

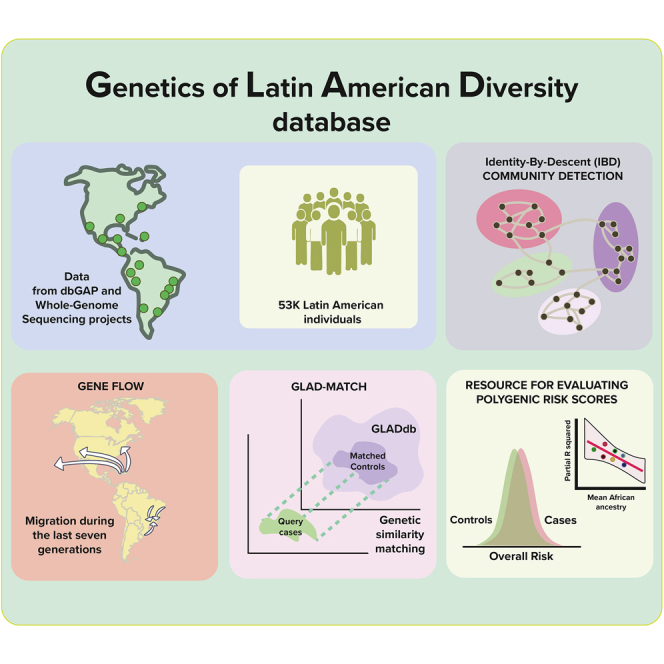
Summary
Latin Americans are underrepresented in genetic studies, increasing disparities in personalized genomic medicine. Despite available genetic data from thousands of Latin Americans, accessing and navigating the bureaucratic hurdles for consent or access remains challenging. To address this, we introduce the Genetics of Latin American Diversity (GLAD) Project, compiling genome-wide information from 53,738 Latin Americans across 39 studies representing 46 geographical regions. Through GLAD, we identified heterogeneous ancestry composition and recent gene flow across the Americas. Additionally, we developed GLAD-match, a simulated annealing-based algorithm, to match the genetic background of external samples to our database, sharing summary statistics (i.e., allele and haplotype frequencies) without transferring individual-level genotypes. Finally, we demonstrate the potential of GLAD as a critical resource for evaluating statistical genetic software in the presence of admixture. By providing this resource, we promote genomic research in Latin Americans and contribute to the promises of personalized medicine to more people.
Keywords: Latin America, GLAD-match, GWAS, migration, imputation, identity-by-descent, local ancestry, population structure
Graphical abstract
Highlights
-
•
We present GLADdb, a collection of 53,000 Latin American individuals
-
•
We identified ancestry-biased migration across the Americas
-
•
We present GLAD-match to identify similarities without sharing individual genotypes
-
•
GLADdb is a valuable resource for evaluating genetic epidemiology software
In this article, Borda, Loech, Guo, and Laboulaye et al. developed the Genetics of Latin American Diversity database (GLADdb), which includes 53,000 Latin Americans. With GLADdb, they explored two gaps in Latino genomics: (1) population structure and recent gene flow and (2) the underrepresentation of Latinos in genetic epidemiology.
Introduction
Latin Americans, as an ethnic label, encompass diverse populations across the Americas with a distinct ancestral composition resulting from admixture between various global populations.1 As such, treating Latin Americans as a homogeneous group oversimplifies their genetic diversity and hinders efforts to improve health and clinical treatment. With a population of 656 million (8.5% of the world’s population),2 Latin Americans represent a significant demographic, particularly in the United States, where they comprise 18% of the population and are the fastest-growing group.3 However, these populations remain understudied and underserved in biomedical research and risk being left behind by the precision medicine revolution. For instance, Latin Americans represent only about 0.38% of participants in genome-wide association studies (GWASs) performed.4
Several essential efforts have been made to understand Latin American (LAm) genetic history and identified genetic variants associated with complex traits.5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26 However, most of these samples are thinly spread across many projects, with few meta-analyses (e.g., Hispanic/Latino Anthropometry Consortium 27) or initiatives to obtain the >100,000 individuals (e.g., the Mexico City Prospective Study28) necessary to have statistical power comparable to other population groups (e.g., Europeans29 and East Asians30,31). To address the underrepresentation of LAms in genomic studies, we have developed the Genetics of Latin American Diversity database (GLADdb), a resource to explore LAm population structure patterns and support epidemiological studies by providing summary statistics for a subset of individuals after genetic matching. In this context, we defined an LAm population as a group of people with heritage from Spanish-speaking or Portuguese-speaking countries in the Americas. We consider that “heritage” encompasses various aspects, from culture and geography to genetics.
We built GLADdb by gleaning LAm individuals through dbGaP (Database of Genotypes and Phenotypes) and whole-genome sequencing (WGS) projects across the Americas. GLADdb includes over 53,000 unrelated individuals, either genotyped and imputed or sequenced, representing 46 geographic groups, labeled based on administrative divisions such as country, state, or city (Figure 1A; Tables S1 and S2). With GLADdb, we addressed two major goals regarding LAm genomics: (1) in population genetics, we uncovered recent, fine-scale patterns of distant relatedness and differentiation across the Americas, providing insights into regions with genetic underrepresentation, and (2) in genetic epidemiology at two levels: (a) by developing GLAD-match, a web tool for matching the genetic background of GLADdb individuals with external pools of samples, providing additional power to discover genotype-phenotype associations, and (b) by demonstrating how GLADdb can be utilized for testing statistical genetic software in diverse LAm cohorts.
Figure 1.

Dimensionality reduction of genetic data and ROH for more than 53,000 unrelated LAms from the GLAD database
(A) Geographic distribution of GLADdb cohorts. Countries represented in GLADdb are highlighted with colors.
(B) PCA of the entire dataset based on high-quality imputed SNPs (Rsq >0.9) showing the sampling spread of LAms. Principal components 2 and 5 were plotted to show the axis of genetic diversity that explains the European-African (EUR-AFR) differentiation (PC2) and the diversity of Indigenous American ancestries from Mexico to Peru (PC5). All principal components are plotted in Figure S9.
(C) Distribution of genome-wide amount of ROHs for LAm groups and reference populations included in GLADdb. The upper part of the plot shows continental reference populations, and the lower part details the distribution in Peru and Brazil. Populations are sorted in a north-to-south pattern. This analysis was restricted to ROH segments >1 Mb. For patterns in ROH segments >8 Mb, see Figure S13. CEU, Utah residents with northern and western European ancestry from CEPH collection; ESN, Esan in Nigeria; EUR, European individuals; FIN, Finnish in Finland; GBR, British from England and Scotland; GWD, Gambian in Western Division-Mandinka; IBS, Iberian populations from Spain; LWK, Luhya in Webuye, Kenya; MSL, Mende in Sierra Leone; NAT, Indigenous American individuals; TSI, Toscani in Italia; USA HI, United States, Hawaii; USA NY, United States, New York; YRI, Yoruba in Ibadan, Nigeria.
First, we started by exploring distant genetic relatedness among LAm countries. Several studies have focused on determining the sources and timing for admixture events that led to the current genetic composition in some LAm countries.6,11,12,13,16,32,33,34 However, understanding LAm genetic diversity goes beyond the initial continental admixture. It involves bottlenecks, founder effects, and migration into and along the Americas, especially concerning fine-scale population structure within the continental sources (i.e., Indigenous American [IA], European [EUR], and African [AFR] groups). We explored population structure and recent migration among LAm regions by analyzing identity-by-descent (IBD) sharing and local ancestry patterns.
