

Abstract
Introduction
Risankizumab has demonstrated superior efficacy compared to other psoriasis treatments, including secukinumab, adalimumab, and ustekinumab; switching to risankizumab from other psoriasis treatments has shown superior clinical and quality of life (QoL) outcomes. We evaluated the efficacy and safety of directly switching patients with moderate-to-severe plaque psoriasis and a suboptimal response to interleukin (IL)-17 inhibitors (secukinumab or ixekizumab) to risankizumab.
Methods
This 52-week, phase 3b study enrolled patients (≥ 18 years) with moderate-to-severe plaque psoriasis who had previously been treated with the recommended dose of secukinumab or ixekizumab for ≥ 6 months but did not achieve an optimal response (static Physician's Global Assessment [sPGA] 2/3; body surface are [BSA] 3– < 10%). Patients received subcutaneous risankizumab (150 mg) without washout. The primary endpoint was the proportion of patients achieving sPGA of 0/1 at week 16. Secondary endpoints included sPGA 0/1 at week 52, sPGA 0, Dermatology Life Quality Index (DLQI) 0/1, and Psoriasis Symptoms Scale (PSS) 0 at weeks 16 and 52. Safety was monitored throughout the study.
Results
The study included 244 patients. sPGA 0/1 was achieved by 57.4% and 62.3% at week 16 and 52. At week 16, sPGA 0, DLQI 0/1, and PSS 0 were achieved by 20.5%, 40.2%, and 20.9%, respectively. At week 52, these proportions increased to 27.1% for sPGA 0, 47.2% for DLQI 0/1, and 27.5% for PSS 0. Most frequently reported adverse events (reported in ≥ 5% of patients) in risankizumab-treated patients were COVID-19 infection (8.6%) and nasopharyngitis (5.7%). No new safety signals were observed.
Conclusions
Directly switching to risankizumab improved outcomes and QoL in patients with moderate-to-severe psoriasis who had suboptimal responses to anti-IL-17 inhibitors (secukinumab or ixekizumab). The safety results are consistent with previously reported safety of risankizumab. This study supports the efficacy of risankizumab in patients previously treated with biologics, including IL-17 inhibitors, and suggests a direct switch to risankizumab for improved clinical outcomes and QoL.
Clinical Trials
ClinicalTrials.gov identifier: NCT04102007.
Graphical Abstract
Supplementary Information
The online version contains supplementary material available at 10.1007/s13555-024-01292-z.
Keywords: Psoriasis, Efficacy, Safety, Risankizumab
Key Summary Points
| Why carry out this study? |
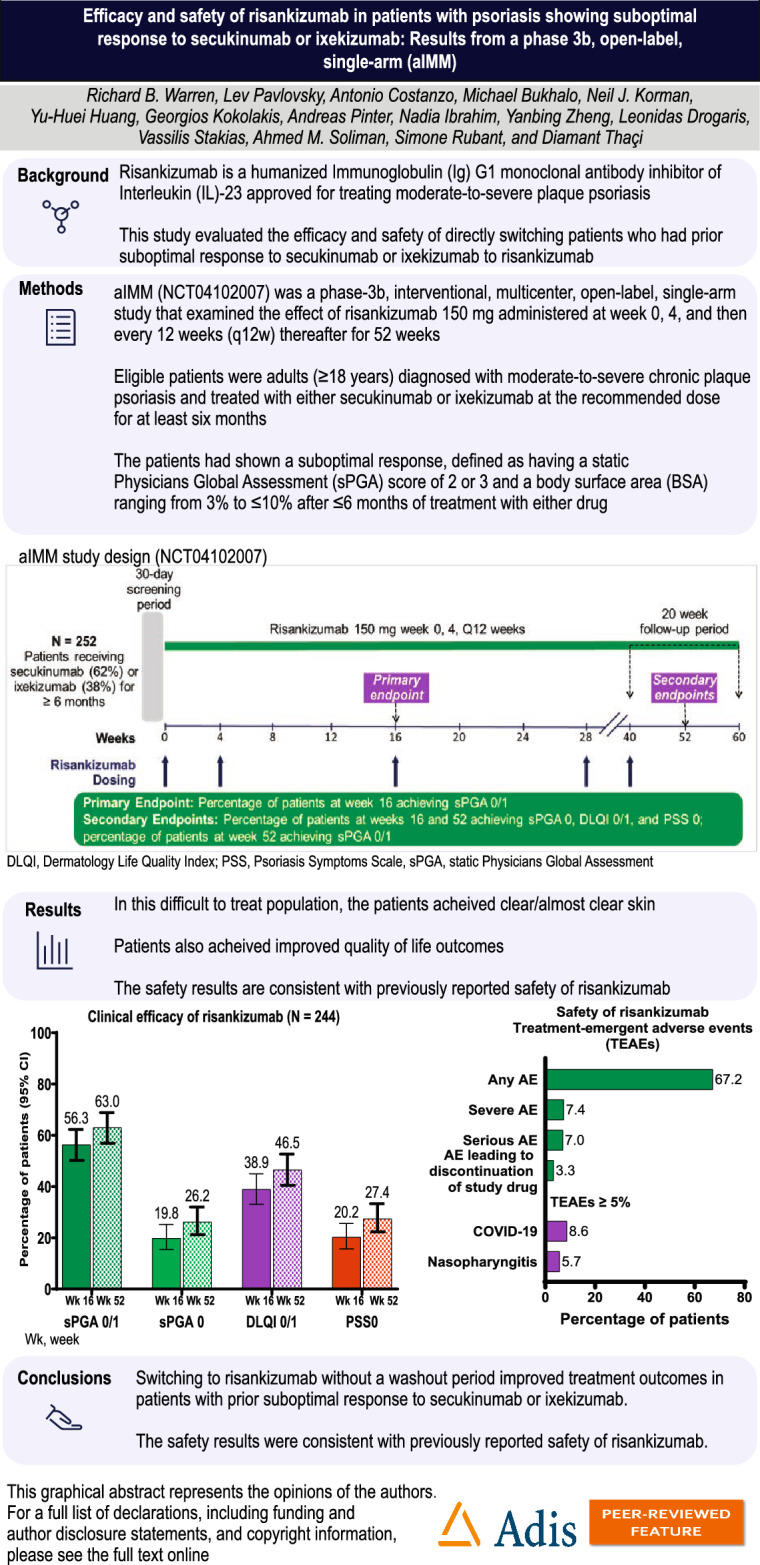
| Risankizumab is a humanized immunoglobulin (Ig) G1 monoclonal antibody inhibitor of interleukin (IL)-23 approved for treating moderate-to-severe plaque psoriasis. This study evaluated the efficacy and safety of directly switching patients who had prior suboptimal response to secukinumab or ixekizumab to risankizumab. |
| What was learned from this study? |
| Among patients with suboptimal response to secukinumab or ixekizumab, switching to risankizumab improved outcomes in patients at weeks 16 and 52. |
| In this difficult-to-treat group, where all patients had prior experience with biologics, 62.3% achieved clear/almost clear skin with risankizumab. |
| Switching to risankizumab without a washout period improved treatment outcomes in patients with prior suboptimal response to secukinumab or ixekizumab. The safety results were consistent with previously reported safety of risankizumab. |
Digital Features
This article is published with digital features, including a graphical abstract, to facilitate understanding of the article. To view digital features for this article go to 10.6084/m9.figshare.27135831.
