

Abstract
Ribonucleic acids (RNAs) are key biomolecules responsible for the transmission of genetic information, the synthesis of proteins, modulation of many biochemical processes, and are often the key components of viruses. Synthetic RNAs or oligoribonucleotides are becoming more widely used as therapeutics. In many cases, RNAs will be chemically modified – either naturally via enzymatic systems within a cell or intentionally during their synthesis. Analytical methods to detect, sequence, identify and quantify RNA and its modifications have demands that far exceed requirements found in the DNA realm. Two complementary platforms have demonstrated their value and utility for the characterization of RNA and its modifications: mass spectrometry (MS) and next-gen sequencing (NGS). This review highlights recent advances in both platforms, examines their relative strengths and weaknesses, and explores some alternative approaches that lie at the horizon.
Keywords: Post-transcriptional modifications, Epitranscriptome, LC-MS/MS, RNA Sequencing, Therapeutic RNAs, Modified nucleosides
Introduction
The discovery of RNA post-transcriptional modifications has been an important aspect of understanding their roles in different biological processes. (1) The challenge for measurement sciences with RNA and its modifications is not an uncommon one – how to identify, quantify and, when necessary, place such modifications into their sequence context. This review will discuss the various approaches for analyzing modified RNA-oligonucleotides and biological RNAs with an emphasis on starting with the simplest analytes and progressing to more complex samples (Figure 1).
Figure 1. Approaches for Analyzing RNA and its Modifications.
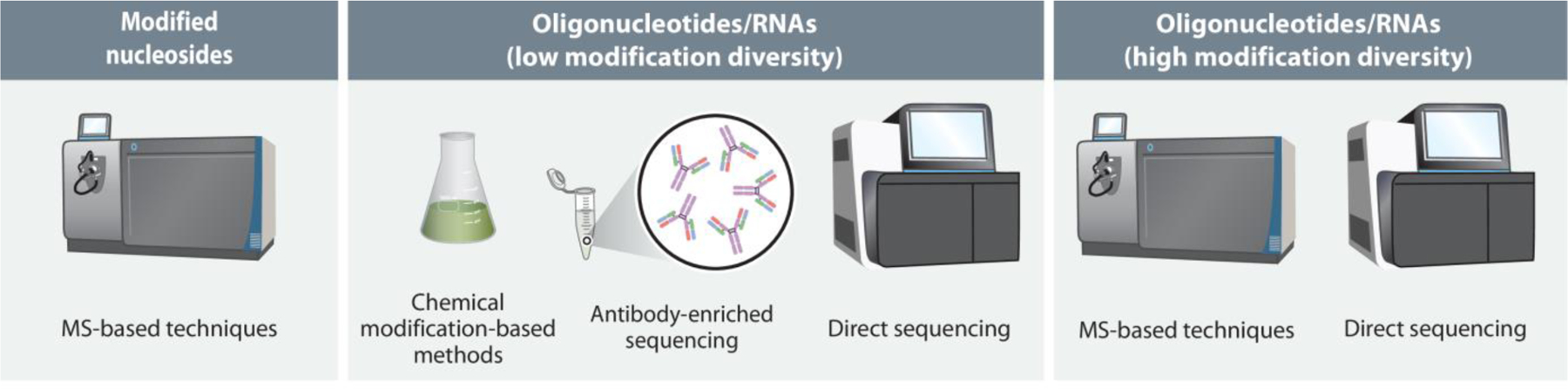
Overview of most common approaches for analyzing oligoribonucleotides and RNA sequences including those that are modified, either biologically or for therapeutic applications. Abbreviations: LC-MS/MS, liquid chromatography tandem mass spectrometry; RNA-Seq, RNA sequencing (next generation sequencing).
Generally, mass spectrometry has proven to be the most powerful approach for identifying, characterizing and quantifying modifications at the nucleobase, nucleoside or nucleotide level. When smaller (~35 nucleotides (nt) or less) RNA-based oligonucleotides need to be characterized, again mass spectrometry approaches have proven most effective. The characterization of larger RNA-based oligonucleotides and naturally occurring RNAs is less settled. While mass spectrometry is still quite appropriate, newer RNA sequencing technologies and approaches have been developed that often focus on characterizing one or a small subset of modifications across a collection of RNAs. In fact, epitranscriptomics – the study of all RNA transcripts in a cell and their variations (quantity, modification status, and fate) is becoming one of the more significant analytical challenges in biomedical sciences.
Nucleoside detection by mass spectrometry
Workflow for analyzing modified nucleosides by liquid chromatography-mass spectrometry
Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) has been and remains the gold standard for the detection, characterization, and quantification of nucleosides, including modified nucleosides such as those from transfer RNA (tRNA) and ribosomal RNA (rRNA). (2–5) This technique involves digesting oligonucleotides or RNAs into constituent nucleosides usually via enzymatic hydrolysis. This mixture of nucleosides is separated – typically by reversed phase high performance liquid chromatography (HPLC) – and then detected by collision-induced dissociation (CID) tandem mass spectrometry. Nucleosides are identified based on precursor m/z values as well as their product ion spectra. Standard analytical methods can be used for the quantification of nucleosides, although calibration curves typical require the presence of modified nucleoside standards which are often difficult to procure commercially.
Advances in the identification and quantification of low abundance modifications
The conventional LC-MS/MS approach has used positive polarity for the detection of modifications which can be problematic for those modified nucleosides that do not ionize efficiently due to their low proton affinities. (6) Borrowing from an old but effective analytical improvement, several solutions based on the chemical derivatization of nucleosides to enhance their detectability have been reported. Dai and co-workers used 2-bromo-1-(3,4- dimethoxyphenyl)-ethanone (BDMOPE) as a derivatization reagent to enhance the detection of modified uridines including 5-hydroxyuridine (ho5U). (7) The Garcia lab has used solid phase permethylation for labeling nucleosides with CD3 groups and monitoring their changes in dynamic multiple reaction monitoring (dMRM) mode. (8) Permethylated nucleosides have increased hydrophobicity versus native nucleosides, which improves their separation and ionization during LC-MS. Another alternative is to eliminate the HPLC step altogether and analyze ribonucleotides via a gas-phase separation method such as ion mobility separation. (9)
While most of the literature documenting the detection of modifications has been focused on tRNA and rRNA, the recent explosion of interest in epitranscriptomics has led to several studies where mass spectrometry is being used for the identification of modifications in messenger RNA (mRNA). However, there are multiple challenges when attempting such analyses including the low cellular abundance of mRNA and ensuring mRNA samples are pure and uncontaminated by other RNAs. Caution and healthy skepticism must be exercised when examining reports claiming the detection of modifications in human mRNAs. (10) More confidence can be placed on those studies using mRNA obtained from single-cell organisms. For example, Feng and co-workers have used polyT-based purification and agarose gel-based purification for extracting high purity mRNA finding both Inosine and 2′-O-methylinosine (Im) in yeast mRNA. (11) They also found that the types and abundances of these modifications varied based on the incubation period. Another report examined Escherichia coli (E. coli) mRNA modifications at early exponential growth (2h) and stationary phase (8h) to understand their biological roles. (12) A significant increase in N6-methyladenosine (m6A), 1-methylguanosine (m1G), Ψ (pseudouridine), and m5C were found in stationary phase samples compared to the levels detected in early exponential phase indicating that changes in RNA modifications are dynamic.
Measuring dynamic changes in post-transcriptional modifications
Nucleic Acid Isotope Labeling with Mass Spectrometry, termed NAIL-MS, has been a significant advance in the utility of using LC-MS/MS to quantify modifications from RNAs isolated from cell culture. (3) NAIL-MS allows one to monitor dynamic changes in both new and mature transcripts of RNA. In two papers by Kellner et al., NAIL-MS was used to study dynamic changes in tRNA and rRNA modifications in yeast (13) and in human cells (Figure 2). (14) They noticed a time-dependent loss of modifications in mature tRNAs, and an increase in adaptation of tRNA modifications in new transcripts but not in mature ones when the cells were stressed by addition of methyl methane sulfonate (MMS).
Figure 2. Capturing dynamic changes in RNA modifications.
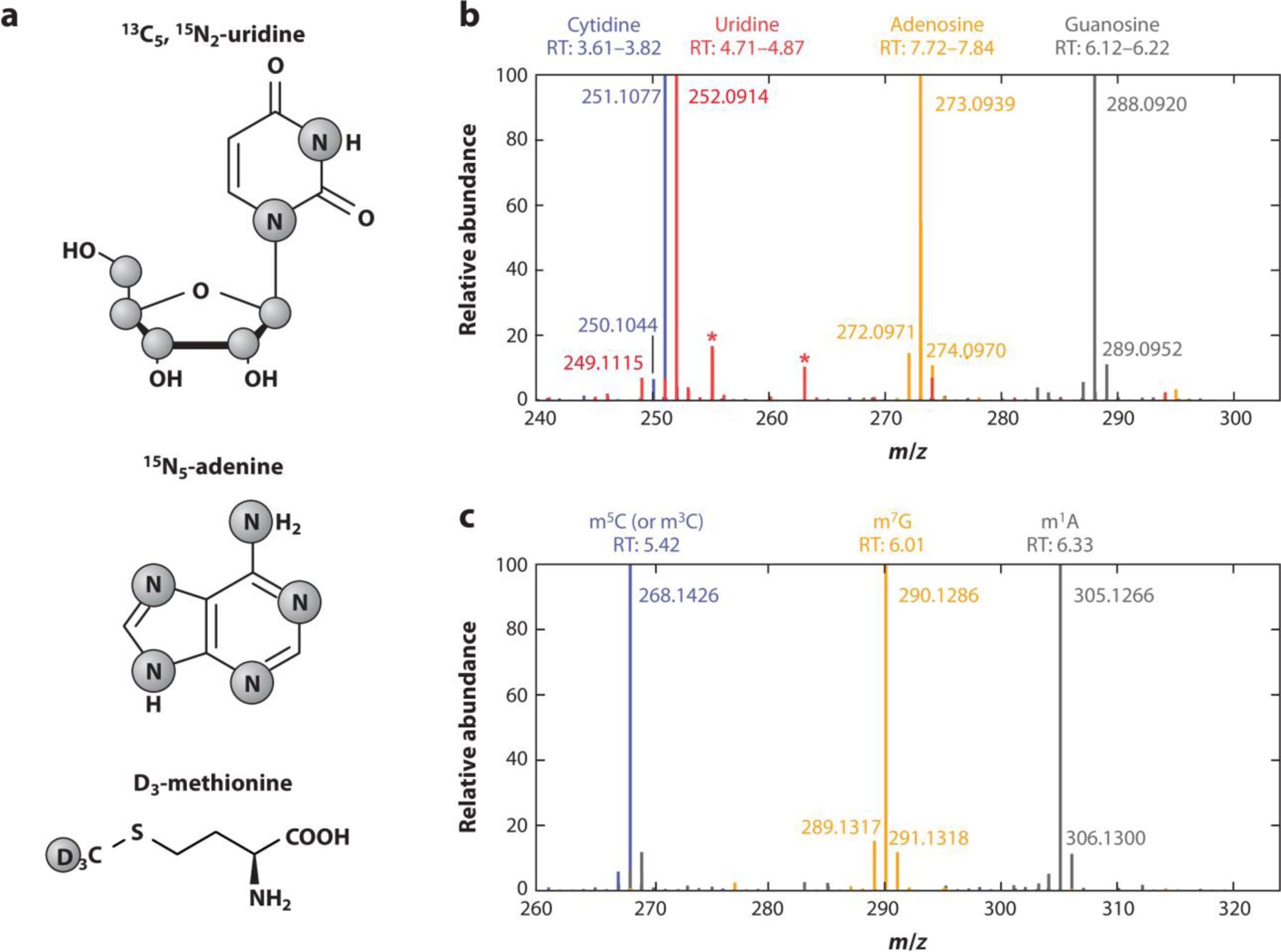
High resolution mass spectra of stable isotope labeled nucleosides from cell culture. (a) Labeling of compounds used for stable isotope labeling in cell culture. Grey circles indicate the positions of isotopes (13C, 15N, or 2H/D). (b) Merged high resolution mass spectra of the four canonical nucleosides of total tRNA after labeling of HEK 293 cells with shown compounds for 7 days. Background signals are marked with asterisks. (c) Merged high resolution mass spectra of three exemplary modifications (m5C, m7G, and m1A) in total tRNA after stable isotope labeling of HEK 293 cells for 7 days. Figure adapted with permission from Reference (14); copyright 2021 The Authors. Abbreviations: tRNA, transfer ribonucleic acid; HEK 293, Human embryonic kidney 293; m5C, 5-methylcytidine; m7G, 7-methylguanosine; m1A, 1-methyladenosine.