Second, we addressed issues about data availability when performing large-scale analyses in LAm populations. Population stratification is a major concern in GWAS studies as it can lead to spurious association signals. Moreover, association studies in LAm populations face additional challenges, such as smaller sample sizes than Europeans and other populations. We introduce here GLAD-match, a matching procedure to share summary statistics based on GLAD individuals to overcome these issues. A matching procedure identifies individuals with similar genetic backgrounds with external data, mitigating genetic control inflation and minimizing spurious associations arising from population structure.35,36 A similar strategy was used in GWASs to identify and increase the number of control individuals.37 GLAD-match explores the principal-component space derived from LAm individuals in GLADdb cohorts, into which we project external samples and match them to GLADdb individuals with no individual-level genotype data transfer needed. From the selected GLADdb individuals, we generate and return summary statistics of genome-wide genotype frequencies and aggregate local ancestry composition to increase the sample size and power of the end-user study for association analyses (i.e., chi-squared test). We demonstrated the effectiveness of GLAD-match compared to another matching algorithm, PCAmatchR,36 using genomic control as a proxy for the quality of matched individuals. Since GLADdb consists of cases and controls for different phenotypes, we also use phenotype filters to select individuals who are useful as controls. We implemented all these features through an interactive web portal (glad.igs.umaryland.edu).
Finally, we demonstrate the potential of GLADdb as a critical resource for evaluating the performance of statistical genetic software in the presence of admixture. We do so by comparing three polygenic risk score (PRS) algorithms for estimating PRS in admixed individuals in a scenario where the ancestries corresponding to the GWAS summary statistics do not match the target cohort. PRS, the linear summation of risk variants weighted by their GWAS effect size, are highly impacted by the European-ancestry bias underlying much of the available GWAS data, and their transferability across populations remains a critical limitation of the approach.38,39 GLADdb is uniquely situated to support methods’ development efforts that help ensure cross-population transferability of statistical genetic applications. GLADdb fills a crucial role by collaboratively combining all available LAm individuals with genomic resources and functions as an ever-evolving and growing database for genetic diversity in LAm individuals worldwide.
Results
Data description and quality control
Our main workflow is described in Figure S1 and STAR Methods. Briefly, we have explored >268,000 samples by gathering data from 39 dbGaP cohorts and other WGS projects that include US Hispanics/LAm individuals5,6,7,8,13,40 (Table S1). For the inclusion criteria, we gathered individuals self-described (GLAD-SD) as “Latino” or “Hispanic” and ADMIXTURE-defined individuals (GLAD-AD). The latter criterion was applied only to US cohorts with no ethnicity information. It was used to identify possible LAm individuals using ADMIXTURE analysis,41 retaining any individuals with >2% IA ancestry (see STAR Methods). For each genotyped cohort (Table S1), we imputed all GLAD-SD and GLAD-AD individuals using the Trans-Omics for Precision Medicine (TOPMed) Imputation server42 and kept imputed variants with Rsq >0.9. After imputation quality control (QC), we kept 43,269 individuals (nGLAD-SD = 25,627 and nGLAD-AD = 17,642), which were combined into a single dataset with sequencing data from the TOPMed Project5 (27,088 individuals) and 1000 Genomes Project high coverage40 (345 individuals). The merged dataset included 3,248,494 biallelic variants, retaining variants displaying high Rsq (>0.95) and empirical Rsq (>0.8) values (see STAR Methods and Figures S2–S6). Higher values of empirical Rsq underscore the high quality of the imputation process. To remove the family structure in GLADdb, we inferred kinship coefficients using IBD segments on the complete dataset, keeping 53,738 unrelated individuals (see STAR Methods). Moreover, to demonstrate the applicability of imputed data for haplotype-based analysis, we performed an IBD analysis on a subset of GLAD cohorts, comparing genotyping-only and imputed-only datasets and examining their overlap (see STAR Methods and Figures S4, S7, and S8). Our results highlighted that utilizing exclusively imputed data for IBD analysis did not introduce bias for segments exceeding 4 cM. This underscores the robustness of GLADdb, a combination of genotyping and imputed data, for haplotype-based methods.
Population structure and levels of genetic diversity within LAm groups
Using our unrelated dataset and ancestry-reference groups (Table S3), we explored the patterns of diversity and differentiation throughout the Americas using principal-component analysis (PCA), uniform manifold approximation and projection, and local ancestry analyses (Figures 1B and S9–S12). These results highlighted some important points. Individuals cluster according to ancestry and not technology or other batch effects (Figures S10 and S11). Notably, GLAD-AD individuals cluster well with other GLAD-SD individuals, which validates our inclusion criteria (Figure S11). By coupling principal-components dispersion with ancestry proportions (Figure S12), we reaffirm the heterogeneous ancestry distribution among LAm individuals with some groups such as those in the United States and Brazil, displaying a majority admixture of EUR and AFR ancestries (Figures S12B and S12C). In contrast, groups in Peru, Mexico, and Guatemala predominantly exhibit IA ancestry (Figure S12D). Regarding sample sizes, the best-represented regions in GLADdb included the United States, Mexico, the Dominican Republic, Costa Rica, Brazil, and Peru.
Although our population structure analyses identified a wide diversity of LAm groups, these groups originated from continental progenitors that suffered a significant drop in effective population size during the colonial period of the Americas.43,44,45 This resulted in a higher level of consanguinity and enrichment of long runs of homozygosity observed in some LAm groups (e.g., CLM [Colombians in Medellin, Colombia] and PEL [Peruvians in Lima, Peru] from 1000 Genomes Project) compared to the Finnish,44 a population notably shaped by a strong founder effect. Based on demographic information available for the cohorts, we organized GLAD-SD individuals into 46 self-described LAm groups, consistent with geographic labels based on administrative division level (e.g., country-, state-, or city-level information) (Table S2). In addition, we included 12 IA populations from the Peruvian Genome Project as well as 5 EUR and 5 AFR populations from the 1000 Genomes Project (see STAR Methods).
We explored the levels of diversity in each group by inferring runs of homozygosity (ROH) (Figure 1C; see STAR Methods). As expected, individuals from Africa showed lower values for total ROH compared to individuals from Europe and Indigenous groups from Peru. In the Americas, Brazilian regions and Afro-Peruvians showed the lowest level of total ROH compared to other LAm regions. However, Peruvian regions, Mexico, and Guatemala showed the highest levels of ROH. This is consistent with a highest proportion of IA ancestry observed in Peru, Mexico, and Guatemala (>60%; Table S2). Interestingly, Central American and Caribbean populations showed the highest density of ROH—>8 Mb (Figure S13)—suggesting that consanguinity is more common in these samples.
Fine-scale population structure revealed by IBD network
To obtain a fine-scale picture of population structure among LAm groups, we built a sample-pair genome-wide total IBD matrix using all IBD segments >5 cM shared among unrelated individuals from LAm groups (n = 51,670). Clusters in this matrix, based on hierarchical clustering (Figure 2) and Louvain algorithm (Figure S14), are mainly consistent with geographic labels, with strong intra-cluster sharing among individuals from Puerto Rico, the Dominican Republic, and Costa Rica. Given the sample size and genetic diversity, finer-scale population structure is observable in clusters representing the United States/Mexico, Peru, and Brazil. To reveal the substructure, we employed an IBD network-based community detection algorithm, Infomap,46,47 to further analyze relatedness patterns. We selected the top 20 IBD network-based communities that accumulated 70% of GLADdb individuals (other communities each have <270 individuals). Several of these communities (labeled as CA1–20 and ordered from largest to most minor) showed enrichment of individuals from a particular country, such as Costa Rica (99.6%, IBD community CA5), Puerto Rico (98%, IBD community CA1), Dominican Republic (95.0%, IBD community CA4), Cuba (89.8%, IBD community CA6), Colombia (89.4%, IBD community CA13), and Chile (84%, IBD community CA22) (Figure 3). In contrast, individuals from Mexico, Peru, and Brazil were grouped in several communities (Mexico: 7, Brazil: 5, Peru: 12 communities enriching ≥1% of individuals in the country). These within-country communities were represented by individuals from particular states or cities, reflecting the extensive sampling performed in these countries (Figure 3). Furthermore, when we analyzed networks for short (Figure S15) and long (Figure S16) IBD segments, we revealed less differentiation in the long-segment network, suggesting higher gene flow between communities in the most recent time frame.
