

Abstract
Background
The only antidepressant drugs that are effective in the treatment of obsessive–compulsive disorder (OCD) are those that effectively block the reuptake of serotonin (5-hydroxytryptamine; 5-HT). In humans, positron emission tomography studies have implicated the orbitofrontal cortex (OFC) in the mediation of OCD symptoms. In animals, administration of selective serotonin reuptake inhibitors (SSRIs) for 8 weeks (but not 3 weeks) led to increased release of 5-HT in the OFC, because of desensitization of the terminal 5-HT autoreceptors. However, the increase in synaptic levels of 5-HT in the OFC after long-term administration of SSRIs might be cancelled out by desensitization of postsynaptic 5-HT receptors. This study was undertaken to investigate if these OFC receptors adapt under such conditions.
Methods
In vivo electrophysiologic techniques were used in this animal study. Male Sprague–Dawley rats received the SSRI paroxetine or vehicle control, delivered by implanted osmotic minipumps, for 3 or 8 weeks. With the rats under anesthesia, neuronal responsiveness to the microiontophoretic application of various drugs was assessed by determining the number of spikes suppressed per nanoampere of ejection current.
Results
After administration of paroxetine for either 3 weeks or 8 weeks, there was no modification in the inhibitory effect of 5-HT, the preferential 5-HT2A receptor agonist (+)-1-(4-iodo-2,5-dimethoxyphenyl)-2-aminopropane hydrochloride (DOI) or the preferential 5-HT2C receptor agonist 3-chlorophenyl piperazine dihydrochloride (mCPP). In contrast, the inhibitory effect of the 5-HT1A receptor agonist 8-hydroxy-2-(di-n-propilamino)-tetralin (8-OH-DPAT) was attenuated in the OFC after both 3 and 8 weeks of paroxetine administration.
Conclusion
These results indicate a desensitization of postsynaptic 5-HT1A receptors in the OFC but a lack of compensatory adaptation of the 5-HT receptor(s) mediating the main effect of 5-HT in this brain region. These observations imply that the activation of normosensitive postsynaptic 5-HT2-like receptors may mediate the effect of enhanced 5-HT release in the OFC.
Medical subject headings: serotonin; prefrontal cortex; receptor, serotonin, 5-HT1A; receptors, serotonin, 5-HT2; paroxetine; obsessive-compulsive disorder
Abstract
Contexte
Seuls les antidépresseurs qui parviennent à bloquer le recaptage de la sérotonine (5-HT) sont efficaces dans le traitement du trouble obsessionnel compulsif (TOC). Chez l'humain, les études fondées sur la tomographie par émission de positrons ont lié le cortex orbitofrontal (COF) avec la médiation des symptômes du TOC. Chez l'animal, l'administration d'inhibiteurs sélectifs du recaptage de la sérotonine (ISRS) pendant huit semaines (plutôt que trois semaines) a conduit à une libération accrue de 5-HT dans le COF, en raison de l'hyposensibilisation des autorécepteurs terminaux de la 5-HT. L'hyposensibilisation des récepteurs post-synaptiques de la 5-HT pourrait toutefois neutraliser l'accroissement des taux synaptiques de 5-HT dans le COF suite à un traitement de longue durée faisant appel aux ISRS. Cette étude visait à étudier la question de savoir si ces récepteurs du COF s'adaptent dans ces conditions.
Méthodes
Des techniques in vivo en électrophysiologie ont été appliqués dans cette étude chez l'animal. Pendant trois ou huit semaines, on a administré à des rats mâles de Sprague–Dawley soit de la paroxétine, un ISRS, soit un véhicule de contrôle, au moyen de minipompes osmotiques implantées. Chez les rats sous anesthésie, la réponse neuronale à l'application microiontophorétique de diverses substances a été évaluée par la détermination du nombre de potentiels d'action supprimés par nanoampère de courant éjecté.
Résultats
Suite à l'administration de paroxétine pendant trois ou huit semaines, il n'y a pas eu de modification de l'effet d'inhibition de la 5-HT, du chlorhydrate (+)-1-(4-iodo-2,5-diméthoxyphényl)-2-aminopropane (DOI), agoniste préférentiel des récepteurs 5-HT2A, ou du dychlorhydrate de pipérazine 3-chlorophényl (mCPP), agoniste préférentiel des récepteurs 5-HT2C. Par ailleurs, l'effet d'inhibition de l'agoniste des récepteurs de la 5-HT1A 8-hydroxy-2-(di-n-propilamino)-tétraline (8-OH-DPAT) s'est amoindri dans le COF suite au traitement à la paroxétine pendant trois ou huit semaines.
Conclusion
Ces résultats indiquent une hyposensibilisation des récepteurs post-sypnaptiques de la 5-HT1A dans le COF et un manque d'adaptation compensatoire des récepteurs de la 5-HT qui entraÎnent la médiation de l'effet principal de la 5-HT dans cette région du cerveau. Ces observations supposent que l'activation des récepteurs post-synaptiques normosensibles semblables aux récepteurs 5-HT2 pourrait entraÎner la médiation de l'effet d'accroissement de la libération de 5-HT dans le COF.