Other types of stresses have also been shown to impact the identity and quantity of modified nucleosides from cellular RNAs. In one example dynamic changes in post-transcriptional modifications of E.coli rRNA under different types of oxidative stress were identified. (15) UV-A radiation exposure generated a significant increase in 5-guanidinohydantoin (Gh), while the Fenton reaction led to an increase in the guanosine oxidation product, 2,6-diamino-4-oxo-5-formamidopyrimidine (Fapy-G). In another case, exposure of tRNAs to UV radiation led to significant variance in modified nucleosides including those arising from photooxidative damage. (16)
Non-standard issues when detecting modified nucleosides by LC-MS
Positional isomers, such as 3-methylcytidine (m3C), 4-methylcytidine (m4C) and 5-methylcytidine (m5C), can be a challenge for mass spectrometry as such isomers have the same precursor mass and often generating the same product ions during CID. This feature is particularly problematic when analyzing modifications on a low-resolution mass spectrometer such as a triple quadrupole mass spectrometer operating in multiple reaction monitoring (MRM) mode (17, 18) especially if these isomers cannot be separated chromatographically or authentic standards are unavailable. Recent work has demonstrated the benefit of examining the product ion spectra of positional isomers at a variety of different collisional energies. (19–21) In one example, the methylated modifications of all four canonicals could be identified by comparing various fragmentation patterns at different normalized CID energy in MS/MS or MS3 spectra. (21) A simplified approach just uses high (80) and low (20) normalized CID energies to differentiate positional isomers in biological samples. (19) In another study, higher energy collisional dissociation (HCD) was found to yield isomer-specific fragmentation fingerprints that enabled the differentiation of positional isomers without requiring multiple energy settings as required for CID-based approaches. (20) These approaches are generally applicable to a number of tandem MS platforms and are good instrumental practices when analyzing any mixture of modified nucleosides.
Another issue that analysts must be aware of has arisen due to advances in mass spectrometry sensitivity. Because modern instrumentation can detect ever decreasing ion signal levels, it is critical to differentiate naturally occurring modifications from artifacts that may arise during sample preparation or analysis. Jora et al. demonstrated that methylsulfomethylisocytidine (msm5isoC) was generated as a minor artifact during ammonium-buffered hydrolysis of tRNA under mild basic conditions. (22) Understanding the chemistry occurring during digestion enables method improvements, such as using a FastAP-digestion procedure, (23) that can then minimize the generation of artifacts. These protocol improvements enhance confidence and accuracy when identifying RNA modifications.
Semi-automated and automated identification of post-transcriptional modifications
Nucleoside data analysis is a time-consuming process, biased and prone to mistakes. Improvements in post-analysis data processing have arisen recently. Nucleos’ID software has been created for the untargeted identification of modified nucleosides. (24) The advantages of this software were illustrated via identification of known as well as unknown modifications in 70S ribosomes from Pseudomonas aeruginosa. Another approach enables semi-automated characterization of ribonucleosides using a spectral matching network (Figure 3). (25) This method was used not only to detect known modifications from different organisms but also to discover new modifications in a retention-time independent manner.
Figure 3. Automating the detection of modified nucleosides by mass spectral networks.
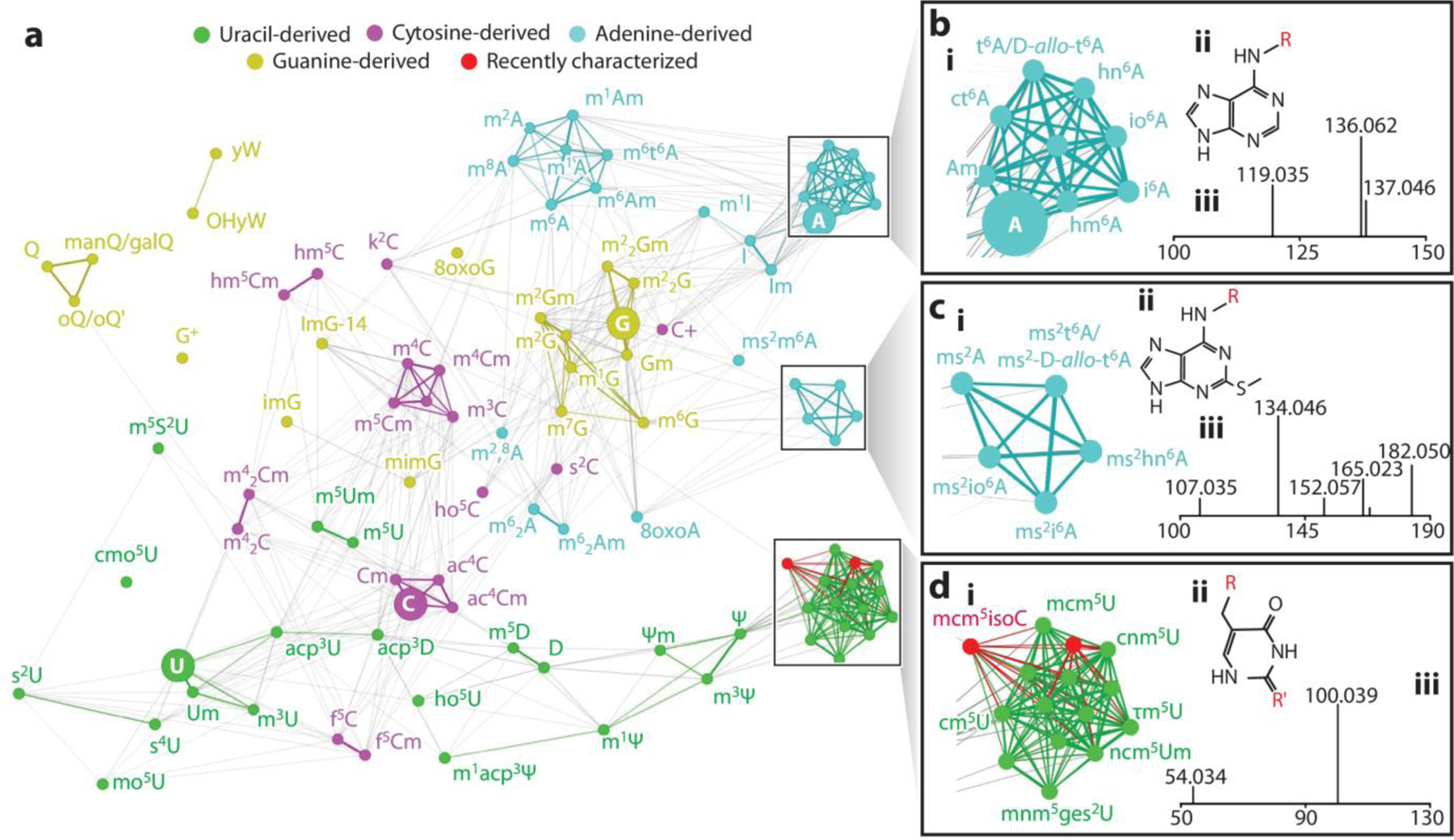
Graph depicting a spectral network of ribonucleosides. For easier identification, canonical ribonucleosides (cytidine, uridine, guanine, and adenosine) are represented by larger nodes. Examples of clusters formed by structurally related modified nucleosides, as well as their shared structural cores and MS/MS spectra, are highlighted on inset A, inset B, and inset C. An interactive version of the spectral network may be found at http://bearcatms.uc.edu/spectral-network-interactive/. Figure adapted with permission from Reference (25); copyright 2022, American Chemical Society. Abbreviations: MS/MS, tandem mass spectrometry.
LC-MS and LC-MS/MS of Oligonucleotides
Recent developments in chromatography
Oligonucleotides are becoming increasingly popular as molecular biology tools and therapeutic agents. For over 25 years, ion-pair reversed phase HPLC has been the gold standard for LC-MS analyses of oligonucleotides. (26) Typically, the combination of triethylamine (TEA) with hexafluoroisopropanol (HFIP) on bridged ethylene hybrid (BEH) or C18 silica columns provides amongst the best separation and compatibility with electrospray ionization methods. (27, 28) Recently, there have been studies that have investigated other alkylamines to enhance the ESI efficiency of oligonucleotides. (29–31) For example, Donegan et al. looked at 13 alkylamines as ion-pairing reagents for the separation of various types and classes of oligonucleotides on a C18 column. (32) Overall, they found that tertiary alkylamines resulted in longer retention time in the presence of 100 mM HFIP buffer compared to ammonium acetate buffer. The observed retention gains were smaller for secondary amines and minimal for primary amines. Interestingly, a high peak capacity was seen in a hydrophobic octylamine system, which resulted in good resolving power for up to 50-mer oligonucleotides (Figure 4). This study builds upon prior work noting that the ion pairing reagent effectiveness depends on the type of oligonucleotide, such as DNA, RNA, and highly modified therapeutic oligonucleotides. (33) Recently, Enmark et al. introduced an ion-pair reagent gradient mode as a new separation technique for oligonucleotides. (34, 35) They tested various alkylamines, but ultimately, dibutylammonium acetate was the best ion-pairing reagent as it led to slightly better selectivity. Additionally, their ion-pair reagent gradient mode could elute oligonucleotides without increasing the co-solvent concentration.
Figure 4. Enhancing the separation of oligonucleotides.