Figure 2.

Clustering of total IBD matrix of unrelated individuals from GLADdb
(A) Heatmap of the square root of sample-pair total IBD shared among unrelated individuals sampled from LAm countries or the United States within GLADdb. Each pixel represents a pair of individuals; the x and y axes indicate individual IDs sorted by unsupervised hierarchical clustering. Annotations within the heatmap represent the most enriched geographic labels (countries or cities) in the indicated blocks. Labels with “USA-NY-country” correspond to self-described US-Hispanic living in New York with a specific country of origin.
(B) Individual-level annotations for the heatmap. The annotations include (1) labels based on agglomerative clustering in the 1st vertical bar, (2) self-described ethnicity in the 2nd bar, and (3) sampling country (combined indicators in the 3rd bar and country-specific indicators in the 4th–14th bars). Each row in these bars corresponds to an individual. Note that the row orders in all label bars are shared with those of (A).
(C) Frequency of labels (log scale) and color keys for agglomerative clustering (bottom), self-described ethnicity (center), and sample country (top), respectively. Note that the “NA” label refers to individuals not assigned any country, self-described ethnicity, or cluster.
Figure 3.

IBD network community detection
We infer the community structure using the Infomap algorithm based on a matrix of IBD segments >5 cM.
(A) Top 20 IBD network communities. Only individuals with connections >30 are included in the layout calculation for visualization purposes. The community labels, such as CA1 and CA2, are named according to the IBD version used and the rank of the community sizes, with CA1 representing the largest community when using all IBD segments, including short (5–9.3 cM) and long (>9.3 cM) segments.
(B) Average IBD sharing among the top 30 inferred communities (ordered by agglomerative clustering; the same order is followed in C and D).
(C) Distribution of IBD shared among individuals in each community.
(D) Enrichment of IBD community membership in the country of origin (i.e., proportions of community labels for individuals born in a given country). Note that for individuals without exact birth country information, broader geographic labels were used when available, such as Central America and South America. To visualize the dynamics before and after the Spanish colonization of the Americas, two different IBD networks were built based on IBD short (Figure S15) and long segments (Figure S16), respectively, which revealed distinct patterns of detected communities.
Long-distance relatedness among LAm groups
To explore recent migration among 46 LAm regions, we restricted our analyses to IBD segments >21.4 cM, representing segments transmitted by shared common ancestors within the past seven generations corresponding to post-colonial times48 and after the admixture process. We reasoned that during this period, the sharing of larger IBD segments could originate predominantly from gene flow among LAm regions. At the interregional level (Figure 4), we detected higher levels of sharing between Caribbean groups (i.e., Puerto Rico and Dominican Republic) with New York, California, and Hawaii groups. Specifically, New York cohorts include several groups of individuals with self-described Caribbean origin. Another tight sub-network of sharing is observed in Brazil (Figure S17), where the southeast region (São Paulo, Rio de Janeiro, and Minas Gerais states) has major connections with other Brazilian populations. Interestingly, there are IBD-sharing connections between Uruguay and southern Brazil. At the intraregional level, Afro-Peruvians from Ica, Peru showed the highest IBD sharing (Figure S17).
Figure 4.

IBD analyses of Latin American groups
We explored the relationship among LAm regions by inferring the average IBD shared among regions (A) and an asIBDscore for AFR (B), EUR (C), and IA ancestries (D). Dots represent LAm regions. Interregional sharing, including <5 pairs, was removed. For IBD sharing (right plot), we removed the intrapopulation sharing in Peru-Ica due to the higher sharing and to improve visualization (for full sharing patterns, see Figure S13).
Considering the multi-way admixed origin of LAm populations, we devised a statistic (ancestry-specific IBD score [asIBDscore]) that quantifies the level of relatedness among two admixed populations for a particular ancestry (AFR, EUR, or IA) (Figure 4). We computed the asIBD score (see STAR Methods) by coupling the IBD and local ancestry inferences. Our asIBD score explains the relationship of asIBD sharing with respect to the global ancestry of the populations. We detected the highest asIBDscore for the IA IBD sharing between Caribbean populations (i.e., Puerto Rico and the Dominican Republic) with New York (Table S4). Interestingly, the Puerto Rico IBD sharing with California and Hawaii showed AFR and IA with the highest value asIBDscore with respect to EUR, suggesting more heterogeneity for the European ancestry (Table S4). The Brazilian populations have higher values of asIBD for the IA ancestry in south and southeast populations, indicating a more homogeneous composition of IA ancestry in those regions (Table S4). In a network for IBD sharing between Peru-Ica and Peru-La-Libertad, the EUR ancestry showed the highest value for the asIBD (Tables S2 and S4).
Supporting external studies through the GLADdb matching algorithm and statistical genetic software benchmarking
One of the ultimate goals of GLADdb is to support GWAS and admixture mapping studies by providing summary statistics (i.e., allele and haplotype counts) of a subset of control individuals from GLAD that match the genetic background of external samples. We addressed this goal by developing a genetic matching algorithm, GLAD-match. Our method, based on nearest-neighbor simulated annealing matching, shown in Figure 5A and outlined in STAR Methods, employs local search to find the optimal cohort from a set of candidates. The algorithm operates on a principal-component space in which the external user-provided query cases can search for controls without needing individual genotypes. The algorithm computes variance-weighted Minkowski distance pairwise between query cases and potential controls, selects the nearest neighbors as candidate controls, samples a set of matches from the candidates, and iteratively resamples and refines the set of matches using simulated annealing, optimizing for the genomic control statistics λ.49,50
Figure 5.

Nearest-neighbor simulated annealing matching algorithm and results
(A) Visual overview of the algorithm.
(B) Comparison with baseline bipartite matching algorithm (x axis), where points below the line y = x indicate our algorithm outperforming the baseline (small box highlights high-density region).
(C) Effect of a number of matches on improvement over the baseline.
We performed the following experiment to evaluate our matching algorithm and the extent to which GLAD cohorts can provide valid control sets. Using 1000 Genomes populations and some GLAD cohorts as cases in which the pseudo-phenotype belongs to the query cohort (see STAR Methods), we ran a greedy bipartite matching baseline used by PCAmatchR,36,51 and our matching algorithm and returned summary statistics (i.e., alternative allele frequency, genotype counts, and haplotype ancestry counts by segment) for various control set sizes. Then, for each pair of cases and controls, we ran a GWAS for which the genomic control λ statistics are reported in Table 1 and more extensively in Figure 5B. For the analyzed cohorts, which represent a variety of admixed groups, the matched controls yield genomic controls close to 1, suggesting that GLAD can provide proper controls for a variety of cohorts, and our matching algorithm shows slight improvements for larger and more varied query cohorts. These improvements narrow progressively as the number of matches required increases (Figure 5C).
Table 1.
