa was excised and subjected to microsequencing at the University of Michigan Protein Core Facility, Ann Arbor, Mich. The sequence of the 15 amino terminal residues was analyzed by BLAST search. Experiments to detect PKR and L18 interaction used the same protocol, except that in addition to COS-1 cell extract, recombinant PKR proteins were also used in the assay. The bound proteins were eluted and subjected to Western blotting with anti-PKR polyclonal antibody (kindly provided by Bryan R. Williams).
Western blotting.
Western immunoblotting was performed with the ECL chemiluminescence kit (Amersham Corp.). Equal amounts of protein were separated by SDS-PAGE and transferred onto nitrocellulose membranes. The nitrocellulose membranes were blocked in TBST (Tris-buffered saline with Tween-20) with 5% nonfat milk, incubated with either anti-PKR primary antibody (provided by Bryan R. Williams), anti-eIF-2α-phosphate antibody (provided by Gary Krause [17]), or anti-eIF-2α monoclonal antibody (provided by C. Henshaw) for 1 to 2 h at room temperature, followed by three washes in TBST. Afterwards the membranes were incubated in TBST containing secondary antibody conjugated to horseradish peroxidase for 45 min, followed by four washes in TBST. Finally, the membranes were incubated in developing solutions (Amersham Corp.) for 1 min and then exposed to film for different time periods to obtain the desired intensity. Band intensities were quantified with the National Institutes of Health Image 1.5b program.
Northern blotting.
Total RNA samples were prepared with Trizol reagent (Gibco-BRL, Gaithersburg, Md.), electrophoresed on formaldehyde-formamide agarose gels, and transferred onto nylon membranes (19). Hybridizations were performed by using a dihydrofolate reductase (DHFR) cDNA probe generated by random priming with [32P]dCTP (>3,000 mCi/mM; Amersham Corp.) and oligonucleotides as described by the supplier (Pharmacia Biotech). Band intensities were quantified with the National Institutes of Health Image 1.5b program.
Kinase assays.
PKR kinase assays were performed by using the protocol described elsewhere (58). Purified human PKR (provided by Michael B. Matthews), purified recombinant eIF-2α (provided by J. W. B. Hershey), GST L18, and GST were incubated with poly(I)-poly(C) (1 μg/ml; Pharmacia Biotech) in the presence of [γ-32P]ATP (Amersham Corp.) in a reaction for 30 min at 30°C. The concentration of poly(I)-poly(C) was optimized to yield maximal activation. The reaction was terminated by adding SDS-PAGE sample buffer, and the products were subjected to SDS-PAGE and autoradiography.
Poly(I)-poly(C) binding assay.
Protein adsorption to poly(I)-poly(C) agarose was performed as described earlier (57). Poly(I)-poly(C) agarose beads were washed three times in binding buffer (20 mM HEPES, pH 7.5; 300 mM NaCl; 5 mM magnesium acetate; 1 mM dithiothreitol; 10% glycerol; 0.5% NP-40) and then incubated with buffer alone, GST, GST-L18, or GST-RNA binding domain of PKR at 30°C for 2 h. The beads were washed four times with binding buffer and then analyzed by SDS-PAGE.
RESULTS
PKR interacts with ribosomal protein L18.
In order to identify novel PKR interacting proteins, affinity chromatography was performed by using GST fusion proteins of wild-type PKR, K296P catalytically inactive mutant PKR, and control GST that were adsorbed to glutathione-Sepharose beads. Proteins in COS-1 cell extract were bound, eluted, subjected to SDS-PAGE, and stained with Coomassie blue. A protein of 22 kDa strongly bound to wild-type GST-PKR, weakly bound to mutant GST-PKR, and did not detectably bind to the control GST (data not shown). The bound protein was transferred onto a PVDF membrane and subjected to microsequencing to obtain the 15 amino terminal residues. The 15 amino terminal residues were entered into a BLAST search (1) that demonstrated identity to a 60S human ribosomal protein, L18.
To confirm the specificity of the interaction between PKR and L18, affinity chromatography with immobilized GST alone or a GST-L18 fusion protein was performed with wild-type and K296P mutant PKR proteins that were obtained by thrombin cleavage of PKR-GST fusion proteins expressed in E. coli. In addition, COS-1 cell extract was directly loaded onto the columns. The columns were washed with 0.1 M NaCl, and the bound proteins were eluted with 1 M NaCl. Eluted proteins were analyzed by SDS-PAGE and immunoblotted with anti-PKR antibody. The results demonstrated a strong interaction between wild-type recombinant PKR and L18 and a slightly weaker interaction between K296P mutant PKR with L18 (Fig. 1, lanes 5 and 6). PKR from COS-1 cell extract was also retained on the GST-L18 column at a significant level (Fig. 1, lane 4). Quantitation of the amount of PKR in the input COS-1 cell extract (Fig. 1, lane 7) to the amount of bound PKR demonstrated that 20% of the input PKR bound GST-L18. None of the proteins were retained on the GST control column (Fig. 1, lanes 1 to 3). These results indicate a specific and direct interaction between PKR and L18.
FIG. 1.

PKR interacts with L18. COS-1 cell extract (1.25 mg), wild-type PKR (20 ng), and K296P PKR (20 ng) were loaded onto glutathione-Sepharose columns bound to either GST or GST-L18 and washed, and then bound proteins were eluted and subjected to SDS-PAGE and Western blot analysis with anti-PKR antibody.
L18 binds to the first dsRNA binding domain in PKR.
To identify the region of PKR that interacts with L18, wild-type, and subdomains of PKR were expressed by transient transfection in COS-1 monkey cells. In addition, a K64E mutant was analyzed that was previously shown to disrupt dsRNA binding within the first dsRNA binding domain (56, 57, 85). The cells were pulse-labeled with [35S]methionine and [35S]cysteine, and protein extracts were prepared. Samples of total cell extract were directly analyzed for expression of the desired polypeptides by SDS-PAGE and autoradiography (Fig. 2A). Whereas synthesis of wild-type PKR (not shown) and K64E mutant PKR (Fig. 2A, lane 3) were not detected because their expression inhibits protein synthesis in the subpopulation of transfected cells, K296P mutant PKR was barely detected migrating just below the 69-kDa marker (Fig. 2A, lane 2). In contrast, the fragments were readily detected as polypeptides migrating at the expected size in the total cell extract that were not observed in mock-transfected cells (Fig. 2A, compare lane 1 to lanes 4 to 7). Radiolabeled cell extracts were then mixed with GST-L18 beads, washed, and resuspended in SDS sample buffer for analysis by SDS-PAGE and autoradiography. The results indicated strong interaction of the dsRNA binding domains 1 and 2 (RBD, residues 1 to 243) and the first dsRNA binding domain (D1, residues 1 to 99) with L18 (Fig. 2B, lanes 11 and 12). K296P mutant PKR also detectably bound to L18 (Fig. 2B, lane 9). In contrast, the K64E mutant D1 fragment and fragment D2 (residues 100 to 243) did not detectably bind L18 (Fig. 2B, lanes 10, 13, and 14). These results identify that the specific interaction between PKR and L18 is mediated through the first dsRNA binding domain in PKR and that the binding site appears to at least partially overlap with determinants required for dsRNA binding.
FIG. 2.