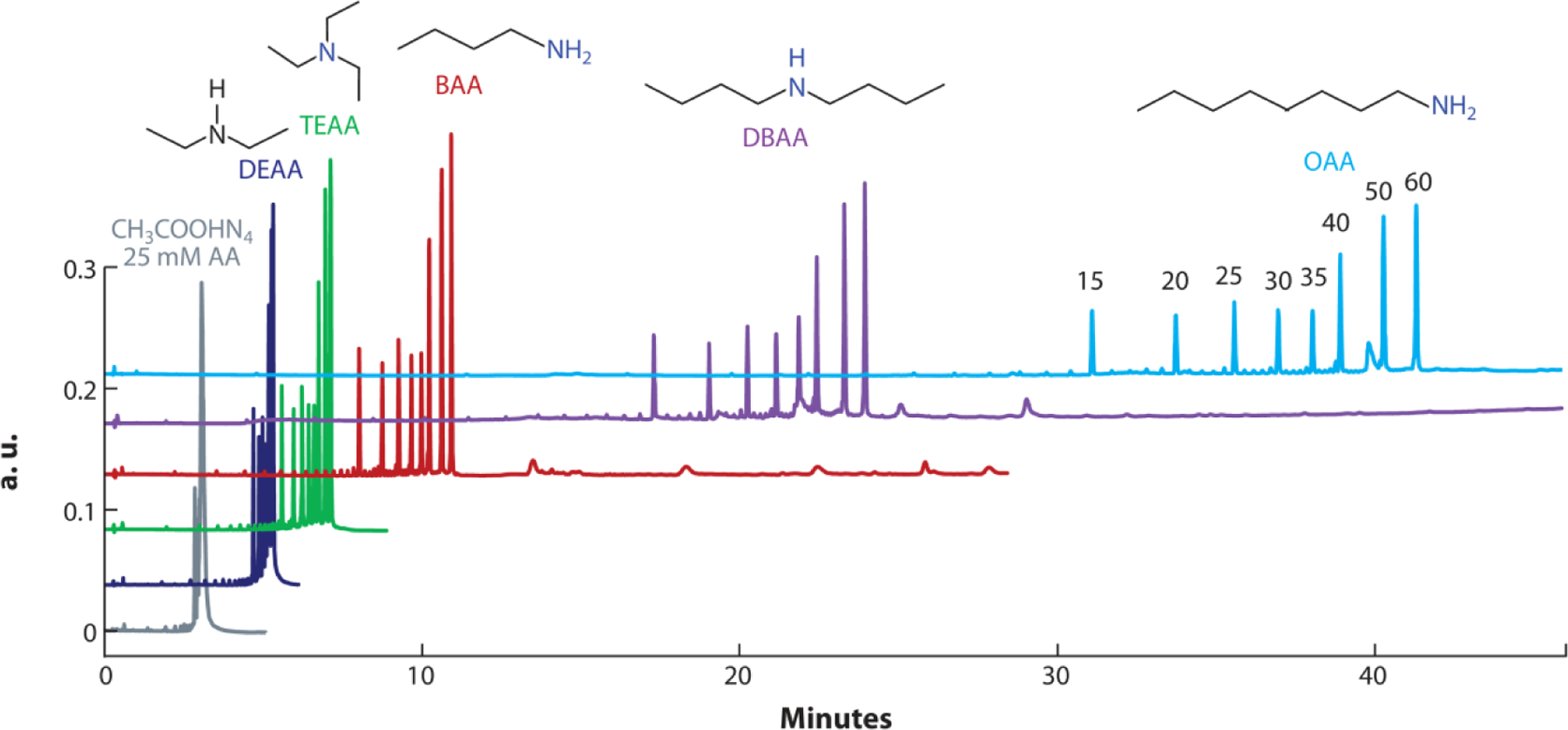
Separation of 15, 20 25, 30, 35, 40, 50, and 60 mer oligodeoxythymidines using selected 100 mM ion-pairing buffers. The data are compared to non-ion-pairing 25 mM ammonium acetate buffer. Longer alkyl-chain ion-pairing buffers lead to enhanced separation of oligonucleotides. Figure adapted with permission from Reference (32); copyright 2022 Elsevier. Abbreviations: AA, ammonium acetate; DEAA, diethylammonium acetate; TEAA, triethylammonium acetate; BAA, butylammonium acetate; DBAA, dibutylammonium acetate; OAA, octylammonium acetate.
It is important to note that TEA and HFIP are often viewed disfavorably by practitioners who do not want to expose their equipment to chemicals that can be hard to eliminate from the system and that are expensive and hazardous to use. Recently, researchers have been exploring and developing new stationary phases that can successfully separate oligonucleotides without the use of ion-pair reagents. (36–38) Lobue and coworkers compared the LC-MS performance of hydrophilic interaction liquid chromatography (HILIC) to conventional ion-pair RP-HPLC for a model mixture of oligonucleotides. (38) While they demonstrated resolving powers for the two approaches were similar, the sensitivity of HILIC remains less than that available using ion-pair reagents. However, the advantage of HILIC is the use of more common mobile phases (e.g., ammonium acetate, acetonitrile, water) that do not lead to the instrumentation challenges of ion pair reagents. Future advances in HILIC stationary phases may enable this technique to replace ion-pairing RP-HPLC in many situations. (39) Beyond HILIC, Kellner et al. used aqueous ammonium acetate buffer and acetonitrile with size exclusion chromatography. (36) They were able to obtain good chromatographic separation and resolution while running MS in positive ion mode. Additionally, their method could be used on most LC-MS set ups. There is no question that continued investigations into new separation modalities that are MS-compatible will be of a great benefit to this field. (40)
Therapeutic Oligonucleotide Analyses
LC-MS/MS has been a useful tool to quantify and analyze therapeutic oligonucleotides such as antisense oligonucleotides (ASOs) and short interfering RNAs (siRNAs) and their metabolites. (41–44) A number of impurities have been identified via LC-MS/MS, pointing to the need for rigorous analytical protocols for their analyses. (45–48) Another recent focal area has been on improving sample recovery and purity when analyzing therapeutics and their metabolites directly from biological matrices. Jiang et al. investigated recovering ASOs by either solid phase extraction (SPE) or hybridization, followed by analytical flow or microflow LC-MS/MS. (49) Overall, a threefold increase in sensitivity (S/N) was seen in the SPE extract and 5.6 fold increase in the hybridization extract. Additionally, microflow LC showed better results compared to analytical LC. The hybridization method provided a cleaner sample extract with lower background compared with the SPE method. Moreover, higher sensitivity was seen with hybridization extract than SPE extract when going from analytical flow LC to microflow LC. One potential limitation of microflow LC for ASO bioanalysis is the column lifetime, which tends to be shorter (few hundred) than that of analytic flow columns (1000 samples). By combining microflow LC-MS/MS with hybridization extraction, a highly sensitive method over the range of 0.100–100 ng/mL was successfully developed and qualified for the quantification of ASO-001 in rat plasma.
RNA modification mapping
Improved bottom-up approaches
Modification mapping by MS can be used to place modifications within RNA sequences. The most common approach is based on enzymatic digestion of one or more RNAs into smaller oligonucleotides amenable to analysis by LC-MS/MS, the so-called “bottom-up” strategy. For RNA, several different enzymes have been used (Table 1). Even though RNases T1, A and U2 are commercially available, the current suite all exhibit limitations impacting RNA modification mapping by LC-MS/MS. (50–52) To increase coverage, several enzymes have been used together generating results that complement one another. (53, 54) For example, digesting samples with RNase T1, cusativin and MC1 improved sequence coverage by generating overlapping digestion products. (55)
Table 1.
Cleavage specificity and limitation of enzymes available for RNA modification mapping.
| Enzyme | Cleavage specificity | Limitations | Ref |
|---|---|---|---|
| RNase T1 | all unmodified guanosine residues and 3’-end of the modified nucleoside N2 -methyl guanosine (m2G) | RNase T1 does not generate high sequence coverage especially when there are G-rich sequence redundancies available | (56) |
| RNase A | canonical pyrimidines and the modified nucleoside Ψ (pseudouridine) | RNase A generates shorter degradation products that are not useful for modification placement | (57) |
| RNase U2 | canonical purines with a slight selectivity towards adenosine | RNase U2 does not increase the sequence coverage of mapped modifications | (58) |
| Human RNase 4 | uridine residues prior to purines (slight preference for UA relative to UG) | RNase 4 best for mRNA and other long RNA substrates/requires addition of T4 polynucleotide kinase to avoid generation of 2’,3’ cyclic phosphates | (59) |
| MC1 | uridine and pseudouridine at the 5’ end | MC1 is commercially unavailable | (53) |
| Cusativin | cytidine and 5-methylcytidine (m5C) at the 3’ end | Cusativin is commercially unavailable | (60) |
When limited to conventional RNases, another strategy has been to modify the enzymatic reaction conditions to minimize cleavage of the RNA to thereby generate longer digestion products. (61, 62) Limited reactivity has been achieved by immobilizing the enzyme (62) and by lowering the incubation temperature. (63) In both cases, longer and more sequence informative digestion products are created which simplify modification mapping. The drawback to these techniques is the greater computational demands at identifying the base composition and number of missed cleavages in the acquired data. (64)
Another approach for increasing sequence coverage is the combination of LC-MS/MS with tRNA-seq to profile tRNA modifications. (65, 66) Such combinations can be done using conventional endonuclease digestions via the RNases listed in Table 1, or can be achieved by digesting samples with RNase H, which cleaves RNA at random positions adjacent to RNA-DNA duplexes in a site-specific manner. (67, 68) This RNase H method could differentiate between positional isomers and provided good coverage for mouse tRNA His-GUG and Val-UAC which were not identified before.
Top-down mass spectrometry
Top-down mass spectrometry is an alternative approach for analyzing RNA and its modifications. The main advantages of the top-down approach are the preservation of the complete sequence information, (69) characterization, and localization of post-transcriptional modifications, (70) and analysis of minor components in complex samples. (71) Because the top-down approach avoids enzymatic digestion, typically specialized instrumentation and methods are required to generate extensive sequence coverage via dissociation of the intact oligonucleotide or RNA in the gas phase. (72) CID can yield nearly full sequence coverage by generating predominantly complementary c- and y-type product ions, (73) but works best for low charge state precursor ions. However, lower charge state precursor ions negatively impact sensitivity and mass resolving power. (74) To overcome this limitation, CID has been coupled with electron detachment dissociation (EDD). (74, 75) EDD generates non-complementary d- and w-type product ions and yields best results for high precursor ion charge states. (75) The limitation of EDD is the low fragment ion yield and internal fragmentation. (74) CID and EDD have been used in combination for the identification, localization, and relative quantification of m5U, m5C, m3U and m6A modifications in intact RNAs. (76)
Efforts have been focused on creating alternative dissociation approaches that would enhance top-down sequencing of nucleic acids including RNAs. These include negative electron transfer dissociation (NETD), activated-ion negative electron transfer dissociation (AI-NETD), (77) ultraviolet photodissociation (UVPD) and activated electron photo detachment dissociation (a-EPD). (78) NETD and AI-NETD generate non-complementary d- and w-type product ions, (77) while UVPD and a-EPD can generate a/w, c/y, b/x, and d/z complementary product ion pairs. (78) The main advantage of NETD and AI-NETD is that they do not require secondary activation. (77) For UVPD and a-EPD dissociation pathways the main advantage is that they reduce generation of base-loss products which are common during CID. (78) One can anticipate that these developments will lead to enhanced capabilities for top-down approaches to mapping RNA modifications, which likely will find high utility in the analysis of longer modified therapeutics including mRNA-based drugs.