Comparison of genomic control results (λ statistics) when returning 500 control individuals from GLAD using greedy bipartite matching and the nearest-neighbor simulated annealing matching algorithm
| Source | 1,000 genomes populations |
GLAD cohorts |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| MXL, n = 60 | CLM, n = 92 | PEL, n = 84 | PUR, n = 104 | HCHS SOL, n = 6,558 | MESA, n = 1,016 | SIGMA, n = 1,145 | LARGE-PD, n = 1,456 | ||
| Greedy bipartite matching | genomic control | 0.9443 ± 0.0008 | 1.0953 ± 0.0021 | 0.9549 ± 0.0012 | 0.9952 ± 0.0019 | 0.9970 ± 0.0057 | 1.0158 ± 0.0155 | 1.0727 ± 0.0072 | 1.0302 ± 0.0165 |
| Nearest-neighbor simulated annealing matching algorithm | 0.9391 ± 0.0001 | 1.0899 ± 0.0010 | 0.9495 ± 0.0009 | 0.9880 ± 0.0004 | 0.9615 ± 0.0043 | 0.9841 ± 0.0044 | 1.0470 ± 0.0063 | 1.0066 ± 0.0061 | |
Genomic controls in the table are the result of a pseudo-GWAS for a dummy binary phenotype representing the belonging to the query cohort. HCHS SOL, Hispanic Community Health Study/Study of Latinos; MXL, Mexican Ancestry in Los Angeles, CA, USA; PUR, Puerto Rican in Puerto Rico; SIGMA, Slim Initiative in Genomic Medicine for the Americas.
In addition to control matching, GLADdb is an optimal resource for benchmarking statistical genetic software in complex, heterogeneous cohorts with a wide range of available traits. We demonstrated this potential by comparing several popular PRS algorithms (Clumping + Thresholding using PRSice-2,52 PRS-CS,53 and PRS-CSx54) using a subset of GLAD-SD (Table S5; STAR Methods) with type 2 diabetes (T2D) status, height, or body mass index (BMI) data under a hypothetical scenario where LAm GWAS data are not available (Table S6). The GLAD-SD subset includes LAm cohorts with different population histories and ancestry proportions (e.g., Afro-Caribbeans, Brazilians, and Peruvians). Although the Bayesian PRS-CS method, in general, outperformed PRSice-2, the inclusion of non-European GWAS data using PRS-CSx yielded the largest increase in PRS predictive performance (Figures 6A–6C and S18). PRS-CSx improved single-ancestry PRS predictive performance (e.g., East Asian PRS from PRS-CSx versus PRS-CS or PRSice-2) in nearly every instance (Table S7). Combining the posterior effect sizes estimated by PRS-CSx further improved models (Figures 6A–6C; Table S7). Note that the best approach for combining PRS information varied by cohort, likely reflecting cohort heterogeneity (Figure S19). Model performance, as measured by partial R2, was negatively associated with mean AFR ancestry (−0.02 per SD AFR ancestry, p = 0.005; Figure 6D). While the percentage of improvement achieved when leveraging non-European GWAS data can be as high as 80% over the Clumping + Thresholding model, the R2 of each PRS still can be modest. For example, in the Alzheimer disease cohort from the Caribbean, the T2D PRS-CSx model improved prediction by nearly 80%, but the R2 of that model was only 0.03 on the observed scale (Figure 6D).
Figure 6.

PRS in select cohorts from GLAD-SD
(A) Comparison of height model performance as percentage of improvement over a EUR-ancestry GWAS Clumping + Thresholding PRS. Models include PRS-CS using EUR-ancestry GWAS, PRS-CSx using EUR and East Asian-ancestry GWAS, and PRS-CSx using EUR, East Asian, and AFR-ancestry GWAS. All models were compared using the correlation between the prediction and the trait.
(B) Comparison of BMI model performance.
(C) Comparison of T2D model performance.
(D) Total R2 of best PRS model by AFR ancestry. Cohorts are labeled by color; traits are labeled by shape. Partial R2 was calculated by squaring Pearson’s r followed by subtracting the full model (PRS + covariates) from the base model (covariates only, see STAR Methods). AFR ancestry proportions were estimated using ADMIXTURE.
Discussion
LAms are underrepresented in genetic and epidemiological research, hindering our knowledge of their genetic diversity and environmental factors. This limitation impacts personalized medicine and our understanding of complex traits.55 GLADdb aims to tackle the underrepresentation of genomic data by gathering genome-wide data of LAm populations into a single resource. Through GLADdb, we have two main contributions to LAm genomics: (1) population genetics: we elucidated population structure and gene flow across LAm regions, and (2) genetic epidemiology: we developed an algorithm and an online portal to provide summary statistics from control individuals from GLADdb with a genetic makeup similar to that of external samples. Also, by assembling a collection of LAm cohorts with different population histories, we created a unique tool for evaluating the performance of statistical genetic software in the presence of admixture and other complexities.
For population genetics, continental migrations were the initial sources of LAm diversity. However, other processes have shaped this diversity and relationships across geographic regions (e.g., internal migration48). Using ROH and IBD inferences, we explored intra- and interpopulation relationships in Latin America. Notably, we observed that Peruvians, despite higher homozygosity, exhibit differentiated groups associated with geographical regions.6,7 Using a similar approach described by Baharian et al.,48 we analyzed long IBD segments (greater than 21.4 cM) to capture the shared ancestry within the last seven generations. Long-IBD-based analysis in LAm regions highlighted recent migration between regions.
We detected two main networks of IBD sharing: the Caribbean–United States (New York, California, and Hawaii) and the Brazilian intragroup sharing. In 20th century Latin America, migrations followed a rural-to-urban or outside-the-country tendency due to regional socioeconomic disparities.56 Notably, in Puerto Rico, during the early 1900s, a migration policy was enacted in response to its social and economic problems.57 Hawaii, the Dominican Republic, and Cuba were the primary destinations during the first stage of this diaspora, followed by a strong migration to New York during the late 1940s.58 Socioeconomic differences characterized each migration stage.59,60 For example, many individuals who migrated from Puerto Rico to Hawaii were recognized as jibaros,60 which are countryside people who traditionally farm the land. However, Puerto Ricans who migrated to New York represented a cross-section of economic and social classes.59 By inferring the ancestral background of IBD segments, we found that the Puerto Rico-Hawaii sharing is characterized by predominant AFR and IA sharing compared to the predominant IA sharing between Puerto Rico and New York. These contrasting patterns reflect the differential composition of the two stages of migration. Interestingly, ancestry-biased migrations like this are not uncommon in the United States, having been observed as far back as the Great Migration.48 Brazil is another example of recent migration due to economic factors. During the 1950s, southeastern Brazil, represented by the states of Rio de Janeiro, São Paulo, and Minas Gerais, experienced huge economic growth that triggered a massive migration to these regions.61 Our IBD analyses showed strong connectivity among and around these southeastern regions (Rio de Janeiro and São Paulo). Moreover, we detected connectivity between southern Brazilian regions and Uruguay, reflecting their recent shared history because Uruguay was annexed to Brazil before its independence,62 and its demographic composition included a significant proportion of Brazilians at that time.63
For genetic epidemiology, our genotype-matching algorithm and subsequent provision of control summary statistics meet a real need in the research community. Groups exploring the genetic architecture of traits in LAm cohorts will be able to increase their sample sizes without further straining budgets. While there are initiatives that significantly increase the representation of LAm subjects in genomics, access to those data remains a concern. In some cases, navigating the bureaucratic maze represents a real barrier, while in other cases, the data are proprietary. By constructing the first version of GLADdb, we acquired and aggregated LAm data from across 39 cohorts. In addition, our matching and data transfer processes only require summary statistics (genotype counts and principal components), thus reducing the exposure of sensitive data. While others have found additional means to abstract external query samples,35 we have utilized individual summaries in the form of principal component values, similar to giving PCA plots in manuscripts, which allows us to improve the matching for groups with substantial population structure, such as LAm groups. This is more abstracted than what is allowed for imputation servers5 because we never have access to individual-level genotypes, nor can we reconstruct them from the subset of principal components included. This means that GLADdb is sufficiently privatized for any cohort consent where PCAs can be shared via publication or imputation can be done through the Michigan or TOPMed imputation servers. Also, our matching algorithm will provide a better set of specific controls that match the genetic background of query samples, reducing the issues due to population structure and finally performing a statistical test that does not require covariates, such as a chi-squared test.