Introduction
Psoriasis is a common chronic, immune-mediated inflammatory skin condition that affects approximately 100 million people worldwide [1]. Patients with plaque psoriasis are impacted by physical, psychological, social, and economic burdens collectively referred to as cumulative life course impairment [1–5].
Patients with moderate-to-severe plaque psoriasis have various treatment options, including topicals, phototherapy, systemic non-biologics, and systemic biologics [6]. Common biologics for treating moderate-to-severe plaque psoriasis include tumor necrosis factor-alpha (TNF-α) inhibitors, interleukin (IL)-17 inhibitors, IL-12/23 inhibitors, and IL-23 specific inhibitors [7, 8].
Advanced biologics have been proven effective in achieving clear skin and reducing psoriasis symptoms in many patients [9]. However, some patients may only experience partial improvement in symptoms and struggle to meet their treatment goals, which may be higher than their physician's [10]. These patients may encounter obstacles in treatment, such as referral challenges, inadequate follow-up, and treatment failure. Additionally, drug response may diminish over time, resulting in clinical inertia and unaddressed treatment goals [10]. To address patient needs and goals, switching to another biologic treatment may benefit patients who do not respond well to their current treatment [11].
Risankizumab is a humanized immunoglobulin (Ig) G1 monoclonal antibody targeting the p19 subunit of IL-23 with high affinity and specificity, inhibiting the activation of the pathogenic T helper 17 cells [12, 13]. Studies have shown that risankizumab is well tolerated in patients with moderate-to-severe psoriasis, and many achieved ≥ 90% improvement in Psoriasis Area and Severity Index (PASI 90) scores [14, 15]. Risankizumab has demonstrated superior efficacy in patients with psoriasis compared to conventional systemic and non-biologic DMARDs, such as methotrexate, fumaric acid esters, and apremilast [14, 16, 17]. It has also demonstrated superior efficacy to other biologic treatments, such as ustekinumab, adalimumab, and secukinumab [15, 18, 19]. Furthermore, research has demonstrated that switching from other psoriasis treatments to risankizumab has clinical benefits on skin outcomes and patients' QoL [14, 20].
Limited evidence exists on switching patients with suboptimal response to an IL-17 inhibitor to risankizumab for psoriasis treatment. Herein, we present findings from the aIMM study, assessing the efficacy and safety of directly switching patients with moderate-to-severe plaque psoriasis from secukinumab or ixekizumab to risankizumab after at least 6 months of treatment.
Methods
Patients
The eligible patients were adults (≥ 18 years) diagnosed with moderate-to-severe chronic plaque psoriasis and treated with either secukinumab or ixekizumab at the recommended dose for at least 6 months. These patients had shown a suboptimal response, defined as having a static Physicians Global Assessment (sPGA) score of 2 or 3 and a body surface area (BSA) ranging from 3% to ≤ 10% after ≤ 6 months of treatment with either drug. Additionally, these patients were eligible for continued biologic therapy as assessed by the investigator. Approvals were obtained from local ethics committees, and the patients provided written informed consent. The study was conducted per the International Conference on Harmonization, Good Clinical Practice (GCP) Guidelines, and the Declaration of Helsinki. Complete eligibility criteria are described in Table S1.
Study Design
aIMM (NCT04102007) was a phase-3b, interventional, multicenter, open-label, single-arm study that examined the effect of risankizumab 150 mg administered at week 0, 4, and then every 12 weeks (q12w) thereafter for 52 weeks in adult patients with moderate-to-severe plaque psoriasis who had been treated with a labeled dose of secukinumab or ixekizumab for ≥ 6 months and experienced a suboptimal response as described above. The patients received two injections of active risankizumab 75 mg (150 mg total dosage) subcutaneously at weeks 0 and 4 and then q12w until the last dose at week 40 (Figure S1).
The study duration was up to 64 weeks, with a 30-day screening period, 52-week open-label study period, and 20-week follow-up period (after week 40). The 52-week open-label period consisted of an initial phase (weeks 0–16) and a maintenance phase (weeks 16–52). The follow-up period consisted of a phone call at week 20 after the last injection of risankizumab (at week 40).
Efficacy endpoints
The primary endpoint was the proportion of patients achieving sPGA 0/1 at week 16. Secondary endpoints included the proportion of patients achieving sPGA 0 at week 16, the proportion of patients achieving sPGA 0/1 and sPGA 0 at week 52, Dermatology Life Quality Index (DLQI) 0 or 1 at weeks 16 and 52, Psoriasis Symptoms Scale (PSS) 0 at weeks 16 and 52, and the time to achieve sPGA 0/1 and sPGA 0.
Exploratory endpoints included proportion of patients achieving absolute PASI (e.g., PASI ≤ 3, ≤ 1, and 0) over time and the proportion of patients who achieved the National Psoriasis Foundation (NPF) treat-to-target (T2T) goal of acceptable (defined as BSA response of ≤ 3% or 75% improvement from baseline) and target response (BSA ≤ 1%).
In addition, we also evaluated the efficacy and health-related quality of life (HRQol) outcomes stratified by patients' number of prior treatments with biologics and their median duration of treatment with secukinumab or ixekizumab prior to enrollment.
Safety endpoints
Safety was monitored throughout the study. Treatment-emergent adverse events (TEAEs), serious adverse events (SAEs), and TEAEs leading to withdrawal were prespecified outcomes. TEAEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) [21]. Prespecified areas of safety interest included major adverse cardiovascular events (MACE), serious infection, tuberculosis, fungal and opportunistic infections excluding herpes zoster, malignancies, hypersensitivity reactions, and hepatic events. Independent cardiovascular and systemic hypersensitivity/anaphylaxis adjudication committees adjudicated all observed cardiovascular, cerebrovascular, thrombotic, hypersensitivity, and anaphylactic events.
Statistical Analysis
Due to limited published literature or clinical trial data, accurate estimation of the primary endpoint sPGA 0/1 response rate at week 16 for patients with suboptimal response to secukinumab or ixekizumab treatment was challenging. However, assuming a response rate between 60% and 70% based on ad hoc analyses self-reported of prior anti-IL-17 failure from phase 3 trials (UltIMMa-1, UltIMMa-2, and IMMhance), the sample size of 250 patients was determined to ensure a half-width of the 95% confidence interval (CI) no greater than 6.2%. This sample size accounted for the maximum variation across all possible response rates (50%). Based on historical data from previous risankizumab psoriasis studies, a screen failure rate of 25% was projected. Hence, it was estimated that screening 333 patients was necessary to enroll the desired 250 patients.
The intent-to-treat population was defined as all patients who received at least one dose of risankizumab and was used for both efficacy and safety analyses. Due to the single-arm design, no statistical tests were conducted. For variables where assessment time was collected, the last non-missing observation gathered on or prior to the date of the first dose of risankizumab was used as the baseline for safety and efficacy analyses. Descriptive statistics were reported. Categorical endpoints were summarized using frequencies, percentages, and 95% CIs. Continuous endpoints were summarized by means, standard deviations, model-based least-square means, and the 95% CIs after accounting for relevant baseline characteristics. Non-responder imputation using multiple imputations handled missing data due to COVID-19 for categorical variable analyses (NRI-C). NRI-C classified patients without an evaluation during a visit window as non-responders; those who were responders before and after the window were considered responders. Additionally, missing data due to a COVID-19 infection or logistical restrictions related to the COVID-19 pandemic were handled through multiple imputations. The data were reported as percentages and events per 100 patient-years for safety.