Introduction
The predominant neurobiologic hypothesis regarding the pathophysiology of obsessive–compulsive disorder (OCD) is that at least some aspects of the disorder are related to abnormal regulation of brain serotonergic function.1 However, this hypothesis was derived mainly from therapeutic studies demonstrating the efficacy of the tricyclic antidepressant and potent serotonin (5-hydroxytryptamine; 5-HT) reuptake inhibitor clomipramine in the treatment of OCD.2,3 These initial observations have been supported by subsequent investigations showing that other drugs with a potent inhibitory effect on 5-HT reuptake not belonging to the tricyclic family, such as paroxetine and fluoxetine, are effective antiobsessional agents, but drugs without potent 5-HT reuptake properties, such as desipramine, are not.4,5,6 On the one hand, clinical studies have shown that treatment with the 5-HT reuptake inhibitors clomipramine and fluoxetine leads to a reduction of activity in human prefrontal cortex areas, particularly the orbitofrontal cortex (OFC)3,7 and the head of caudate nucleus.8,9 Provocative stimuli inducing OCD symptoms increase regional cerebral blood flow in the OFC and the head of caudate nucleus. It is noteworthy that the reduction of OFC and caudate nucleus activity has been associated with clinical improvement in patients with OCD who responded to either pharmacotherapy or psychotherapy.9,10 On the other hand, preclinical investigations designed to assess 5-HT function have found that guinea pigs treated with selective serotonin reuptake inhibitors (SSRIs) have increased 5-HT release occurring through terminal 5-HT1D autoreceptor desensitization in the OFC but not in the caudate nucleus. In addition, it has been shown that 5-HT release is enhanced in the OFC after an 8-week but not a 3-week treatment with a different SSRI, such as paroxetine or fluoxetine, but not by the monoamine oxidase type A inhibitor moclobemide or electroconvulsive shocks, the latter 2 of which are ineffective in OCD.11,12
It is important to emphasize not only that alterations of presynaptic neurotransmitter components can underlie the effectiveness of drug treatment but also that the responsiveness of postsynaptic 5-HT receptors may contribute to, as well as altering, overall 5-HT transmission. Various classes of antidepressant treatments enhance 5-HT neurotransmission in the rat hippocampus with a time course that is consistent with their delayed therapeutic effect. This action would be mediated by different mechanisms such as postsynaptic sensitization to 5-HT, desensitization of the somatodendritic or terminal 5-HT autoreceptors, or both, or a desensitization of α2-adrenergic heteroreceptors located on 5-HT terminals.13,14 Long-term administration of SSRIs results in the desensitization of somatodendritic 5-HT1A autoreceptor function in the dorsal raphe, thereby allowing the firing rate of the 5-HT neurons to recover in the presence of the drugs, which in turn leads to an increase in 5-HT neurotransmission in the hippocampus.13,15 Although similar regimens result in the desensitization of physiologic responses mediated by postsynaptic 5-HT1A receptors in some brain regions such as the hypothalamus,16,17,18 electrophysiologic studies indicate that the sensitivity of postsynaptic 5-HT1A receptor-mediated responses in the hippocampus is not changed by SSRIs.13
The present study aimed to assess the sensitivity of postsynaptic 5-HT receptors in the OFC after 3- and 8-week treatments with paroxetine. The OFC is part of the circuitry implicated in OCD, and it is already known that 5-HT release in this structure is increased after 8 weeks of SSRI administration. The responsiveness to 5-HT itself was examined, as well as the responsiveness to the 5-HT1A receptor agonist 8-hydroxy-2-(di-n-propilamino)-tetralin (8-OH-DPAT), the preferential 5-HT2A receptor agonist (+)-1-(4-iodo-2,5-dimethoxyphenyl)-2-aminopropane hydrochloride (DOI) and the preferential 5-HT2C receptor agonist 3-chlorophenyl piperazine dihydrochloride (mCPP).
Materials and methods
Animals
Male Sprague–Dawley rats, weighing 250–350 g, were obtained from Charles River (St. Constant, Que.) and were kept on a 12:12 hour light–dark cycle under controlled conditions with regular indoor temperature and humidity. The animals had free access to standard rodent food (Charles River) and water during the study.
Treatments
The rats were anesthetized with halothane, and osmotic minipumps (Alza, Palo Alto, Calif.) were implanted subcutaneously; the minipumps delivered paroxetine (10 mg/kg per day) or the vehicle used to dilute this drug (50:50 mixture of ethanol and water) (in the controls). For the 8-week treatment, a new minipump was installed 4 weeks after the first implantation. The electrophysiology experiments were performed with the minipumps in place. For each treated rat, a control rat from the same batch was also studied on the same day, to prevent any variations in the effects of the drugs. All experiments were performed with the approval of the University Animal Care Committee and complied with rules set forth in the Guide to the Care and Use of Experimental Animals (1993) of the Canadian Council on Animal Care.
