

Summary
Single-cell cytometry data are crucial for understanding the role of the immune system in diseases and responses to treatment. However, traditional methods for annotating cytometry data face challenges in scalability, robustness, and accuracy. We propose a cytometry masked autoencoder (cyMAE), which automates immunophenotyping tasks including cell type annotation. The model upholds user-defined cell type definitions, facilitating interpretability and cross-study comparisons. The training of cyMAE has a self-supervised phase, which leverages large amounts of unlabeled data, followed by fine-tuning on specialized tasks using smaller amounts of annotated data. The cost of training a new model is amortized over repeated inferences on new datasets using the same panel. Through validation across multiple studies using the same panel, we demonstrate that cyMAE delivers accurate and interpretable cellular immunophenotyping and improves the prediction of subject-level metadata. This proof of concept marks a significant step forward for large-scale immunology studies.
Keywords: high-dimensional cytometry, mass cytometry, automated gating, machine learning, deep learning, representation learning, immunophenotyping
Graphical abstract
Highlights
-
•
Masked cytometry modeling learns relationships among proteins without cell identity
-
•
We develop cytometry masked autoencoder (cyMAE) to automate immunophenotyping
-
•
cyMAE improves both cell-level and subject-level immune profiling
Kim et al. introduces cyMAE, a cytometry masked autoencoder that automates immune cell profiling from single-cell cytometry data. By leveraging unlabeled data for pre-training and fine-tuning on specific tasks, the model improves immune profiling accuracy and enhances prediction of subject-level clinical data, advancing large-scale immune studies.
Introduction
High-throughput single-cell protein expression data, acquired through flow and mass cytometry, are essential to understanding the role of the immune system in infectious diseases, autoimmunity, cancer, and the response of immune cells post-treatment. Cytometry assays are designed to profile millions of cells from a biological sample, precisely quantifying biomarkers specific to various cell types. In contrast, single-cell RNA sequencing (scRNA-seq) approaches are generally on the scale of thousands of cells. This substantial difference in scale grants cytometry significant advantages for the identification and characterization of rare cell population and enhances the overall comprehensiveness of the data collected. For example, cytometry can pinpoint cell populations that are differentially abundant or proteins that are differentially expressed between subject groups. This process of immune profiling effectively delineates both similarities and diversities within the immune landscape of different subjects, contributing significantly to precision medicine by enabling predictions at an individual level.
The most prevalent approach for analyzing cytometry data is manual gating, a process involving user-applied sequential filters to bivariate plots of protein markers, thereby isolating specific cell subsets for focused analysis.1 These bivariate plots visually represent the distribution of protein markers, allowing a human analyst to manually identify and select cells based on their prior knowledge of these distributions. Despite widespread use of this approach, manual gating faces several significant challenges. Firstly, it is a time-intensive process, particularly for panels with over a dozen markers,2,3 as the number of biaxial plots to consider increases quadratically with the number of parameters measured. Secondly, manual gating is prone to subjectivity and bias.2,3 Each analysis is influenced by pre-existing knowledge, which can lead to a bias toward anticipated results. Subjectivity also enters through the selection of the order of marker combinations and the definition of gate boundaries. Thirdly, results from manual gating can be challenging to replicate.2,3 Different studies may employ varied gating strategies, including distinct gating sequences, shapes, and boundaries for gates, impacting the robustness and consistency of identified cell subsets. Moreover, the level of gating stringency also varies between individual analysts, contributing to inconsistent results.
The ability to simultaneously measure multiple protein markers has significantly increased the complexity of cytometry data. This complexity has led to the development of automated analysis techniques, particularly unsupervised clustering methods like FlowSOM,4 PhenoGraph,5 Scaffold Maps,6 and X-shift.7 Although these clustering approaches address some limitations of manual gating, they also introduce their own set of constraints. Notably, while unsupervised clustering methods can detect data variability, they struggle to differentiate between biological or technical sources of this variability. This limitation makes these methods susceptible to batch effects, shifts in data distribution, and non-specific binding of antibodies.8 Another challenge arises in cross-study comparisons, where minor variations in panel selection, sample collection, measurement noise, or random seeding can lead to abrupt changes in cluster boundaries. For example, CD4 T cells might be clustered differently in studies based on memory or functional subtypes, complicating direct comparisons between even highly overlapping datasets.
To remedy these limitations and provide cell annotations that conform to predictable cell type ontologies, supervised or semi-supervised methods have been proposed, such as ACDC,9 SCINA,10 LDA,11 and CyAnno.12 ACDC and SCINA use information about marker proteins across cell types to inform clustering, while LDA and CyAnno directly train supervised models using training data in which individual cells have been annotated. However, the approaches used by ACDC and SCINA have the drawback of overly relying on prior knowledge. If the manual gating is not comprehensive or contains errors, these will propagate through the automated process. CyAnno, which relies on the original Flow Cytometry Standard (FCS) feature space, may fail to create robust cell representations that are less sensitive to noise and technical variations. Additionally, given the trend of increasing data sizes nowadays, methods that heavily depend on manual gating results are not scalable. Therefore, there is a need for supervised methods that can effectively utilized large, unlabeled datasets. In this study, we aim to leverage large-scale unlabeled cytometry data to improve cell representation learning, ultimately achieving automated immunophenotyping and enhancing predictive accuracy. To this end, we draw inspiration from recent advances in computer vision and natural language processing.
In the broader context of big data and advanced computational models, artificial intelligence (AI) has achieved great success in fields like computer vision and natural language processing. The effort needed to manually label data makes it extremely difficult to fully leverage the vast amounts of existing unlabeled data in the supervised learning paradigm. However, the revolution of self-supervised learning techniques, particularly in the pre-training phase, empowers models to more accurately learn data distributions and utilize unlabeled data effectively. The core concept behind self-supervised pre-training is randomly masking a portion of the input data and training the model to reconstruct masked information using context clues from the surrounding data. This approach allows the pre-trained model to be fine-tuned for specific downstream tasks or to function as generative AI. Coupling the transformer13 architecture, known for its high expressiveness and scalability, has led to significant synergistic effects. Notable examples include masked language modeling as seen in BERT14 and GPT15 and masked image modeling in models like DINOv2,16 BeiT,17 and masked autoencoder (MAE)18 in computer vision.
The success of the masking approach has reverberated within the biomedical field as well. For example, protein language models (pLMs) are a set of AI models trained on extensive sets of unlabeled protein sequences.19,20,21 pLMs have steadily gained traction across diverse applications for protein design, including antibody engineering22 and drug discovery.23,24 In addition, AI models trained on unlabeled scRNA-seq data have been published and used for cell annotation purposes.25,26,27,28,29,30,31 Thus, masking models have proven to substantially outperform previous conventional methods in effectiveness and show great potential in biomedical applications. Similarly, we apply these techniques for immunophenotyping, as cytometry data can be structured in a similar way.
In this proof-of-concept study, we develop an accurate and interpretable automated immunophenotyper for single-cell cytometry data, using a technique we call masked cytometry modeling (MCM). This approach employs self-supervised pre-training on single-cell cytometry data. During MCM, our model learns the relationships and dependencies among markers on immune cells by analyzing expression patterns in the massive amount of datasets, without requiring additional information about cellular identity. The resulting pre-trained model can then be fine-tuned for various downstream tasks, surpassing the utility of the original data. We demonstrate that our model not only overcomes the challenges of manual gating and clustering methods but also provides accurate results even on independent datasets that were never seen during its training. Crucially, this study utilizes the exact same panel across all datasets, highlighting the necessity of panel consistency for applying the model to new datasets. By validating across multiple datasets, we showed that our model can accurately identify complex cell types, interpret which crucial protein markers predict targets, and enhance precision in subject-level phenotyping. Moreover, our model demonstrates fast, scalable, reproducible modeling of cytometry data through pre-training and fine-tuning approach. For instance, pre-training and fine-tuning only took 2–3 days each with large datasets, and cell type annotation can process approximately 15,000 cells per second. This approach in immunophenotyping promises to broaden the impact of existing cytometry data and enhance immunological knowledge by more accurately phenotypes at both cellular and subject levels.