L18 interacts with RBD D1 in PKR. (A) Expression of PKR and subdomains in COS-1 cells. COS-1 cells were transfected with the indicated PKR expression vectors. The transfected cells were metabolically labeled with [35S]methionine and [35S]cysteine and harvested in NP-40 lysis buffer as described in Materials and Methods. The total cell extracts were analyzed by SDS-PAGE and autoradiography. Arrows indicate the proteins expressed from plasmid DNA. The film is overexposed to identify low-molecular-weight proteins. (B) Binding of PKR and subdomains to L18. COS-1 cell extracts from cells transfected with different PKR expression vectors were tested for their ability to interact with GST-L18 as described in Materials and Methods. The arrows identify proteins specifically adsorbed in lanes 9, 11, and 12.
L18 and dsRNA compete for binding to the first dsRNA binding domain of PKR.
Because the L18 interacting domain appeared to overlap with the dsRNA binding domain of PKR, we determined whether dsRNA and L18 compete for binding to PKR. To test whether dsRNA prevents the RBD interaction with L18, extracts were prepared from [35S]methionine- and [35S]cysteine-labeled COS-1 cells that were transiently transfected with the RBD expression construct. Extracts were incubated in the presence or absence of poly(I)-poly(C) prior to affinity chromatography with GST-L18 beads. The beads were washed and analyzed by SDS-PAGE. RBD in the total cell extract bound effectively to GST-L18 (Fig. 3A, lane 1) as previously observed (Fig. 2B). Prior incubation of cell extract with poly(I)-poly(C) (5 and 15 μg/ml) significantly reduced the amount of RBD bound to GST-L18 (Fig. 3A, lanes 2 and 3). We then asked whether dsRNA could displace RBD bound to GST-L18. [35S]methionine- and [35S]cysteine-labeled RBD in COS-1 cell extract was adsorbed to GST-L18, washed, and then eluted with either buffer alone or buffer containing poly(I)-poly(C) (2 μg/ml). Nineteen percent of the RBD bound to GST-L18 was eluted with poly(I)-poly(C) but not with buffer alone, indicating that dsRNA displaced RBD from GST-L18 (Fig. 3A, lanes 4 and 5). We also tested whether L18 could displace RBD bound to poly(I)-poly(C). 35S-labeled RBD from COS-1 cell extract was adsorbed to poly(I)-poly(C) agarose beads, washed three times, and then eluted with either GST or GST-L18 recombinant proteins. The amount of GST and GST-L18 were quantitated by SDS-PAGE and Coomassie staining (Fig. 3B, bottom panel). Approximately 23% of RBD was eluted with GST-L18 but not with GST, suggesting that L18 could displace RBD from poly(I)-poly(C) agarose beads (Fig. 3B, lanes 1 to 3).
FIG. 3.

L18 and dsRNA compete for binding to PKR. (A) dsRNA prevents interaction of RBD with GST-L18 and displaces GST-L18 bound to RBD. [35S]methionine and [35S]cysteine metabolically labeled RBD from COS-1 cell extract was adsorbed onto GST-L18 beads in a protein binding assay as described in Materials and Methods. The 35S-labeled RBD in COS-1 cell extract was incubated with 5 and 15 μg/ml of poly(I)-poly(C) and then incubated with GST-L18 beads for 2 h and washed, and then the beads were resuspended in SDS-PAGE sample buffer and analyzed by SDS-PAGE and autoradiography (Bound). The RBD bound to GST-L18 beads was washed and eluted with either buffer or poly(I)-poly(C) (Eluted). Migration of RBD is indicated by an arrow. (B) L18 displaces poly(I)-poly(C)-bound RBD. 35S-labeled RBD was adsorbed onto poly(I)-poly(C) agarose beads as described in Materials and Methods, and bound RBD was eluted either with GST or with GST-L18. Equal proportionate volumes of samples were analyzed by SDS-PAGE and autoradiography (lane 1, total RBD bound to poly(I)-poly(C); lane 2, GST-eluted RBD; lane 3, GST-L18-eluted RBD). The arrow indicates the migration of RBD. Bacterially expressed GST and GST-L18 proteins used for the elution were prepared as described in Materials and Methods and analyzed by SDS-PAGE and staining with Coomassie blue (bottom). (C) L18 does not bind to dsRNA. GST-RBD, GST-L18, and GST proteins were adsorbed onto poly(I)-poly(C) agarose beads as described in Materials and Methods. After the beads were washed three times, the bound proteins were subjected to Western blot analysis with anti-GST antibody. The proteins bound to poly(I)-poly(C) agarose beads (Bound) and the amount of each protein loaded on the beads (Input) are indicated. The arrows represent the migration of the indicated proteins.
Since the PKR requirements for binding to dsRNA appear similar to those for binding to L18, we tested the hypothesis that dsRNA mediates the interaction between PKR and L18. The ability for L18 to directly bind poly(I)-poly(C) was tested. Poly(I)-poly(C) agarose beads were incubated with recombinant GST-L18 fusion protein, GST alone for control, and GST-RBD (GST fused to the dsRNA binding domain of PKR). The bound proteins were eluted and subjected to SDS-PAGE for Western immunoblot analysis with an anti-GST antibody. The results demonstrated no binding of GST or GST-L18 to poly(I)-poly(C) agarose beads (Fig. 3C, lanes 3 and 4) under conditions where 65% of input GST-RBD was retained on the beads (Fig. 3C, lane 2). These data indicate that L18 does not significantly bind directly to dsRNA, although we cannot rule out that L18 binds some other RNA structure or molecule. Therefore, L18 does not appear to interact with PKR through a dsRNA bridge.
L18 inhibits both PKR autophosphorylation and PKR-mediated eIF-2α phosphorylation.
Since L18 competes with dsRNA for binding to PKR, it seemed feasible that L18 would inhibit PKR activation and phosphorylation of eIF-2α. An in vitro kinase assay was performed with purified PKR and eIF-2α in the presence of increasing concentrations of purified GST-L18 or GST as a control. The purified GST-L18 fusion protein inhibited both PKR autophosphorylation and PKR-mediated eIF-2α phosphorylation in a dose-dependent manner (Fig. 4). Fifty percent inhibition occurred at approximately stoichiometric levels of PKR (0.5 μg/ml) and L18 (approximately 0.6 μg/ml). This would represent approximately three molecules of L18 for each molecule of PKR. Over this concentration range, GST alone had no effect on PKR activity (Fig. 4). These results demonstrate that L18 can inhibit both PKR autophosphorylation and phosphorylation of eIF-2α.
FIG. 4.

L18 inhibits PKR autophosphorylation and eIF-2α phosphorylation. Reactions (50 μl) containing purified PKR (25 ng), 1 μg of poly(I)-poly(C) per ml, and eIF-2α (30 ng) were incubated in the presence of increasing amounts of either GST or GST-L18 in a kinase assay as explained under Materials and Methods. Migration of PKR and eIF-2α are indicated by arrows.
L18 rescues PKR-mediated inhibition of protein synthesis.