Single-unit recording and microiontophoresis
The rats were anesthetized with chloral hydrate (400 mg/kg i.p., followed by doses of 100 mg/kg as necessary to maintain complete anesthesia) and placed in a stereotaxic frame. Body temperature was maintained at 36°C–37°C with a thermostatically controlled heating pad. Extracellular unitary activity was amplified and displayed on an oscilloscope. Action potentials were discriminated from background noise by means of a differential amplitude discriminator, and the frequency of discharge was integrated over 10-second intervals with a ratemeter. Five-barrel micropipettes used for microiontophoresis were pulled with a vertical electrode puller and were broken back under a microscope to a diameter of approximately 5–9 μm. The stereotaxic coordinates for the OFC were as follows: A, 3.7 mm; L, 2.0 mm from the bregma; H, 2.5–4 mm below the cortical surface.19 The coordinates for the OFC in rats correspond to those for humans, which delineate Brodman area 47.20 At the end of the experiment, the final recording site was marked by passing a cathodal current through the recording barrel to deposit fast green dye such that the recording sites could be confirmed histologically. Because most OFC neurons are not spontaneously active under chloral hydrate anesthesia, a leak or a small ejection current of quisqualate (QUIS) was used to activate them within their physiologic firing range.21 The central barrel contained 2 mmol/L NaCl for recording, as did one of the side barrels for current balancing. Another side barrel contained QUIS (1.5 mmol/L in 400 mmol/L NaCl, pH 8; Tocris Cookson, Ballwin, Mo.). The remaining barrels were filled with 2 of the following drugs, all in 200 mmol/L NaCl adjusted to pH 4.0: 5-hydroxytryptamine creatinine sulfate (5-HT, 20 mmol/L; Sigma, St. Louis, Mo.), 8-OH-DPAT (20 mmol/L; RBI, Natick, Mass.), DOI (50 mmol/L; RBI, Natick, Mass.), mCPP (20 mmol/L; Bristol-Myers Squibb, Wallingford, Conn.). Neuronal responsiveness to the microiontophoretic application of various drugs was assessed by determining the number of spikes suppressed per nanoampere of ejection current.
Statistics
All results are expressed as mean (and standard error of the mean [SEM]). The magnitude of inhibition of different agonists was compared between control and treated rats with the 2-tailed Student's t test (nonpaired). Differences were considered significant at p < 0.05.
Results
OFC discharge rates were not significantly different in control and treated rats (control rats, 10.3 [SEM 0.5] spikes/s, n = 104; treated rats, 11.1 [SEM 0.8] spikes/s, n = 112; p > 0.3), and no significant alteration in the ejection current of QUIS, used to activate these neurons, was observed in any group of rats (control rats, –80 [SEM 11] nA, treated rats, –70 [SEM 15] nA; p > 0.5).
It has previously been demonstrated that the in vivo microiontophoretic application of 5-HT, DOI and mCPP onto OFC neurons produces a suppressant effect on firing activity in rats,21 guinea pigs22 and mice.23 In the present study, all OFC neurons tested with 5-HT, 8-OH-DPAT, DOI and mCPP induced a reduction of firing activity (Fig. 1 and Fig. 2). This suppressant effect occurred in the absence of any alteration in the shape of the action potentials.
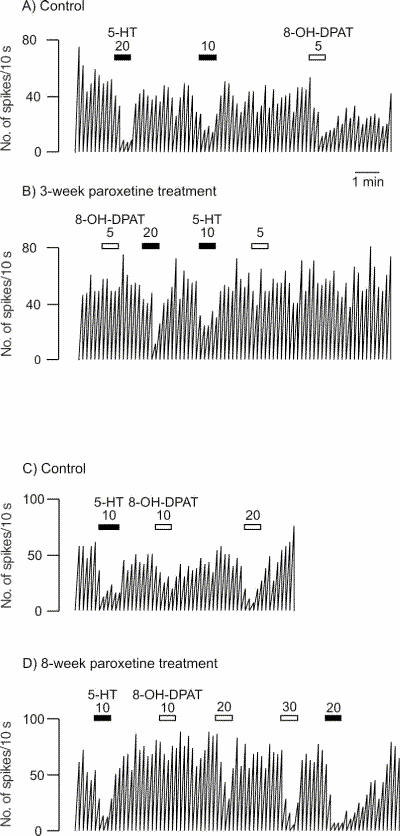
Fig. 1: Integrated firing rate histogram of an orbitofrontal cortex neuron, showing its responsiveness to microiontophoretic application of serotonin (5-HT) and 8-OH-DPAT in control animals and rats treated with paroxetine for 3 and 8 weeks. Horizontal bars indicate the duration of the applications (currents given in nanoamperes). The time base in A applies to all traces. Traces in the control and treated rats were obtained from animals from the same shipment that were treated simultaneously after administration of vehicle or paroxetine. These traces were all obtained on the same day. Note the decreased responses to 8-OH-DPAT and the unaltered response to 5-HT in the treated rats.