In addition to supporting genetic studies through control matching, GLADdb presents a valuable resource for evaluating the performance of genetic epidemiology software for methods development and benchmarking. Such software needs to be evaluated in the presence of admixture in addition to the more homogeneous cohorts. This is particularly evident for PRS estimation, where the impact of long-standing biases in GWAS data is well documented.38,39,64 In our test case, we evaluated three popular PRS algorithms: Clumping + Thresholding implemented in PRSice-2, PRS-CS, and PRS-CSx. We found that PRS-CSx, which can model multiple GWAS populations simultaneously, significantly improved predictive performance over single ancestry methods. This was true despite not using GWAS data from any LAm cohorts for this example. Variability in model performance likely reflected population heterogeneity across the different cohorts, and model performance was negatively associated with mean AFR ancestry. The sample sizes of the AFR ancestry GWAS cohorts used for this study were smaller by an order of magnitude than the East Asian and EUR ancestry GWAS cohorts. It is clear that well-powered, diverse GWAS is critical for equitable PRS performance. In the meantime, methodological innovation is required to improve cross-population portability for GWAS traits lacking adequate representation.65 In addition to PRS-CSx, several methods such as LDPred-funct and PolyPred include functional data, and TL-Multi utilizes transfer learning.66,67,68 The robustness of existing and new PRS methods to admixture can be evaluated using the heterogeneous cohorts represented in GLADdb.
Limitations of the study
While GLADdb offers valuable insights, it does have limitations. First, ADMIXTURE-defined LAms are restricted to individuals with more than 2% IA ancestry. This definition could be very restrictive, considering some LAm groups might have no IA ancestries (i.e., European descendants in Brazil). Still, it ensures that the maximum number of individuals is collected without strong bias. Second, GLADdb is restricted to case-control studies without covariate control. To enhance the capabilities of GLAD, we are expanding its scope to encompass a wider range of phenotypes, including age and BMI, as potential covariates. This expansion will facilitate the provision of summary statistics for continuous variables. Third, regarding the absence of covariates, it’s important to highlight that this limitation is not solely attributable to the sharing approach but is due to the availability of phenotypic data in the original cohorts. The heterogeneity of the external sample, while present, does not pose a significant challenge during the matching process. We have successfully identified matched individuals within heterogeneous cohorts, such as LARGE-PD. However, it is important to stress that allele and haplotype counts will still be derived from this diverse pool of matched individuals. Thus, we recommend a separate matching process when appropriate.
In LAm genomics, another challenge lies in the underrepresentation of IA ancestries in public datasets, primarily consisting of a few isolated populations that may introduce limitations in global and local ancestry inferences. IA ancestry closely relates to local Indigenous groups in admixed LAm populations, as several studies have shown.6,11,16 We used a reference panel of Indigenous Peruvians and Guatemalans to address IA ancestry challenges. These populations have larger effective population sizes compared to other native groups,69 reducing issues related to higher levels of genetic drift. In this way, we can get around the problem of IA inferences in Brazilians or US individuals with some level of IA ancestry (i.e., individuals with ancestry related to tribal nations in which genetic studies have not been allowed). Still, better ethically aware representation in genomics is preferred. Furthermore, GLADdb highlights better-represented regions like Brazil, Mexico, and Peru, but ethnic diversity remains unbalanced (predominantly EUR ancestry). Urgent inclusion of regions like Bolivia and Paraguay, as well as diverse ethnicities (AFR and Asian ancestries in the Americas), is imperative.
In conclusion, through GLADdb, we highlighted the heterogeneous ancestry composition across Latin America and inferred ancestry differences in recent gene flow events. Also, by sharing summary statistics, we contribute to improving global equity in genomic research, specifically in epidemiological research in which GWAS is performed routinely. This is one more step to ensuring that health disparities arising from genetic studies do not become pervasive in admixed and non-European populations.
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Timothy D. O’Connor (timothydoconnor@gmail.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
No data were generated for this study. The code utilized for this study is publicly available on GitHub at the following:
-
•
IBD analysis: https://github.com/umb-oconnorgroup/ibdtools. All original code has been deposited at Zenodo and is publicly available at https://doi.org/10.5281/zenodo.13851024 as of the date of publication.
-
•
ROH and asIBD: https://github.com/umb-oconnorgroup/GLAD_DemographicAnalysis. All original code has been deposited at Zenodo and is publicly available at https://doi.org/10.5281/zenodo.13850990 as of the date of publication.
-
•
PRS estimation and evaluation: https://github.com/dloesch/PRSHelpDesk. All original code has been deposited at Zenodo and is publicly available at https://doi.org/10.5281/zenodo.13851588 as of the date of publication.
-
•
GLADdb: https://github.com/umb-oconnorgroup/gladprep and https://github.com/umb-oconnorgroup/gladdb. All original code has been deposited at Zenodo and is publicly available at https://doi.org/10.5281/zenodo.13851635 as of the date of publication.
Consortia
National Institute of Neurological Disorders and Stroke (NINDS) Stroke Genetics Network (SiGN) Consortium: Stephen J. Kittner, Braxton D. Mitchell, and Jordi Jimenez-Conde. TOPMed Population Genetics Working Group: Sebastian Zoellner. Latin American Research Consortium on the Genetics of Parkinson’s Disease (LARGE-PD): Emilia Gatto, Grace Letro, Jorge Luis Orozco, Carlos Velez-Pardo, Marlene Jimenez-Del-Rio, Francisco Lopera, Patricio Olguin, Andrew Sobering, Alex Medina, Daniel Martinez, Mayela Rodriguez, Sarael Alcauter, Alejandra Medina, Mario Cornejo-Olivas, Angel Medina Colque, Julia Rios Pinto, Ivan Cornejo Herrera, Edward Ochoa-Valle, Nicanor Mori Quispe, and Angel Viñuela.