Results
cyMAE algorithm
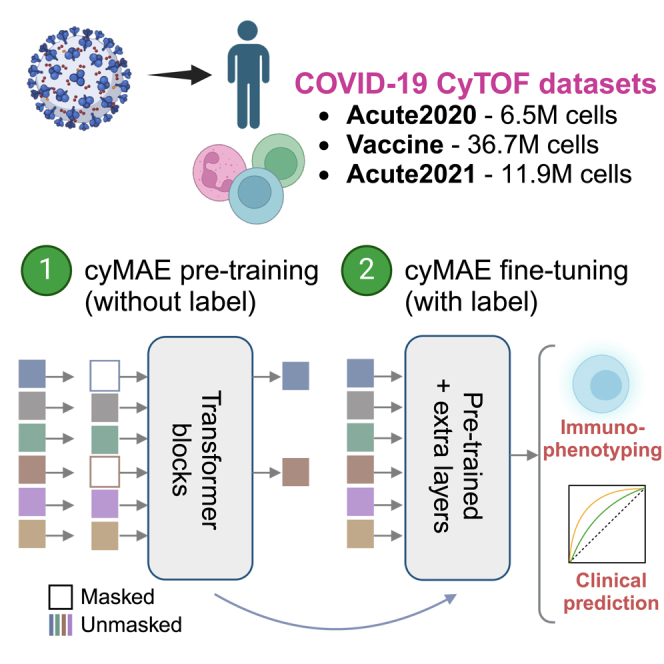
To address the challenges of time-consuming and labor-intensive immunophenotyping in cytometry data, we propose cyMAE, a cytometry masked autoencoder model. This innovative model constructs and employs latent embeddings of single-cell cytometry data to obtain state-of-the-art performance on various cell-level and subject-level tasks. cyMAE is built upon an MAE18 architecture, featuring stacked transformer blocks in both the encoder and decoder. Inspired by successful methodologies in computer vision and natural language processing, cyMAE undergoes a two-phase training process: self-supervised pre-training followed by supervised fine-tuning, as illustrated in Figure 1A. The main advantage of this approach is its ability to use large-scale, easily obtainable unlabeled data during the initial self-supervised pre-training phase, thus reducing the reliance on scarce and labor-intensive labeled data in the subsequent fine-tuning phase. During pre-training, a randomly selected subset of the protein expression data is masked and fed to an encoder, which produces latent embeddings of the masked data. In turn, these embeddings are processed by a decoder that attempts to reconstruct the unmasked, original data (Figures 1B and S1). Through this process, the encoder-decoder system learns to optimize the embeddings to minimize reconstruction error, effectively enabling the model to obtain informative data embeddings without requiring explicit ground truth labels. During the second fine-tuning stage, the model employs the full, unmasked protein expression data to generate latent cell representations using the encoder that was pre-trained in the first stage. These representations are applicable to a range of downstream tasks, whether they involve labeled data or not. Cell representations generated by the pre-trained encoder can be used for unsupervised tasks or plugged into another classifier to solve tasks through supervised fine-tuning. Specifically, we evaluated the pre-trained cyMAE’s performance on two cell-level tasks: cell type annotation and imputation. Moreover, for subject-level tasks, we tested SARS-CoV-2 infection prediction, secondary immune response prediction against COVID-19, and prediction of the infection stage in the COVID-19 progression.
Figure 1.

Overview of cyMAE pre-training and fine-tuning process
(A) In the pre-training step, protein expression data are randomly masked for each cell. Only the unmasked protein identities undergo dimension expansion to create learnable unmasked protein embeddings. These embeddings are then concatenated with the unmasked protein expressions and fed into the encoder. This encoder generates unmasked latent representations, which are merged with learnable mask embeddings and fed to the decoder for reconstruction of the masked values. In the fine-tuning step, the pre-trained encoder produces latent representations for both cells and subjects, facilitating cell-level and subject-level downstream tasks, respectively. The fine-tuning datasets need to be designed using the exact same panel as the pre-training dataset.
(B) From left to right, masked, imputed (reconstructed), and original data. Each row represents a marker protein, and each column represents a randomly sampled cell. Initially, 25% of the original data are randomly masked, shown in white in the masked data visualization. cyMAE effectively reconstructs these masked regions, demonstrating the model’s accuracy.
We analyzed cytometry by time of flight (CyTOF) data from three distinct COVID-19 studies conducted at the University of Pennsylvania, referred to as the Acute2020 dataset, Vaccine dataset, and Acute2021 dataset. For all datasets, whole blood was stained with the same 30-marker panel. Each of the datasets underwent a routine manual gating practice executed by domain experts to extract single, intact cells in preparation for downstream analysis. The Acute2020 dataset consists of single-time-point samples from 13 patients hospitalized with acute SARS-CoV-2 infection in 2020 and 13 healthy controls, comprising a total of 6.5 M cells. The Vaccine dataset includes 37 healthy adults followed longitudinally before and after (7 days after second dose) SARS-CoV-2 mRNA vaccine, for a total of 150 FCS files. This dataset is composed of 36.7 M cells. Lastly, the Acute2021 dataset contains longitudinal samples from 42 SARS-CoV-2-infected individuals who were enrolled in the I-SPY COVID-19 trial32 in 2021. Samples were collected at the time of hospital admission and 7 days later. This dataset includes 11.9 M cells from 56 FCS files. Prior analysis of flow cytometry data from the Acute2020 dataset revealed heterogeneous peripheral blood profiles among patients hospitalized with SARS-CoV-2, capturing both common and uncommon cells and cell phenotypes compared to healthy individuals.33 Thus, the Acute2020 dataset was chosen for pre-training, while all three datasets were used in the downstream evaluations. Despite being stained with the same panel and acquired on the same instrument, the three datasets were processed in different years with different reagent lots and slightly different protocols. Because of this, there are moderate batch effects that distinguish the datasets, especially for the neutrophil population (Figure S2). This provides an opportunity to test the robustness of cyMAE to realistic amounts of technical variability between the datasets used for training and inference.
cyMAE learns antibody co-occurrence patterns
The pre-trained cyMAE effectively learns the patterns of co-occurrence among antibodies targeting specific proteins, purely from data, without relying on any prior knowledge. This capability is demonstrated by the way cyMAE groups proteins based on their co-localization on particular cell types, as seen in Figure 2A. For example, proteins that appear mostly on T cells (CD3, CD4, CD8, CD28, CD183, etc.) cluster together, while proteins that appear on natural killer (NK) cells and some T cells (CD56, CD57) are nearby. Similarly, proteins abundantly expressed on one cell type, such as neutrophils (CD16, CD66b), basophils (CD123, CD294), and B cells (CD20, immunoglobulin D, CD185, HLA-DR), are grouped in cell type-specific regions of the protein embedding space. One exception is the B cell marker CD19, which is far away from the other B cell proteins. This is likely because of background CD19 expression in the most abundant cell type, neutrophils, which pulls the CD19 expression away from the B cell region and toward the neutrophil region. Because it is unsupervised, the pre-training phase of cyMAE does not know that CD19 expression on neutrophils is a technical artifact, and this is reflected in the protein embedding. Overall, these results illustrate that cyMAE can effectively capture the contextual relationships and dependencies between immune cell marker expression levels. Thus, this model can be learnable for data patterns, enabling it to successfully perform subsequent downstream tasks.
Figure 2.

Evaluation of cyMAE protein embeddings and cell type annotation across various datasets
(A) Principal-component analysis plot of the cyMAE protein embeddings, demonstrating how the model, through unsupervised pre-training, effectively learns protein embeddings that represent the spatial closeness of antibody probes.
(B) Model comparisons in the 46 cell type annotation with balanced accuracy (Bacc). The internal test set is Vaccine dataset after train-test split, the external set 1 is Acute2021, and the external set 2 is Acute2020. GBDT is a gradient boosting decision tree. Static gating is a method to aggregate into a single consensus gate for each gate in the hierarchy (see STAR Methods). Deep neural network (DNN) denotes a fully connected neural network, used as a cell type annotator in methods like DGCyTOF34 and DeepCyTOF35 for cytometry data analysis. Convolutional neural network (CNN) denotes a model that uses the same convolution layers as Deep CNN8 without pooling layers for the cell-level task.
(C) Accuracy of cell type annotation for both 5 abundant and 15 rare cell types.
(D) The few-shot learning for cell type annotation. cyMAE (from scratch) refers to the same model architecture as cyMAE but without pre-training. Each green dashed line represents the performance of the full fine-tuned cyMAE from (B).
cyMAE is an accurate cell immunophenotyper
Cell type annotation, traditionally achieved through manual gating and clustering methods, is now efficiently automated by our cyMAE model. By fine-tuning with cell type labels, cyMAE accurately annotates cell types in single-cell datasets. Ground truth labels for 46 cell types, obtained from manual gating, were used (Figure S3). We used 60% of the Vaccine dataset for fine-tuning the model, 20% as validation, and the remaining 20% as an internal test set. We further evaluated cyMAE using the Acute2021 dataset and the Acute2020 dataset as external validation sets (external set 1 and 2, respectively). We compared cyMAE with a gradient boosting decision tree (GBDT),36 a fully connected deep neural network, and a convolutional neural network (CNN) (see STAR Methods) as well as cytometry-specific analysis methods: CyAnno, static gating, and unsupervised clustering with FlowSOM.
As a baseline, we took the gating strategy developed on the training dataset and applied it statically to the testing datasets, without adjustments for inter-sample variability (Figure S4). This approach is equivalent to manually constructing a decision tree and then applying it on the testing data. The other supervised models used here can be seen as refinements of this idea: they attempt to learn a more robust encoding of the gating information by using multivariate rather than bivariate expression patterns. Alongside the supervised classification methods, we included FlowSOM, a popular unsupervised clustering method for cytometry. To match our supervised paradigm, we add an inference mode to FlowSOM by mapping each unseen test datapoint to the nearest SOM node (see STAR Methods).
Given the imbalanced distribution of cell type, with neutrophils comprising over 60% of cells, we used balanced accuracy (Bacc) to assess model performance fairly. The experimental results showed consistently high Bacc on both internal test sets and two external sets, with the internal test set achieving 93.1% Bacc and the external sets 81.9% and 82.6% Bacc, respectively (Figure 2B). When we examined performance by cell type, our model was found to be more accurate than others for most cell types (Figures 2C and S5). Notably, the model performed particularly well on rare cell types. Accurate prediction of rare cell types is difficult because it is easy for a model to be trained with a bias toward more frequent cell types. However, when comparing performance on cells with a frequency of less than 0.1% in Figure 2C, both the internal test set and external sets show more accurate predictions for rare cell types than the comparison models in most cases. In addition, cyMAE’s performance benefits from pre-training, outperforming the cyMAE model from scratch (non-pre-trained), demonstrating the value of leveraging large-scale unlabeled data for pre-training (Table S1).