Since PKR activation and subsequent eIF-2α phosphorylation correlates with inhibition of translation initiation in vivo, we next asked whether L18 expression can affect translation in response to PKR activation in vivo. To characterize the in vivo significance of the PKR-L18 interaction, the L18 cDNA was cloned into the mammalian cell expression vector, pMTVA−. To detect expression of L18, COS-1 cells were transfected with either pMTVA-L18 or pMTVA− vector in the presence of pPUR, a vector encoding a Streptomyces alboniger protein, puromycin–N-acetyltransferase, that confers resistance to the antibiotic puromycin (45). At 30 h posttransfection, cells were treated with puromycin for 30 h. Under these conditions, approximately 30% of cells transfected with pPUR survived the selection, whereas mock-transfected cells did not survive the puromycin selection. These results indicate a significant selection for a relatively homogeneous population of transfected cells that received plasmid DNA. At 60 h posttransfection, cells were metabolically pulse-labeled with [35S]methionine and [35S]cysteine, and cell extracts were prepared for analysis by SDS-PAGE and autoradiography. L18 was detected as a protein migrating at 22 kDa in COS-1 cells transfected with the expression construct pMTVA-L18 that was absent in extracts prepared from vector-transfected cells (Fig. 5A, lanes 1 and 2). The pMTVA- vector-transfected cells express significant amounts of DHFR from the vector (Fig. 5A, lane 1). Overexpression of L18 did not detectably alter affect the spectrum of translated polypeptides by this analysis on a one-dimensional gel.
FIG. 5.
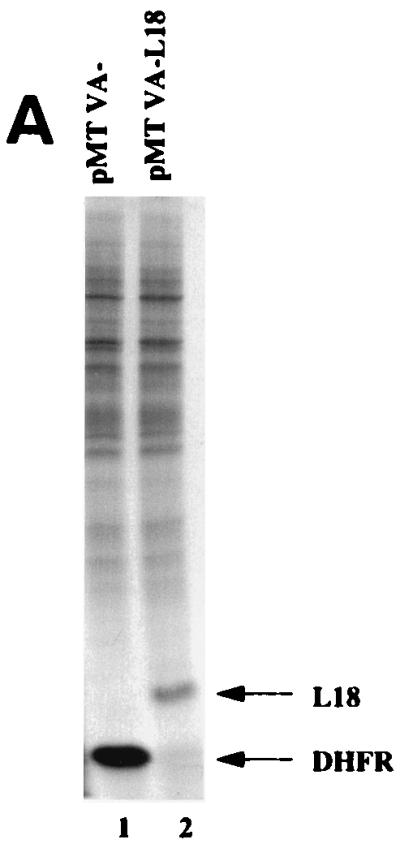


L18 rescues PKR-mediated translational repression and inhibits phosphorylation of eIF-2α in vivo. (A) Expression of L18 in COS-1 cells. COS-1 cells were transfected with vector pMTVA- (lane 1, pMTVA-) or L18 expression construct, pMTVA-L18 (lane 2, pMTVA-L18), in the presence of pPUR. After 30 h the cells were treated with 10 μg of puromycin per ml for an additional 30 h. Cells were then pulse-labeled with [35S]methionine and [35S]cysteine, and extracts were prepared for analysis by SDS-PAGE and autoradiography. (B) Expression plasmids used to study PKR activation. The pETFVA- and pMTVA- expression vectors contain the simian virus 40 (SV40) origin of replication and enhancer element (SV40ori/enh), the adenovirus major late promoter (AdMLP), the adenovirus tripartite leader (TPL), a small intron (IVS), a polycloning site for insertion of PKR or L18, and either the DHFR cDNA (in MTVA−) or encephalomyocarditis internal ribosomal entry site with tissue factor cDNA (TF) (in pETFVA-), and the SV40 early polyadenylation signal. p9A contains the ADA coding sequence and differs from pETFVA- and pMTVA- primarily in vector backbone sequences (pUC18 in pETFVA- and pMTVA-; pBR322 in p9A) that result in the activation of PKR to inhibit translation of p9A-derived mRNA. The structures of the derived mRNAs are depicted below. (C) L18 rescues PKR-mediated translational repression. COS-1 cells were cotransfected with the ADA expression vector p9A in the presence of vector pMTVA-, the L18 expression vector pMTVA-L18, or with the wild-type PKR or K296P PKR pETFVA- expression vectors as indicated. The cells were labeled and harvested with NP-40 lysis buffer as described in Materials and Methods. Total protein synthesis was analyzed by SDS-PAGE and autoradiography. ADA synthesis is indicated by the arrow (top panel). In parallel, RNA was isolated for Northern blot analysis to quantitate the ADA mRNA levels as described in Materials and Methods. Quantitation demonstrated that the levels of ADA mRNA varied by less than 15% between the different lanes.
To study the functional significance of L18 on PKR activity in vivo, we used a transfection system described previously (37). This system exploits the ability of certain plasmid DNAs to activate PKR in transiently transfected cells to selectively inhibit in a cis-acting manner the translation of mRNAs derived from that plasmid (Fig. 5B). This translational repression is dependent on PKR-mediated phosphorylation of eIF-2α (37, 38). The plasmid p9A produces an mRNA encoding adenosine deaminase (ADA) that is poorly translated as a consequence of PKR activation. In the presence of a PKR inhibitor, the mRNA derived from p9A is efficiently translated. The unique feature of this assay system is that when the p9A vector is cotransfected with another vector, pMTVA- or pETFVA-, the mRNA from p9A is inefficiently translated, whereas the mRNAs from pMTVA- or pETFVA- are efficiently translated (Fig. 5B) (36). This system permits characterization of gene products for their ability to inhibit PKR activation because it is possible to synthesize the desired gene product expressed from the pMTVA- or pETFVA- vectors. COS-1 cells were cotransfected with the ADA expression vector p9A and the pMTVA- vector alone or pMTVA- harboring the L18 coding region. In addition, another vector derived from pMTVA-, pETFVA-, was used to demonstrate the effect of wild-type PKR expression and of K296P mutant PKR expression. Cells were pulse-labeled with [35S]methionine and [35S]cysteine at 60 h posttransfection, and total extracts were analyzed for the synthesis of ADA by SDS-PAGE and autoradiography. At the same time, RNA was isolated to measure mRNA levels in the transfected cells. The synthesis of ADA was detected in p9A cells cotransfected with pMTVA- vector alone as a polypeptide migrating just below actin. ADA synthesis was inhibited by cotransfection with the wild-type PKR expression vector. Significantly, ADA synthesis was increased in cells cotransfected with either the K296P mutant PKR expression vector or the L18 expression vector compared to cells cotransfected with the pMTVA- vector alone (Fig. 5C, lane 2 versus lanes 4 and 5). When L18 and wild-type PKR were cotransfected together with p9A, ADA synthesis as well as L18 synthesis were not detected, presumably due to PKR-mediated inhibition of translation prior to accumulation of L18 (data not shown).
To determine whether the increased ADA synthesis was due to increased translation, the ADA mRNA levels in the transfected cells were measured by Northern blot hybridization analysis. The DHFR probe used in this analysis hybridizes to a DHFR sequence present in the 3′-untranslated region of both the p9A vector (ADA mRNA) and the pMTVA- vector (i.e., L18 and DHFR mRNAs) (Fig. 5B). The levels of ADA mRNA were similar, within 15% of each other, in each population of cotransfected cells (Fig. 5C, lower panel). The results demonstrate that L18 stimulates the ADA expression at the translational level to a similar degree as expression of the trans-dominant negative K296P mutant PKR.
L18 inhibits phosphorylation of eIF-2α in vivo.