Acknowledgments
We would like to thank Evangeline “Eevee” O’Connor for assistance in providing an acronym that is both accurate and contributes to generally uplifting our research. T.D.O., V.B., R.L., and D.V.-O. were supported by the National Human Genome Research Institute of the NIH under award no. R35HG010692. E.T.-S. was supported by FAPEMIG (Fundação de Amparo à Pesquisa do Estado de Minas Gerais) RED 00314-16, Programa Nacional de Genômica e Saúde de Precisão – Genomas Brasil from the Brazilian Ministry of Health (CNPq Process 403502/2020-9) and Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq. R.D.H. is supported by the NIH under award no. R01 GM142112. D.P.L. was supported by the National Heart, Lung, and Blood Institute of the NIH under award no. T32 HL007698-25. J.N.F. was supported by Research Training in the Epidemiology of Aging, funded by the National Institute on Aging under award no. T32 AG000262. The Latin American Research Consortium on the Genetics of Parkinson’s Disease (LARGE-PD) is funded by the NIH/NINDS through award no. R01 NS112499. The NINDS-sponsored Stroke Genetics Network (SiGN) is funded by the NIH/NINDS under award nos. R01 NS105150 and R01 NS100178. The Genome Sequencing for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung, and Blood Institute (NHLBI). Genome sequencing for “NHLBI TOPMed: The Genetic Epidemiology of Asthma in Costa Rica” (phs000988.v3p1) was performed at Northwest Genomics Center (HHSN268201600032I/3R37HL066289-13S1). Genome sequencing for “NHLBI TOPMed: San Antonio Family Heart Study (SAFHS)” (phs001215.v4.p2) was performed at Illumina (3R01HL113323-03S1 and R01HL113322). Genome sequencing for “NHLBI TOPMed: Women’s Health Initiative (WHI)” (phs001237.v3.p1) was performed at Broad Institute Genomics Platform (HHSN268201500014C). Genome sequencing for “NHLBI TOPMed: Hispanic Community Health Study - Study of Latinos (HCHS/SOL)” (phs001395.v2.p1) was performed at Baylor College of Medicine Human Genome Sequencing Center (HHSN268201600033I). Genome sequencing for “NHLBI TOPMed: Multi-Ethnic Study of Atherosclerosis” (MESA) (phs001416.v2.p1) was performed at Broad Institute Genomics Platform (3U54HG003067-13S1). Genome sequencing for “NHLBI TOPMed: Severe Asthma Research Program (SARP)” (phs001446.v2.p1) was performed at New York Genome Center Genomics (3U54HG003067-13S1). Genome sequencing for “NHLBI TOPMed: Recipient Epidemiology and Donor Evaluation Study-III Brazil Sickle Cell Disease Cohort (REDS-BSCDC)” (phs001468.v3.p1) was performed at Baylor College of Medicine Human Genome Sequencing Center (HHSN268201600033I/HHSN268201500015C). Genome sequencing for “NHLBI TOPMed: My Life Our Future (MLOF) Research Repository of Patients with Hemophilia A (Factor VIII Deficiency) or Hemophilia B (Factor IX Deficiency)” (phs001515.v2.p2) was performed at New York Genome Center Genomics (HHSN268201500016C). Genome sequencing for “NHLBI TOPMed: Boston-Brazil Sickle Cell Disease (SCD) Cohort” (phs001599.v1.p1) was performed at Baylor College of Medicine Human Genome Sequencing Center (HHSN268201600033I, HHSN268201500015C, and HHSN268201600033). Genome sequencing for “NHLBI TOPMed: Children’s Health Study (CHS) Integrative Genomics and Environmental Research of Asthma (IGERA)” (phs001603.v2.p1) was performed at Northwest Genomics Center (HHSN268201600032I). Genome sequencing for “NHLBI TOPMed: Children’s Health Study (CHS) Effects of Air Pollution on the Development of Obesity in Children (Meta-AIR)” (phs001604.v2.p1) was performed at Northwest Genomics Center (HHSN268201600032I). Genome sequencing for “NHLBI TOPMed: NHGRI CCDG: The BioMe Biobank at Mount Sinai” (phs001644.v2.p2) was performed at Baylor College of Medicine Human Genome Sequencing Center (HHSN268201600033I) and the McDonnell Genome Institute (HHSN268201600037I). Genome sequencing for “NHLBI TOPMed: Lung Tissue Research Consortium (LTRC)” (phs001662.v2.p1) was performed at Broad Institute Genomics Platform (HHSN268201600034I). Genome sequencing for “NHLBI TOPMed: Childhood Asthma Management Program (CAMP)” (phs0017265.v2.p1) was performed at Northwest Genomics Center (HHSN268201600032I). Core support, including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering, were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support, including phenotype harmonization, data management, sample-identity QC, and general program coordination, were provided by the TOPMed Data Coordinating Center (R01HL-120393 and U01HL-120393; contract HHSN268201800001I). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. The full study-specific acknowledgments are included in Data S1.
Author contributions
T.D.O., V.B., D.P.L., B.G., and R.L. developed the concepts in this study. T.D.O. and V.B. supervised the study. Analyses were performed by V.B., D.P.L., B.G., R.L., D.V.-O., J.N.F., and T.D.O. The manuscript was written by V.B., D.P.L., B.G., R.L., and T.D.O., with contributions and revisions from all authors. The STAR Methods section was written by V.B., D.P.L., B.G., R.L., and T.D.O., with contributions from all authors. The bioinformatic resources were created by B.G., R.L., T.P.L., V.B., and T.D.O.
Declaration of interests
The authors declare no competing interests. D.P.L. is now an employee of AstraZeneca. This is unrelated to the work of this paper.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Reference panel | 1000 Genomes Project High Coverage40 | https://www.internationalgenome.org/ |
| Native American Genomes | Harris et al.6 | https://ega-archive.org/studies/EGAS00001004995 |
| EPIGEN | Kehdy et al.12 | https://ega-archive.org/studies/EGAS00001001245 |
| LARGE-PD | Loesch et al.8 | Data available on request due to privacy/ethical restrictions. |
| Mega-GWAS ALS I | dbGAP | phs000101 |
| Northwestern NUgene Project: Type 2 Diabetes | dbGAP | phs000237.v1.p1 |
| GENEVA Prostate Cancer | dbGAP | phs000306.v4.p1 |
| eMERGE-I Genome Wide Association Studies of Network Phenotypes | dbGAP | phs000360.v3.p1 |
| IPM BioBank GWAS | dbGAP | phs000388.v1.p1 |
| Columbia University Study of Caribbean Hispanics with Familial and Sporadic Late Onset Alzheimer’s disease | dbGAP | phs000496.v1.p1 |
| GWAS of Breast Cancer in the Multiethnic Cohort | dbGAP | phs000517.v3.p1 |
| The Two Sister Study: A Family-Based Study of Genes and Environment in Young-Onset Breast Cancer | dbGAP | phs000678.v1.p1 |
| PAGE: The Charles Bronfman Institute for Personalized Medicine (IPM) BioMe Biobank | dbGAP | phs000864.v1.p1 |
| PAGE: Global Reference Panel | dbGAP | phs000925.v1.p1 |
| International Age-Related Macular Degeneration Genomics Consortium | dbGAP | phs001033.v1.p1 |
| Population Genetics Analysis Program: Immunity to Vaccines/Infections (NIAID/NIH) | dbGAP | phs001039.v1.p1 |
| Hispanic Colorectal Cancer Study | dbGAP | phs001057.v1.p1 |
| Ancestry Admixture among Chileans | dbGAP | phs001193.v1.p1 |
| SIGMA Diabetes in Mexico Study | dbGAP | phs001385.v1.p1 |
| Uncovering the Genetic Architecture of Colorectal Cancer with Focus of Rare and Less Frequent Variants | dbGAP | phs001388.v1.p1 |
| eMERGE Network Phase III | dbGAP | phs001415.v1.p1.c12 |
| Gene-Environment Interactions in COCCaINE Use Disorder: Collaborative Case-Control Initiative in Cocaine Addiction | dbGAP | phs001584.v1.p1 |
| LIMAA | Luo et al.70 | phs002025.v1.p1 |
| NCI_Guatemala | National Cancer Institute | Data available on request due to privacy/ethical restrictions. |
| NHLBI TOPMed: The Genetic Epidemiology of Asthma in Costa Rica | Taliun et al.5 | phs000988 |
| NHLBI TOPMed: San Antonio Family Heart Study (SAFHS) | Taliun et al.5 | phs001215 |
| NHLBI TOPMed: Women’s Health Initiative (WHI) | Taliun et al.5 | phs001237 |
| NHLBI TOPMed - NHGRI CCDG: Hispanic Community Health Study/Study of Latinos (HCHS/SOL) | Taliun et al.5 | phs001395 |
| NHLBI TOPMed: MESA and MESA Family AA-CAC | Taliun et al.5 | phs001416 |