Software development
Manual interpretation of oligonucleotide MS/MS data is challenging and time-consuming. These complications arise due to the complexity of the product ion spectrum generated during CID or other dissociation methods. (79) To simplify data interpretation, a variety of software programs have been developed (Table 2). Most of these software packages work on the same principal – based upon certain input data (e.g., expected sequence, nucleoside composition, presence of modification(s)), a theoretical (“in silico) MS/MS spectrum is generated and the experimental data is then compared against this theoretical construct. (80–83) A variety of different scoring and evaluation metrics are then used to determine whether the experimental data is considered the “correct” match with the theoretical MS/MS data. These programs also differ in output formatting, batch processing of multiple sequences, and workflow. Alternative approaches include the de novo interpretation of MS/MS data of oligonucleotides that can contain multiple modifications, (84) or creating spectral databases from oligonucleotide standards and then using spectral matching for sequence identification. (85) Callout #1
Table 2.
Summary of software programs freely available for RNA modification mapping.
| Software | Advantages | Limitations | Ref |
|---|---|---|---|
| RoboOligo | • de novo sequencing of complex and multiple modifications | Inability to distinguish between: • positional isomers of the same nucleobase, • uridine and pseudouridine, • precursor ions with the same or nearly identical m/z values. |
(84) |
| RAMM | • in-silico database • Fixed and variable sequencing |
• Cannot differentiate between methyl and sulfur modifications of the same canonical nucleobase. • Does not completely eliminate manual interpretation of the data. |
(80, 81) |
| NIST spectral software | • Detects presence or absence of a specific modification | • Can be used only for known oligonucleotides • Cannot differentiate between cytidine and uridine |
(85) |
| NASE | • open MS software • false discovery rate (FDR) parameter through target/decoy search strategy • corrects precursor mass values and cation adductions |
• High false discovery rate • Cannot differentiate between positional isomers and between uridine and pseudouridine. • Does not completely eliminate manual interpretation of the data. |
(82) |
| Pytheas | • open-source software • FDR through target/decoy strategy • isotope labelled sequences • differentiation between positional isomers, uridine and pseudouridine |
• Computationally intensive • Manual interpretation of the data is still required. |
(83) |
Callouts.
Callout #1: Where to find information on RNA sequences and RNA modifications?
RNA databases are expanding with the new discoveries in RNA and RNA modifications. For RNA-Sequences, databases like GtRNAdB (2) is a collection of predicted tRNAs and tRNA genes including the programs tRNAscanSE (3) which allow the user to find tRNA genes within the genome of interest. For RNA modifications, MODOMICS (4) offers information on types of modifications- structures, tRNA-modifying enzyme pathways, presence in domains of life, and modified sequences. Recently updated, RNACentral, offers one of the largest compilations of RNA databases and information regarding protein- coding and non-coding RNAs.(5) Along with enhanced 2D structure information, RNACentral provides access to 44 RNA resources including the ones listed above. For similarity searches, a tool powered by the nhmmer software can compare query sequencings to a large collection of non-coding RNAs in parallel reducing search time. In addition, Sequence Ontology allows for the user to annotate RNA species type with higher precision. This collection of tools and databases specifically for RNA enables easier access to resources for RNA analysis.
Callout #2: Reducing Errors in Short-Read Sequencing
While methods for modification-calling take advantage of these traits, errors in cDNA synthesis greatly hinder traditional RNA-Seq used for transciptomics or relative quantification of RNAs. The major reason for this is incomplete or erroneous strands are formed for sequencing. Shorter reads make it difficult to confidently align the strand to the original sequence impairing the qualitative and quantitative analysis. To overcome issues with cDNA synthesis due to naturally occurring modifications in RNA, other enzymes or chemical treatments are used to remove these modifications. The methods, ARM-Seq- developed by the Lowe Lab (106) and DM-tRNA-Seq developed by the Pan Lab (107), implement the use of enzymes to remove methyl groups on bases that interrupt base pairing leading to incomplete cDNA strands. Variants of the commonly used enzyme AlkB has been show to provide higher selectivity in the removal of methylations on specific bases, such as m1G. (108) The future of this field will investigate the development or discovery of enzymes with a higher specificity or chemicals to selectively target modifications leading to highly predicable and reproducible results.
Structural analysis by Mass Spectrometry
Over the years, it has become apparent that sequence information alone is unable to give a proper look into the function of RNA. However, information on the higher order structure of RNA may identify potential intra- and inter-molecular interactions revealing conformational dynamics and functional characteristics. Structural analysis approaches generally use probes (86) or enzymatic digestion (87) with predictable and reproducible reactivity to gain insights into substrate surface or internal interactions of the biomolecule of interest. For example a class of alkylating agents, Bis-3-chloropiperidines (B-CePs), were used to investigate the dimerization initiation site domain of the HIV-1 genome. (88) B-CePs have a high reactivity to DNA causing depurination and strand cleavage at guanine residues. However, it was found that B-CePs form adducts with RNA without strand cleavage and bridge across adjacent guanines. To confirm the formation of the G-G bridges, dsDNA was reacted with B-CeP probes and solution thermal melting with electrospray ionization mass spectrometry (STHEM-ESI) was employed to denature the dsDNA in sample solution before entering the gas phase. At high temperature, conjugated products were observed indicating the crosslinking at the two G sites was preventing strand disassociation. These probes were then tested on the highly structured dimerization initiation site of the HIV-1 genome. It was found that the two guanine residues within the loop structure were able to cross-link leading to insight into the structure of the loop.
A top-down method that has become a widely used tool in structural analysis of RNA is native mass spectrometry. Native mass spectrometry is predominantly used in proteomics, however, more work has recently been done to study RNA complexes. (40) Native mass spectrometry is the control of electrospray-ionization parameters to form gas-phase ions of the biomolecule while still retaining as much of its native structure in solution as possible. (89) It has been shown that native mass spectrometry can be a valuable tool in determining protein–RNA binding sites through CID. (90) One elegant example is the work examining the binding of an arginine rich motif (ARM) peptide from rev protein to rev response element RNA from HIV-1. (91) It was found that the RNA constructs and ARM formed 1:1 and 1:2 RNA-peptide complexes (Figure 5). Through CID, specific binding sites were elucidated from fragmentation along the RNA phosphodiester backbone. It was revealed that the first and second rev ARM peptides bind in regions including the internal loop. Using stoichiometric ratios of RNA-peptide, the binding sites were identified up to 1:5 RNA-peptide complexes. Due to the continuous recruitment of rev ARM peptide to the upper stem loop, a mechanism was proposed where the first ARM peptide binds to a region but is relayed to a different region so that additional ARM peptides may bind. Native spectrometry analysis revealed structural information and conformation dynamics of RRE/rev in this study paving the way for potential drug design and substrate binding studies in the future.
Figure 5. The use of native mass spectrometry to understand RNA binding.
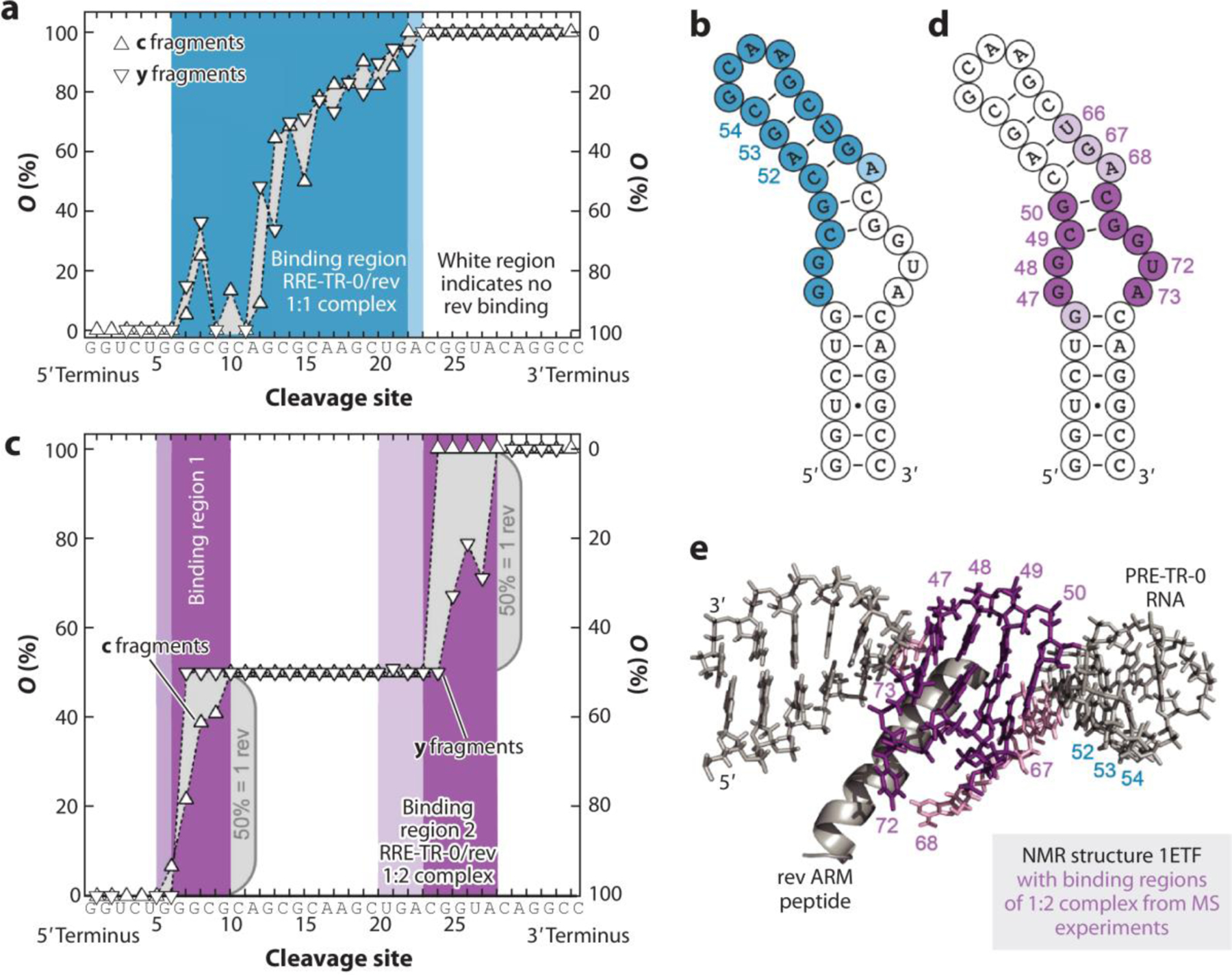
Binding site mapping of RRE-TR- 0/rev complexes by CAD MS. (a) Site-specific occupancies (O) of c (left axis) and y (right axis) fragments with rev ARM peptide from CAD of 1:1 complex ions, (RRE-TR-0+ 1·rev - 14H)14−, at 137.2 eV and the corresponding binding region (blue) mapped onto the predicted secondary structure of RRE-TR-0 (b) show poor agreement with binding sites in the NMR structure (e). (c) Occupancies of fragments from CAD of 1:2 complex ions, (RRE-TR-0+ 2·rev - 14H)14−, at 175.5 eV and corresponding binding sites (violet) mapped onto the RRE-TR-0 structure (d) show good agreement with binding sites in the NMR structure (e). Darker and lighter colors in b, d, and e stand for stronger and weaker binding, respectively. Panel adapted with permission from Reference (91); copyright 2020 Nature Communications. Abbreviations: CAD, collision activated disassociation mass spectrometry; ARM, arginine-rich motif; NMR, nuclear magnetic resonance.