During the training and validation processes described earlier, cyMAE and the other methods did not use all events present in the FCS files, but only the subset of CD45-positive single cells, which were assigned a terminal label by manual gating. Some ungated or partially gated events were left out: debris, doublets, or events in the space left out between the gate boundaries (for example, a small number of T cells which do not belong to either of the CD4, CD8, double-negative, or double-positive gates). However, we are interested in a real-world setting where cell labels from manual gating are not available, so leaving out ungated events is not an option. Therefore, we asked the same pre-trained and fine-tuned cyMAE model to annotate all events in a file, both gated and ungated.
The model assigned each event to the best matching label among the 46 that it learned during training (Figure S6A). Partially gated cells were mostly assigned to a more specific cell type from the same lineage: for example, partially gated T cells that were not captured in downstream gates were assigned more detailed naive or memory T cell phenotypes. The vast majority of manually gated debris events were assigned the CD66bnegCD45lo label, which was originally included in the gating hierarchy to capture debris or platelet events which escaped the cleanup gates. Among events that were manually classified as doublets, cyMAE labeled many as CD45hiCD66bpos (a catch-all class for heterotypic granulocyte-lymphocyte doublets), but a significant number of events were labeled as individual cell types (homotypic doublets of that cell type). For example, neutrophil-neutrophil doublets were classified as neutrophils, the closest available match. We hypothesize that including more doublet labels in the training data would help cyMAE to better distinguish these homotypic doublets from singlets. Finally, many events manually labeled as doublets were classified by cyMAE as eosinophils. Based on multivariate protein expression, these events are very similar to those manually labeled as eosinophils (Figures S6B and S6C). We hypothesize that these events are true eosinophils, which bound high levels of DNA intercalator, and, because of this, they were erroneously excluded from the single cell gate.
While it is difficult to give an objective assessment of model performance when ungated cells are included, three versions of accuracy measurements were computed for cyMAE in this setting (Table S2). First, ungated cells were included, and a strict scoring was used that evaluates any prediction different from the training label as wrong, despite the fact that the model has no way of predicting classes not seen during training (Accuracy = 0.89, Bacc = 0.61). Second, ungated cells were included, and a lax scoring was used that evaluates any prediction which is a descendant of the training label in the gating hierarchy as correct as well as accepts the prediction CD66bnegCD45lo for debris events and the prediction CD45hiCD66bpos for doublet events (Accuracy = 0.95, Bacc = 0.81). Third, the evaluation without ungated events, as in Figure 2, was recapitulated (Accuracy = 0.99, Bacc = 0.90). In summary, the inclusion of ungated events decreases cyMAE’s performance, but a majority of this deterioration can be attributed to the presence of classes not known to the model, which are impossible to predict without re-training with updated training data.
These results show that our cyMAE model is robust to technical variation between datasets, showcasing superior performance across different collection and processing protocols. For example, despite being trained on the Vaccine dataset from cryopreserved samples of healthy subjects in 2021, cyMAE outperformed all other methods on the Acute2020 dataset, which comprised fresh samples from subjects with acute COVID in 2020. These results underline cyMAE’s potential as a reliable tool for cell immunophenotyping across diverse datasets.
cyMAE is a few-shot learner
Unlike full fine-tuning, few-shot learning trains a model with a limited amount of training data. N-shot uses only N samples for each class in the classification problem. A pre-trained large language model, developed through self-supervised learning, is recognized for its effectiveness as a few-shot learner.15 Similarly, we evaluated our model, cyMAE, in a few-shot learning context for cell type annotation, conducting experiments with 5-shot, 10-shot, 15-shot, and 20-shot settings. Training, validation, testing, and external testing sets are the same as in the previous cell type annotation tasks.
As expected, the performance of cyMAE, when pre-trained, approached that of training with the full training set as the number of N-shots increased (Figure 2D). On the other hand, since the cyMAE from scratch (non-pre-trained) has many parameters and no pre-trained information, this method does not learn with small sample size. It is worth noting that GBDT also performed reasonably well, but cyMAE outperformed GBDT based on the pre-trained knowledge. This analysis shows that cyMAE, once pre-trained, can effectively adapt to new tasks even with sparse labeled data, guiding learning in the appropriate direction.
cyMAE enhances regression imputation for cytometry data
Current technology for flow and mass cytometry only allows a few dozen markers, and sometimes cost considerations may reduce the number even further. This limitation is unlike scRNA-seq or other similar single-cell techniques, which can capture thousands of parameters. Despite these limitations, cytometry remains a powerful tool in single-cell biology due to its widespread use, ease of application, clinical implementation, and its capacity to analyze significantly more cells (typically millions versus thousands in single-cell genomics). This latter point means that cytometric approaches are much more robust for interrogating rare cell types often sparsely sampled or missed altogether by single-cell sequencing approaches. Fully exploiting these advantages of cytometric approaches through advanced computational methods, for example, allowing measurements on small panel sizes to yield analytical results similar to those on larger panel sizes, would be a major advance for the field. To investigate this feasibility, Becht et al.37 proposed Infinity Flow, applying a gradient boosting tree model36 to impute the expression of over 300 markers from merely 15. We assessed whether cyMAE’s cell latent representations could further enhance regression imputation. In our experiments, we masked 7 markers associated with memory subsets in T cells (CD27, CD28, CD45RA, CD45RO, CD127, CD197), using the remaining data to predict the masked marker expressions with both Infinity Flow and cyMAE, the latter fine-tuned for imputation. We used the Acute2020 data for training, and the Vaccine dataset and Acute2021 dataset as external sets (external set 1 and 2, respectively). R-squared values were used for evaluation.
The cyMAE model achieved imputation performances with R-squared values ranging from 0.297 to 0.664 for the external set 1 and 0.164 to 0.583 for the external set 2 (Figure 3A), despite being limited to 23 markers not directly indicative of T cell memory states and their associated masked markers. Notably, cyMAE outperformed Infinity Flow for all seven markers in the external set 1 and four of the seven markers in the external set 2. Beyond identifying patterns of universally expressed proteins, such as CD45RA in NK cells and CD45RO in neutrophils, cyMAE also showed high correlations between true and predicted values, specifically within T cells or for CD27 expression in B cells (Figures 3B, S7, S8, and S9). These results suggest that cyMAE can infer information about the memory states of T and B cells, even in the absence of the standard memory markers.
Figure 3.

Comparison of imputation performance between cyMAE and Infinity Flow
(A) R-squared comparison between Infinity Flow and cyMAE for the imputation task. A total 7 markers were masked and then predicted by the two models.
(B) Plots of actual versus predicted expression levels for each marker in the external set (Vaccine dataset). The dashed line represents the ideal relationship, serving as a reference to assess the performance.
cyMAE is an interpretable immunophenotyper
A key challenge in deploying machine learning models for clinical or biological analyses is their “black box” nature, which means that the rationale for cell type prediction decisions is difficult to interpret. Unlike these models, cyMAE incorporates a multi-head self-attention mechanism within its transformer architecture, enabling interpretable predictions for downstream tasks. The attention scores generated by this model indicate the importance of specific marker information and their interrelations in the context of prediction tasks, with higher attention score indicating greater reliance on a maker’s information relative to other markers. In our analysis, we first measured the attention scores attributed to each feature across different cell types during cell type annotation. Then, we highlighted relatively higher scores (those exceeding 1.5 standard deviations above the mean) for each marker in a yellow box (Figure 4A). Notably, CD45 consistently emerged as the marker with the highest attention score across all cell types, serving as a key discriminator between major immune cell lineages, such as granulocytes and mononuclear cells. Aside from CD45, most markers were highly attended in cell types in which they are highly expressed: for example, CD19 in B cells, CD123 in basophils and pDCs, and CD294 in basophils and eosinophils.
Figure 4.

Interpretation in cell type annotation and imputation tasks by the attention scores
(A) For the Acute2021 dataset (external set 1), the heatmap shows protein markers with high attention score as bright red for each cell type and highlights the relatively higher scores on each marker in yellow box.
(B) From 23 markers to impute the other 7 markers, the attention score measures which input features have high attention from the other features during prediction. For the Vaccine dataset (external set 1), the heatmap shows the protein markers with high attention score as bright white or red for each cell type with highlighting the relatively higher scores on each marker in a yellow box. For the left figure in (A) and (B), we used Bertvis38 for visualization of attention weights.
Similarly, we assessed the attention score of 23 markers for each cell type when processing the inputs that included masking of 7 masked markers during the imputation task (Figure 4B). For cell types with constitutive expression or non-expression of masked markers, the model primarily focused on markers indicative of cell type identity (e.g., CD294, CD66b, and CD45 for eosinophils; CD16 and CD45 for neutrophils). In the case of T and B cells, the masked markers have variable as opposed to constitutive expression, and the level of expression is commonly used to define memory subtypes. Since the detailed T and B cell phenotype is so strongly related to the marker variability we are trying to predict, we examined the model’s performance and attention scores at the level of major cell type.
We found that the model attended to the T cell marker protein CD3 and other proteins like CD57. For example, CD3 turned out to be predictive of high values of the masked protein CD197, while CD57 was predictive of low values (Figure S10). The correlation between CD3 and CD197 in T cells was not expected, but cyMAE found and exploited it to improve imputation performance.