The effect of L18 overexpression on in vivo phosphorylation of eIF-2α was studied by cotransfecting cells with an eIF-2α expression plasmid alone or in the presence of increasing amounts of the L18 expression vector or the K296P mutant PKR expression vector. In this way it is possible to detect small changes in the level of eIF-2α phosphorylation in the subpopulation of transfected cells that overexpress eIF-2α. Previously, the level of phosphorylation of the overexpressed eIF-2α subunit was shown to correlate with the level of phosphorylation in heterotrimeric eIF-2 (11). Transfected cells were harvested at 60 h posttransfection for analysis by SDS-PAGE and Western immunoblotting by using an antibody that reacts with only phosphorylated eIF-2α (17). Subsequently, the filter was stripped and reprobed with an antibody that reacts with total eIF-2α. Analysis of the amount of total eIF-2α demonstrated that the transfected cells express significant amounts of eIF-2α over the endogenous nondetectable level in mock-transfected cells (Fig. 6, bottom; compare lane 1 with lanes 2 to 7). Cotransfection of either the L18 expression vector or the K296P mutant PKR expression vector did not significantly affect the steady-state level of eIF-2α. However, cotransfection of either L18 or K296P mutant PKR significantly reduced the amount of phosphorylated eIF-2α detected. Quantitation demonstrated a 2.6- and 2.7-fold reduction in the proportion of phosphorylated eIF-2α when corrected for the total amount of eIF-2α in the presence of 15 μg of L18 or K296P mutant PKR expression vectors, respectively. These results demonstrate that either L18 or mutant PKR can inhibit eIF-2α phosphorylation to a similar degree.
FIG. 6.

L18 inhibits phosphorylation of eIF-2α in vivo. COS-1 cells were cotransfected with the pD61 eIF-2α expression vector (7.5 μg) in the presence of a constant amount of DNA (15 μg) composed of control vector pETFVA− and/or L18 or K296P PKR as indicated. The cells were harvested at 60 h posttransfection and analyzed by Western immunoblot analysis with antibody specific for phosphorylated eIF-2α (top). The filter was stripped and reprobed with antibody specific to total eIF-2α (bottom). The relative band intensities of eIF-2α-P to eIF-2α were determined by using the National Institutes of Health image 1.5b program to show that 7.5 μg of L18 and K296P PKR reduced the fraction of phosphorylated eIF-2α to 67 and 38%, respectively, and that 15 μg of L18 and K296P PKR reduced the amount of phosphorylated eIF-2α to 38 and 37%, respectively.
DISCUSSION
The rate of protein synthesis is tightly correlated with the growth state of the cell. Because subtle alterations in the level of eIF-2α phosphorylation have dramatic effects on the rate of polypeptide chain initiation, cells have precise mechanisms to regulate eIF-2α kinases. The dsRNA-activated protein kinase was originally identified as a fundamental pathway for the IFN antiviral response through its transcription induction by type 1 IFNs and its activation by dsRNA synthesized from viral genomes. However, more recent observations suggest that PKR is also activated in response to numerous environmental conditions, such as inducers of the heat shock response, growth factor deprivation, treatment with tumor necrosis factor α, and release of calcium from the endoplasmic reticulum (8, 30, 67, 78). For this reason, there must exist cellular regulators that modulate PKR activity in the absence of a viral infection. To identify potential regulators, we have used affinity chromatography to isolate PKR-interacting proteins. In this study, we describe how the large ribosomal subunit protein L18 inhibits PKR activation by dsRNA. In addition, PKR can reverse dsRNA binding to PKR and may relieve the cell from dsRNA-dependent activation of PKR.
Taken together, the following observations show that L18 is a direct negative regulator of PKR activity. (i) A GST-L18 fusion protein directly bound to PKR. (ii) The L18 binding site on PKR was localized to the first dsRNA binding domain (amino acid residues 1 to 123). (iii) dsRNA and L18 competed for binding to PKR, although L18 did not bind dsRNA directly. (iv) Mutation of K64E within the first dsRNA binding domain of PKR destroyed both dsRNA interaction (56, 57, 85) and L18 interaction. (v) L18 released a portion of the PKR bound to dsRNA. (vi) L18 prevented PKR autophosphorylation and eIF-2α phosphorylation by dsRNA. (vii) Approximately stoichiometric levels of L18 were sufficient to inhibit PKR autophosphorylation. (viii) Finally, overexpression of L18 increased translation of a reporter mRNA and reduced phosphorylation of eIF-2α in transfected cells. The most direct interpretation of these data is that L18 and dsRNA compete for binding to overlapping sites on PKR. From these observations, we propose that L18 prevents also reverses dsRNA-mediated activation of PKR. Jeffrey et al. concluded that there is approximately one molecule of PKR for every five ribosomes before induction by IFN (32). Therefore, under these conditions, PKR would be effectively inhibited by L18. However, after IFN induction, the increased level of PKR would titrate out L18 and would therefore be more susceptible to activation.
PKR was originally characterized as a ribosomal protein that could be washed away with high-salt buffers (46, 51, 74). We have found that L18, a protein from the large ribosomal subunit, specifically binds and inhibits PKR activation. Based on these observations, we were curious that recent localization studies showed that human PKR expressed in yeast cells is associated with the 40S ribosomal subunit through an interaction mediated through the dsRNA binding domain (91). In contrast to these observations in yeast cells, we have recently demonstrated that the PKR expressed in COS-1 monkey cells is associated with the 60S ribosomal subunit and that this interaction is mediated through two independent interactions with the dsRNA binding domain and the kinase domain (86). We believe the discrepancy reflects differences between yeast and mammalian ribosomes in their interaction with PKR. Comparison of the primary sequences of yeast and human L18 proteins indicates significant divergence (58% identity), so it would not be unexpected that the yeast counterpart may not conserve the PKR interacting residues. Overexpression of L18 did not displace PKR from the ribosomes, suggesting that other contacts, possibly mediated by the kinase domain, may be necessary to maintain ribosome association. We propose that the dsRNA binding domain-dependent interaction with the ribosome is mediated by L18. Previous studies suggested that ribosome-associated PKR is a monomer, whereas free PKR is a dimer (46). Activation of PKR correlates with its dimerization. Indeed, recently it was shown that the HIV-1 transactivating region RNA (TAR), an RNA molecule that can activate PKR, promotes dimerization of PKR (9). We propose that L18 is responsible for facilitating ribosome association of PKR and for ensuring that it is in a monomeric inactive state until an insult arises that permits its release from L18, dimerization, and subsequent activation (Fig. 7).
FIG. 7.

PKR inhibitors and their mechanism of action. PKR is depicted as a monomer bound to and inhibited by ribosomal protein L18. Upon accumulation of excess dsRNA, PKR is released from L18 and is susceptible to dsRNA-induced dimerization, autophosphorylation, and activation of the eIF-2α kinase activity. Several viral inhibitors that act through different mechanisms are depicted. Adenovirus VAI RNA binds and inhibits PKR and also displaces it from the ribosome (86). Numerous viral proteins, such as vaccinia virus E3L, bind and sequester dsRNA, thereby reversing the equilibrium from dsRNA activation to L18-mediated inhibition. Vaccinia virus encodes a protein, K3L, that acts as a pseudosubstrate for PKR (77). Protein phosphatase PP1 acts to dephosphorylate phosphorylated eIF-2α as well as phosphorylated PKR. Herpes simplex virus type 1 encodes a protein that facilitates activation of PP1 (26). Finally, two cellular inhibitors, P58IPK and TRBP, prevent activation of PKR by interfering with the activation process through inhibition of PKR dimerization. Other cellular inhibitors of PKR, including Alu RNA (14), La antigen (88), and p67 (87), are not depicted here.