Results
Patient disposition and baseline characteristics
In total, 252 patients were enrolled from eight countries: Australia, Germany, Italy, Israel, Spain, Taiwan, UK, and the USA. However, due to several instances of non-compliance to GCP standards from one site, data provided from that site (N = 8) were excluded from the analysis.
The results included data from 244 patients. The median (range) age of the patients was 50 (21–81) years; 74.2% were male, and 87.7% were White (Table 1). The mean (SD) duration of plaque psoriasis was 21.3 (13.6) years. All patients had received prior treatment with a biologic, and 41.8% received ≥ 2 biologic treatments prior to enrollment. At baseline, 12 (4.9%) patients had a history of psoriatic arthritis, and 1 (0.4%) had inflammatory bowel disease.
Table 1.
Demographics and baseline disease characteristics
| Risankizumab (N = 244) | |
|---|---|
| Age (years), median (range) | 50 (21–81) |
| Age categories (years), n (%) | |
| < 40 | 65 (26.6) |
| 40 to 65 | 150 (61.5) |
| ≥ 65 | 29 (11.9) |
| Sex, n (%) | |
| Female | 63 (25.8) |
| Male | 181 (74.2) |
| Ethnicity, n (%) | |
| Hispanic or Latino | 17 (7.0) |
| Not Hispanic or Latino | 227 (93.0) |
| Race1, n (%) | |
| American Indian or Alaska Native | 1 (0.4) |
| Asian | 27 (11.1) |
| Black or African-American | 1 (0.4) |
| Multiple race2 | 1 (0.4) |
| Native Hawaiian or Pacific Islander | 0 |
| White | 214 (87.7) |
| Weight (kg), mean (SD) | 93.4 (22.1) |
| Weight (kg), n (%) | |
| ≤ 100 | 173 (70.9) |
| > 100 | 71 (29.1) |
| BMI (kg/m2), mean (SD) | 30.9 (6.7) |
| BMI (kg/m2), n (%) | |
| < 25 | 42 (17.2) |
| ≥ 25 to < 30 | 86 (35.2) |
| ≥ 30 | 116 (47.5) |
| sPGA category, n (%) | |
| 2 | 129 (52.9) |
| 3 | 115 (47.1) |
| PASI, mean (SD) | 6.5 (2.8) |
| BSA, mean (SD) | 6.1 (2.3) |
| PSS, mean (SD) | 6.7 (3.8) |
| DLQI, mean (SD) | 8.7 (6.7) |
| Duration of plaque psoriasis (in years), mean (SD) | 21.3 (13.6) |
| Immediate prior treatment, n (%) | |
| Secukinumab | 149 (61.1) |
| Ixekizumab | 95 (38.9) |
| Duration of last treatment prior to switching to risankizumab (in years), median (range) | |
| Secukinumab | 2.4 (0.2–9.4) |
| Ixekizumab | 1.6 (0.4–13.6) |
| Number of prior biologic therapies, n (%) | |
| ≤ 2 | 142 (58.2) |
| > 2 | 102 (41.8) |
| Prior biologic therapy (inadequate response), n (%) | 217 (88.9) |
| History of psoriatic arthritis | 12 (4.9) |
| History of IBD | 1 (0.4) |
BMI body mass index, BSA body surface area; DLQI Dermatology Life Quality Index; IBD inflammatory bowel disease; PASI Psoriasis Area and Severity Index; SD standard deviation. 1Data for race and ethnicity were self-reported. 2Patients who chose more than one category were placed in the 'multiple' category
Before study enrollment, 61.1% received secukinumab, and 38.9% received ixekizumab. The median (range) duration of treatment before switching to risankizumab was 2.4 (0.2–9.4) years for secukinumab and 1.6 (0.4–13.6) years for ixekizumab.
In total, 233 (95.5%) patients completed week 16, and 205 (84.0%) completed the entire 52-week study; 39 patients discontinued the study (withdrew consent, 13; adverse events, 8; lost to follow-up, 4; other, 21). Risankizumab treatment was discontinued by 28 (11.5%) patients (withdrew consent, 8; adverse event, 8; other, 19).
Efficacy
The median (range) duration of risankizumab treatment was 364 (84–393) days. The primary endpoint of clear or almost clear skin, measured by sPGA 0/1, was achieved by 57.4% at week 16 (Fig. 1).
Fig. 1.
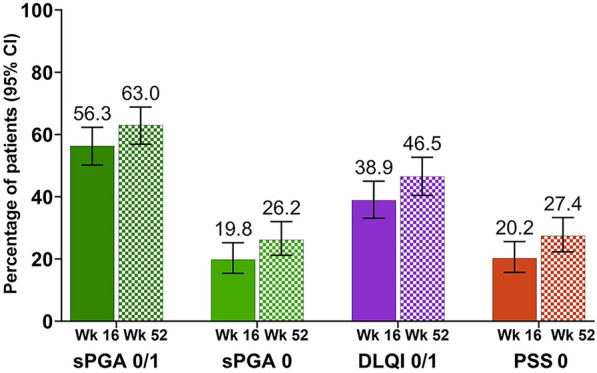
Primary and secondary efficacy endpoints. CI confidence interval; DLQI Dermatology Life Quality Index; PSS Psoriasis Symptoms Scale, sPGA static Physicians Global Assessment; Wk week. Non-responder imputation incorporating multiple imputations to handle missing data due to COVID-19 was used; CIs for response rates were based on the Wilson score method
At week 16, the secondary endpoint of sPGA 0 was achieved by 20.5%; 40.2% achieved DLQI 0/1, and 20.9% achieved PSS 0.
At week 52, the secondary endpoint of sPGA 0/1 was achieved by 62.3%, with an increase in response rates for sPGA 0 (27.1%), DLQI 0/1 (47.2%), and PSS 0 (27.5%, Fig. 2).
Fig. 2.

Achievements of efficacy endpoints over time: a sPGA, b DLQI 0/1, c PSS 0. CI confidence interval; DLQI Dermatology Life Quality Index; sPGA static Physicians Global Assessment; PSS Psoriasis Symptoms Scale. Non-responder imputation incorporating multiple imputations to handle missing data due to COVID-19 was used; CIs for response rate were based on the Wilson score method
Additionally, 47.9% of those who achieved sPGA 0/1 response at week 16 maintained their response at week 52.
At week 52, the sPGA 0/1 response rates among patients with immediate prior treatment with secukinumab and ixekizumab were 69.1% and 51.6%, respectively. In the overall population, only two patients experienced a deterioration in their skin response from sPGA 2 to sPGA 3 between baseline and week 52.
The patients reported a mean change of ≥ 4 points in DLQI scores from baseline at week 16 (Δ = − 4.2) that was maintained through week 52 (Δ = − 4.8, Figure S2). Among 161 patients who had a DLQI score ≥ 4 at baseline, 97 (60.2%) and 94 (58.4%) patients achieved ≥ 4 point reduction in DLQI at week 16 and 52, respectively.