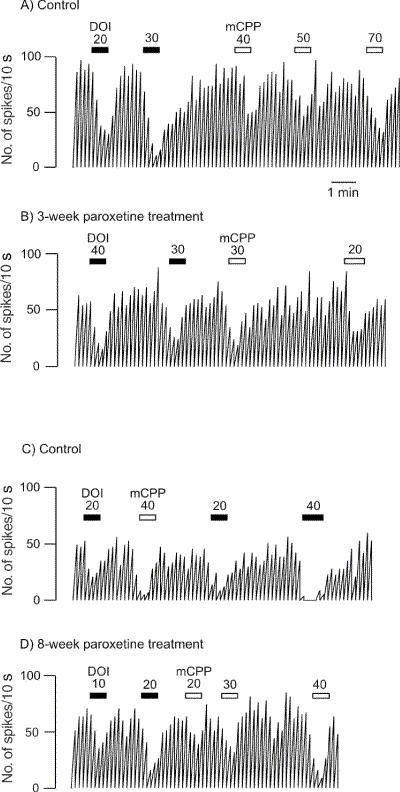
Fig. 2: Integrated firing rate histogram of an orbitofrontal cortex neuron, showing its responsiveness to microiontophoretic application of DOI and mCPP in control animals and rats treated with paroxetine for 3 and 8 weeks. Horizontal bars indicate the duration of the applications (currents given in nanoamperes). The time base in A applies to all traces. Traces in the control and the treated rats were obtained from animals from the same shipment that were treated simultaneously after administration of vehicle or paroxetine. These traces were all obtained on the same day.
After 3 weeks of administration of paroxetine, there was no modification in the inhibitory effect of 5-HT, DOI or mCPP (Fig. 1, Fig. 2 and Fig. 3) in the OFC, whereas the inhibitory effect of 8-OH-DPAT was significantly attenuated (t24 = 4.05, p < 0.005; Figs. 1 and 3). This same attenuation of the inhibitory response of 8-OH-DPAT in the OFC was obtained after 8 weeks of paroxetine treatment (t42 = 5.18, p < 0.001; Fig. 1 and Fig. 3), whereas the responsiveness to 5-HT, DOI and mCPP remained unchanged (Fig. 1, Fig. 2 and Fig. 3).
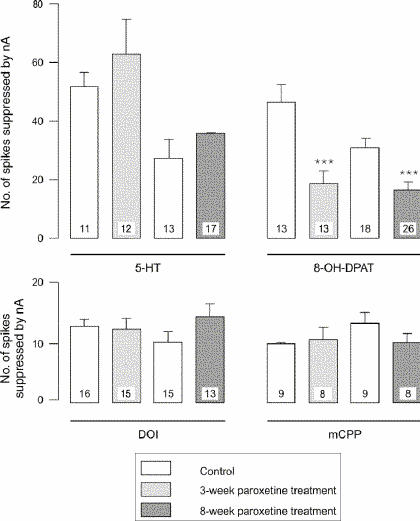
Fig. 3: Responsiveness to 5-HT, 8-OH-DPAT, DOI and mCPP in control animals and rats treated with paroxetine, expressed as the number of spikes suppressed by current (nA); the number in each column represents the number of neurons tested. ***p < 0.001.
It is noteworthy that the data for the 3-week treatment group were generated in part by assessment of neuronal responsiveness using the same micropipette in pairs of rats, a control rat and a treated rat taken from the same shipment, housed together and receiving the vehicle or the SSRI, respectively, for the same period of time. Use of the same micropipette in assessing neuronal responsiveness in such pairs of animals was designed to ascertain whether the difference, or lack of difference, between rats did not result merely from variations in the properties of the micropipettes. In 3 such pairs, 2 pipettes filled with 5-HT and 8-OH-DPAT or with mCPP and DOI were used, and the results from these experiments did not differ from the others.