The growth-inhibitory properties of PKR require that the cell tightly regulates its activity. Therefore, cells and viruses have evolved numerous mechanisms to prevent PKR activation that are depicted in Fig. 7. IFN-resistant viruses circumvent PKR activation through multiple mechanisms that include: (i) inhibition of activation by viral encoded RNA inhibitors that compete with binding to and block activation by dsRNA (e.g., adenovirus VAI RNA [54]), (ii) expression of viral proteins that trap or sequester dsRNA molecules capable of activating PKR (e.g., vaccinia virus E3L protein and reovirus sigma 3 protein [10, 22]), (iii) expression of viral proteins that mimic the PKR substrate eIF-2α (e.g., vaccinia virus K3L protein and baculovirus PK2 [16, 20]), (iv) degradation of PKR (e.g., poliovirus [7]), (v) activation of phosphatase activities to dephosphorylate eIF-2α (e.g., herpes simplex virus [25]), and finally (vi) activation of cellular inhibitors of PKR to prevent PKR activation. The best-characterized example of the latter is the P58IPK cellular inhibitor that was originally identified in cells infected with influenza virus (49). P58IPK is a member of the tetratricopeptide family that contains several 34 amino acid repeats thought to effect protein-protein interactions. P58IPK is a member of the tetratricopeptide family containing several 34-amino-acid repeats that are thought to effect protein-protein interactions. In addition, P58IPK also contains a “J domain” of the DnaJ molecular chaperone family. Recently, it was shown that P58IPK can inhibit PKR activation by preventing its dimerization (80). We propose that L18 provides a similar inhibitory role and that multiple inhibitors are required to ensure that PKR activity is tightly regulated. In addition, the different inhibitors may dissociate from PKR by different mechanisms, thereby providing multiple signaling pathways capable of PKR activation.
Given the potential importance of tight regulation of PKR activity in growth control, it is not surprising that numerous, less-well-characterized, cellular inhibitors of PKR have been identified. A dsRNA binding protein, the human immunodeficiency virus TAR RNA binding protein (TRBP), is a dsRNA binding protein that inhibits PKR activation (63). TRBP can bind dsRNA as well as form heterodimers with endogenous PKR, and its overexpression induces a transformed phenotype in NIH 3T3 cells (5, 15). Expression of the transforming Harvey ras oncogene induces a 100-kDa cellular protein inhibitor of PKR in transformed NIH 3T3 cells (60). A 15-kDa protein is produced in association with growth arrest of murine 3T3-F442A cells that are induced to differentiate into adipocytes (34). The La antigen inhibits PKR by sequestering and unwinding dsRNA (88). Finally, a 67-kDa protein in reticulocyte lysate can inhibit phosphorylation of eIF-2α by activated heme-regulated inhibitor kinase and PKR, although the importance of this regulator for PKR activity in vivo is not known (87). Recently, it was also suggested that Alu RNA may serve to inhibit PKR activation in response to a variety of stress conditions (14). Our studies add ribosomal protein L18 to this catalogue of known cellular PKR inhibitors and implicate the importance of tight control of the PKR activation status.
Most evidence supports that PKR acts as a negative growth regulator and that its activation can induce an apoptotic cell death (18, 48, 78). Inactivation of the PKR pathway results in deregulated cell growth (43, 44, 83). For example, expression of a trans-dominant-negative mutant PKR induces a transformed phenotype in NIH 3T3 cells (43, 44). Given the crucial role of PKR in growth control, it is not surprising that overexpression of cellular inhibitors of PKR can deregulate cell growth. For example, overexpression of either P58IPK or TRBP elicits transformation of NIH 3T3 cells (5, 35). Therefore, we expect that overexpression of L18 should also exhibit a similar transforming phenotype in NIH 3T3 cells. In this respect it is intriguing that L18 was originally characterized as a protein that was overexpressed in colorectal cancer tissue and not in normal colorectal tissue (4, 69). Further studies should elucidate whether overexpression of L18 has growth-promoting properties.
ACKNOWLEDGMENT
Portions of this work were supported by NIH grant AI/CA 42394 (R.J.K.).
REFERENCES
- 1.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 2.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: John Wiley and Sons; 1987. [Google Scholar]
- 3.Barber G N, Jagus R, Meurs E F, Hovanessian A G, Katze M G. Molecular mechanisms responsible for malignant transformation by regulatory and catalytic domain variants of the interferon-induced enzyme RNA-dependent protein kinase. J Biol Chem. 1995;270:17423–17428. doi: 10.1074/jbc.270.29.17423. [DOI] [PubMed] [Google Scholar]
- 4.Barnard G F, Staniunas R J, Mori M, Puder M, Jessup M J, Steele G D, Jr, Chen L B. Gastric and hepatocellular carcinomas do not overexpress the same ribosomal protein messenger RNAs as colonic carcinoma. Cancer Res. 1993;53:4048–4052. [PubMed] [Google Scholar]
- 5.Benkirane M, Neuveut C, Chun R F, Smith S M, Samuel C E, Gatignol A, Jeang K T. Oncogenic potential of TAR RNA binding protein TRBP and its regulatory interaction with RNA-dependent protein kinase PKR. EMBO J. 1997;16:611–624. doi: 10.1093/emboj/16.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bevilacqua P C, Cech T R. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry. 1996;35:9983–9994. doi: 10.1021/bi9607259. [DOI] [PubMed] [Google Scholar]
- 7.Black T L, Safer B, Hovanessian A, Katze M. The 68,000 Mr protein kinase is highly autophosphorylated and activated yet significantly degraded during poliovirus infection: implications for translational regulation. J Virol. 1989;63:2244–2251. doi: 10.1128/jvi.63.5.2244-2251.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brostrom C O, Prostko C R, Kaufman R J, Brostrom M A. Inhibition of translational initiation by activators of the glucose-regulated stress protein and heat shock protein stress response systems. Role of the interferon-inducible double-stranded RNA-activated eukaryotic initiation factor 2 alpha kinase. J Biol Chem. 1996;271:24995–25002. doi: 10.1074/jbc.271.40.24995. [DOI] [PubMed] [Google Scholar]
- 9.Carpick B W, Graziano V, Schneider D, Maitra R K, Lee X, Williams B R G. Characterization of the solution complex between the interferon-induced, double-stranded RNA-activated protein kinase and HIV-I trans-activating region RNA. J Biol Chem. 1997;272:9510–9516. doi: 10.1074/jbc.272.14.9510. [DOI] [PubMed] [Google Scholar]
- 10.Chang H-W, Watson J C, Jacobs B L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci USA. 1992;89:4825–4829. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi S Y, Scherer B J, Schnier J, Davies M V, Kaufman R J, Hershey J W B. Stimulation of protein synthesis in COS cells transfected with variants of the a-subunit of initiation factor eIF-2. J Biol Chem. 1992;267:286–293. [PubMed] [Google Scholar]