PASIs ≤ 3 and ≤ 1 were achieved by 62.7% and 33.2% at week 16 and by 66.0% and 37.5% at week 52, respectively. Additionally, at weeks 16 and 52, 20.1% and 26.7% achieved PASI 0 (Fig. 3).
Fig. 3.
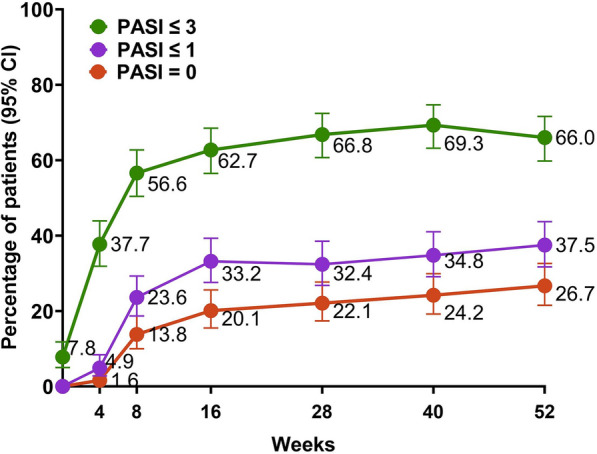
Change in absolute PASI scores from baseline to week 52. CI confidence interval; PASI Psoriasis Area and Severity Index. Non-responder imputation incorporating multiple imputations to handle missing data due to COVID-19 was used; CIs for response rate were based on the Wilson score method
Overall, 58.6% and 70.2% achieved an acceptable NPF T2T goal at week 16 and week 52, and 36.1% and 48.4% achieved the target NPF T2T goal (≤ 1% BSA) at week 16 and week 52, respectively (Figure S3).
Patients in this difficult-to-treat group had clinical responses regardless of their prior biologic treatments (≤ or > 2). A numerically higher proportion of patients who received ≤ 2 biologic treatments achieved a higher proportion of sPGA 0/1 at weeks 16 and 52 (63.8%, and 67.0%) compared to patients who received > 2 biologic treatments (35.7% and 46.4%), respectively, and 17.9% achieved sPGA 0 at both week 16 and 52, respectively (Fig. 4a, b). Patients receiving ≤ 2 or > 2 biologic treatments before enrollment also reported improved psoriasis symptoms and HrQoL. Among patients who received ≤ 2 biologic treatments, the PSS 0 was 22.9% and 29.9%, and DLQI 0/1 scores were 45.2 % and 53.8% at weeks 16 and 52, respectively. Among patients who received > 2 biologic treatments, PSS 0 scores were 14.3% and 19.6%, and DLQI 0/1 scores were 23.2% and 25.0% at weeks 16 and − 52, respectively, (Fig. 4c, d).
Fig. 4.

Clinical efficacy stratified by baseline body weight and the number of biologic treatments prior to enrollment. a sPGA 0/1, b sPGA 0, c PSS 0, and d DLQI 0/1. CI confidence interval; DLQI Dermatology Life Quality Index; sPGA static Physicians Global Assessment; PSS Psoriasis Symptoms Scale
We observed numerical differences in clinical outcomes in patients irrespective of immediate prior treatment history and duration of treatment. In patients who were extensively pretreated (≥ median duration of last treatment cycle) with secukinumab, sPGA 0/1 response was observed in 74.3% at week 16 and 73.0% at week 52, while in patients previously treated with ixekizumab (≥ median duration of last treatment cycle), sPGA 0/1 response rates were 46.8% and 57.4% at week 16 and 52, respectively, Figure S4 a. The efficacy of patients as determined by sPGA 0, DLQI 0/1, and PSS 0 based on prior treatment with secukinumab or ixekizumab is shown in Figure S4 b–d.
Safety
Treatment-emergent adverse effects (TEAEs) were reported by 164 (67.2%) patients, of which 48 (19.7%) were possibly related to the study treatment (Table 2). Severe and serious AEs were reported in 18 (7.4%) and 17 (7.0%) patients. Of the 17 patients who experienced treatment-emergent serious adverse events (SAEs), one patient experienced two SAEs (muscular weakness and aphasia) with the reasonable possibility of being related to the study treatment as assessed by the investigator. Eight patients (3.3%) had AEs that led to discontinuation of study treatment, of which five experienced TEAEs that the investigator considered to have a reasonable possibility of being related to the study drug. No deaths were reported. The most frequently reported TEAEs reported in ≥ 5% of patients were COVID-19 (8.6%) and nasopharyngitis (5.7%).
Table 2.
Treatment-emergent adverse events
| Risankizumab (N = 244) n (%) |
Risankizumab (PY = 265.2) E/100 PY |
|
|---|---|---|
| AE | 164 (67.2) | 408 (153.8) |
| AE with a reasonable possibility of being drug-related1 | 48 (19.7) | 93 (35.1) |
| Severe AE | 18 (7.4) | 24 (9.0) |
| Serious AE | 17 (7.0) | 21 (7.9) |
| AE leading to discontinuation of study drug | 8 (3.3) | 9 (3.4) |
| Adjudicated MACE2 | 3 (1.2) | 3 (1.1) |
| Serious infection | 3 (1.2) | 3 (1.1) |
| Tuberculosis3 | 0 | 0 |
| Malignant tumor | 4 (1.6) | 4 (1.5) |
| Malignant tumor excluding NMSC | 2 (0.8) | 2 (0.8) |
| Serious hypersensitivity | 0 | 0 |
| AE leading to death | 0 | 0 |
| Any COVID-19 related AE | 31 (12.7) | 33 (12.4) |
| All deaths4 | 0 | 0 |
| TEAE ≥ 5% | ||
| COVID-19 | 21 (8.6) | 21 (7.9) |
| Nasopharyngitis | 14 (5.7) | 18 (6.8) |
AE adverse events; PY patient-years; E/100PY = events per 100 PY; MACE major adverse cardiovascular events; NMSC non-melanoma skin cancer; TEAE treatment-emergent adverse events. TEAEs were defined as any event with an onset that was after the first dose of the study drug and with an onset date within 20 weeks (140 days) after the last dose of the study drug. Patients were counted once in each row, regardless of the number of events they may have had
1As assessed by the investigator, 2MACE was defined as cardiovascular death, non-fatal myocardial infarction, and non-fatal stroke. The time lapse between the initiation of risankizumab and every MACE event is as follows: patient 1, MI occurred on day 356; patient 2, MI occurred on day 174; patient 3, lacunar infarct on day 176. 3Based on CMQ of active tuberculosis, 4includes non-treatment-emergent deaths
Among TEAEs of special interest (Table 3), adjudicated MACE was reported in three patients (myocardial infarction, 2; stroke, 1); none of the three events were considered to have a reasonable possibility of being related to risankizumab treatment. Both patients with myocardial infarction had pre-existing cardiac risk factors that included previous myocardial infarctions at a younger age with previous stent placement (n = 1), obesity (n = 1), sleep apnea (n = 1), hypertension, diabetes, hypercholesterolemia, tobacco, and alcohol use. One patient experienced right-sided facial numbness and right ulnar sensory decrease post-COVID-19 vaccination and prior to study enrollment. Due to COVID-19-related access issues, obtaining magnetic resonance imaging was delayed until approximately 5 months after the start of the study drug, at which time a finding of lacunar infarct was noted. Per the investigator, the patient had no significant disability despite facial numbness symptoms and was able to carry out usual duties and activities. The patient completed the study with no new related AEs. Of note, the nonserious event of lacunar infarct was adjudicated with onset date prior to study drug administration. All three adjudicated MACE events were considered to have no reasonable possibility of relationship to the study drug.