RNA-Seq
Next Generation Sequencing (NGS) – based RNA Sequencing (RNA-Seq) relies on the use of reverse transcriptase – polymerase chain reaction (RT-PCR) to prepare a library from the RNA sample of interest and amplifying it for downstream sequencing. This library is sequenced on an NGS platform (92) where the cDNA library is bound to primers on the flow cell and sequenced through images taken with fluorescently labelled bases. This allows information to be gathered on the base composition and the length of the RNA within the sample. New technologies have been developed to sequence RNA from single cells (93) or nuclei (94) allowing information to be uncovered differentiating heterogeneous cell composition within tissue samples. Recently, a study by Alvarez et al. combined single cell (sc) and single nuclei (sn) techniques to identify cell type in hepatocellular carcinoma (HCC) affecting survival. (95) Using snRNA-seq data and preexisting scRNA-seq data from HCC and non-tumor liver biopsies, the authors performed a pathway enrichment analysis to look for upregulated genes for each liver cell type. A new proliferative cell type (Prol) was found to be enriched in oxidative phosphorylation and cell cycle genes suggesting that their functions are mainly associated to cell division and growth. Applying this knowledge to bulk RNA-Seq data, the authors were able to associate this cell type’s marker genes with lower overall survival and progression-free in 361 HCC patients.
DART-Seq for m6A detection in mRNA
In the field of epitranscriptomics, the RNA modification that has attracted the most attention is N6-methyladenosine (m6A). Most methods for identification of m6A modifications use immunoprecipitation. (96, 97) While these methods have been proven to be useful and robust, the limitation of using antibodies is their promiscuity towards modifications with similar moieties. DART-Seq, developed by the Meyer lab, uses an m6A-binding YTH domain tethered to a cytidine deaminase, APOBEC1, which directs C-to-U editing in the base adjacent to the m6A in the cDNA strand to indicate the presence of a m6A (Figure 6). (98) Recently, a YTH domain variant has been developed to enhance m6A recognition in DART-Seq. (99) DART protein variant APO1-YTHD422N demonstrated higher C-to-U editing rate compared to the wildtype and the mutant protein in cellular studies as well as in vitro. Overall, the DART protein variant demonstrated overall increased selectivity and sensitivity towards m6A. Additionally, the authors demonstrated the ADAR catalytic domain with the YTH variant can be used in place of APO1 identifying m6A sites through A-to-I editing. The ADAR-YTHD422N protein variant identified more m6A sites showing increased sensitivity and enhanced resolution as compared to the APO1 containing variant. Using both protein variants in future studies could be used as orthogonal methods raising the confidence of m6A calling in sequencing studies.
Figure 6. Enhanced detection of m6A sites in RNA.
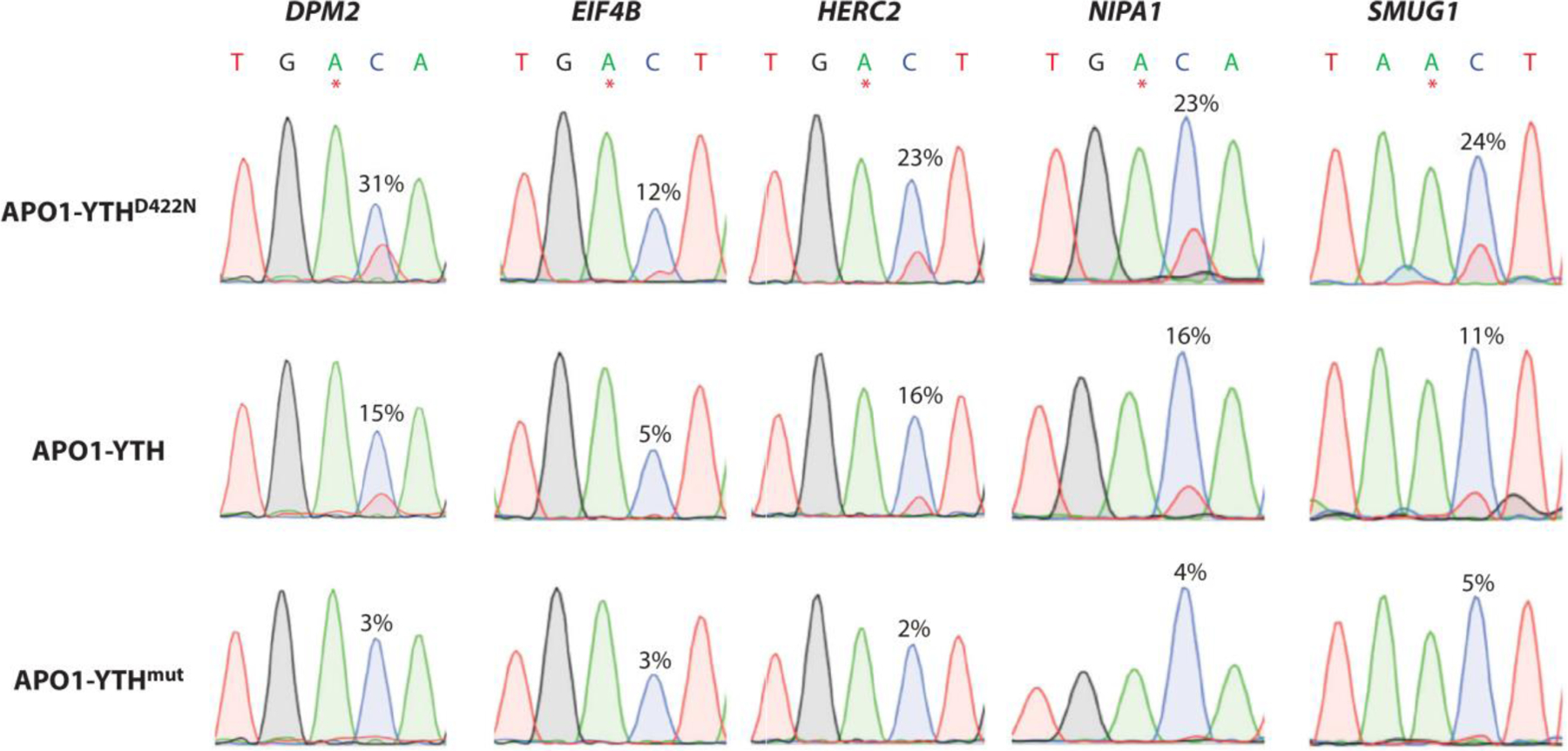
Sanger sequencing traces showing C-to-U editing adjacent to m6A sites in cells expressing APO1-YTHD422N, APO1-YTH, and APO1-YTHmut for five mRNAs previously shown to contain m6A: DPM2, EIF4B, HERC2, NIPA1, and SMUG1. m6A sites are indicated by asterisks. C-to-U editing rate (%U) is indicated above the adjacent cytidine. Data are representative of three biological replicates. Panel adapted with permission from Reference (98) copyright 2022; Frontiers in Cell and Developmental Biology. Abbreviations: mRNA, messenger RNA.
Alternative RNA-Seq methods for modified RNAs
While RNA-Seq was developed primarily for analysis of mRNA transcripts, it has since been applied to non-coding RNAs. (100, 101) Non-coding RNAs, such as tRNAs, contain many naturally occurring modifications. Due to the nature of reverse transcriptases used for cDNA synthesis, these modifications can interrupt base incorporation, lead to missed incorporation sites, or generate incorrect base incorporation in the resulting cDNA strand. (102) Several methods have been developed to take advantage of these characteristics for identifying the sequence location of modified nucleosides (i.e., modification mapping or modification calling). (103–105) Techniques such as ARM-Seq (106) and DM-tRNA-Seq (107) implement the use of enzymes to remove methyl groups on bases that interrupt base pairing leading to incomplete cDNA strands. Variants of the commonly used enzyme AlkB can provide higher specificity in the removal of methylations on specific bases, such as m1G. (108)
Chemical treatment may be used to produce signatures for modifications that are otherwise ‘signature silent’. Periodate-dependent analysis of queuosine and sulfur modification sequencing (PACS-Seq) uses chemical treatment to produce cDNA synthesis signatures for Queuosine containing modifications and 2-thio modifications. (109) After tRNA is treated with periodate, Queuosine and the Queuosine derivatives, manQ and galQ, were found to produce a deletion signature while 2-thio modifications were found to produce both mutational and deletion signatures. Using this method these signatures can be used for relative quantification or modification mapping of queuosine and its derivatives in RNA-Seq data. Callout #2
Nanopore Sequencing and Modification Analysis
Biological vs. Solid-State Nanopores
Nanopore sequencing is third generation sequencing (NNGS) developed by Oxford Nanopore Technologies. The composition of the analytes is detected through the modulation of ionic current as the biomolecule passes through a nanopore. This process is either electrophoretically driven or requires using an engineered motor protein. In comparison to NGS, nanopore sequencing has the advantages of having a high turn-around time with real-time data analysis. However, the error rate for NNGS short-read sequencing (5–15%) is still high compared to Illumina sequencing (0.1%). (110)
Nanopore sequencing was first developed by using current to pull the substrate of interest through the pores. However, it was difficult to control the rate at which the oligonucleotides would enter the pore causing low resolution. (111) Advances in nanopore technology used pore-forming enzymes such as Staphylococcus aureus alpha-hemolysin (α-HL) or Mycobacterium smegmatis porin A (MspA) (110) which have the ability to translocate the analyte of interest at a constant rate. This allows for proper identification of the biopolymer with higher resolution. Solid-state nanopores are currently being investigated as an alternative to enzymes. Alessio et al. compared three biological nanopores, MspA, α-HL, and Fragaceatoxin C -the mutant of FraC with a positively charged constriction (ReFraC), against two solid-state nanopores, SiNx and MoS2. (112) It was found that SiNx had the highest signal modulation and signal-to-noise ratio when comparing all of the nanopores tested. However, these experiments used electrophoretically-driven translocation. When a helicase is used for the biological nanopores, the time-controlled translocation speed raised the signal-to-ratios up to ~650, 2 orders of magnitude higher. Consequently, current technology of biological nanopores has so far been proven to be superior due to the enzymatically controlled translocation speed. However, the future of nanopore sequencing, DNA and RNA alike, may be improved with solid-state nanopores after the issue of time control has been resolved.
Nanopore sequencing of modified RNAs
Native nanopore sequencing retains the modification on the sequenced strand. This allows for modifications to be identified or mapped through modulations in current intensity that differ from unmodified bases. Smith and co-workers explored the use of native nanopore long-read sequencing to look at modified and canonical bases in Escherichia coli 16S rRNA. (113) They found that the accuracy in alignment increased from 67.9% for short reads to 96.9% for long reads. Additionally, the modification m7G, known to be present at positions 527 and 1405, was identified. It was found to have reproducible base-call errors producing a decrease or alteration in ion current. An E. coli enzymatic knockdown of the RNA modifying enzyme RsmG, known to methylate G527 in rRNA further confirmed the presence of m7G in WT E. coli by erasing the base-call error. However, the aminoglycoside resistant strain BL21 (RmtB+) contains an m7G at position 1405 which produced a deletion that was not present in the wild type. The authors speculate that the presence of a modifications N4-methylcytidine (m4C) and N4,2-O-dimethylcytidine (m4Cm) down and upstream of the m7G modification, respectively, may contribute to the difference in base-call error produced. The impact of modifications on signal modulation is a new field of research that may open many doors in the RNA modification mapping world.