Discussion
The present results show that sustained paroxetine administration, over 3 and 8 weeks, induced a decrease in the inhibitory effect of the 5-HT1A receptor agonist 8-OH-DPAT, which indicates a desensitization of postsynaptic 5-HT1A receptors in the OFC. Such desensitization had already been reported in at least 2 brain structures. In the amygdala, postsynaptic 5-HT1A receptor-mediated feedback, which regulates serotonergic neuronal firing rate in the dorsal raphe nucleus and 5-HT release, is diminished after 2 weeks of treatment with the SSRI citalopram.24 In addition, long-term treatment with paroxetine and fluoxetine reduces the neuroendocrine response to 8-OH-DPAT, which provides evidence of desensitization of hypothalamic postsynaptic 5-HT1A receptors.16,17,18 In vivo and in vitro electrophysiologic studies indicate, however, that in the hippocampus the sensitivity of postsynaptic 5-HT1A receptors is unchanged after administration of various SSRIs, including paroxetine, for 2–3 weeks.25,26 Interestingly, in constitutional 5-HT transporter knockout, a small but significant 5-HT1A receptor desensitization was observed in the hippocampus.27 Similarly, after a 3-week treatment with the selective monoamine oxidase type A inhibitor clorgyline, the responsiveness of pyramidal neurons to 5-HT itself, which is mediated by 5-HT1A receptors, is attenuated.28 Consequently this population of 5-HT1A receptors does have the capacity to desensitize following exposure to enhanced levels of synaptic 5-HT, but the desensitization does not occur with sustained administration of SSRIs.
The desensitization of 5-HT1A receptors in the OFC contrasts with results obtained by Bobula et al29 for the frontal cortex; in that study, the inhibitory effect of 8-OH-DPAT was enhanced in slices prepared from rats and treated with citalopram or imipramine for 14 days. It is noteworthy, however, that these experiments did not assess 5-HT1A receptor responsiveness under physiologic conditions, but rather under epileptic activity triggered by picrotoxin. Interestingly, the 5-HT1A receptor response to 8-OH-DPAT in the OFC appears atypical.22 Indeed, the inhibitory effect of 8-OH-DPAT was not blocked by the 5-HT1A receptor antagonists WAY 100635 or BMY7378. Also, the inhibitory effect of the 5-HT1A receptor agonist gepirone on OFC neurons was not antagonized by WAY 100635. Since 8-OH-DPAT but not gepirone is endowed with 5-HT7 receptor affinity, these observations have excluded a contribution of the latter receptor subtype to the action of 8-OH-DPAT and gepirone. These results would still be compatible with the existence of different 5-HT1A receptor subtypes or a different mechanism modulating the function of this receptor. A regional difference in the functional and regulatory properties of 5-HT1A sites has already been reported within the same brain structure, i.e., CA1 versus dentate gyrus in the hippocampus.30 This difference in properties is supported by the existence of 3 separate mRNA bands coding for 5-HT1A receptors in the rat brain.31
The desensitization of 5-HT1A receptors in the OFC could account for the lack of beneficial effect of the addition of the 5-HT1A receptor agonist buspirone to the regimen of patients with SSRI-resistant OCD.32,33 Indeed, it would be difficult for buspirone to increase the activation of 5-HT1A receptors in the OFC if the latter were desensitized. Consistent with this possibility is the report of Lesch et al34 showing that long-term exposure to fluoxetine attenuated the adrenocorticotropic hormone response to the 5-HT1A receptor agonist ipsapirone in patients with OCD; this finding suggests that long-term exposure to fluoxetine results in hyposensitivity of that 5-HT1A receptor function, in accordance with the desensitization of 5-HT1A receptors observed in the rat hypothalamus after long-term treatment with paroxetine and fluoxetine.16,17,18
In the presence of desensitized 5-HT1A receptors in the OFC, the inhibitory effect of 5-HT was unchanged in rats treated with paroxetine, which suggests that another 5-HT receptor may be mediating most of the effect of the endogenous neurotransmitter. Indeed, it is possible that 5-HT exerts its action via 5-HT2 receptors, since the present data show that these receptors are not desensitized after 3 and 8 weeks of treatment with paroxetine. These results contrast with earlier data showing attenuation of the excitatory effect on 5-HT2 receptors with application of DOI, based on recordings from slices of frontal cortex.29 Indeed, excitatory 5-HT2-mediated responses have generally been reported in in vitro experiments using bath application of agonists. It is thus possible that different populations of 5-HT2 receptors are activated in these experimental paradigms: 5-HT2 receptors located on both the cell body and the dendritic tree are activated when 5-HT agonists are used in a bath application, whereas 5-HT2 receptors located primarily on the cell body are activated when agonists are ejected by microiontophoresis. Because of a lack of variety of specific ligands, the exact receptor subtype mediating the effect of DOI and mCPP in the OFC remains controversial. Nevertheless, combinations of different strategies have led to the conclusion that 5-HT2 receptors in the OFC are pharmacologically distinct from those in other regions of the cerebral cortex. Studies from our laboratory have documented that the inhibitory effect of DOI and mCPP is attenuated by the 5-HT2 receptor antagonists ritanserin and risperidone in the medial prefrontal cortex but is not antagonized by ritanserin and the 5-HT1/2 antagonist metergoline in the OFC, indicating that 5-HT2 receptor responses may be mediated by an atypical receptor in the OFC of rats and guinea pigs.21,22 On the other hand, using 5-HT2C receptor null mutant mice, Rueter et al23 showed that DOI may be acting predominantly via a 5-HT2A receptor and that the effect of mCPP is mediated by both 5-HT2A and 5-HT2C receptors in the OFC.