| NHLBI TOPMed: Severe Asthma Research Program (SARP) | Taliun et al.5 | phs001446 |
| NHLBI TOPMed: Recipient Epidemiology and Donor Evaluation Study-III Brazil Sickle Cell Disease Cohort (REDS-BSCDC) | Taliun et al.5 | phs001468 |
| NHLBI TOPMed: My Life Our Future (MLOF) Research Repository of Patients with Hemophilia A (Factor VIII Deficiency) or Hemophilia B (Factor IX Deficiency) | Taliun et al.5 | phs001515 |
| NHLBI TOPMed: Boston-Brazil Sickle Cell Disease (SCD) Cohort | Taliun et al.5 | phs001599 |
| NHLBI TOPMed: Children’s Health Study (CHS) Integrative Genomics and Environmental Research of Asthma (IGERA) | Taliun et al.5 | phs001603 |
| NHLBI TOPMed: Children’s Health Study (CHS) Effects of Air Pollution on the Development of Obesity in Children (Meta-AIR) | Taliun et al.5 | phs001604 |
| NHLBI TOPMed - NHGRI CCDG: The BioMe Biobank at Mount Sinai | Taliun et al.5 | phs001644 |
| NHLBI TOPMed: Lung Tissue Research Consortium (LTRC) | Taliun et al.5 | phs001662 |
| NHLBI TOPMed: Childhood Asthma Management Program (CAMP) | Taliun et al.5 | phs001726 |
| Software and algorithms | ||
| Picard | Broad Institute71 | https://broadinstitute.github.io/picard/; RRID:SCR_006525 |
| PLINK | Purcell et al.72 | https://www.cog-genomics.org/plink/1.9/; RRID:SCR_001757 |
| ADMIXTURE | Alexander et al.41 | https://dalexander.github.io/admixture/ ; RRID:SCR_001263 |
| bcftools | Danecek et al.73 | https://samtools.github.io/bcftools/ ; RRID:SCR_002105 |
| Shapeit ver4 | Delaneau et al.74 | https://odelaneau.github.io/shapeit4/; RRID: SCR_024355 |
| hap-ibd | Zhou et al.75 | https://github.com/browning-lab/hap-ibd |
| Ibdtools | This study | https://doi.org/10.5281/zenodo.13851024 |
| IBDKin | Zhou et al.76 | https://github.com/YingZhou001/IBDkin |
| NAToRA | Leal et al.77 | https://github.com/ldgh/NAToRA_Public |
| SNPRelate | Zheng et al.78 | https://github.com/zhengxwen/SNPRelate |
| PCAir | Conomos et al.79 | https://rdrr.io/bioc/GENESIS/man/pcair.html |
| RFMix ver2 | Maples et al.80 | https://github.com/slowkoni/rfmix |
| GAfIS | This study | https://doi.org/10.5281/zenodo.13850990 |
| python-igraph | Csardi and Nepusz47 | https://python.igraph.org/en/stable/ |
| GCTA | Yang et al.81 | https://yanglab.westlake.edu.cn/software/gcta/ |
| PRSHelpDesk | This study | https://doi.org/10.5281/zenodo.13851588 |
| PRSice-2 | Choi and O’Reilly52 | https://choishingwan.github.io/PRSice/ |
| PRS-CS | Ge et al.53 | https://github.com/getian107/PRScs |
| PRS-CSx | Ruan et al.54 | https://github.com/getian107/PRScsx |
| PCAmatchR | Brown et al.36 | https://github.com/machiela-lab/PCAmatchR |
| gladprep | This study | https://doi.org/10.5281/zenodo.13851635 |
Experimental model and subject details
Data description
For this research, we defined a Latin American population as a group of people with heritage from Spanish-speaking or Portuguese-speaking countries in the Americas. We chose this definition because "heritage" encompasses various aspects, from culture and geography to genetics. Furthermore, our definition does not include the Indigenous populations because this ethnicity label sometimes has a country-specific legal background.
With this delimitation, we gathered datasets for the GLADdb by combining accessible genomic information from Whole-Genome Sequencing (WGS) and microarray genotyping chip sources. We requested and received access to 39 dbGaP cohorts, including the eMERGE,82 PAGE,83 SIGMA,9 and LIMAA70 projects (Table S1) and the WGS projects in TOPMed.5 Especially regarding the TOPMed cohorts incorporated into this study, we obtained approval from both TOPMed’s institutional review boards and the corresponding institutional review boards of each TOPMed cohort study.
Other important sources were the EPIGEN,13 LARGE-PD,8 and Peruvian Genome Project (PGP).6 We have explored over 268K samples in detail to find 70,702 Latin American subjects for this initial set.
Method details
Figure S1 shows our general workflow. For each non-WGS dataset (Table S1), we converted their genome coordinates (liftover) from the original reference (NCBI36/hg18 or GRCh37/hg19) to the genome reference GRCh38/hg38 using Picard.71 After a first liftover run, we used the strand flip option of PLINK72 on the rejected variants and performed a second liftover run. Furthermore, variants were filtered using PLINK for 5% missingness, a p-value less than 1x10−6 on the Hardy Weinberg exact test (HWE), keeping only biallelic autosomal variants with a minimum minor allele frequency (MAF) of 1%. Samples were filtered for 5% missingness and heterozygosity exceeding three times the standard deviation from the mean. Also, a linkage disequilibrium (LD) pruned dataset was created using PLINK’s indep-pairwise algorithm using the parameters 50 10 0.1.
For each dataset for which we acquired genomic information and appropriate consent, we evaluated self-described demographic variables such as an ethnic designation of Hispanic/Latino. We included the entire cohort if the primary study design was focused on Latin American individuals, e.g., SIGMA.9 For the remaining datasets, many without demographic information provided via dbGaP, we identified possible Latin American individuals using genetic clustering analysis.41 We merged each of these remaining datasets (the LD pruned data) with a custom panel of 361 individuals to assess genome-wide ancestry proportions for European, African, East Asian, and Indigenous American ancestries. This custom panel included 100 each for European, African, and East Asian from the 1000 Genomes Project high coverage data40 (Table S3). In addition, we included 61 unrelated, previously estimated as near 100% Indigenous American high-coverage genomes from the PGP.6 Each dataset was combined with this reference panel; then, we ran a supervised ADMIXTURE analysis.41 These results were then evaluated for admixture proportions, and any sample found to have greater than 2% Indigenous American ancestry was extracted and included for additional analyses. These samples were then designated as ADMIXTURE-defined, which will persist in our evaluations of the database as to their utility as matches or exclusions.
After we collected all self-described and ADMIXTURE-defined individuals in each dataset, we imputed the non-LD pruned data against the TOPMed Imputation server.42 The TOPMed imputation panel contained over 90K individuals and was shown to accurately impute Latin Americans.5 After imputation, for each cohort, we selected genotyped and high-quality imputed variants based on the Rsq threshold (Rsq >0.9). Rsq represents the squared correlation between imputed and true genotypes. As true genotypes remain unobserved, estimating Rsq relies on the concept of poorly imputed genotype counts shrinking toward their expected population allele frequencies. After Rsq filtering per cohort, we merged all datasets, including the non-imputed TOPMed WGS data, and removed variants with missing information in more than 0.1% of the final dataset using bcftools73:
bcftools filter -e 'F_MISSING >0.001' ${mergedGLAD} -O b -o $QC1
and normalized and kept biallelic SNPs with the following command line:
bcftools norm -m +any -s $QC1 | bcftools view -m2 -M2 -v snps | bcftools sort -O b -o $GLAD
Our initial freeze of GLADdb consists of 3,248,494 biallelic SNPs (Rsq >0.9) and 63,589 individuals (R0.9 dataset).
To assess the imputation quality variants included after merging, we compared the distribution of two imputation metrics, Rsq (for all genotyped and imputed variants) and Empirical Rsq statistics (for genotyped variants) included on GLAD for each non-WGS dataset (Figures S2–S26). Empirical Rsq (EmpRsq) reflects the correlation between the true genotyped values and the imputed dosages obtained by hiding all known genotypes for the specific SNP. Higher values of this correlation reflect the higher imputation accuracy.