The attention scores not only demonstrated a consistent pattern across external datasets (Figure S11) but also showed minimal variance between samples (Figures S12 and S13), underscoring the model’s interpretability and reliability in identifying critical biomarkers for cell type predictions.
cyMAE improves subject status predictions
In a typical flow cytometry or CyTOF analysis, hundreds of thousands of single cells are generally obtained from an individual sample, aiming to understand cellular-level immunophenotypes, like cell type identification. However, extending these analyses to achieve phenotypic precision at the individual level is also critical. While manual gating is a sophisticated method for extracting subject-level features using expert knowledge, it may overlook complex co-expression patterns indicative of cellular states like activation, senescence, or exhaustion in T cells due to the high-dimensional nature of accurate definitions of these T cell states. Ideally, our aim is to leverage the comprehensive global distribution of cell information to gain deeper biological insights.
A key requirement for this goal is ensuring the method’s predictions remain consistent regardless of the cells’ order in the dataset, a property known as permutation invariance. This property ensures that the method is robust and not reliant on the specific ordering of cells. Additionally, the method should adaptively focus on marker cell types relevant to the study, such as leukemic blast cells in an acute myeloid leukemia study39 or CTLA4+ or PD1+ cells in cancer immunotherapy study.40 To address these needs, we aggregated cyMAE representations of all cells from each subject into a subject-level representation, exploring several pooling methods to find the most effective one for each task (see STAR Methods).
We compared the cyMAE with global pooling to manual gating, FlowSOM, and Deep CNN8 across three prediction tasks (Figure 5). Using 5-fold cross-validation with 10 repetitions for each task, we first assessed the model ability to distinguish between COVID-19 patients and healthy subjects. Manual gating and FlowSOM demonstrated high accuracy, with mean area under the receiver operating characteristic curve (AUROC) 0.975 (standard deviation [SD] 0.042) and mean AUROC 0.936 (SD 0.096) on the test set, respectively, whereas Deep CNN showed a lower mean AUROC 0.543 (SD 0.256). Notably, cyMAE with global max pooling achieved the highest performance, with a mean AUROC 0.989 (SD 0.033) (Table S3). The second task was to predict whether a secondary or recall immune response was triggered by SARS-CoV-2 infection or by SARS-CoV-2 vaccination. The performance of manual gating, FlowSOM, and Deep CNN were characterized by mean AUROC values of 0.641 (SD 0.154), 0.579 (SD 0.151), and 0.520 (SD 0.163), respectively. In this task, cyMAE with global minimum pooling attained a mean AUROC of 0.668 (SD 0.157) (Table S4). Finally, we evaluated the ability to distinguish the pre- and post-treatment status of COVID-19 patients. Manual gating, FlowSOM, and Deep CNN showed mean AUROC values of 0.796 (SD 0.124), 0.869 (SD 0.112), and 0.531 (SD 0.201), respectively. In comparison, cyMAE with global max pooling achieved a mean AUROC of 0.887 (SD 0.114) (Table S5). The results of the three experiments suggest that cyMAE captures critical information overlooked by manual gating, FlowSOM, or Deep CNN, thereby enhancing the prediction of subject status across various tasks.
Figure 5.

Classification and prediction of COVID-19 outcomes using the cyMAE subject representations
From left to right, COVID-19 patient and healthy subject classification using the Acute2020 and Acute2021 dataset, secondary immune response against COVID-19 prediction using the Vaccine dataset, and COVID-19 pre- and post-treatment classification using the Acute2021 dataset. The number in parentheses is the sample size. All the experiments are conducted by 5-fold cross-validation repeating 10 times. The shade for each curve represents the variance of these experiments. Green dashed lines stand for performance of a random classifier.
cyMAE immunophenotypes provide immunological insights in subject status prediction
In addition to achieving performance improvements in subject status prediction, we aim to understand which cell immunophenotypes contribute to differentiating the subject groups. Our focus is on the COVID-19 pre- and post-treatment classification, where cyMAE showed notable performance gains over manual gating. We investigated which information, potentially missed by manual gating, led to these improvements. Initially, we examined the Pearson correlation between the cyMAE subject representations and manually gated features (cell population proportion) and found no noticeable correlations (Figure S14). Furthermore, when both cyMAE subject representations and manual gating information were used together for the prediction task, there was no significant performance improvement, suggesting that the cyMAE subject representations capture more than just cell proportional information, contributing to the enhanced performance.
To delve deeper into the subject representation at the cell level, we tracked the cell identities involved in the global pooling process (Figure 6A, see STAR Methods). Initially, we identified key components in the subject representations using Shapley additive explanations (SHAP).41 Subsequently, we traced back the cells contributing to these components and marked each relevant cell with a star. Cells contributing to post-treatment predictions were marked in blue, while those contributing to pre-treatment predictions were marked in red. We examined the distribution of these starred cells and randomly selected background cells (1,000 cells per FCS samples) in the cyMAE cell embedding space (Figures 6C–6E), exploring the ratio of starred to background cells across different regions (Figure 6F). The top 10 regions with the highest ratios were designated as regions of interest (ROIs) for further exploration (Figure 6G).
Figure 6.

Analysis of cell contributions to subject representations and their impact on COVID-19 pre- and post-treatment classification
(A) The process of tracking back from subject representations to cell-level contributions using global maximum or minimum pooling in cyMAE.
(B) Identification of key components in the subject representation using SHAP, differentiating post-treatment and pre-treatment predictions.
(C) Uniform manifold approximation and projection (UMAP)42 plots showing the distribution of the starred cells and randomly selected background cells in the cyMAE cell embedding space. Post-treatment associated cells (blue stars) are labeled as “pred = 1,” and pre-treatment associated cells (red cells) are labeled as “pred = 0.”
(D) The distribution of the all starred cells.
(E) The distribution of the background cells.
(F) A heatmap showing the ratios between the starred cells and background cells, with the top 10 ROIs highlighted.
(G) For each ROI, predominantly representing one or more specific cell types, the ratio of blue stars to red stars is analyzed using using the Fisher’s exact test, with false discovery rate-corrected p values. (∗∗∗) indicates p value <0.001; (ns) indicates p value >0.05.
The cells in the ROIs mainly contributed to forming the subject representations in our model and were primarily used for pre- and post-treatment predictions. Notably, the ROIs consisted of cell types such as eosinophils, neutrophils (in some areas), pDCs, plasmablasts, CD66negCD45lo, and IgGnegMemB. For a more detailed exploration, we found that in the ROI 1 and 4 (predominantly eosinophils), the counts of post-treatment associated cells (blue starred cells) and pre-treatment associated cells (red starred cells) are significantly different, with a higher number of blue starred cells. This finding indicates that a greater presence of eosinophils is more likely associated with post-treatment in cyMAE’s prediction.
Next, we analyzed the distribution differences in protein expression between blue starred cells and red starred cells within each ROI (Figure S15). We observed a few marker expression differences between pre-treatment and post-treatment predictions using the Kolmogorov-Smirnov test: (1) CD4 expression is increased in eosinophil/CD66negCD45lo cells (in ROI 4) after COVID-19 treatment and (2) CD3 expression is increased in some neutrophils (in ROI 10) after COVID-19 treatment. These observations indicate that our model can effectively highlight the significance of specific cell states, including cell types and protein expression, in predicting patient status in the context of COVID-19 treatment.
Discussion
Due to the popularity, ease, and relative affordability of cytometry experiments, there is an abundance of high-dimensional cytometry data compared to other single-cell modalities. Although manual gating remains the preferred classification approach, it becomes impractical for the expansive datasets of multi-cohort and/or multi-institutional studies due to its time-consuming and labor-intensive nature. Additionally, some clustering methods, which require loading all the data simultaneously, are not suitable for large-scale datasets due to memory constraints. On the other hand, the cyMAE method uses a mini-batch approach for processing large-scale datasets, where it breaks down the data into small, manageable segment. This approach reduces memory demands and improves training efficiency on large-scale data (see STAR Methods). For instance, cyMAE pre-training requires 3,184 MB of memory and 1,286 MB of GPU memory (with a batch size of 4,096), taking 64.7 h, while cyMAE fine-tuning requires 10,053 MB of memory and 7,916 MB of GPU memory (with a batch size of 16,384), taking 67.2 h. Despite taking up to longer than other models, cyMAE uses less memory (Figure S16). Even with larger training datasets, the mini-batch approach in cyMAE keeps memory usage similar, whereas the other models require more memory as data size increases. Also, cyMAE can quickly and accurately make inferences on new datasets designed with the exact same panel as the pre-training dataset, processing up to 15,276 cells per second for cell type annotation (see STAR Methods, Figure S16). In summary, we demonstrate that cyMAE is a fast, scalable, and efficient solution superior to existing methods for analyzing large-scale data.
To make the model directly applicable, we adopted a paradigm of training models and then using them to make inferences on a new dataset. In contrast, manual gating usually imports historical gates, which are then manually adjusted when necessary for each sample, a time-consuming and often error-prone approach. The alternative of simply using clustering approaches to discover sources of variability in each dataset independently can be difficult to scale and does not use a priori information on cell types. Supervised and semi-supervised learning approaches, including cyMAE, excel within train-inference paradigm. The main advantages of the train-inference paradigm are scalability and reproducibility: any investigator can apply the exact same model to any dataset that uses the same panel as the pre-training dataset, obtaining results that are easily interpretable within the biologically established framework of immunology. These results show that cyMAE outperforms alternative models within this paradigm.
Directly comparing learning without pre-training (from scratch) and with pre-training, performance improved not only in cell type annotation but also in the few-shot setting (Table S1; Figure 2D). This model was able to learn stably, while the from-scratch model was unable to learn effectively with little training data. While not a dramatic performance improvement, in the other experiments using the pre-trained cell embeddings, it was encouraging to see that the pre-trained embedding was good at learning antibody co-localization patterns, imputing unavailable protein expressions, and contributes meaningful performance gains for the subject-level predictions.