If the antiobsessional effect of SSRIs depends on ongoing enhancement of serotonergic transmission, administration of a potent 5-HT antagonist might be expected to provoke symptom exacerbation in patients with a response to SSRIs. Indeed, the administration of the 5-HT1/2 antagonist metergoline over 4 days causes an exacerbation of OCD symptoms in patients with this condition.2,35 This result is in accordance with a report that the subacute administration of metergoline does attenuate the responsiveness of DOI in the caudate nucleus.22 Consequently, attenuation of inhibitory 5-HT2 receptor neurotransmission at any point in the neuronal OCD circuitry, whether it be the 5-HT2 receptors in the caudate nucleus or those in the OFC, should reinstate the initial hyperactivity in the neuronal loop and trigger symptom exacerbation in patients with OCD whose condition has improved with SRI therapy.
Both 5-HT2A and 5-HT2C receptor antagonism have been postulated to play a role in the generation of obsessive–compulsive symptoms in patients with a psychotic disorder. Although low doses of the atypical antipsychotic risperidone have clearly shown a therapeutic effect in SSRI-resistant OCD,36,37 exacerbation of OCD with high doses of clozapine and risperidone may be due to the antagonism of 5-HT2 receptors in the OFC. Indeed, with low doses of risperidone, which block 5-HT2 receptor response in the medial prefrontal cortex, the responsiveness of 5-HT2 receptors in the OFC is unaffected. In contrast, high doses of the same drug attenuate the responsiveness of 5-HT2 receptors in the OFC.21
Consistent with the possibility that enhanced 5-HT2 receptor activation might underlie the beneficial action of SSRIs, case studies have reported that the use of hallucinogens with 5-HT2 receptor agonistic properties, such as LSD and psilocybin, results in relief of OCD symptoms.38,39,40 Moreover, it was demonstrated in an electrophysiology study that LSD enhances the inhibitory effect of 5-HT in the OFC, possibly through 5-HT2 receptors, but not in the hippocampus, where 5-HT exerts most of its action in these conditions via 5-HT1A receptors.41 Taken together, these clinical observations indicate that by enhancing the activation of 5-HT2 receptors with 5-HT agonists, a beneficial effect might be obtained in OCD, whereas by decreasing transmission at these sites with high doses of antagonists, OCD symptoms may be exacerbated in patients whose condition has previously been improved by an SSRI or produced de novo in patients with schizophrenia.
Acknowledgments
This work was supported by a Canadian Institutes for Health Research grant and by salary support from the University of Ottawa Institute of Mental Health Research to both authors, as well as a Research Chair in Psychopharmacology from the Canadian government to Dr. Blier.
Footnotes
Contributors: Drs. El Mansari and Blier conceived and designed the study, collected and interpreted the data, drafted and revised the article, and gave final approval for it to be published.
Competing interests: None declared.
Correspondence to: Dr. Mostafa El Mansari, University of Ottawa Institute of Mental Health Research, Royal Ottawa Hospital, LG 2050, 1145 Carling Ave., Ottawa ON K1Z 7K4; fax 613 792-3935; melmansa@rohcg.on.ca
Submitted Jan. 26, 2005; Revised Mar. 28, 2005; Accepted Apr. 11, 2005
References
- 1.Insel TR, Mueller EA, Alterman I, Linnoila M, Murphy DL. Obsessive-compulsive disorder and serotonin: Is there a connection? Biol Psychiatry 1985;20:1174-88. [DOI] [PubMed]
- 2.Benkelfat C, Murphy DL, Zohar J, Hill JL, Grover G, Insel TR. Clomipramine in obsessive–compulsive disorder. Further evidence for a serotonergic mechanism of action. Arch Gen Psychiatry 1989;46:23-8. [DOI] [PubMed]