Sequencing methods, Nanopore and RNA-Seq alike, have improved significantly in resolution and accuracy since their development. Similarly, the scope of the knowledge gained from performing these analyses has increased to include multiple RNA species and can identify modification placement within a sequence. RNA-Seq has the advantage of higher accuracy and high throughput, however, PCR-based amplification is required for analysis. This caveat causes the identity of RNA modifications to be lost. With Nanopore sequencing, the error rate is higher in comparison to RNA-Seq and requires a higher sample amount but native RNA strands may be sequenced recovering RNA modification information that is lost in PCR-based amplification. Developments in both fields will include higher accuracy in alignment with sequencing programs, methods targeting the identification of specific RNA modifications with increased accuracy and sensitivity, single cell/nuclei sequencing and progression towards improvements in native strand sequencing.
Future Approaches
As the world of RNA expands, alternative methods are now being explored for the sequencing and modification mapping of natural RNAs. Cryo-EM has been a popular method used for structural analysis of RNA and other biomolecules. (114) This method is done by vitrifying the sample containing the biomolecule of interest and imaging them with electron microscopy. The 3D structure of the molecule is reconstructed from averaging thousands of collected images through computational methods. This allows for the structure of the biomacromolecule to be evaluated in nearly native buffer conditions. Due to heterogeneity within the macromolecule, ab initio modelling using cryo-EM has struggled to produce high resolution mapping. (105) However, recent studies are showing improvement of this limitation. Previously, the human 80S ribosomal subunit was visualized using cryo-EM. (115) However, the obtained resolution reached a maximum of approximately 2.5 Å. Very recently cryo-EM was used to reconstruct the human 40S ribosomal subunit at an average of 2.15 Å resolution. (116) At this resolution the authors could visualize 73 out of 91 known rRNA modifications in the 40S ribosomal subunit. Additionally, modifications were found that had not been previously assigned or modelled: 2’-O-methylations, Am and Gm at positions 590 and 1447 respectively, and pseudouridine (ψ) at position 572, 863 and 1136. This high-resolution structural modeling allows for inference on residues and modifications that may play a major role in the translation of proteins.
Another imaging based approach using CRISPR/dCas9-MS2-based RNA fluorescence in situ hybridization assay (RCasFISH) was recently reported. (117) Targeting the oncogenic transcript human epidermal growth factor receptor 2 (HER2), the authors used a sgRNA scaffold guide that binds to fluorescently labeled MCP dimers to quantify the mRNA in fixed cells with enhanced signal. To estimate detection efficiency, they co-labeled HER2 mRNA with RCasFISH and Stellaris smFISH and compared the fraction of dots labeled with the fluorescence intensity and found the efficiency to be ≥85%. To prove this method would work for tissue samples they tested RCasFISH on formalin-fixed, paraffin-embedded (FFPE) mouse xenograft models by intradermal injections of the cell lines: MCF-7, MDA-MB-453 and SK-BR-3. They found that true positive rates were >81% for all FFPE samples and the signal provided adequate sensitivity (>38.9 average detected mRNA dots). Overall, this method was proven to have a higher signal-to-noise ratio than traditional smFISH and may be done in experiment and clinical FFPE samples.
Outlook
Many research questions have been directed to changes in nucleoside abundances between subsets of samples. An example of this this type of question would be- do oxidized RNA modifications increase in cancer or certain diseases and what oxidized species are they? To answer this question, it would be advantageous to perform nucleoside analysis via LCMS/MS. Compared to RNAseq and NNGS, nucleosides are identified and quantified using their specific mass to charge ratio whereas the mentioned methods indirectly identify modifications using characteristic signatures. These signatures may be used to infer the the position of a modification in its subsequent sequence however the identity of the modification is not directly known. This makes it difficult to relatively quantify the nucleosides of interest.
Another type of question would be concerning the differential expression of mRNA between subsets of samples. For example, are certain mRNAs more highly expressed in exposed vs unexposed samples? To answer this question, it would be preferable to use RNAseq or NNGS. As previously mentioned, NNGS currently has a higher error rate than illumina-based sequencing. However, it has been successfully used for differential expression as well. Contrarily, it would be difficult to gain this type of information using LCMS/MS. Differentiation between intact RNAs from each other based on mass to charge ratio is especially difficult when they are similar in composition and length. Normally to combat this, bottom-up approaches are used to identify different RNAs, however, it would be difficult to assess differential expression unless the RNA had a distinctive region which is not always the case.
Some questions require a combination of these methods to answer them. One specifically interesting question is the modification status of an RNA. What RNAs are differentially expressed and are they fully modified? To answer this question, information on the global RNA and modification abundances as well as the modification identity are required. RNAseq and NNGS may be used to answer this question if the modification position is known and has a characteristic signature. Lack of this signature between samples may indicate an unmodified status. However, using LCMS/MS to complement sequencing methods may provide higher confidence on the modification identity and status at a specific position. This is especially beneficial for modifications that are hypermodified to identify the specific precursor modifications that may be present.
The various technologies described in this review illustrate the breadth of approaches that exist for the analytical characterization of RNA and its modifications. Beyond some of the existing challenges and opportunities noted above, it is expected that future advances will enable more single transcript studies that can more effectively reveal the heterogeneity of RNA and its modifications in a variety of biological and therapeutic contexts. In addition to needed scientific and technological advances, the promise of this scientific domain will be more fully realized as additional analytical standards, improved databases for aggregating sequence and modification information on specific RNA transcripts, and consensus protocols around RNA isolation, characterization and quantification are developed.
Literature Cited
- 1.Nachtergaele S, He C. 2017. The emerging biology of RNA post-transcriptional modifications. RNA Biol 14: 156–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thuring K, Schmid K, Keller P, Helm M. 2016. Analysis of RNA modifications by liquid chromatography-tandem mass spectrometry. Methods 107: 48–56 [DOI] [PubMed] [Google Scholar]
- 3.Heiss M, Borland K, Yoluc Y, Kellner S. 2021. Quantification of Modified Nucleosides in the Context of NAIL-MS. Methods Mol Biol 2298: 279–306 [DOI] [PubMed] [Google Scholar]
- 4.Sarkar A, Gasperi W, Begley U, Nevins S, Huber SM, et al. 2021. Detecting the epitranscriptome. Wiley Interdiscip Rev RNA 12: e1663. [DOI] [PubMed] [Google Scholar]
- 5.Deng L, Kumar J, Rose R, McIntyre W, Fabris D. 2022. Analyzing RNA posttranscriptional modifications to decipher the epitranscriptomic code. Mass Spectrom Rev: e21798. [DOI] [PubMed] [Google Scholar]
- 6.Liguori A, Napoli A, Sindona G. 1994. Determination of substituent effects on the proton affinities of natural nucleosides by the kinetic method. Rapid Commun Mass Spectrom 8: 89–93 [DOI] [PubMed] [Google Scholar]
- 7.Dai Y, Qi CB, Feng Y, Cheng QY, Liu FL, et al. 2021. Sensitive and Simultaneous Determination of Uridine Thiolation and Hydroxylation Modifications in Eukaryotic RNA by Derivatization Coupled with Mass Spectrometry Analysis. Anal Chem 93: 6938–46 [DOI] [PubMed] [Google Scholar]
- 8.Xie Y, Janssen KA, Scacchetti A, Porter EG, Lin Z, et al. 2022. Permethylation of Ribonucleosides Provides Enhanced Mass Spectrometry Quantification of Post-Transcriptional RNA Modifications. Anal Chem 94: 7246–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kenderdine T, Nemati R, Baker A, Palmer M, Ujma J, et al. 2020. High-resolution ion mobility spectrometry-mass spectrometry of isomeric/isobaric ribonucleotide variants. J Mass Spectrom 55: e4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiener D, Schwartz S. 2021. The epitranscriptome beyond m6A. Nature Reviews Genetics 22: 119–31 [DOI] [PubMed] [Google Scholar]
- 11.Feng YJ, You XJ, Ding JH, Zhang YF, Yuan BF, Feng YQ. 2022. Identification of Inosine and 2’-O-Methylinosine Modifications in Yeast Messenger RNA by Liquid Chromatography-Tandem Mass Spectrometry Analysis. Anal Chem 94: 4747–55 [DOI] [PubMed] [Google Scholar]
- 12.Petrov DP, Kaiser S, Kaiser S, Jung K. 2022. Opportunities and Challenges to Profile mRNA Modifications in Escherichia coli. Chembiochem 23: e202200270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoluç Y, van de Logt E, Kellner-Kaiser S. 2021. The Stress-Dependent Dynamics of Saccharomyces cerevisiae tRNA and rRNA Modification Profiles. Genes 12: 1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heiss M, Hagelskamp F, Marchand V, Motorin Y, Kellner S. 2021. Cell culture NAIL-MS allows insight into human tRNA and rRNA modification dynamics in vivo. Nat Commun 12: 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Estevez M, Valesyan S, Jora M, Limbach PA, Addepalli B. 2021. Oxidative Damage to RNA is Altered by the Presence of Interacting Proteins or Modified Nucleosides. Front Mol Biosci 8: 697149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun C, Limbach PA, Addepalli B. 2020. Characterization of UVA-Induced Alterations to Transfer RNA Sequences. Biomolecules 10: 1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin X, Zhang Q, Qin Y, Zhong Q, Lv D, et al. 2022. Potential Misidentification of Natural Isomers and Mass-Analogs of Modified Nucleosides by Liquid Chromatography-Triple Quadrupole Mass Spectrometry. Genes (Basel) 13: 878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodell R, Tsao N, Ganguly A, Mosammaparast N. 2022. Use of High-Performance Liquid Chromatography-Mass Spectrometry (HPLC-MS) to Quantify Modified Nucleosides. Methods Mol Biol 2444: 125–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espadas G, Morales-Sanfrutos J, Medina R, Lucas MC, Novoa EM, Sabidó E. 2022. High-performance nano-flow liquid chromatography column combined with high- and low-collision energy data-independent acquisition enables targeted and discovery identification of modified ribonucleotides by mass spectrometry. J Chromatogr A 1665: 462803. [DOI] [PubMed] [Google Scholar]