Here, we introduced cyMAE, a masked autoencoder model which builds latent embeddings of single-cell cytometry data and uses them to achieve good performance across a range of cell-level and subject-level tasks. Especially, the fine-tuned cyMAE is as accurate as manual gating, with the labor-free advantages of automated analysis. This study is a proof of concept for applying a combination of unsupervised and supervised analysis in the training-inference paradigm to multiple COVID-19 cytometry datasets that use the same panel. We envision creating a highly powerful model by including a more diverse population including multiple diseases in our training data in the future. This approach promises scalability across thousands of samples from multiple studies, providing robust and interpretable results while minimizing manual analysis.
Limitations of the study
In this study, we pre-trained our model using only one of the three available cohorts to evaluate the performance on several downstream tasks using all three cohorts. Future research will expand this approach by pre-training on a broader array of data from multiple studies, including more diverse subject phenotypes. This expansion is expected to enhance the model’s power and robustness, enabling it to more effectively distinguish between biological variations and gain a deeper understanding of protein functions and protein expression patterns. This, in turn, will lead to more accurate predictions in various downstream tasks. However, it is important to note that the fine-tuning of independent datasets must be designed using the exact same panel as the pre-training dataset to ensure consistency and accuracy.
This study has several limitations. First, while these models are trained only on CyTOF data, their application to flow cytometry data might not be recommended due to inherent technical differences. Specifically, the methodologies used in flow and mass cytometry yield disparate patterns of protein expression. Yet, a model like cyMAE, if pre-trained on flow cytometry data from scratch, could indeed become a viable approach for flow cytometry datasets. Second, we assume that cell type information from manual gating is the ground truth. However, this may not be the case in practice. Even the most skilled experts are prone to subjectivity and bias, which might lead to a bias toward “expected” results. This claim can be reinforced by our experimental results of the subject status prediction, where the pre-trained cyMAE showed higher predictive power than the manual gated features in some tasks (Figure 5). This observation raises the possibility that there may be information that manual gating misses. Related to this, the evaluation of cyMAE and the other computational methods was performed using only the subset of cells which have a well-defined, terminal label in the gating hierarchy, while ungated or partially gated cells were left out. This setting differs from a more realistic one in which subsets of cells cannot be left out but was chosen because it facilitates unambiguous evaluation of model predictions. Finally, the size and type of cytometry panels used in practice vary widely depending on the research purpose. This iteration of cyMAE was developed using one of the only pre-made, commercially available high-dimensional cytometry panels. While immunologists often change their cytometry panels, this is a known panel that does not change and can be used by any researcher. While our model was trained to work on data with fixed markers, it shows the potential for robust performance. For more meaningful research, it should work robustly for different panels in future studies, for examples, to accommodate data where only a subset of the markers has been measured. A related limitation is that cyMAE has only been validated on data acquired with one panel, due to the difficulty of obtaining multiple datasets that use the same panel and that are also manually annotated. This is also likely to be a limitation for users who wish to use cyMAE with their own choice of panel: the cost of manually annotating enough samples, spanning enough inter-subject variability, and then using them to train a new model make the most sense for users who intend to acquire data repeatedly with a fixed panel. Despite these limitations, this study demonstrates the high potential of pre-training in single-cell cytometry, both because an approach like ours has not been applied to cytometry data analysis before and because it shows advantages over previous methods.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dokyoon Kim (dokyoon.kim@pennmedicine.upenn.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The data presented within this study are available (see key resources table).
-
•
All original code has been deposited at GitHub and is publicly available as of the date of publication: https://github.com/JaesikKim/cyMAE. DOIs are listed in the key resources table (software and algorithms section of the key resources table).
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Acknowledgments
This work was partially supported by NIH AI082630, QuantumLeap Healthcare Collaborative fund, the University of Pennsylvania Perelman School of Medicine COVID Fund. We thank Takuya Ohtani and the Penn CyTOF Core at the University of Pennsylvania for data acquisition.
Author contributions
J.K., M.I., A.R.G., and D.K. conceived the study. J.K. and M.I. conducted analysis with the help of M.L., M.L.M., Y.N., M.M.P., J. Wagenaar., S.A.A., P.O., S.-H.J., and J. Woerner., and C.C., M.L., and M.L.M. provided critical comments for model development. A.P. conducted sample preparation, and D.T.N. annotated the data by manual gating. The following researchers recruited subjects, collected clinical data, and obtained and shipped biosamples for the I-SPY clinical trial: C.A.G.I., A.P.T., M.E., T.G.D., N.S.M., J.P.R., and N.J.M. from Perelman School of Medicine, University of Pennsylvania; C.S.C., K.D.L., M.A.M., and L.B.S. from the University of California, San Francisco, School of Medicine; E.L.B. and J.M. from the University of Colorado School of Medicine; S.G. and D.W.R. from the University of Alabama at Birmingham; and D.C.F., K.W.G., K.W.T., and H.B. from Wake Forest School of Medicine. J.K., M.I., A.R.G., and Y.N. provided graphical design of the figures. J.K. and M.I. wrote the manuscript, and D.M., V.T., A.R.G., D.K., and E.J.W. provided critical comments and valuable edits. A.R.G., D.K., and E.J.W. supervised the study.
Declaration of interests
E.J.W. is a member of the Parker Institute for Cancer Immunotherapy, which supports cancer immunotherapy research in his laboratory. E.J.W. is an advisor for Arsenal Biosciences, Coherus, Danger Bio, IpiNovyx, NewLimit, Marengo, Pluto Immunotherapeutics, Related Sciences, Santa Ana Bio, and Synthekine. E.J.W. is a founder of and holds stock in Coherus, Danger Bio, Prox Biosciences, and Arsenal Biosciences.
STAR★Methods
Key resources table
Experimental model and subject details
All subjects consented and enrolled with approval of the University of Pennsylvania Institutional Review Board (Vaccine IRB no. 844642; Acute2020 IRB no. 808542; Acute2021 IRB no. 843758). All participants or their surrogates provided informed consent in accordance with protocols approved by the regional ethical research boards and the Declaration of Helsinki.
For the Vaccine dataset, 43 individuals were enrolled for longitudinal monitoring of response to SARS-CoV-2 mRNA vaccine beginning in December 2020 through March 2021. All subjects received either Pfizer (BNT162b2) or Moderna (mRNA-1273) mRNA vaccines. Samples were collected at six time points: baseline, ∼2 weeks after primary immunization, day of secondary immunization, ∼1 week after secondary immunization, ∼3 months after primary immunization, and ∼6 months after primary immunization. Participants were self-reported healthy without ongoing chronic health conditions. In the Vaccine dataset, the definition of secondary immune response was defined as follows. We labeled a secondary immune response as “Yes” if it occurred after a healthy person received two vaccines, or after a person with COVID-19 received one vaccine, or after a person with COVID-19 received two vaccines. If a healthy person received a single vaccine, we labeled it “No”.
For the Acute2020 dataset, patients were consented and enrolled within 3 days of admission to the Hospital of the University of Pennsylvania with a positive SARS-CoV-2 PCR test, regardless of the oxygen support needed. Clinical data were abstracted from the electronic medical record into standardized case report forms. All subjects in this dataset were consented and enrolled between March and December 2020 at the University of Pennsylvania. Subjects in the Acute2021 dataset were enrolled in the I-SPY COVID-19 Trial.32 Hospitalized participants at 5 trial sites (Penn, University of Alabama Birmingham, University of California San Francisco, University of Colorado, and Wake Forest University Atrium Health) with confirmed SARS-CoV-2 PCR or antigen testing and requiring greater than 6 L per minute oxygen flow (including high flow nasal oxygen, high flow face mask oxygen, non-invasive ventilation, or invasive mechanical ventilation consistent with World Health Organization ordinal scale ≥5) for fewer than 72 h were enrolled in this trial. Patients or their legally authorized representatives consented to be randomized to receive a backbone treatment (remdesivir and dexamethasone) alone versus backbone with one of 12 investigational treatments. Details of the trial inclusion and exclusion criteria, and the non-backbone treatment arms have been published at https://clinicaltrials.gov/study/NCT04488081. Whole blood was collected at time of admission and 7 days later. Samples from subjects enrolled at the University of Pennsylvania were processed on the day of collection. Samples from subjects enrolled at the University of Alabama at Birmingham, University of Colorado, University of California at San Francisco, and Wake Forest University were shipped to the University of Pennsylvania and processed the day of arrival.
Method details
Mass cytometry
For all samples, 270μL of whole blood were stained using the MaxPar Direct Immune Profiling Assay (Standard BioTools, Inc, South San Francisco, CA).47 For the Acute2020 dataset, samples were stained in accordance with manufacturer protocols. Briefly, whole blood was added to a 5mL tube containing a pellet of lyophilized antibodies. Blood was incubated at room temperature for 30 min and then lysed with Cal-Lyse lysing solution Standard BioTools, Inc, South San Francisco, CA). Cells were washed, followed by fixation with 1.6% PFA. Cells sat at 4°C over night prior to staining with Cell-ID Intercalator-Ir. These samples are referred to as “fresh” because they did not undergo cryopreservation and thawing. Vaccine and Acute2021 datasets underwent a similar workflow as described above. However, after incubating for 30 min in the tube of lyophilized antibodies, stained whole blood was fixed with PROT1 buffer (Smart Tube Inc, Las Vegas, NV) and cryopreserved. Lyse, wash, and intercalator staining were performed as above after thaw. Stained samples were collected on a CyTOF 2 instrument with EQ4 beads (four element calibration beads, Standard BioTools, Inc).