- 3.Benkelfat C, Nordahl TE, Semple WE, King AC, Murphy DL, Cohen RM. Local cerebral glucose metabolic rates in obsessive–compulsive disorder. Patients treated with clomipramine. Arch Gen Psychiatry 1990;47:840-8. [DOI] [PubMed]
- 4.Fontaine R, Chouinard G. Fluoxetine in the treatment of obsessive compulsive disorder. Prog Neuropsychopharmacol Biol Psychiatry 1985;9:605-8. [DOI] [PubMed]
- 5.Goodman WK, Price LH, Delgado PL, Palumbo J, Krystal JH, Nagy LM, et al. Specificity of serotonin reuptake inhibitors in the treatment of obsessive–compulsive disorder. Comparison of fluvoxamine and desipramine. Arch Gen Psychiatry 1990;47:577-85. [DOI] [PubMed]
- 6.Denys D, van Megen HJ, van der Wee N, Westenberg HG. A double-blind switch study of paroxetine and venlafaxine in obsessive–compulsive disorder. J Clin Psychiatry 2004;65:37-43. [DOI] [PubMed]
- 7.Swedo SE, Pietrini P, Leonard HL, Schapiro MB, Rettew DC, Goldberger EL, et al. Cerebral glucose metabolism in childhood-onset obsessive compulsive disorder. Revisualization during pharmacotherapy. Arch Gen Psychiatry 1992;49:690-4. [DOI] [PubMed]
- 8.Baxter LR Jr, Phelps ME, Mazziotta JC, Guze BH, Schwartz JM, Selin CE. Local cerebral glucose metabolic rates in obsessive–compulsive disorder. A comparison with rates in unipolar depression and in normal controls. Arch Gen Psychiatry 1987;44:211-8. [DOI] [PubMed]
- 9.Baxter LR Jr. Neuroimaging studies of obsessive compulsive disorder. Psychiatr Clin North Am 1992;15:871-84. [PubMed]
- 10.Brody AL, Saxena S, Schwartz JM, Stoessel PW, Maidment K, Phelps ME, et al. FDG-PET predictors of response to behavioral therapy and pharmacotherapy in obsessive compulsive disorder. Psychiatry Res 1998;84:1-6. [DOI] [PubMed]
- 11.El Mansari M, Bouchard C, Blier P. Alteration of serotonin release in the guinea pig orbito-frontal cortex by selective serotonin reuptake inhibitors. Relevance to treatment of obsessive–compulsive disorder. Neuropsychopharmacology 1995;13:117-27. [DOI] [PubMed]
- 12.Bergqvist PB, Bouchard C, Blier P. Effect of long-term administration of antidepressant treatments on serotonin release in brain regions involved in obsessive–compulsive disorder. Biol Psychiatry 1999;15:164-74. [DOI] [PubMed]
- 13.Blier P, de Montigny C. Current advances and trends in the treatment of depression. Trends Pharmacol Sci 1994;15:220-6. [DOI] [PubMed]
- 14.Mongeau R, Blier P, de Montigny C. The serotonergic and noradrenergic systems of the hippocampus: their interactions and the effects of antidepressant treatments. Brain Res Brain Res Rev 1997;23:145-95. [DOI] [PubMed]
- 15.Le Poul E, Laaris N, Doucet E, Laporte AM, Hamon M, Lanfumey L. Early desensitization of somato-dendritic 5-HT1A autoreceptors in rats treated with fluoxetine or paroxetine. Naunyn Schmiedebergs Arch Pharmacol 1995;352:141-8. [DOI] [PubMed]
- 16.Li Q, Muma NA, Battaglia G, Van de Kar LD. A desensitization of hypothalamic 5-HT1A receptors by repeated injections of paroxetine: reduction in the levels of G(i) and G(o) proteins and neuroendocrine responses, but not in the density of 5-HT1A receptors. J Pharmacol Exp Ther 1997;282:1581-90. [PubMed]
- 17.Li Q, Muma NA, Van de Kar LD. Chronic fluoxetine induces a gradual desensitization of 5-HT1A receptors: reductions in hypothalamic and midbrain Gi and G(o) proteins and in neuroendocrine responses to a 5-HT1A agonist. J Pharmacol Exp Ther 1996;279:1035-42. [PubMed]
- 18.Raap DK, Evans S, Garcia F, Li Q, Muma NA, Wolf WA, et al. Daily injections of fluoxetine induce dose-dependent desensitization of hypothalamic 5-HT1A receptors: reductions in neuroendocrine responses to 8-OH-DPAT and in levels of Gz and Gi proteins. J Pharmacol Exp Ther 1999;288:98-106. [PubMed]
- 19.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic Press; 1986.