Table 3.
Treatment-emergent adverse events of special interest among patients
| Risankizumab (N = 244) n (%) |
Risankizumab (N = 244) (PY = 265.2) E/100 PY |
|
|---|---|---|
| MACE1 | 3 (1.2) | 3 (1.1) |
| Myocardial infarction | 2 (0.8) | 2 (0.8) |
| Stroke | 1 (0.4) | 1 (0.4) |
| Extended MACE2 | 4 (1.6) | 4 (1.5) |
| Coronary revascularization procedures | 1 (0.4) | 1 (0.4) |
| Myocardial infarction | 2 (0.8) | 2 (0.8) |
| Stroke | 1 (0.4) | 1 (0.4) |
| Serious infections | 3 (1.2) | 3 (1.1) |
| Appendicitis | 1 (0.4) | 1 (0.4) |
| Diverticulitis | 1 (0.4) | 1 (0.4) |
| Postoperative wound infection | 1 (0.4) | 1 (0.4) |
| Tuberculosis | 0 | 0 |
| Opportunistic infections, excluding tuberculosis and herpes zoster | 0 | 0 |
| Malignant tumors | 4 (1.6) | 4 (1.5) |
| Basal cell carcinoma | 1 (0.4) | 1 (0.4) |
| Non-small cell lung cancer | 1 (0.4) | 1 (0.4) |
| Prostate cancer stage I | 1 (0.4) | 1 (0.4) |
| Squamous cell carcinoma of skin | 1 (0.4) | 1 (0.4) |
| NMSC | 2 (0.8) | 2 (0.8) |
| Basal cell carcinoma | 1 (0.4) | 1 (0.4) |
| Squamous cell carcinoma of skin | 1 (0.4) | 1 (0.4) |
| Malignant tumors excluding NMSC | 2 (0.8) | 2 (0.8) |
| Non-small cell lung cancer | 1 (0.4) | 1 (0.4) |
| Prostate cancer stage I | 1 (0.4) | 1 (0.4) |
| Hypersensitivity | 9 (3.7) | 10 (3.8) |
| Hepatic events | 5 (2.0) | 7 (2.6) |
E events; MACE major adverse cardiovascular events; NMSC non-melanoma skin cancer; PY patient-years. Treatment-emergent adverse events (TEAEs) were defined as any event with an onset that was after the first dose of the study drug and with an onset date within 20 weeks (140 days) after the last dose of the study drug. Patients were counted once in each row, regardless of the number of events they may have had
1MACE was defined as cardiovascular death, non-fatal myocardial infarction, and non-fatal stroke. 2Extended MACE was defined as events identified as MACE along with hospitalizations for unstable angina and coronary revascularization procedures and included all patients with adjudicated MACE and any patient with revascularization
Three events of serious infections were reported, including a single event of appendicitis, diverticulitis, and postoperative wound infection; four patients experienced malignancies, and none were considered as having a reasonable possibility of being related to the study drug. Hypersensitivity and hepatic events were reported by nine and five patients, respectively. All were nonserious except for one SAE of esophageal varices in a patient with a history of portal hypertension and esophageal varices. No TEAEs of active tuberculosis, opportunistic infections excluding tuberculosis and herpes zoster, serious hypersensitivity or adjudicated anaphylaxis, or death were reported.
Discussion
In this cohort of patients with moderate-to-severe psoriasis, who were refractory to treatment and had prior experience with biologic therapies, directly switching to risankizumab, an IL-23 inhibitor, from secukinumab or ixekizumab was found to be effective and resulted in clinically relevant improvements. These findings are meaningful, considering that the enrolled patients had previously exhibited inadequate responses to two efficacious IL-17 inhibitors, secukinumab or ixekizumab, after at least 6 months of treatment with a median exposure of 2.4 years and 1.6 years, respectively. The improvements in clinical symptoms and HRQoL were achieved in the short term (16 weeks) and increased with 1 year of risankizumab treatment.
Risankizumab treatment improved patients’ HRQoL. This was evident as more patients experienced improvements in their DLQI and PSS scores over time. We observed a noticeable decrease of at least 4 points in DLQI scores at week 16, and this improvement continued until week 52, demonstrating considerable benefits to the patient's HRQoL. Furthermore, about a quarter of patients (27.5%) achieved PSS 0 at week 52, indicating a substantial reduction in the most burdensome symptoms for patients. Considering the impact of psoriasis on the overall quality of life, risankizumab is a valuable and effective treatment option for patients who do not respond well to IL-17 inhibitors (IL-17i) like secukinumab and ixekizumab.
IL-23/IL-17A/F immune axis has been shown to be central to psoriasis pathogenesis, and IL-23 has emerged as a master regulator in psoriasis [22]. IL-23 promotes terminal differentiation, expansion, and maintenance of IL-17-producing cells (Th17), expressing CD4 + or CD8 + T cells [23]. It has also been reported to impair the function of regulatory T cells (Treg) and to promote the differentiation of Treg into TH17-like cells [24, 25]. In addition, IL-23 appears to be involved in the differentiation and survival of pathogenic tissue-resident memory T cells (TRM), which seem to have a role in the recurrence of psoriatic lesions [26, 27]. Therefore, inhibiting the regulatory cytokine IL-23 is hypothesized to lead to long-lasting therapeutic effects by restoring a physiological Treg/TRM balance. This contrasts with the blockade of an effector cytokine, which reduces inflammatory cells but has a limited effect on relative numbers of pro- and anti-inflammatory T cells [28]. Blocking the activity of IL-23 with a specific antibody directly reduces the IL-17-induced inflammation and therefore offers an attractive therapeutic intervention for psoriasis.
There is growing evidence that two different types of Th17 cells exist in vivo in murine models and seem to be important in the pathogenesis of psoriasis and other IMIDs: non-pathogenic and pathogenic TH17 cells [29]. In psoriasis, targeting the IL-23p19 subunit can restore favorable ratios of T cell populations in lesional skin compared with IL-17A inhibition. The durable, long-lasting therapeutic effect observed with selective targeting of the IL-23p19 subunit is hypothesized to derive from the suppression of TRM cells. IL-23 inhibition has been shown to shift the relative ratio of CD8 + TRM cells and Treg cells favorably in psoriasis lesions compared with IL-17A inhibition [30]. Treg cells help to maintain immune homeostasis and self-tolerance by suppressing inflammation and effector T cells. IL-23 has been shown to suppress the differentiation of Treg cells and promote differentiation, survival, and expansion of pathogenic Th17 cells [31]. Clinical studies have shown superior long-term maintenance of response with IL-23 inhibition over IL-17A blockade; IL-17A inhibitors, in essence targeting an effector cytokine, have less impact on the relative numbers of pro- and anti-inflammatory T cells compared to IL-23 inhibitors [19]. Thus, risankizumab may be effective for patients who have not responded well to IL-17 inhibitors because of its binding to the p19 subunit of IL-23.