- 12.Chong K L, Feng L, Schappert K, Meurs E, Donahue T F, Friesen J D, Hovanessian A G. Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. EMBO J. 1992;11:1553–1562. doi: 10.1002/j.1460-2075.1992.tb05200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chong K L, Feng L, Schappert K, Meurs E, Donahue T F, Friesen J D, Hovanessian A G, Williams B R G. Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. EMBO J. 1992;11:1553–1562. doi: 10.1002/j.1460-2075.1992.tb05200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu W M, Ballard R, Carpick B W, Williams B R, Schmid C W. Potential Alu function: regulation of the activity of double-stranded RNA-activated kinase PKR. Mol Cell Biol. 1998;18:58–68. doi: 10.1128/mcb.18.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cosentino G P, Venkatesan S, Serluca F C, Green S R, Mathews M B, Sonenberg N. Double-stranded RNA-dependent protein kinase and TAR RNA-binding protein form homo- and heterodimers in vivo. Proc Natl Acad Sci USA. 1995;92:9445–9449. doi: 10.1073/pnas.92.21.9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies M V, Chang H-W, Jacobs B L, Kaufman R J. The E3L and K3L vaccinia virus gene products stimulate translation through inhibition of the double-stranded RNA-activated protein kinase by different mechanisms. J Virol. 1993;67:1688–1692. doi: 10.1128/jvi.67.3.1688-1692.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeGracia D J, Sullivan J M, Neumar R W, Alousi S S, Hikade K R, Pittman J E, White B C, Rafols J A, Krause G S. Effect of brain ischemia and reperfusion on the localization of phosphorylated eukaryotic initiation factor 2 alpha. J Cereb Blood Flow Metab. 1997;17:1291–1302. doi: 10.1097/00004647-199712000-00004. [DOI] [PubMed] [Google Scholar]
- 18.Der S D, Yang Y L, Weissmann C, Williams B R. A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:3279–3283. doi: 10.1073/pnas.94.7.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derman E, Krauter K, Walling L, Weinberger C, Ray M, Darnell J E., Jr Transcriptional control in the production of liver-specific mRNAs. Cell. 1981;23:731–739. doi: 10.1016/0092-8674(81)90436-0. [DOI] [PubMed] [Google Scholar]
- 20.Dever T E, Sripriya R, McLachlin J R, Lu J, Fabian J R, Kimball S R, Miller L K. Disruption of cellular translational control by a viral truncated eukaryotic translation initiation factor 2 alpha kinase homolog. Proc Natl Acad Sci USA. 1998;95:4164–4169. doi: 10.1073/pnas.95.8.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donze O, Jagus R, Koromilas A E, Hershey J W, Sonenberg N. Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J. 1995;14:3828–3834. doi: 10.1002/j.1460-2075.1995.tb00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giantini M, Shatkin A J. Stimulation of chloramphenicol acetyltransferase mRNA translation by reovirus capsid polypeptide sigma 3 in cotransfected COS cells. J Virol. 1989;63:2415–2421. doi: 10.1128/jvi.63.6.2415-2421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green S R, Manche L, Mathews M B. Two functionally distinct RNA-binding motifs in the regulatory domain of the protein kinase DAI. Mol Cell Biol. 1995;15:358–364. doi: 10.1128/mcb.15.1.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guan K L, Dixon J E. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- 25.He B, Gross M, Roizman B. The γ34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA. 1997;94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He B, Gross M, Roizman B. The γ34.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J Biol Chem. 1998;273:20737–20743. doi: 10.1074/jbc.273.33.20737. [DOI] [PubMed] [Google Scholar]
- 27.Hershey J W. Translational control in mammalian cells. Annu Rev Biochem. 1991;60:717–755. doi: 10.1146/annurev.bi.60.070191.003441. [DOI] [PubMed] [Google Scholar]
- 28.Hovanessian A G. Interferon-induced RNA-activated protein kinase (PKR): antiproliferative, antiviral and anti-tumoral functions. Semin Virol. 1993;4:237–245. [Google Scholar]
- 29.Hunter T, Hunt T, Jackson R J, Robertson H D. The characteristics of inhibition of protein synthesis by double-stranded ribonucleic acid in reticulocyte lysates. J Biol Chem. 1975;250:409–417. [PubMed] [Google Scholar]
- 30.Ito T, Jagus R M, May W S. Interleukin-3 stimulates protein synthesis by regulating dsRNA-dependent protein kinase (PKR) Proc Natl Acad Sci USA. 1994;91:7455–7459. doi: 10.1073/pnas.91.16.7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jagus R, Gray M M. Proteins that interact with PKR. Biochimie. 1994;76:779–791. doi: 10.1016/0300-9084(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 32.Jeffrey I W, Kadereit S, Meurs E F, Metzger T, Bachmann M, Schwemmle M. Nuclear localization of the interferon-inducible protein kinase PKR in human cells and transfected mouse cells. Exp Cell Res. 1995;218:17–27. doi: 10.1006/excr.1995.1126. [DOI] [PubMed] [Google Scholar]
- 33.Jimenez-Garcia L F, Green S R, Matthews M B, Spector D L. Organization of the double-stranded RNA-activated protein kinase DAI and virus-associated VA RNA1 in adenovirus-2-infected HeLa cells. J Cell Sci. 1993;106:11–22. doi: 10.1242/jcs.106.1.11. [DOI] [PubMed] [Google Scholar]
- 34.Judware R, Petryshyn R. Mechanism of action of a cellular inhibitor of the dsRNA-dependent protein kinase from 3T3-F442A cells. J Biol Chem. 1992;267:21685–21690. [PubMed] [Google Scholar]
- 35.Katze M G. Regulation of the interferon-induced PKR: can viruses cope? Trends Microbiol. 1995;3:75–78. doi: 10.1016/s0966-842x(00)88880-0. [DOI] [PubMed] [Google Scholar]
- 36.Kaufman R J. DNA transfection to study translational control in mammalian cells. Methods. 1997;11:361–370. doi: 10.1006/meth.1996.0434. [DOI] [PubMed] [Google Scholar]
- 37.Kaufman R J, Davies M V, Pathak V K, Hershey J W B. The phosphorylation state of eukaryotic initiation factor 2 alters translational efficiency of specific mRNAs. Mol Cell Biol. 1989;9:946–958. doi: 10.1128/mcb.9.3.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaufman R J, Murtha-Riel P. Translational control mediated by eukaryotic initiation factor-2 is restricted to mRNA derived from plasmid DNA in transiently transfected cells. Mol Cell Biol. 1987;7:1568–1571. doi: 10.1128/mcb.7.4.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kharrat A, Macias M J, Gibson T J, Nilges M, Pastore A. Structure of the dsRNA binding domain of E. coli RNase III. EMBO J. 1995;14:3572–3584. doi: 10.1002/j.1460-2075.1995.tb07363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kibler K V, Shors T, Perkins K B, Zeman C C, Banaszak M P, Biesterfeldt J, Langland J O, Jacobs B L. Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J Virol. 1997;71:1992–2003. doi: 10.1128/jvi.71.3.1992-2003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koromilas A E, Roy S, Barber G N, Katze M G, Sonenberg N. Malignant transformation by a mutant of the interferon-inducible double-stranded RNA dependent protein-kinase. Science. 1992;257:1685–1689. doi: 10.1126/science.1382315. [DOI] [PubMed] [Google Scholar]
- 42.Kostura M, Mathews M B. Purification and activation of the double-stranded RNA-dependent eIF-2 kinase DAI. Mol Cell Biol. 1989;9:1576–1586. doi: 10.1128/mcb.9.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar A, Hague J, Lacoste J, Hiscott J, Williams B R G. Double-stranded RNA-dependent protein kinase activates transcription factor NF-κB by phosphorylating I-κB. Proc Natl Acad Sci USA. 1994;91:6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar A, Yang Y L, Flati V, Der S, Kadereit S, Deb A, Haque J, Reis L, Weissmann C, Williams B R. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-κB. EMBO J. 1997;16:406–416. doi: 10.1093/emboj/16.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44a.Kumar, K. U. Unpublished observations.