- 20.Jora M, Burns AP, Ross RL, Lobue PA, Zhao R, et al. 2018. Differentiating Positional Isomers of Nucleoside Modifications by Higher-Energy Collisional Dissociation Mass Spectrometry (HCD MS). J Am Soc Mass Spectrom 29: 1745–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Zhou J, Yuan G. 2020. Discrimination of common isomerides of methyl nucleosides by collision-induced dissociation tandem mass spectrometry. J Mass Spectrom 56: e4594. [DOI] [PubMed] [Google Scholar]
- 22.Jora M, Borland K, Abernathy S, Zhao R, Kelley M, et al. 2021. Chemical Amination/Imination of Carbonothiolated Nucleosides During RNA Hydrolysis. Angew Chem Int Ed Engl 60: 3961–66 [DOI] [PubMed] [Google Scholar]
- 23.Richter F, Plehn JE, Bessler L, Hertler J, Jörg M, et al. 2022. RNA marker modifications reveal the necessity for rigorous preparation protocols to avoid artifacts in epitranscriptomic analysis. Nucleic Acids Res 50: 4201–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gosset-Erard C, Didierjean M, Pansanel J, Lechner A, Wolff P, et al. 2023. Nucleos’ID: A New Search Engine Enabling the Untargeted Identification of RNA Post-transcriptional Modifications from Tandem Mass Spectrometry Analyses of Nucleosides. Anal Chem 95: 1608–17 [DOI] [PubMed] [Google Scholar]
- 25.Jora M, Corcoran D, Parungao GG, Lobue PA, Oliveira LFL, et al. 2022. Higher-Energy Collisional Dissociation Mass Spectral Networks for the Rapid, Semi-automated Characterization of Known and Unknown Ribonucleoside Modifications. Anal Chem 94: 13958–67 [DOI] [PubMed] [Google Scholar]
- 26.Apffel A, Chakel JA, Fischer S, Lichtenwalter K, Hancock WS. 1997. Analysis of Oligonucleotides by HPLC-Electrospray Ionization Mass Spectrometry. Anal Chem 69: 1320–5 [DOI] [PubMed] [Google Scholar]
- 27.Bartlett MG, Omuro S. 2021. Evaluation of alkylamines and stationary phases to improve LC-MS of oligonucleotides. Biomed Chromatogr 35: e5045. [DOI] [PubMed] [Google Scholar]
- 28.Studzinska S, Bocian S, Siecinska L, Buszewski B. 2017. Application of phenyl-based stationary phases for the study of retention and separation of oligonucleotides. J Chromatogr B Analyt Technol Biomed Life Sci 1060: 36–43 [DOI] [PubMed] [Google Scholar]
- 29.Li N, El Zahar NM, Saad JG, van der Hage ERE, Bartlett MG. 2018. Alkylamine ion-pairing reagents and the chromatographic separation of oligonucleotides. J Chromatogr A 1580: 110–19 [DOI] [PubMed] [Google Scholar]
- 30.Studzinska S, Cywoniuk P, Sobczak K. 2019. Application of ion pair chromatography coupled with mass spectrometry to assess antisense oligonucleotides concentrations in living cells. Analyst 144: 622–33 [DOI] [PubMed] [Google Scholar]
- 31.Chen X, Liu Z, Gong L. 2021. Evaluating the interplay among stationary phases/ion-pairing reagents/sequences for liquid chromatography mass spectrometry analysis of oligonucleotides. Anal Biochem 625: 114194. [DOI] [PubMed] [Google Scholar]
- 32.Donegan M, Nguyen JM, Gilar M. 2022. Effect of ion-pairing reagent hydrophobicity on liquid chromatography and mass spectrometry analysis of oligonucleotides. J Chromatogr A 1666: 462860. [DOI] [PubMed] [Google Scholar]
- 33.McGinnis AC, Grubb EC, Bartlett MG. 2013. Systematic optimization of ion-pairing agents and hexafluoroisopropanol for enhanced electrospray ionization mass spectrometry of oligonucleotides. Rapid Commun Mass Spectrom 27: 2655–64 [DOI] [PubMed] [Google Scholar]
- 34.Enmark M, Samuelsson J, Fornstedt T. 2023. Development of a unified gradient theory for ion-pair chromatography using oligonucleotide separations as a model case. J Chromatogr A 1691: 463823. [DOI] [PubMed] [Google Scholar]
- 35.Enmark M, Harun S, Samuelsson J, Örnskov E, Thunberg L, et al. 2021. Selectivity limits of and opportunities for ion pair chromatographic separation of oligonucleotides. J Chromatogr A 1651: 462269. [DOI] [PubMed] [Google Scholar]
- 36.Hagelskamp F, Kellner S. 2021. Analysis of the epitranscriptome with ion-pairing reagent free oligonucleotide mass spectrometry. Methods Enzymol 658: 111–35 [DOI] [PubMed] [Google Scholar]
- 37.Kaczmarkiewicz A, Nuckowski Ł, Studzińska S, Buszewski B. 2019. Analysis of Antisense Oligonucleotides and Their Metabolites with the Use of Ion Pair Reversed-Phase Liquid Chromatography Coupled with Mass Spectrometry. Crit Rev Anal Chem 49: 256–70 [DOI] [PubMed] [Google Scholar]
- 38.Lobue PA, Jora M, Addepalli B, Limbach PA. 2019. Oligonucleotide analysis by hydrophilic interaction liquid chromatography-mass spectrometry in the absence of ion-pair reagents. J Chromatogr A 1595: 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Studzińska S, Nuckowski Ł, Buszewski B. 2022. Oligonucleotides Isolation and Separation-A Review on Adsorbent Selection. Int J Mol Sci 23: 9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kenderdine T, Fabris D. 2023. The multifaceted roles of mass spectrometric analysis in nucleic acids drug discovery and development. Mass Spectrom Rev 42: 1332–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li P, Gong Y, Kim J, Liu X, Gilbert J, et al. 2020. Hybridization Liquid Chromatography-Tandem Mass Spectrometry: An Alternative Bioanalytical Method for Antisense Oligonucleotide Quantitation in Plasma and Tissue Samples. Anal Chem 92: 10548–59 [DOI] [PubMed] [Google Scholar]
- 42.Mahajan S, Zhao H, Kovacina K, Lachacz E, Hoxha S, et al. 2022. High-sensitivity quantification of antisense oligonucleotides for pharmacokinetic characterization. Bioanalysis 14: 603–13 [DOI] [PubMed] [Google Scholar]
- 43.Guimaraes G, Yuan L, Li P. 2022. Antisense Oligonucleotide In Vitro Protein Binding Determination in Plasma, Brain, and Cerebral Spinal Fluid Using Hybridization LC-MS/MS. Drug Metab Dispos 50: 268–76 [DOI] [PubMed] [Google Scholar]
- 44.Yuan L, Dupuis JF, Mekhssian K. 2023. A Novel Hybridization LC-MS/MS Methodology for Quantification of siRNA in Plasma, CSF and Tissue Samples. Molecules 28: 1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pourshahian S 2021. THERAPEUTIC OLIGONUCLEOTIDES, IMPURITIES, DEGRADANTS, AND THEIR CHARACTERIZATION BY MASS SPECTROMETRY. Mass Spectrom Rev 40: 75–109 [DOI] [PubMed] [Google Scholar]
- 46.Roussis SG, Cedillo I, Rentel C. 2019. Semi-quantitative determination of co-eluting impurities in oligonucleotide drugs using ion-pair reversed-phase liquid chromatography mass spectrometry. J Chromatogr A 1584: 106–14 [DOI] [PubMed] [Google Scholar]
- 47.Guimaraes GJ, Sutton JM, Gilar M, Donegan M, Bartlett MG. 2022. Impact of Nonspecific Adsorption to Metal Surfaces in Ion Pair-RP LC-MS Impurity Analysis of Oligonucleotides. J Pharm Biomed Anal 208: 114439. [DOI] [PubMed] [Google Scholar]
- 48.Demelenne A, Servais AC, Crommen J, Fillet M. 2021. Analytical techniques currently used in the pharmaceutical industry for the quality control of RNA-based therapeutics and ongoing developments. J Chromatogr A 1651: 462283. [DOI] [PubMed] [Google Scholar]
- 49.Jiang D, Yuan L. 2022. Microflow LC-MS/MS to improve sensitivity for antisense oligonucleotides bioanalysis: critical role of sample cleanness. Bioanalysis 14: 1365–76 [DOI] [PubMed] [Google Scholar]
- 50.Herbert C, Dzowo YK, Urban A, Kiggins CN, Resendiz MJE. 2018. Reactivity and Specificity of RNase T(1), RNase A, and RNase H toward Oligonucleotides of RNA Containing 8-Oxo-7,8-dihydroguanosine. Biochemistry 57: 2971–83 [DOI] [PubMed] [Google Scholar]
- 51.Douthwaite S, Kirpekar F. 2007. Identifying modifications in RNA by MALDI mass spectrometry. Methods Enzymol 425: 3–20 [DOI] [PubMed] [Google Scholar]
- 52.Solivio B, Yu N, Addepalli B, Limbach PA. 2018. Improving RNA modification mapping sequence coverage by LC-MS through a nonspecific RNase U2-E49A mutant. Anal Chim Acta 1036: 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Addepalli B, Lesner NP, Limbach PA. 2015. Detection of RNA nucleoside modifications with the uridine-specific ribonuclease MC1 from Momordica charantia. Rna 21: 1746–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Durairaj A, Limbach PA. 2008. Improving CMC-derivatization of pseudouridine in RNA for mass spectrometric detection. Anal Chim Acta 612: 173–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thakur P, Estevez M, Lobue PA, Limbach PA, Addepalli B. 2020. Improved RNA modification mapping of cellular non-coding RNAs using C- and U-specific RNases. Analyst 145: 816–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greiner-Stöffele T, Foerster HH, Hahn U. 2000. Ribonuclease T1 cleaves RNA after guanosines within single-stranded gaps of any length. Nucleosides Nucleotides Nucleic Acids 19: 1101–9 [DOI] [PubMed] [Google Scholar]
- 57.Prats-Ejarque G, Lu L, Salazar VA, Moussaoui M, Boix E. 2019. Evolutionary Trends in RNA Base Selectivity Within the RNase A Superfamily. Front Pharmacol 10: 1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Houser WM, Butterer A, Addepalli B, Limbach PA. 2015. Combining recombinant ribonuclease U2 and protein phosphatase for RNA modification mapping by liquid chromatography-mass spectrometry. Anal Biochem 478: 52–8 [DOI] [PubMed] [Google Scholar]
- 59.Wolf EJ, Grünberg S, Dai N, Chen TH, Roy B, et al. 2022. Human RNase 4 improves mRNA sequence characterization by LC-MS/MS. Nucleic Acids Res 50: e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Addepalli B, Venus S, Thakur P, Limbach PA. 2017. Novel ribonuclease activity of cusativin from Cucumis sativus for mapping nucleoside modifications in RNA. Anal Bioanal Chem 409: 5645–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jones JD, Simcox KM, Kennedy RT, Koutmou KS. 2023. Direct sequencing of total S. cerevisiae tRNAs by LC-MS/MS. Rna [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vanhinsbergh CJ, Criscuolo A, Sutton JN, Murphy K, Williamson AJK, et al. 2022. Characterization and Sequence Mapping of Large RNA and mRNA Therapeutics Using Mass Spectrometry. Anal Chem 94: 7339–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jones JD, Simcox KM, Kennedy RT, Koutmou KS. 2023. Direct sequencing of total Saccharomyces cerevisiae tRNAs by LC-MS/MS. Rna 29: 1201–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pomerantz SC, Kowalak JA, McCloskey JA. 1993. Determination of oligonucleotide composition from mass spectrometrically measured molecular weight. J Am Soc Mass Spectrom 4: 204–9 [DOI] [PubMed] [Google Scholar]
- 65.Puri P, Wetzel C, Saffert P, Gaston KW, Russell SP, et al. 2014. Systematic identification of tRNAome and its dynamics in Lactococcus lactis. Mol Microbiol 93: 944–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yan TM, Pan Y, Yu ML, Hu K, Cao KY, Jiang ZH. 2021. Full-Range Profiling of tRNA Modifications Using LC-MS/MS at Single-Base Resolution through a Site-Specific Cleavage Strategy. Anal Chem 93: 1423–32 [DOI] [PubMed] [Google Scholar]