- 20.Uylings HB, Sanz Arigita E, de Vos K, Smeets WJ, Pool CW, Amunts K, et al. The importance of a human 3D database and atlas for studies of prefrontal and thalamic functions. Prog Brain Res 2000;126:357-68. [DOI] [PubMed]
- 21.Bergqvist PB, Dong J, Blier P. Effect of atypical antipsychotic drugs on 5-HT2 receptors in the rat orbito-frontal cortex: an in vivo electrophysiological study. Psychopharmacology (Berl) 1999;143:89-96. [DOI] [PubMed]
- 22.El Mansari M, Blier P. In vivo electrophysiological characterization of 5-HT receptors in the guinea pig head of caudate nucleus and orbitofrontal cortex. Neuropharmacology 1997;36:577-88. [DOI] [PubMed]
- 23.Rueter LE, Tecott LH, Blier P. In vivo electrophysiological examination of 5-HT2 responses in 5-HT2C receptor mutant mice. Naunyn Schmiedebergs Arch Pharmacol 2000;361:484-91. [DOI] [PubMed]
- 24.Bosker FJ, Cremers TI, Jongsma ME, Westerink BH, Wikstrom HV, den Boer JA. Acute and chronic effects of citalopram on postsynaptic 5-hydroxytryptamine(1A) receptor-mediated feedback: a microdialysis study in the amygdala. J Neurochem 2001;76:1645-53. [DOI] [PubMed]
- 25.Haddjeri N, de Montigny C, Blier P. Long-term antidepressant treatments result in a tonic activation of forebrain 5-HT1A receptors. J Neurosci 1998;18:10150-6. [DOI] [PMC free article] [PubMed]
- 26.Le Poul E, Boni C, Hanoun N, Laporte AM, Laaris N, Chauveau J, et al. Differential adaptation of brain 5-HT1A and 5-HT1B receptors and 5-HT transporter in rats treated chronically with fluoxetine. Neuropharmacology 2000;39:110-22. [DOI] [PubMed]
- 27.Gobbi G, Murphy DL, Lesch K, Blier P. Modifications of the serotonergic system in mice lacking serotonin transporters: an in vivo electrophysiological study. J Pharmacol Exp Ther 2001;296:987-95. [PubMed]
- 28.Blier P, De Montigny C, Azzaro AJ. Modification of serotonergic and noradrenergic neurotransmissions by repeated administration of monoamine oxidase inhibitors: electrophysiological studies in the rat central nervous system. J Pharmacol Exp Ther 1986;237:987-94. [PubMed]
- 29.Bobula B, Tokarski K, Zahorodna A, Hess G. Adaptive changes in the reactivity of 5-HT1A and 5-HT2 receptors induced in rat frontal cortex by repeated imipramine and citalopram. Naunyn Schmiedebergs Arch Pharmacol 2003;367:444-50. [DOI] [PubMed]
- 30.Radja F, Daval G, Hamon M, Verge D. Pharmacological and physicochemical properties of pre- versus postsynaptic 5-hydroxytryptamine1A receptor binding sites in the rat brain: a quantitative autoradiographic study. J Neurochem 1992;58:1338-46. [DOI] [PubMed]
- 31.Albert PR, Zhou QY, Van Tol HH, Bunzow JR, Civelli O. Cloning, functional expression, and mRNA tissue distribution of the rat 5-hydroxytryptamine1A receptor gene. J Biol Chem 1990;265:5825-32. [PubMed]
- 32.Pigott TA, L'Heureux F, Hill JL, Bihari K, Bernstein SE, Murphy DL. A double-blind study of adjuvant buspirone hydrochloride in clomipramine-treated patients with obsessive–compulsive disorder. J Clin Psychopharmacol 1992;12:11-8. [DOI] [PubMed]
- 33.McDougle CJ, Goodman WK, Leckman JF, Holzer JC, Barr LC, McCance-Katz E, et al. Limited therapeutic effect of addition of buspirone in fluvoxamine-refractory obsessive–compulsive disorder. Am J Psychiatry 1993;150:647-9. [DOI] [PubMed]
- 34.Lesch KP, Hoh A, Schulte HM, Osterheider M, Muller T. Long-term fluoxetine treatment decreases 5-HT1A receptor responsivity in obsessive–compulsive disorder. Psychopharmacology (Berl) 1991;105:415-20. [DOI] [PubMed]
- 35.Greenberg BD, Benjamin J, Martin JD, Keuler D, Huang SJ, Altemus M, et al. Delayed obsessive–compulsive disorder symptom exacerbation after a single dose of a serotonin antagonist in fluoxetine-treated but not untreated patients. Psychopharmacology (Berl) 1998;140:434-44. [DOI] [PubMed]
- 36.McDougle CJ, Epperson CN, Pelton GH, Wasylink S, Price LH. A double-blind, placebo-controlled study of risperidone addition in serotonin reuptake inhibitor-refractory obsessive–compulsive disorder. Arch Gen Psychiatry 2000;57:794-801. [DOI] [PubMed]
- 37.Erzegovesi S, Guglielmo E, Siliprandi F, Bellodi L. Low-dose risperidone augmentation of fluvoxamine treatment in obsessive–compulsive disorder: a double-blind, placebo-controlled study. Eur Neuropsychopharmacol 2005;15:69-74. [DOI] [PubMed]
- 38.Leonard HL, Rapoport JL. Relief of obsessive compulsive symptoms by LSD and psilocin. Am J Psychiatry 1987;144:1239-40. [DOI] [PubMed]
- 39.Hanes KR. Serotonin, psilocybin, and body dysmorphic disorder: a case report. J Clin Psychopharmacol 1996;16:188-9. [DOI] [PubMed]
- 40.Moreno FA, Delgado PL. Hallucinogen-induced relief of obsessions and compulsions. Am J Psychiatry 1997;154:1037-8. [DOI] [PubMed]
- 41.Zghoul T, Blier P. Enhancing action of LSD on neuronal responsiveness to serotonin in a brain structure involved in obsessive–compulsive disorder. Int J Neuropsychopharmacol 2003;6:13-21. [DOI] [PubMed]