- 45.Lacalle R A, Pulido D, Vara J, Zalacain M, Jimenez A. Molecular analysis of the pac gene encoding a puromycin N-acetyltransferase from Streptomyces alboniger. Gene. 1989;79:375–380. doi: 10.1016/0378-1119(89)90220-5. [DOI] [PubMed] [Google Scholar]
- 46.Langland J O, Jacobs B L. Cytosolic double-stranded RNA-dependent protein kinase is likely a dimer of partially phosphorylated Mr = 66,000 subunits. J Biol Chem. 1992;267:10729–10736. [PubMed] [Google Scholar]
- 47.Lee S B, Esteban M. The interferon-induced double-stranded RNA-activated protein kinase induces apoptosis. Virology. 1994;199:491–496. doi: 10.1006/viro.1994.1151. [DOI] [PubMed] [Google Scholar]
- 48.Lee S B, Rodriguez D, Rodriguez J R, Esteban M. The apoptosis pathway triggered by the interferon-induced protein kinase PKR requires the third basic domain, initiates upstream of Bcl- 2, and involves ICE-like proteases. Virology. 1997;231:81–88. doi: 10.1006/viro.1997.8494. [DOI] [PubMed] [Google Scholar]
- 49.Lee T G, Tang N, Thompson S, Miller J, Katze M G. The 58,000-dalton cellular inhibitor of the interferon-induced double-stranded RNA-activated protein kinase (PKR) is a member of the tetratricopeptide repeat family of proteins. Mol Cell Biol. 1994;14:2331–2342. doi: 10.1128/mcb.14.4.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lengyel P. Double-stranded RNA and interferon action. J Interferon Res. 1987;7:511–519. doi: 10.1089/jir.1987.7.511. [DOI] [PubMed] [Google Scholar]
- 51.Levin D, London I M. Regulation of protein synthesis: activation by double-stranded RNA of a protein kinase that phosphorylates eukaryotic initiation factor 2. Proc Natl Acad Sci USA. 1978;75:1121–1125. doi: 10.1073/pnas.75.3.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manche L, Green S R, Schmedt C, Mathews M B. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol Cell Biol. 1992;12:5238–5248. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marcus P I, Sekellick M J. Interferon induction by viruses. XVI. 2-Aminopurine blocks selectively and reversibly an early stage in interferon induction. J Gen Virol. 1988;69:1637–1645. doi: 10.1099/0022-1317-69-7-1637. [DOI] [PubMed] [Google Scholar]
- 54.Mathews M B. Interactions between viruses and the cellular machinery for protein synthesis. In: Hershey J W B, Mathews M, Sonenberg N, editors. Translational control. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 505–548. [Google Scholar]
- 55.McCormack S J, Ortega L G, Doohan J P, Samuel C E. Mechanism of interferon action motif I of the interferon-induced, RNA-dependent protein kinase (PKR) is sufficient to mediate RNA-binding activity. Virology. 1994;198:92–99. doi: 10.1006/viro.1994.1011. [DOI] [PubMed] [Google Scholar]
- 56.McCormack S J, Thomis D C, Samuel C E. Mechanism of interferon action: identification of a RNA binding domain within the N-terminal region of the human RNA-dependent P1/eIF-2α protein kinase. Virology. 1992;188:47–56. doi: 10.1016/0042-6822(92)90733-6. [DOI] [PubMed] [Google Scholar]
- 57.McMillan N A, Carpick B W, Hollis B, Toone W M, Zamanian-Daryoush M, Williams B R. Mutational analysis of the double-stranded RNA (dsRNA) binding domain of the dsRNA-activated protein kinase, PKR. J Biol Chem. 1995;270:2601–2606. doi: 10.1074/jbc.270.6.2601. [DOI] [PubMed] [Google Scholar]
- 58.Mellits K H, Pe’ery T, Manche L, Robertson H D, Mathews M B. Removal of double-stranded contaminants from RNA transcripts: synthesis of adenovirus VA RNAI from a T7 vector. Nucleic Acids Res. 1990;18:5401–5406. doi: 10.1093/nar/18.18.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meurs E F, Galabru J, Barber G N, Katze M G, Hovanessian A G. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA. 1993;90:232–236. doi: 10.1073/pnas.90.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mundschau L, Faller D V. Oncogenic ras induces an inhibitor of dsRNA-dependent eIF2α kinase activation. J Biol Chem. 1992;267:23092–23098. [PubMed] [Google Scholar]
- 61.Mundschau L J, Faller D V. Endogenous inhibitors of the dsRNA-dependent eIF-2 alpha protein kinase PKR in normal and ras-transformed cells. Biochimie. 1994;76:792–800. doi: 10.1016/0300-9084(94)90083-3. [DOI] [PubMed] [Google Scholar]
- 62.Pain V M. Initiation of protein synthesis in eukaryotic cells. Eur J Biochem. 1996;236:747–771. doi: 10.1111/j.1432-1033.1996.00747.x. [DOI] [PubMed] [Google Scholar]
- 63.Park H, Davies M V, Langland J O, Chang H-W, Nam Y S, Tartaglia J, Paoletti E, Jacobs B L, Kaufman R J, Vankatesan D. TAR RNA-binding protein is an inhibitor of the interferon-induced protein kinase PKR. Proc Natl Acad Sci USA. 1994;91:4713–4717. doi: 10.1073/pnas.91.11.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel R C, Stanton P, Sen G C. Role of the amino-terminal residues of the interferon-induced protein kinase in its activation by double-stranded RNA and heparin. J Biol Chem. 1994;269:18593–18598. [PubMed] [Google Scholar]
- 65.Petryshyn R, Chen J-J, London I M. Growth-related expression of a double-stranded RNA-dependent protein kinase in 3T3 cells. J Biol Chem. 1984;259:14736–14742. [PubMed] [Google Scholar]
- 66.Petryshyn R, Chen J-J, London I M. Detection of activated double-stranded RNA-dependent protein kinase in 3T3-F442A cells. Proc Natl Acad Sci USA. 1988;85:1427–1431. doi: 10.1073/pnas.85.5.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prostko C R, Dholakia J N, Brostrom M A, Brostrom C O. Activation of the double-stranded RNA-regulated protein kinase by depletion of endoplasmic reticular calcium stores. J Biol Chem. 1995;270:6211–6215. doi: 10.1074/jbc.270.11.6211. [DOI] [PubMed] [Google Scholar]
- 68.Proud C G. PKR: a new name and new roles. Trends Biochem Sci. 1995;20:241–246. doi: 10.1016/s0968-0004(00)89025-8. [DOI] [PubMed] [Google Scholar]
- 69.Puder M, Barnard G F, Staniunas R J, Steele G D, Jr, Chen L B. Nucleotide and deduced amino acid sequence of human ribosomal protein L18. Biochim Biophys Acta. 1993;1216:134–136. doi: 10.1016/0167-4781(93)90050-n. [DOI] [PubMed] [Google Scholar]