After data acquisition, .fcs files were gated to remove beads, debris, doublets, and dead cells using the OMIQ platform (Boston, MA); representative gates are shown in Figure S18. After gating, DNA intercalator, viability, Gaussian and bead channels were dropped, and the remaining protein expression channels were transformed using inverse hyperbolic sine with a cofactor of 5.
Model details
Transformer block
The transformer block consists of alternating layers of multihead self-attention (MSA) and multilayer perceptron (MLP) blocks (Equations 1 and 2). Layer norm (LN)48 is applied before every block, and Drop path (DP)49 is applied after every block. The MLP contains two linear layers with GELU activation function.
| (Equation 1) |
| (Equation 2) |
where denotes output embeddings of the -th layer and input embeddings of the -th layer at the same time.
Multi-head self-attention
In the multi-head self-attention (MSA) layer, we compute query, key, and value matrix () from the input embeddings () for each head (Equation 3) and compute heads by weighted sum of all values by attention weight for each head, where attention weight is calculated by the pairwise similarity between two elements of the input and their respective query and key representations (Equation 4). Finally, heads are concatenated, and the output is linearly projected (Equation 5).
| (Equation 3) |
where is input embeddings is learnable weight matrix, and is set to .
| (Equation 4) |
| (Equation 5) |
where , and is linear weight matrix.
Cytometry masked autoencoder
The whole structure consists of an encoder and a decoder, which are used in the pre-training step. The encoder is only then used with a single linear layer in the downstream supervised fine-tuning. The encoder () consists of 6 layers of transformer blocks, each with 6 heads and 30 hidden dimensions, totaling 67,890 parameters. In contrast, the decoder () is smaller than the encoder. It consists of 2 layers of transformer blocks with 3 heads and 15 hidden dimensions for a total of 5,806 parameters. The dimension sizes of latent cell or subject representations for the downstream tasks are 900 and 30 each.
Kaplan et al.50 provided several insights into the training of neural language models, one of which is particularly interesting: if infinite training data is available, the model’s capacity increases with its size. However, with limited training resources, performance changes with model size are not as significant. Our cytometry data is notably smaller, approximately million tokens, compared to existing large language models (normally over billion tokens). Thus, beyond a certain size, increasing our model’s size offers minimal performance advantage. Empirically, we explored the model size needed to achieve sufficient performance in the cell type annotation. We varied the latent dimension size of a marker’s expression and the layer depth, comparing these under the cell type annotation task (Figure S19). Consistent with Kaplan et al.'s findings, we discovered that beyond 69,316 parameters (for the encoder and classifier), performance stabilizes. Additionally, overall model parameters had stronger correlation with performance than detailed parameters like layer depth and latent dimension size.
Training details
Masked Cytometry Modeling (MCM)
cyMAE learns to maximize
| (Equation 6) |
for cell , where denotes masked protein expressions of cell , and denotes masked protein embeddings after masking. is a masking ratio, is the number of proteins in the data, and is a hidden dimension size. Likewise, denotes unmasked protein expressions of cell , and denotes unmasked protein embeddings.
The encoder () generates a latent representation of the cell. The unmasked latent representation of cell is defined as as the following,
| (Equation 7) |
where is sine-cosine positional embeddings for masked proteins. The idea of the concatenation () of protein embeddings with expression values was inspired from MET.51
The decoder () reconstructs the masked values as followings,
| (Equation 8) |
Let denote a learnable mask token embedding represented as a row vector . We construct a matrix by stacking this vector times, such that the resulting matrix has dimensions . is sine-cosine positional embeddings. To calculate the reconstruction loss, we use Mean squared error (MSE) loss for all cells,
| (Equation 9) |
Why positional embedding is necessary
It might seem that positional embedding is not necessary because the input is a tabular data. However, the position serves as an index to indicate which protein’s expression value should be reconstructed by the decoder during MCM. For example, 2nd, 3rd, and 7th proteins of 10 proteins are masked, positional embedding provides information to reconstruct the expression of the 2nd, 3rd, and 7th proteins. Therefore, when using cyMAE, users make sure to match the order of the proteins.
Cell representation
After pre-training, the cell representations of cell is obtained through the pre-trained encoder without masking protein expressions (masking ratio ). We propose two versions of cell representations: and as follows:
| (Equation 10) |
| (Equation 11) |
| (Equation 12) |
where is an output of the encoder, is the flatten version of , and is the mean pooling of . Depending on downstream analysis, these cell representations are used as input of a linear layer for cell-level downstream tasks.
Subject representation
The subject representation is obtained by applying multiple global pooling methods across the cell representations (full version) , where is the number of cells in a single subject data:
-
•
Global mean pooling
| (Equation 13) |
-
•
Global sum pooling
| (Equation 14) |
-
•
Global max pooling
| (Equation 15) |
-
•
Global min pooling
| (Equation 16) |
Then, this subject representation is used as input of a linear layer for subject-level supervised downstream tasks.
| (Equation 17) |
Supervised learning in downstream tasks
Cross entropy loss is employed for classification tasks and Mean squared error (MSE) loss is employed for regression tasks.
In the cell type annotation task,
| (Equation 18) |
| (Equation 19) |
where and indicate the ground truth cell type and the predicted probability for cell type of cell , respectively.
In the imputation task,
| (Equation 20) |
| (Equation 21) |
where and denote the ground truth expression value and the predicted value of masked protein , respectively.
In the subject-level prediction tasks,
| (22) |
| (23) |
where and are the ground truth label and predicted probability for label of subject , respectively.
Impact of masking ratio during pre-training
To test if masking ratio affects cyMAE training, we trained three different versions of the model with masking ratios of 0.25, 0.5, and 0.75. The result was there was no big significant difference in performance except for ratio = 0.5 on the cell type annotation task (Figure S19). Therefore, all the cyMAE experiments were performed with a 0.25 masking ratio.
Training setting
The configuration of pre-training includes a batch size of 4,096, drop path regularization of 0.1, AdamW optimizer with momentum of 0.9 and weight decay of 0.05, learning rate of 1.5e−5 with a cosine scheduler and masking ratio of 0.25. The fine-tuning (for cell type annotation) is also the same except for a batch size of 16,384, learning rate of 1e−4, and label smoothing.
Computational cost in training and inference
A cost comparison with other models is available in Figure S16. The cyMAE pre-training required around 3 days with a single NVIDIA A100 Tensor Core GPU to process 6.5 million cells through 200 epochs. Fine-tuning cyMAE for cell type annotation took 2 to 3 days on the same machine to process 29.4 million cells through 200 epochs. Although cyMAE pre-training and fine-tuning may take longer, they require considerably less memory compared to other models such as GBDT. Memory usage depends on the batch size. Specifically, cyMAE pre-training, with a batch size of 4,096, used 3,184 MB of CPU memory and 1,286 MB of GPU memory, compared to GBDT, which uses over 25,000 MB of memory. The fine-tuning process, with a batch size of 16,384, used 10,053 MB of CPU memory and 7,916 MB of GPU memory. The batch size can be adjusted according to the GPU performance available.
For inference, the runtime was 569 s for 7.3 million cells under the Vaccine test dataset, 1,066 s for 11.9 million cells under the Acute2021 dataset, and 721 s for 6.5 million cells under the Acute2020 dataset on the same GPU machine. This indicates that cyMAE, when fine-tuned, processes up to 15,276 cells per second for cell type annotation, demonstrating its efficiency in practical applications. While cyMAE might be slightly slower than some other models during inference (Figure S17), it is not expected to cause significant inconvenience in practical use. The efficiency and scalability of cyMAE during inference make it a valuable tool for large-scale cytometry data analysis.
If a different panel were used for pre-training from scratch, similar costs in terms of memory and time would be expected for datasets of similar size. Larger datasets would require more time, scaling linearly, while memory usage would remain similar due to the mini-batch approach. Systems with less powerful GPUs might experience longer processing times.
Benchmarking models
Manual gating
Each sample from all datasets was manually gated using the OMIQ platform to obtain the 46 terminal populations used as ground truth labels. Representative gates from our strategy are shown in Figure S3.
Static gating
For each gate in our hierarchy, we aggregated the candidate gate positions from all training samples in the Vaccine dataset into one consensus gate. By definition, a point is in the consensus gate if it falls into at least 30% of all the candidate gates (Figure S4). We then created a consensus hierarchy out of all consensus gates and applied it statically to all test samples.
FlowSOM clustering
The same 60% of the Vaccine data samples were used to train an unsupervised FlowSOM clustering model. Version 2.6.0 of the FlowSOM R package was used with default parameters, except for the total number of metaclusters, which we set to 46 to match the number of ground truth labels. As an unsupervised clustering algorithm, FlowSOM does not have an inference mode. We performed inference on testing datasets (20% of the Vaccine dataset as an internal test set, and the two external test sets) by assigning each datapoint to the nearest SOM node from the trained model, and preserving the assignment of nodes to metaclusters from the training phase. Evaluation of accuracy and balanced accuracy required the extra information of a bipartite matching between the 46 FlowSOM clusters and the 46 ground truth labels. Following Weber, L. M. et al.,52 we obtained the matching using the Hungarian algorithm, implemented in the function solve_LSAP of the R package clue.