Real-world studies support the safety of long-term use of IL-23 inhibitors in patients with psoriasis, including those for whom multiple biologics have failed [32]. In a recent study, risankizumab demonstrated a favorable long-term safety profile with no new safety concerns [33]. The safety profile of risankizumab in this study was also consistent with previous trials [14, 15, 18]. No new safety signals were observed when directly switching from secukinumab or ixekizumab without a washout period to risankizumab.
A head-to-head trial with the IL 17 inhibitors secukinumab versus risankizumab showed that risankizumab treatment was able to maintain superior efficacy at week 48 and -52, respectively, where secukinumab gradually started to lose the level of efficacy after 24 weeks of treatment [18]. These findings suggest that IL-23 treatment offers a sustained and robust response over the long term. This study demonstrated that risankizumab, as a durable treatment option, benefits patients with psoriasis who experience inadequate treatment efficacy or encounter safety and tolerability concerns [34, 35].
Recent studies have shown that switching within the same biologic class may be less beneficial than switching to another mode of action [11, 36, 37]. Two small real-world single-center studies evaluating the efficacy and safety of switching to risankizumab in patients who previously failed anti-IL 17 inhibitors found that switching to risankizumab was beneficial [38, 39]. In addition, real-world data analyzing the differences between modes of action regarding switching demonstrated that IL-23 inhibitors, and especially risankizumab, were shown to have the lowest switch rates relative to all other biologics [11, 40].
International guidelines recommend clear or almost clear skin (PASI > 90 or absolute PASI ≤ 2) as a treatment target in managing psoriasis [41–46]. This study demonstrated that most patients benefited from switching from an IL-17i to risankizumab. Only two patients' disease worsened from sPGA2 to 3 within the study period.
In this study, patients had a baseline PASI of 6.5, and at week 52, 66.0% and 37.5% achieved absolute PASI ≤ 3 and ≤ 1, respectively, representing meaningful clinical improvement for this hard-to-treat patient group. Furthermore, 26.7% of patients achieved complete clearance (PASI0) at week 52, demonstrating the potent efficacy of risankizumab in this refractory population.
Patients had a mean baseline BSA of 6.1%, and 70.2% and 48.4% achieved an acceptable and target NPF T2T goal at week 52, demonstrating the efficacy of risankizumab among this hard-to-treat patient population who had suboptimal responses to prior treatment with secukinumab or ixekizumab.
A potential limitation of the study was the open-label single-arm design that includes a lack of blinding, which can introduce selection bias and influence the subjective assessment of outcomes, limiting the generalizability of the results. Another limitation of the study was the low participation rate of diverse ethnicities, a recurring challenge in numerous psoriasis clinical trials that limits the generalizability of the study results [47, 48]. To ensure a more comprehensive representation of populations affected by psoriasis, future studies should aim to include patients from diverse ethnic backgrounds and various geographic locations.
Conclusion
In conclusion, the demonstrated efficacy, improvements in quality of life, and favorable safety profile of risankizumab support its use as a beneficial treatment option for patients with moderate-to-severe plaque psoriasis who exhibit a suboptimal response to secukinumab or ixekizumab.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgments
Medical Writing and Editorial Assistance
Medical writing support was provided by Dalia Majumdar, PhD, and editorial support by Angela T. Hadsell, BA, both employees of AbbVie.
Author Contribution
Richard B. Warren, Lev Pavlovsky, Antonio Costanzo, Michael Bukhalo, Neil J. Korman, Yu-Huei Huang, Georgios Kokolakis, Andreas Pinter, Nadia Ibrahim, Yanbing Zheng, Leonidas Drogaris, Vassilis Stakias, Ahmed M. Soliman, Simone Rubant, and Diamant Thaçi contributed to acquisition of data (acquired and managed patients, provided facilities, etc.), analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis), commented on the drafts of the manuscript, and approved the final manuscript.
Funding
Risankizumab was developed in collaboration between AbbVie and Boehringer Ingelheim. This research was funded by AbbVie (NCT04102007) and supported by the NIHR Manchester Biomedical Research Centre (NIHR203308). AbbVie participated in the design, study conduct, analysis, and interpretation of the data, as well as in the writing, review, and approval of this publication. All authors had access to relevant data and participated in the drafting, review, and approval of this manuscript. No honoraria or payments were made for authorship. Funding for the journal’s Rapid Service Fee was provided by AbbVie. AbbVie and the authors thank the participants, study sites, and investigators who participated in this clinical trial.
Data Availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymised, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://vivli.org/ourmember/abbvie/ then select "Home".
Declarations
Conflict of Interest
Richard B. Warren has received grants from AbbVie, Almirall, Amgen, Celgene, Janssen, Lilly, LEO Pharma, Novartis, and Pfizer. He has received personal fees from AbbVie, Almirall, Amgen, Arena Pharmaceuticals, Avillion, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Janssen, Lilly, LEO Pharma, Novartis, Pfizer, Sanofi, and UCB. Lev Pavlovsky has served as an investigator for AbbVie, Coherus, Novartis Pharmaceuticals Corporation, Janssen Biotech, Eli Lilly, Bristol Myers Squibb and as an adviser, consultant, and/or invited lecturer for AbbVie, Janssen Biotech, Novartis Pharmaceuticals Corporation, Pfizer Inc., Dexcel Pharma, Eli Lilly, and Boehringer Ingelheim. Antonio Costanzo has served as a clinical study investigator for AbbVie, has received research grants and/or speaker/ad board honoraria from AbbVie, Eli Lilly, Amgen, Almirall, Leo Pharma, Novartis, UCB, and Pfizer, and is currently affiliated with ESDR (member), Sidemast (member), and EDF (board member). Michael Bukhalo has received honoraria or fees for serving on advisory boards, as a speaker, as a consultant, and grants as an investigator from Allergan, Boehringer Ingelheim, Celgene, Centocor, DUSA Pharmaceuticals, Eli Lilly, Galderma, Leo Pharma, MedImmune, Merck, and Novartis. Neil J. Korman has served as an investigator, speaker, adviser, or consultant for AbbVie, Abcentra, Amgen, Argenx, AstraZeneca, Biogen, Boehringer Ingelheim, Bristol Myers Squibb, Castle Biosciences, Celgene, Chemocentryx, Dermavant, Eli Lilly, Galderma, Janssen, Kyowa Hakko Kirin Pharma, Leo Pharma, Menlo Therapeutics, Novartis, OliX Pharma, Pfizer, Prothena, Principia, Regeneron, Rhizen, Sanofi, Soligenix, Sun Pharma, Syntimmune, Trevi, UCB, and XBiotech. Yu-Huei Huang has conducted clinical trials while serving as a principal investigator for Galderma, Eli Lilly, Novartis Pharmaceuticals Corporation, and Janssen-Cilag Pharmaceutica; received honoraria for serving as an advisory board member for Pfizer Limited, AbbVie, and Celgene; and received speaking fees from AbbVie, Eli Lilly, and Novartis Pharmaceuticals Corporation. Georgios Kokolakis has received travel grants or honoraria, has been a consultant member of advisory boards and speaker bureaus, or has served as an investigator for AbbVie, Actelion, Almirall, Amgen, Basilea, Biogen, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Hexal-Sandoz, Janssen-Cilag, LEO Pharma, Eli Lilly, MSD, Novartis, Pfizer, Sanofi-Aventis, Stada, Takeda and UCB. Andreas Pinter is an investigator and/or speaker and/or adviser for the following pharmaceutical companies: AbbVie, Almirall-Hermal, Amgen, Biogen Idec, Boehringer Ingelheim, Celgene, GSK, Eli Lilly, Galderma, Hexal, Janssen, LEO Pharma, MC2, Medac, Merck Serono, Mitsubishi, MSD, Novartis, Pascoe, Pfizer, Tigercat Pharma, Regeneron, Roche, Sandoz Biopharmaceuticals, Sanofi Genzyme, Schering-Plough, UCB Pharma and Zuellig Pharma. Simone Rubant, Nadia Ibrahim, Yanbing Zheng, Leonidas Drogaris, Ahmed M. Soliman, and Vassilis Stakias are full-time employees of AbbVie and may own stock/options or patents. Diamant Thaçi is an adviser, speaker, and/or consultant for AbbVie, Almirall, Amgen, Asana Biosciences, Boehringer Ingelheim, Bristol Myers Squibb, Celltrion, Galderma, Janssen, Kyowa Kirin, LEO Pharma, Lilly, New Bridge, Novartis, Pfizer, Regeneron, Sanofi/Genzyme, Tamro, UCB, and Zuellig Pharma. He is also involved in research for AbbVie, LEO Pharma, and Novartis.