- 70.Romano P R, Green S R, Barber G N, Mathews M B, Hinnebusch A G. Structural requirements for double-stranded RNA binding, dimerization, and activation of the human eIF-2 alpha kinase DAI in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:365–378. doi: 10.1128/mcb.15.1.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saito S, Kawakita M. Inhibitor of interferon-induced double-stranded RNA-dependent protein kinase and its relevance to alteration of cellular protein kinase activity level in response to external stimuli. Microbiol Immunol. 1991;35:1105–1114. doi: 10.1111/j.1348-0421.1991.tb01632.x. [DOI] [PubMed] [Google Scholar]
- 72.Samuel C E. Antiviral actions of interferon: interferon-regulated cellular proteins and their surprisingly selective antiviral activities. Virology. 1991;183:1–11. doi: 10.1016/0042-6822(91)90112-o. [DOI] [PubMed] [Google Scholar]
- 73.Samuel C E. The eIF-2α protein kinases, regulators of translations in eucaryotes from yeasts to humans. J Biol Chem. 1993;268:7063–7066. [PubMed] [Google Scholar]
- 74.Samuel C E, Knutson G S, Berry M J, Atwater J A, Lasky S R. Purification of double-stranded RNA-dependent protein kinase from mouse fibroblasts. Methods Enzymol. 1986;119:499–516. doi: 10.1016/0076-6879(86)19070-7. [DOI] [PubMed] [Google Scholar]
- 75.Schmedt C, Green S R, Manche L, Taylor D R, Ma Y, Mathews M B. Functional characterization of the RNA-binding domain and motif of the double-stranded RNA-dependent protein kinase DAI (PKR) J Mol Biol. 1995;249:29–44. doi: 10.1006/jmbi.1995.0278. [DOI] [PubMed] [Google Scholar]
- 76.Schwemmle M, Clemens M J, Hilse K, Pfeifer K, Troster H, Muller W E, Bachmann M. Localization of Epstein-Barr virus-encoded RNAs EBER-1 and EBER-2 in interphase and mitotic Burkitt lymphoma cells. Proc Natl Acad Sci USA. 1992;89:10292–10296. doi: 10.1073/pnas.89.21.10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sharp T V, Witzel J E, Jagus R. Homologous regions of the alpha subunit of eukaryotic translational initiation factor 2 (eIF2alpha) and the vaccinia virus K3L gene product interact with the same domain within the dsRNA-activated protein kinase (PKR) Eur J Biochem. 1997;250:85–91. doi: 10.1111/j.1432-1033.1997.00085.x. [DOI] [PubMed] [Google Scholar]
- 78.Srivastava S P, Kumar K U, Kaufman R J. Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J Biol Chem. 1998;273:2416–2423. doi: 10.1074/jbc.273.4.2416. [DOI] [PubMed] [Google Scholar]
- 79.Takizawa T, Ohashi K, Nakanishi Y. Possible involvement of double-stranded RNA-activated protein kinase in cell death by influenza virus infection. J Virol. 1996;70:8128–8132. doi: 10.1128/jvi.70.11.8128-8132.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tan S L, Gale M J, Jr, Katze M G. Double-stranded RNA-independent dimerization of interferon-induced protein kinase PKR and inhibition of dimerization by the cellular P58IPK inhibitor. Mol Cell Biol. 1998;18:2431–2443. doi: 10.1128/mcb.18.5.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thomis D C, Samuel C E. Mechanism of interferon action: evidence for intermolecular autophosphorylation and autoactivation of the interferon-induced, RNA-dependent protein kinase PKR. J Virol. 1993;67:7695–7700. doi: 10.1128/jvi.67.12.7695-7700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tiwari R K, Kusari J, Kumar R, Sen G C. Gene induction by interferons and double-stranded RNA: selective inhibition by 2-aminopurine. Mol Cell Biol. 1988;8:4289–4294. doi: 10.1128/mcb.8.10.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wong A H, Tam N W, Yang Y L, Cuddihy A R, Li S, Kirchhoff S, Hauser H, Decker T, Koromilas A E. Physical association between STAT1 and the interferon-inducible protein kinase PKR and implications for interferon and double-stranded RNA signaling pathways. EMBO J. 1997;16:1291–1304. doi: 10.1093/emboj/16.6.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu S, Kaufman R J. Double-stranded RNA binding is required and dimerization is not sufficient for the activation of the dsRNA-dependent protein kinase (PKR) J Biol Chem. 1996;271:1756–1763. doi: 10.1074/jbc.271.3.1756. [DOI] [PubMed] [Google Scholar]
- 85.Wu S, Kaufman R J. A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J Biol Chem. 1997;272:1291–1296. doi: 10.1074/jbc.272.2.1291. [DOI] [PubMed] [Google Scholar]
- 86.Wu S, Kumar K, Kaufman R J. Identification and requirements of three ribosome binding domains in dsRNA-dependent protein kinase (PKR) Biochemistry. 1998;37:13816–13826. doi: 10.1021/bi981472h. [DOI] [PubMed] [Google Scholar]
- 87.Wu S, Rehemtulla A, Gupta N K, Kaufman R J. A eukaryotic translation initiation factor 2-associated 67 kDa glycoprotein partially reverses protein synthesis inhibition by activated double-stranded RNA-dependent protein kinase in intact cells. Biochemistry. 1996;35:8275–8280. doi: 10.1021/bi953028+. [DOI] [PubMed] [Google Scholar]
- 88.Xiao Q, Sharp T V, Jeffrey I W, James M C, Pruijn G J, van Venrooij W J, Clemens M J. The La antigen inhibits the activation of the interferon-inducible protein kinase PKR by sequestering and unwinding double-stranded RNA. Nucleic Acids Res. 1994;22:2512–2518. doi: 10.1093/nar/22.13.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang Y-L, Reis L F, Pavlovic J, Aguzzi A, Schäfer R, Kumar A, Williams B R G, Aguet M, Weissmann C. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 1995;14:6095–6106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yeung M C, Liu J, Lau A S. An essential role for the interferon-inducible, double-stranded RNA-activated protein kinase PKR in the tumor necrosis factor-induced apoptosis in U937 cells. Proc Natl Acad Sci USA. 1996;93:12451–12455. doi: 10.1073/pnas.93.22.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu S, Romano P R, Wek R C. Ribosome targeting of PKR is mediated by two double-stranded RNA-binding domains and facilitates in vivo phosphorylation of eukaryotic initiation factor-2. J Biol Chem. 1997;272:14434–14441. doi: 10.1074/jbc.272.22.14434. [DOI] [PubMed] [Google Scholar]
- 92.Zinn K, Keller A, Whittemore L A, Maniatis T. 2-Aminopurine selectively inhibits the induction of beta-interferon, c-fos, and c-myc gene expression. Science. 1988;240:210–213. doi: 10.1126/science.3281258. [DOI] [PubMed] [Google Scholar]