CyAnno
CyAnno is a semi-automated machine learning approach for annotating cell types in mass cytometry datasets, specifically addressing the challenge of ungated cells—those not classified during manual gating. By integrating these ungated cells into its modeling process, CyAnno significantly enhances the precision of cell type predictions compared to existing methods like DeepCyTOF and LDA, which often misclassify ungated cells. We used the CyAnno implementation at https://github.com/abbioinfo/CyAnno for our analysis.
Gradient boosting decision tree (GBDT)
We used XGBoost36 python package for GBDT. We ran XGBoost classifier with 100 estimators. For hyperparameter tuning, we searched across following ranges:
max depth: [3, 4, 5], learning rate: [0.01, 0.03, 0.1], and subsampling ratio: [0.8, 0.9, 1]. Also, we set early stopping based on the performance change for the validation set.
Fully connected deep neural network (DNN)
DGCyTOF (Cheng, L. et al.34) is a method that sequentially utilizes deep learning classification, graphic clustering, and dimension reduction to discover new cell populations. In this process, DNN is employed for cell type identification. DeepCyTOF (Li, H et al.35) is a cell type annotator that first performs denoising and domain adaptation to transform the data into a new feature space. It then uses a single labeled sample to train a cell annotation model. It also utilizes a DNN as the cell classifier. Given that DNNs have been actively used as cell type annotators, we used a 3-layer DNN as a comparative model for cell type annotation.
Convolutional neural network (CNN)
Hu. Z et al.8 proposed Deep CNN using convolutional neural network for cytomegalovirus (CMV) classification. As the authors guided, we first subsampled 10,000 cells per fcs file and ran the CNN model with the hyperparameters provided by the authors to the subject-level tasks. Also we compared it to the cell type annotation task with some modifications in model architecture. Since it uses a CNN structure to draw cell representations and pool them, we modified to the same architecture without the pooling layer as a comparison model.
Protein embeddings
After pre-training through MCM, the trained in cyMAE represents protein embeddings for proteins. It is expected that they have protein information about the heterogeneous, complex, and dynamic immune cells without any prior information, only through learning on the data itself. This was confirmed by the PCA 2-dimensional plot in Figure 2A.
Few-shot learning setting in the cell type annotation
For N-shots, we trained using only the first s samples per class in the training and validation sets and then evaluated on the entire test set. We compared performance for 5, 10, 15, and 20 shots.
Imputation
We masked CD45RO, CD45RA, CD27, CD28, TCRgd, CD197, and CD127 expressions and used the remaining markers to predict the expression of these seven marker expressions. Infinity Flow used GBDT as the imputer. Similarly, the unmasked cell representations were first generated through the pre-trained cyMAE and used as input to GBDT to train and then evaluated on the external test sets (not end-to-end).
Attention score
From Equation 4, we first obtain the output of function for the interpretation for cell (Equation 24) and calculate the attention score by averaging over the query axis (Equation 25).
| (Equation 24) |
| (Equation 25) |
In our experiments, we sampled 2% of all cells for each dataset. To calculate the attention score, we only used the information from the first layer, because the first layer is the most influential in determining which inputs to give attention to, and there was no significant difference in attention between inputs after the second layer.
Uniform Manifold Approximation and Projection for dimension reduction (UMAP)
In Figures 6C–6G, the cyMAE cell representation was visualized in two dimensions by reducing the dimensionality using UMAP. The UMAP space is represented as
| (Equation 26) |
Identifying the most contributing cells to subject representations
We obtained cell representations for each cell in a subject FCS file using the cyMAE encoder. Subsequently, we derived subject representations by applying global max/min pooling. By tracing back the pooling process, we can easily determine which cells significantly contribute to the subject representation (Figure 6A). Specifically, for global max pooling, we identified the cell with the maximum value for each component and retain that cell. Similarly, for global min pooling, we found the cell with the minimum value for each component and retain that cell. This process identifies the relevant cells for each of the 900 components in the subject representation. We focused on the top 30 task-specific important components (identified via SHAP) in the subject representation and identified the cells that contributed to these components during the pooling process. For example, If the identified cells do not overlap, we obtain 30 cells from a single subject. We then use this approach to the COVID-19 pre-treatment or post-treatment classification task to identify post-treatment associated cells and pre-treatment associated cells.
SHapley additive exPlanations (SHAP)
SHAP41 is a well-known and popular explainable method for machine learning models based on a game theoretic approach. SHAP values measure the relative contribution of features to specific model outputs on a per-instance basis. We applied SHAP to the test set of each fold in a subject status prediction task to determine which components of the subject representation were important for the given task (Figure 6B).
Quantification and statistical analysis
Metrics
Balanced accuracy (Bacc)
For a multi-class imbalanced dataset, we used Balanced accuracy (Bacc) instead of Accuracy. Balanced accuracy is defined as a macro-average of recall scores per class in a multi-class classification.
A Recall score is defined as:
| (Equation 27) |
where TP is true positive, and FN is false negative.
R-squared
In a regression task, if is the predicted value of the -th sample and is the corresponding true value for total samples, the R-squared is defined as:
| (Equation 28) |
where .
AUROC
A receiver operating characteristic (ROC) curve is widely used for evaluating prediction models. It plots True Positive Rate (TPR) against False Positive Rate (FPR).
| (Equation 29) |
| (Equation 30) |
Where TP, FP, TN, and FN are the number of true positives, false positives, true negatives, and false negatives respectively. AUROC stands for the area under the ROC curve.
Fisher’s exact test
Fisher’s exact test was used to determine if the counts of blue starred cells (post-treatment associated cells) and red starred cells (pre-treatment associated cells) are significantly different based on the total blue and red starred cells in Figure 6G. The p-values for all tests were corrected using false discovery rate (FDR) correction.
Kolmogorov-Smirnov test
The Kolmogorov-Smirnov test was used to determine if the distributions of each marker for blue starred cells and red starred cells are drawn from the same underlying distribution in Figure S15. The p-values for all tests were corrected using false discovery rate (FDR) correction.
Published: November 7, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101808.
Contributor Information
Allison R. Greenplate, Email: allie.greenplate@pennmedicine.upenn.edu.
E. John Wherry, Email: wherry@pennmedicine.upenn.edu.
Dokyoon Kim, Email: dokyoon.kim@pennmedicine.upenn.edu.
Supplemental information
References
- 1.Maecker H.T., McCoy J.P., Nussenblatt R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 2012;12:191–200. doi: 10.1038/nri3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mair F., Hartmann F.J., Mrdjen D., Tosevski V., Krieg C., Becher B. The end of gating? An introduction to automated analysis of high dimensional cytometry data. Eur. J. Immunol. 2016;46:34–43. doi: 10.1002/eji.201545774. [DOI] [PubMed] [Google Scholar]
- 3.Olsen L.R., Leipold M.D., Pedersen C.B., Maecker H.T. The anatomy of single cell mass cytometry data. Cytometry. 2019;95:156–172. doi: 10.1002/cyto.a.23621. [DOI] [PubMed] [Google Scholar]
- 4.Van Gassen S., Callebaut B., Van Helden M.J., Lambrecht B.N., Demeester P., Dhaene T., Saeys Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A. 2015;87:636–645. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 5.Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir E.a.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R., et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spitzer M.H., Gherardini P.F., Fragiadakis G.K., Bhattacharya N., Yuan R.T., Hotson A.N., Finck R., Carmi Y., Zunder E.R., Fantl W.J., et al. IMMUNOLOGY. An interactive reference framework for modeling a dynamic immune system. Science. 2015;349 doi: 10.1126/science.1259425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samusik N., Good Z., Spitzer M.H., Davis K.L., Nolan G.P. Automated mapping of phenotype space with single-cell data. Nat. Methods. 2016;13:493–496. doi: 10.1038/nmeth.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Z., Tang A., Singh J., Bhattacharya S., Butte A.J. A robust and interpretable end-to-end deep learning model for cytometry data. Proc. Natl. Acad. Sci. USA. 2020;117:21373–21380. doi: 10.1073/pnas.2003026117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee H.C., Kosoy R., Becker C.E., Dudley J.T., Kidd B.A. Automated cell type discovery and classification through knowledge transfer. Bioinformatics. 2017;33:1689–1695. doi: 10.1093/bioinformatics/btx054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z., Luo D., Zhong X., Choi J.H., Ma Y., Wang S., Mahrt E., Guo W., Stawiski E.W., Modrusan Z., et al. SCINA: A Semi-Supervised Subtyping Algorithm of Single Cells and Bulk Samples. Genes. 2019;10 doi: 10.3390/genes10070531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdelaal T., van Unen V., Höllt T., Koning F., Reinders M.J.T., Mahfouz A. Predicting Cell Populations in Single Cell Mass Cytometry Data. Cytometry A. 2019;95:769–781. doi: 10.1002/cyto.a.23738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaushik A., Dunham D., He Z., Manohar M., Desai M., Nadeau K.C., Andorf S. CyAnno: a semi-automated approach for cell type annotation of mass cytometry datasets. Bioinformatics. 2021;37:4164–4171. doi: 10.1093/bioinformatics/btab409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser Ł., Polosukhin I. Attention is All you Need. Adv. Neural Inf. Process. Syst. 2017;30:6000–6010. [Google Scholar]
- 14.Devlin J., Chang M.-W., Lee K., Toutanova K. In: Human Language Technologies. Burstein J., Doran C., Soloria T., editors. Vol. 1. Association for Computational Linguistics; 2019. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics; pp. 4171–4186. [DOI] [Google Scholar]
- 15.Brown T., Mann B., Ryder N., Subbiah M., Kaplan J.D., Dhariwal P., Neelakantan A., Shyam P., Sastry G., Askell A., et al. Language Models are Few-Shot Learners. Adv. Neural Inf. Process. Syst. 2020;33:1877–1901. [Google Scholar]
- 16.Oquab M., Darcet T., Moutakanni T., Vo H., Szafraniec M., Khalidov V., Fernandez P., Haziza D., Massa F., El-Nouby A., et al. DINOv2: Learning Robust Visual Features without Supervision. arXiv. 2023 doi: 10.48550/arXiv.2304.07193. [DOI] [Google Scholar]
- 17.Bao, H., Dong, L., Piao, S., and Wei, F. (2022). BEiT: BERT Pre-Training of Image Transformers. International Conference on Learning Representations.