Ethical Approval
Approvals were obtained from local ethics committees, and the patients provided written informed consent. The study was conducted in accordance with the International Conference on Harmonisation, Good Clinical Practice Guidelines, and the Declaration of Helsinki.
Footnotes
Prior presentation: The results were partially presented at a Late-Breaking Research session during the 2023 American Academy of Dermatology (AAD) Annual Meeting in New Orleans, Louisiana.
References
- 1.Organization WH. 2016 Global report on psoriasis [Internet]. [cited 2023 Feb 13]. Available from: https://apps.who.int/iris/bitstream/handle/10665/204417/9789241565189_eng.pdf.psoriasis?sequence=1
- 2.Boehncke W-H, Menter A. Burden of disease: psoriasis and psoriatic arthritis. Am J Clin Dermatol. 2013;14:377–88. [DOI] [PubMed] [Google Scholar]
- 3.Blackstone B, Patel R, Bewley A. Assessing and improving psychological well-being in psoriasis: considerations for the clinician. Psoriasis Targets Ther. 2022;12:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.von Stülpnagel CC, Augustin M, Düpmann L, da Silva N, Sommer R. Mapping risk factors for cumulative life course impairment in patients with chronic skin diseases—a systematic review. J Eur Acad Dermatol Venereol. 2021;35:2166–84. [DOI] [PubMed] [Google Scholar]
- 5.Bulat V, Šitum M, Aždajić MD, Lovrić I, Dediol I. Study on the impact of psoriasis on quality of life: psychological. Soc Financ Implic Psychiatr Danub. 2020;32:553–61. [PubMed] [Google Scholar]
- 6.Kim WB, Jerome D, Yeung J. Diagnosis and management of psoriasis. Can Fam phys Med Fam Can. 2017;63:278–85. [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang Y, Chen Y, Yu Q, Shi Y. Biologic and small-molecule therapies for moderate-to-severe psoriasis: focus on psoriasis comorbidities. BioDrugs. 2023;37:35–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahil SK, Ezejimofor MC, Exton LS, Manounah L, Burden AD, Coates LC, et al. Comparing the efficacy and tolerability of biologic therapies in psoriasis: an updated network meta-analysis. Br J Dermatol. 2020;183:638–49. [DOI] [PubMed] [Google Scholar]
- 9.Brownstone ND, Hong J, Mosca M, Hadeler E, Liao W, Bhutani T, et al. Biologic treatments of psoriasis: an update for the clinician. Biol Targets Ther. 2021;15:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strober BE, van der Walt JM, Armstrong AW, Bourcier M, Carvalho AVE, Chouela E, et al. Clinical goals and barriers to effective psoriasis care. Dermatol Ther. 2019;9:5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Armstrong AW, Patel M, Li C, Garg V, Mandava MR, Wu JJ. Real-world switching patterns and associated characteristics in patients with psoriasis treated with biologics in the United States. J Dermatol Treat. 2023;34:2200870. [DOI] [PubMed] [Google Scholar]
- 12.Singh S, Kroe-Barrett RR, Canada KA, Zhu X, Sepulveda E, Wu H, et al. Selective targeting of the IL23 pathway: generation and characterization of a novel high-affinity humanized anti-IL23A antibody. MAbs. 2015;7:778–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gold LFS, Bagel J, Tyring SK, Hong HC, Pavlovsky L, Vender R, et al. Comparison of risankizumab and apremilast for the treatment of adults with moderate plaque psoriasis eligible for systemic therapy: results from a randomized, open-label, assessor-blinded phase IV study (IMMpulse). Br J Dermatol. 2023;189(5):540–52. [DOI] [PubMed] [Google Scholar]
- 15.Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392:650–61. [DOI] [PubMed] [Google Scholar]
- 16.Cestari TF, daSilva Souza C, Azulay-Abulafia L, Fabricio L, Kalabic J. 26197 Efficacy and safety of risankizumab vs methotrexate in patients with moderate-to-severe plaque psoriasis: results from the 28-week randomized, double-blind period of an ongoing phase 3 study in Brazil. J Am Acad Dermatol. 2021;85:AD88. [Google Scholar]
- 17.Thaçi D, Eyerich K, Pinter A, Sebastian M, Unnebrink K, Rubant S, et al. Direct comparison of risankizumab and fumaric acid esters in systemic therapy–naïve patients with moderate-to-severe plaque psoriasis: a randomized controlled trial*. Br J Dermatol. 2022;186:30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warren RB, Blauvelt A, Poulin Y, Beeck S, Kelly M, Wu T, et al. Efficacy and safety of risankizumab vs. secukinumab in patients with moderate-to-severe plaque psoriasis (IMMerge): results from a phase III, randomized, open-label, efficacy–assessor-blinded clinical trial*. Br J Dermatol. 2021;184:50–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reich K, Gooderham M, Thaçi D, Crowley JJ, Ryan C, Krueger JG, et al. Risankizumab compared with adalimumab in patients with moderate-to-severe plaque psoriasis (IMMvent): a randomised, double-blind, active-comparator-controlled phase 3 trial. Lancet. 2019;394:576–86. [DOI] [PubMed] [Google Scholar]
- 20.Strober B, Armstrong A, Rubant S, Patel M, Wu T, Photowala H, et al. Switching to risankizumab from ustekinumab or adalimumab in plaque psoriasis patients improves PASI and DLQI outcomes for sub-optimal responders. J Dermatol Treat. 2022;33:2991–6. [DOI] [PubMed] [Google Scholar]
- 21.Services US Department of Health and Human. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.03. 2018; Available from: https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf
- 22.Hawkes JE, Yan BY, Chan TC, Krueger JG. Discovery of the IL-23/IL-17 signaling pathway and the treatment of psoriasis. J Immunol. 2018;201:1605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan TC, Hawkes JE, Krueger JG. Interleukin 23 in the skin: role in psoriasis pathogenesis and selective interleukin 23 blockade as treatment. Ther Adv Chronic Dis. 2018;9:111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kannan AK, Su Z, Gauvin DM, Paulsboe SE, Duggan R, Lasko LM, et al. IL-23 induces regulatory T cell plasticity with implications for inflammatory skin diseases. Sci Rep. 2019;9:17675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soler DC, McCormick TS. The dark side of regulatory T cells in psoriasis. J Investig Dermatol. 2011;131:1785–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheuk S, Wikén M, Blomqvist L, Nylén S, Talme T, Ståhle M, et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol. 2014;192:3111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark RA. Resident memory T cells in human health and disease. Sci Transl Med. 2015;7:269rvl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eyerich K, Weisenseel P, Pinter A, Schäkel K, Asadullah K, Wegner S, et al. IL-23 blockade with guselkumab potentially modifies psoriasis pathogenesis: rationale and study protocol of a phase 3b, randomised, double-blind, multicentre study in participants with moderate-to-severe plaque-type psoriasis (GUIDE). BMJ Open. 2021;11: e049822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13:991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehta H, Mashiko S, Angsana J, Rubio M, Hsieh Y-CM, Maari C, et al. Differential changes in inflammatory mononuclear phagocyte and T-cell profiles within psoriatic skin during treatment with Guselkumab vs Secukinumab. J Investig Dermatol. 2021;141:1707–17189. [DOI] [PubMed] [Google Scholar]
- 31.McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17–producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blauvelt A, Chiricozzi A, Ehst BD, Lebwohl MG. Safety of IL-23 p19 inhibitors for the treatment of patients with moderate-to-severe plaque psoriasis: a narrative review. Adv Ther. 2023;40:3410–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gordon KB, Blauvelt A, Bachelez H, Coates LC, den Bosch FEV, Kaplan B, et al. Long-term safety of risankizumab in patients with psoriatic disease: a comprehensive analysis from clinical trials. Dermatol Ther. 2024;14:2523–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menter A, Strober BE, Kaplan DH, Kivelevitch D, Prater EF, Stoff B, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol. 2019;80:1029–72. [DOI] [PubMed] [Google Scholar]
- 35.Kerdel F, Zaiac M. An evolution in switching therapy for psoriasis patients who fail to meet treatment goals. Dermatol Ther. 2015;28:390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ortonne J-P, Chimenti S, Reich K, Gniadecki R, Sprøgel P, Unnebrink K, et al. Efficacy and safety of adalimumab in patients with psoriasis previously treated with anti-tumour necrosis factor agents: subanalysis of BELIEVE. J Eur Acad Dermatol Venereol. 2011;25:1012–20. [DOI] [PubMed] [Google Scholar]
- 37.Graier T, Weger W, Jonak C, Sator P, Zikeli C, Prillinger K, et al. Real-world effectiveness of anti-interleukin-23 antibodies in chronic plaque-type psoriasis of patients from the Austrian Psoriasis Registry (PsoRA). Sci Rep. 2022;12:15078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Megna M, Fabbrocini G, Ruggiero A, Cinelli E. Efficacy and safety of risankizumab in psoriasis patients who failed anti-IL-17, anti-12/23 and/or anti IL-23: preliminary data of a real-life 16-week retrospective study. Dermatol Ther. 2020;33: e14144. [DOI] [PubMed] [Google Scholar]
- 39.Bonifati C, Morrone A, Cristaudo A, Graceffa D. Effectiveness of anti-interleukin 23 biologic drugs in psoriasis patients who failed anti-interleukin 17 regimens. a real-life experience. Dermatol Ther. 2021;34: e14584. [DOI] [PubMed] [Google Scholar]
- 40.Tada Y, Soliman AM, Ishii K, Sakuma R, Puig L, Davis M, et al. Real-world discontinuation and switching patterns for interleukin-inhibitor treatments in patients with moderate-to-severe psoriasis in Japan. Dermatol Ther. 2024;14:99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith CH, Yiu ZZN, Bale T, Burden AD, Coates LC, Edwards W, et al. British association of dermatologists guidelines for biologic therapy for psoriasis 2020: a rapid update. Br J Dermatol. 2020;183:628–37. [DOI] [PubMed] [Google Scholar]
- 42.Cristina; E, Kogan, Nora. Argentine Guidelines for the systemic treatment of moderate-to-severe psoriasis. [Internet]. [cited 2023 Sep 19]. Available from: https://www.soarpso.org/recursos/archivos/AG_psoriasis_2020_english.pdf
- 43.Nast A, Smith C, Spuls PI, Valle GA, Bata-Csörgö Z, Boonen H, et al. EuroGuiDerm guideline on the systemic treatment of Psoriasis vulgaris – Part 1: treatment and monitoring recommendations. J Eur Acad Dermatol Venereol. 2020;34:2461–98. [DOI] [PubMed] [Google Scholar]
- 44.Nast A, Smith C, Spuls PI, Valle GA, Bata-Csörgö Z, Boonen H, et al. EuroGuiDerm guideline on the systemic treatment of Psoriasis vulgaris – Part 2: specific clinical and comorbid situations. J Eur Acad Dermatol Venereol. 2021;35:281–317. [DOI] [PubMed] [Google Scholar]
- 45.Carrascosa JM, Puig L, Romero IB, Salgado-Boquete L, del Alcázar E, Lencina JJA, et al. Actualización práctica de las recomendaciones del Grupo de Psoriasis de la Academia Española de Dermatología y Venereología (GPS) para el tratamiento de la psoriasis con terapia biológica. Parte 2 «Manejo de poblaciones especiales, pacientes con comorbilidad y gestión del riesgo. Actas Dermo-Sifiliogr. 2022;113:583–609. [DOI] [PubMed] [Google Scholar]
- 46.Carrascosa JM, Puig L, Romero IB, Salgado-Boquete L, del Alcázar E, Lencina JJA, et al. [Translated article] practical update of the recommendations published by the psoriasis group of the spanish academy of dermatology and venereology (GPs) on the treatment of psoriasis with biologic therapy. part 1. concepts and general management of psoriasis with biologic therapy. Actas Dermo-Sifiliogr. 2022;113:261–77. [DOI] [PubMed] [Google Scholar]
- 47.Sevagamoorthy A, Sockler P, Akoh C, Takeshita J. Racial and ethnic diversity of US participants in clinical trials for acne, atopic dermatitis, and psoriasis: a comprehensive review. J Dermatol Treat. 2022;33:3086–97. [DOI] [PubMed] [Google Scholar]
- 48.Reddy VD, Myers BA, Chan SY, Thibodeaux QG, Brownstone ND, Bhutani T, et al. A review of current phase III clinical trials of plaque psoriasis: under-representation of nonwhite participants and need for reform. Br J Dermatol. 2021;184:348–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymised, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://vivli.org/ourmember/abbvie/ then select "Home".