- 67.Inoue H, Hayase Y, Iwai S, Ohtsuka E. 1987. Sequence-dependent hydrolysis of RNA using modified oligonucleotide splints and RNase H. FEBS Lett 215: 327–30 [DOI] [PubMed] [Google Scholar]
- 68.Lima WF, Crooke ST. 1997. Cleavage of single strand RNA adjacent to RNA-DNA duplex regions by Escherichia coli RNase H1. J Biol Chem 272: 27513–6 [DOI] [PubMed] [Google Scholar]
- 69.Catherman AD, Skinner OS, Kelleher NL. 2014. Top Down proteomics: facts and perspectives. Biochem Biophys Res Commun 445: 683–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Siuti N, Kelleher NL. 2007. Decoding protein modifications using top-down mass spectrometry. Nat Methods 4: 817–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jebanathirajah JA, Pittman JL, Thomson BA, Budnik BA, Kaur P, et al. 2005. Characterization of a new qQq-FTICR mass spectrometer for post-translational modification analysis and top-down tandem mass spectrometry of whole proteins. J Am Soc Mass Spectrom 16: 1985–99 [DOI] [PubMed] [Google Scholar]
- 72.Huang TY, Liu J, McLuckey SA. 2010. Top-down tandem mass spectrometry of tRNA via ion trap collision-induced dissociation. J Am Soc Mass Spectrom 21: 890–8 [DOI] [PubMed] [Google Scholar]
- 73.Riml C, Glasner H, Rodgers MT, Micura R, Breuker K. 2015. On the mechanism of RNA phosphodiester backbone cleavage in the absence of solvent. Nucleic Acids Res 43: 5171–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taucher M, Breuker K. 2010. Top-down mass spectrometry for sequencing of larger (up to 61 nt) RNA by CAD and EDD. J Am Soc Mass Spectrom 21: 918–29 [DOI] [PubMed] [Google Scholar]
- 75.Taucher M, Breuker K. 2012. Characterization of modified RNA by top-down mass spectrometry. Angew Chem Int Ed Engl 51: 11289–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Glasner H, Riml C, Micura R, Breuker K. 2017. Label-free, direct localization and relative quantitation of the RNA nucleobase methylations m6A, m5C, m3U, and m5U by top-down mass spectrometry. Nucleic Acids Res 45: 8014–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peters-Clarke TM, Quan Q, Brademan DR, Hebert AS, Westphall MS, Coon JJ. 2020. Ribonucleic Acid Sequence Characterization by Negative Electron Transfer Dissociation Mass Spectrometry. Anal Chem 92: 4436–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Santos IC, Lanzillotti M, Shilov I, Basanta-Sanchez M, Roushan A, et al. 2022. Ultraviolet Photodissociation and Activated Electron Photodetachment Mass Spectrometry for Top-Down Sequencing of Modified Oligoribonucleotides. J Am Soc Mass Spectrom 33: 510–20 [DOI] [PubMed] [Google Scholar]
- 79.Gaston KW, Limbach PA. 2014. The identification and characterization of non-coding and coding RNAs and their modified nucleosides by mass spectrometry. RNA Biol 11: 1568–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lobue PA, Yu N, Jora M, Abernathy S, Limbach PA. 2019. Improved application of RNAModMapper - An RNA modification mapping software tool - For analysis of liquid chromatography tandem mass spectrometry (LC-MS/MS) data. Methods 156: 128–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yu N, Lobue PA, Cao X, Limbach PA. 2017. RNAModMapper: RNA Modification Mapping Software for Analysis of Liquid Chromatography Tandem Mass Spectrometry Data. Anal Chem 89: 10744–52 [DOI] [PubMed] [Google Scholar]
- 82.Wein S, Andrews B, Sachsenberg T, Santos-Rosa H, Kohlbacher O, et al. 2020. A computational platform for high-throughput analysis of RNA sequences and modifications by mass spectrometry. Nat Commun 11: 926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.D’Ascenzo L, Popova AM, Abernathy S, Sheng K, Limbach PA, Williamson JR. 2022. Pytheas: a software package for the automated analysis of RNA sequences and modifications via tandem mass spectrometry. Nat Commun 13: 2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sample PJ, Gaston KW, Alfonzo JD, Limbach PA. 2015. RoboOligo: software for mass spectrometry data to support manual and de novo sequencing of post-transcriptionally modified ribonucleic acids. Nucleic Acids Res 43: e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Paulines MJ, Wetzel C, Limbach PA. 2019. Using spectral matching to interpret LC-MS/MS data during RNA modification mapping. J Mass Spectrom 54: 906–14 [DOI] [PubMed] [Google Scholar]
- 86.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. 2005. RNA Structure Analysis at Single Nucleotide Resolution by Selective 2’-Hydroxyl Acylation and Primer Extension (SHAPE). Journal of the American Chemical Society 127: 4223–31 [DOI] [PubMed] [Google Scholar]
- 87.Scalabrin M, Siu Y, Asare-Okai PN, Fabris D. 2014. Structure-Specific Ribonucleases for MS-Based Elucidation of Higher-Order RNA Structure. Journal of the American Society for Mass Spectrometry 25: 1136–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sosic A, Göttlich R, Fabris D, Gatto B. 2021. B-CePs as cross-linking probes for the investigation of RNA higher-order structure. Nucleic Acids Research 49: 6660–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Karch KR, Snyder DT, Harvey SR, Wysocki VH. 2022. Native Mass Spectrometry: Recent Progress and Remaining Challenges. Annual Review of Biophysics 51: 157–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schneeberger E-M, Breuker K 2017. Native Top-Down Mass Spectrometry of TAR RNA in Complexes with a Wild-Type tat Peptide for Binding Site Mapping. Angewandte Chemie (International Ed. in English) 56: 1254–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schneeberger E-M, Halper M, Palasser M, Heel SV, Vušurović J, et al. 2020. Native mass spectrometry reveals the initial binding events of HIV-1 rev to RRE stem II RNA. Nature Communications 11: 5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. 2008. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Research 18: 1509–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, et al. 2009. mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods 6: 377–82 [DOI] [PubMed] [Google Scholar]
- 94.Grindberg RV, Yee-Greenbaum JL, McConnell MJ, Novotny M, O’Shaughnessy AL, et al. 2013. RNA-sequencing from single nuclei. Proceedings of the National Academy of Sciences of the United States of America 110: 19802–07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alvarez M, Benhammou JN, Darci-Maher N, French SW, Han SB, et al. 2022. Human liver single nucleus and single cell RNA sequencing identify a hepatocellular carcinoma-associated cell-type affecting survival. Genome Medicine 14: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. 2015. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 12: 767–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149: 1635–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Meyer KD. 2019. DART-seq: an antibody-free method for global m6A detection. Nature Methods 16: 1275–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhu H, Yin X, Holley CL, Meyer KD. 2022. Improved Methods for Deamination-Based m6A Detection. Frontiers in Cell and Developmental Biology 10: 888279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang Y, Feng F, Zheng P, Wang L, Wang Y, et al. 2022. Dysregulated lncRNA and mRNA may promote the progression of ischemic stroke via immune and inflammatory pathways: results from RNA sequencing and bioinformatics analysis. Genes & Genomics 44: 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Su Z, Monshaugen I, Klungland A, Ougland R, Dutta A. 2022. Characterization of novel small non-coding RNAs and their modifications in bladder cancer using an updated small RNA-seq workflow. Frontiers in Molecular Biosciences 9: 887686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Potapov V, Fu X, Dai N, Corrêa IR, Tanner NA, Ong JL. 2018. Base modifications affecting RNA polymerase and reverse transcriptase fidelity. Nucleic Acids Research 46: 5753–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Behm-Ansmant I, Helm M, Motorin Y. 2011. Use of specific chemical reagents for detection of modified nucleotides in RNA. Journal of Nucleic Acids 2011: 408053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Incarnato D, Anselmi F, Morandi E, Neri F, Maldotti M, et al. 2017. High-throughput single-base resolution mapping of RNA 2΄-O-methylated residues. Nucleic Acids Research 45: 1433–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kuksa PP, Leung YY, Vandivier LE, Anderson Z, Gregory BD, Wang L-S. 2017. In Silico Identification of RNA Modifications from High-Throughput Sequencing Data Using HAMR. Methods in Molecular Biology (Clifton, N.J.) 1562: 211–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cozen AE, Quartley E, Holmes AD, Hrabeta-Robinson E, Phizicky EM, Lowe TM. 2015. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nature Methods 12: 879–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zheng G, Qin Y, Clark WC, Dai Q, Yi C, et al. 2015. Efficient and quantitative high-throughput tRNA sequencing. Nature Methods 12: 835–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang Y, Katanski CD, Watkins C, Pan JN, Dai Q, et al. 2020. A high-throughput screening method for evolving a demethylase enzyme with improved and new functionalities. Nucleic Acids Research 49: e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Katanski CD, Watkins CP, Zhang W, Reyer M, Miller S, Pan T. 2022. Analysis of queuosine and 2-thio tRNA modifications by high throughput sequencing. Nucleic Acids Research 50: e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang Y, Zhao Y, Bollas A, Wang Y, Au KF. 2021. Nanopore sequencing technology, bioinformatics and applications. Nature Biotechnology 39: 1348–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Barkova DV, Andrianova MS, Komarova NV, Kuznetsov AE. 2020. Channel and Motor Proteins for Translocation of Nucleic Acids in Nanopore Sequencing. Moscow University Chemistry Bulletin 75: 149–61 [Google Scholar]
- 112.Fragasso A, Schmid S, Dekker C. 2020. Comparing Current Noise in Biological and Solid-State Nanopores. ACS Nano 14: 1338–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Smith AM, Jain M, Mulroney L, Garalde DR, Akeson M. 2019. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLoS ONE 14: e0216709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ma H, Jia X, Zhang K, Su Z. 2022. Cryo-EM advances in RNA structure determination. Signal Transduction and Targeted Therapy 7: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Natchiar SK, Myasnikov AG, Kratzat H, Hazemann I, Klaholz BP. 2017. Visualization of chemical modifications in the human 80S ribosome structure. Nature 551: 472–77 [DOI] [PubMed] [Google Scholar]
- 116.Pellegrino S, Dent KC, Spikes T, Warren AJ. 2023. Cryo-EM reconstruction of the human 40S ribosomal subunit at 2.15 Å resolution. Nucleic Acids Research: gkad194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang M, Chen K, Wu Q, Peng R, Zhang R, Li J. 2020. RCasFISH: CRISPR/dCas9-Mediated in Situ Imaging of mRNA Transcripts in Fixed Cells and Tissues. Analytical Chemistry 92: 2468–75 [DOI] [PubMed] [Google Scholar]