- 18.He K., Chen X., Xie S., Li Y., Dollár P., Girshick R. Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR); 2022. Masked Autoencoders Are Scalable Vision Learners; pp. 16000–16009. [DOI] [Google Scholar]
- 19.Madani A., Krause B., Greene E.R., Subramanian S., Mohr B.P., Holton J.M., Olmos J.L., Jr., Xiong C., Sun Z.Z., Socher R., et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 2023;41:1099–1106. doi: 10.1038/s41587-022-01618-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elnaggar A., Heinzinger M., Dallago C., Rehawi G., Wang Y., Jones L., Gibbs T., Feher T., Angerer C., Steinegger M., et al. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. Ieee T Pattern Anal. 2022;44:7112–7127. doi: 10.1109/Tpami.2021.3095381. [DOI] [PubMed] [Google Scholar]
- 21.Lin Z., Akin H., Rao R., Hie B., Zhu Z., Lu W., Smetanin N., Verkuil R., Kabeli O., Shmueli Y., et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science. 2023;379:1123–1130. doi: 10.1126/science.ade2574. [DOI] [PubMed] [Google Scholar]
- 22.Hie B.L., Shanker V.R., Xu D., Bruun T.U.J., Weidenbacher P.A., Tang S., Wu W., Pak J.E., Kim P.S. Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol. 2024;42:275–283. doi: 10.1038/s41587-023-01763-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shanehsazzadeh A., McPartlon M., Kasun G., Steiger A.K., Sutton J.M., Yassine E., McCloskey C., Haile R., Shuai R., Alverio J., et al. Unlocking de novo antibody design with generative artificial intelligence. bioRxiv. 2023 doi: 10.1101/2023.01.08.523187. [DOI] [Google Scholar]
- 24.Eguchi R.R., Choe C.A., Parekh U., Khalek I.S., Ward M.D., Vithani N., Bowman G.R., Jardine J.G., Huang P.-S. Deep Generative Design of Epitope-Specific Binding Proteins by Latent Conformation Optimization. bioRxiv. 2022 doi: 10.1101/2022.12.22.521698. [DOI] [Google Scholar]
- 25.Cui H., Wang C., Maan H., Pang K., Luo F., Wang B. scGPT: Towards Building a Foundation Model for Single-Cell Multi-omics Using Generative AI. bioRxiv. 2023 doi: 10.1101/2023.04.30.538439. [DOI] [PubMed] [Google Scholar]
- 26.Gong J., Hao M., Zeng X., Liu C., Ma J., Cheng X., Wang T., Zhang X., Song L. xTrimoGene: An Efficient and Scalable Representation Learner for Single-Cell RNA-Seq Data. bioRxiv. 2023 doi: 10.1101/2023.03.24.534055. [DOI] [Google Scholar]
- 27.Yang F., Wang W., Wang F., Fang Y., Tang D., Huang J., Lu H., Yao J. scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data. Nat. Mach. Intell. 2022;4:852–866. doi: 10.1038/s42256-022-00534-z. [DOI] [Google Scholar]
- 28.Chen J., Xu H., Tao W., Chen Z., Zhao Y., Han J.D.J. Transformer for one stop interpretable cell type annotation. Nat. Commun. 2023;14:223. doi: 10.1038/s41467-023-35923-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen H., Liu J., Hu J., Shen X., Zhang C., Wu D., Feng M., Yang M., Li Y., Yang Y., et al. Generative pretraining from large-scale transcriptomes for single-cell deciphering. iScience. 2023;26 doi: 10.1016/j.isci.2023.106536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minsheng H., Jing G., Zeng X., Liu C., Guo Y., Cheng X., Wang T., Ma J., Song L., View ORCID ProfileXuegong Zhang Large Scale Foundation Model on Single-cell Transcriptomics. bioRxiv. 2023 doi: 10.1101/2023.05.29.542705. [DOI] [PubMed] [Google Scholar]
- 31.Theodoris C.V., Xiao L., Chopra A., Chaffin M.D., Al Sayed Z.R., Hill M.C., Mantineo H., Brydon E.M., Zeng Z., Liu X.S., Ellinor P.T. Transfer learning enables predictions in network biology. Nature. 2023;618:616–624. doi: 10.1038/s41586-023-06139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.I-SPY COVID Consortium Report of the first seven agents in the I-SPY COVID trial: a phase 2, open label, adaptive platform randomised controlled trial. EClinicalMedicine. 2023;58 doi: 10.1016/j.eclinm.2023.101889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D'Andrea K., et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369 doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng L., Karkhanis P., Gokbag B., Liu Y., Li L. DGCyTOF: Deep learning with graphic cluster visualization to predict cell types of single cell mass cytometry data. PLoS Comput. Biol. 2022;18 doi: 10.1371/journal.pcbi.1008885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H., Shaham U., Stanton K.P., Yao Y., Montgomery R.R., Kluger Y. Gating mass cytometry data by deep learning. Bioinformatics. 2017;33:3423–3430. doi: 10.1093/bioinformatics/btx448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen T., Guestrin C. XGBoost: A Scalable Tree Boosting System. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. Association for Computing Machinery. 2016 doi: 10.1145/2939672.293978. [DOI] [Google Scholar]
- 37.Becht E., Tolstrup D., Dutertre C.A., Morawski P.A., Campbell D.J., Ginhoux F., Newell E.W., Gottardo R., Headley M.B. High-throughput single-cell quantification of hundreds of proteins using conventional flow cytometry and machine learning. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abg0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vig J. A Multiscale Visualization of Attention in the Transformer Model. Proceedings of the 57th Annual Meeting of the Association for Computational Linguistics: System Demonstrations. 2019:37–42. [Google Scholar]
- 39.Lowenberg B., Downing J.R., Burnett A. Acute myeloid leukemia. N. Engl. J. Med. 1999;341:1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 40.Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lundberg S.M., Lee S.-I. Curran Associates Inc; 2017. A Unified Approach to Interpreting Model Predictions. Proceedings of the 31st International Conference on Neural Information Processing Systems; pp. 4768–4777. [Google Scholar]
- 42.McInnes L., Healy J., Saul N., Großberger L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018;3:861. doi: 10.21105/joss.00861. [DOI] [Google Scholar]
- 43.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Müller A., Nothman J., Louppe G., et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. doi: 10.48550/arXiv.1201.0490. [DOI] [Google Scholar]
- 44.Yurtsev E., Friedman J., Gore J. 2015. FlowCytometryTools. Zenodo. [DOI] [Google Scholar]
- 45.Seabold S., Perktold J. Statsmodels: econometric and statistical modeling with python. SciPy. 2010;7:1. [Google Scholar]
- 46.Wickham H., Averick M., Bryan J., Chang W., François R., François R., Grolemund G., Hayes A., Henry L., Hester J., et al. Welcome to the tidyverse. J. Open Source Softw. 2019;4:1686. doi: 10.21105/joss.01686. [DOI] [Google Scholar]
- 47.Geanon D., Lee B., Gonzalez-Kozlova E., Kelly G., Handler D., Upadhyaya B., Leech J., De Real R.M., Herbinet M., Magen A., et al. A streamlined whole blood CyTOF workflow defines a circulating immune cell signature of COVID-19. Cytometry A. 2021;99:446–461. doi: 10.1002/cyto.a.24317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ba J.L., Kiros J.R., Hinton G.E. Layer Normalization. arXiv. 2016 doi: 10.48550/arXiv.1607.06450. [DOI] [Google Scholar]
- 49.Larsson G., Maire M., Shakhnarovich G. FractalNet: Ultra-Deep Neural Networks without Residuals. International Conference on Learning Representations. 2016 doi: 10.48550/arXiv.1605.07648. [DOI] [Google Scholar]
- 50.Kaplan J., McCandlish S., Henighan T., Brown T.B., Chess B., Child R., Gray S., Radford A., Wu J., Amodei D. Scaling Laws for Neural Language Models. arXiv. 2020 doi: 10.48550/arXiv.2001.08361. [DOI] [Google Scholar]
- 51.Majmundar K.A., Goyal S., Netrapalli P., Jain P. MET: Masked Encoding for Tabular Data. NeurIPS 2022 First Table Representation Workshop. 2022 doi: 10.48550/arXiv.2206.08564. [DOI] [Google Scholar]
- 52.Weber L.M., Robinson M.D. Comparison of clustering methods for high-dimensional single-cell flow and mass cytometry data. Cytometry A. 2016;89:1084–1096. doi: 10.1002/cyto.a.23030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The data presented within this study are available (see key resources table).
-
•
All original code has been deposited at GitHub and is publicly available as of the date of publication: https://github.com/JaesikKim/cyMAE. DOIs are listed in the key resources table (software and algorithms section of the key resources table).
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.