Our initial threshold for Rsq was 0.9, but after merging, most cohorts kept a distribution of variants between 0.95 and 1, retaining very high-quality variants (Figures S2–S6). Notably, all cohorts showed EmpRsq values higher than 0.8, and most of the cohort included variants with EmpRsq higher than 0.96.
Importantly, GLADdb includes 31,523 individuals with non-ambiguous geographical information (Table S2). This means that we have country-level or, in some cases, state or city-level information like Peru, Brazil, and the USA. For the latter three groups, we did not include individuals without state-level information. A particular case is the Rio Grande do Sul state in South Brazil. Two of the three cohorts sampled in this state corresponded to specific cities (Porto Alegre and Pelotas) and were considered independent groups. To support the clustering of individuals of different projects into groups of similar geographical regions (e.g., USA-Wisconsin, Chile, Brazil-São Paulo), we performed an FST analysis. We calculated the FST among individuals sampled by different projects but of the same sample region. No regional cluster showed an Fst value above 0.07 (Table S2). Finally, these 31K individuals were organized into 46 regions (Table S2). We used this information for ROH and IBD analyses. Furthermore, this clustering was supported by our IBD clustering (See below).
To avoid any phase issues during the merging process, we infer the haplotype phase for the complete GLADdb using SHAPEIT ver474 using the TOPMed freeze9 dataset5 (130K individuals) as a reference panel. We ran SHAPEIT with the following parameters:
shapeit4 --input $GLAD --map $map --thread 60 --region chr${chr} --reference $TOPMEDRef --output $Phased_GLAD --log phased_chr${chr}.log --mcmc-iterations 10b,1p,1b,1p,1b,1p,1b,1p,10m.
Identity-by-descent and relatedness analyses
Before running haplotype-based inferences on GLADdb, we assess the quality of IBD (Identity-By-Descent) analyses using imputed data by identifying the overlapping between the IBD inferences on genotyping (original data) and imputed data.
By selecting five cohorts with different ancestry profiles.
-
(1)
EPIGEN: Includes 3 Brazilian cohorts with predominant European and African genome-wide ancestry.
-
(2)
Peruvian Genome Project: Includes 641 individuals with predominant Indigenous American genome-wide.
-
(3)
Uncovering the Genetic Architecture of Colorectal Cancer with Focus of Rare and Less Frequent Variants: This study includes a cohort that includes individuals from Hawaii and other US states.
We performed the following experiment: We ran IBD inferences using hap-ibd75 on genotyped variants included on GLADdb for each cohort. A second run of IBD was performed on imputed variants selected on the following criteria.
-
(1)
A qualified, imputed variant should be physically close (in a 0.2 cM window) to and highly linked (LD r2 ≥0.8) with the corresponding genotyped variant. LD patterns were determined for each cohort.
-
(2)
If multiple imputed variants are present, the one with the highest linkage is selected; if none is found, the genotype variant is excluded for both runs.
Using these criteria, we generated genotyped and imputed datasets with the same SNP density and similar site frequency spectrum. After IBD inferences and removal of IBD in low-SNP density regions (<3 SNPs per cM), we calculated the overlapping of IBD segments inferred from genotyped variants (used as true segments) with those from imputed variants (used as inferred segments) within each pair of haplotypes of different individuals in the form of false positive and false negative ratios.84 We call a false positive the portion of an inferred segment that is not overlapped by true segments from the same sample pair and a false negative the portion of a true segment that is not overlapped by inferred segments.
For IBD segments greater than 4 cM, our analyses showed false positive and negative rates lower than 4% (Figure S7). The highest level of false positives and false negatives is observed for small segments (<4 cM). Interestingly, these rates for the 3–4 cM intervals are observed in EPIGEN - Salvador and the Peruvian Genome Project. Predominant African and Indigenous American ancestries, respectively, characterize these cohorts. These ancestries have a poor representation on imputation panels.
Moreover, we determined the relationship between total IBD between pairs inferred on genotyped and imputed data (Figure S8). We demonstrated that for all cohorts except PGP, the range of the differences between both IBD distributions (i.e., IBD amount Genotyped = IBD amount Imputed) is minimal (between −1 and 1cM) for more than 80% of the pairs (after excluding pairs that do not share IBD with genotyped and imputed data at the same time).
This comparison between genotyped and only imputed data represents the worst-case scenario. Our analyses suggest that imputed data from individuals with predominant Indigenous American ancestry have a poor inference for IBD-based methods. Since our GLADdb includes a combination of imputed and genotyped variants, we expected lower rates of error for IBD inferences. Also, we restricted our downstream analysis to IBD with a length greater than 5cM for lower error rates.
After demonstrating the feasibility of haplotype-based methods on imputed data, we perform IBD inferences for the entire GLADdb together with HapMap genetic maps (GRCh38) as input for inferences of IBD segments using hap-ibd.75 For hap-ibd, we set the parameters “min-seed = 3” and “min-output = 3” to reduce the rate of false positiveness; defaults were used for all the other parameters. Given IBD coverage is dramatically increased by the paucity of SNP markers, we defined low SNP density regions as 1-cM windows with a number of SNPs less than 30 and processed all IBD segments overlapping with these regions by splitting them and removing the parts within the low SNP density regions. The processed IBD segments were then used as input for ancestry-specific downstream analysis. For non-ancestry-specific analyses, we further merged and flattened the processed IBD segments for each sample-pair when two segments were either overlapping or close (gap no longer than 0.6cM and the number of phasing-informative discordant markers no more than 1).85 The flattened and merged IBD segments were kept if the segment length ≥ 5cM. The genome-wide total IBD length of all segments shared by each sample pair was then calculated and organized into an IBD matrix, with each element representing the relatedness between a pair of individuals. For agglomerative clustering, we transformed the matrix into a dissimilarity matrix by the formula X = (max-min)/(X-min+1e−9). The IBD post-processing steps, including encoding, removing low SNP density regions, decoding, sorting, merging, filtering, and matrix-building, were implemented in a C++ toolkit ibdtools (https://github.com/umb-oconnorgroup/ibdtools) to accelerate the computation for large IBD datasets, for instance, hundreds of billions of IBD segments.
We estimated the kinship coefficient for each pair of individuals in GLADdb with IBDkin.76 After kinship coefficient inferences, we pruned for relatedness in GLADdb using NAToRA77 to exclude the minimum number of related individuals while removing the main kinship relationships in the dataset. We used 0.0442 as the kinship coefficient threshold, which is the lower bound for the theoretical kinship coefficient expected for a 3rd-degree relationship.
Continental population structure
PCA and UMAP
To explore population structure, first, we used SNPRelate78 to generate an LD-pruned dataset and a matrix of relatedness using the snpgdsLDpruning and snpgdsIBDKING functions, respectively. Then, principal component analysis was performed using PCAir79 on LD pruned data using the KING matrix, keeping variants with MAF higher than 1% and kin.thresh and div.thresh parameters equal to 2−9/2 and -2−9/2, respectively.
We kept the top 50 components. To help with cluster visualization, we reduced the 50 principal components to 2 dimensions by applying the UMAP algorithm, using the umap-learn package,86 with n_neighbors set to 10 and min_dist set to 0.5.
Runs of homozygosity (ROH)
We inferred the ROH segments for our 46 Latin American groups and 24 reference populations to explore the level of homogenization in each group. For each group, we used PLINK to apply an LD filter ( --indep-pairwise parameters 50 10 0.9) and to perform ROH analysis (--homozyg flag). Two runs of ROH were performed using 1 and 8 Mb as the minimum threshold for ROH segment detection.
Processing and plotting scripts are available at https://github.com/umb-oconnorgroup/GLAD_DemographicAnalysis.
Local ancestry inferences
We ran local ancestry inference using RFMix ver280 on GLADdb. We inferred local ancestry for the phased dataset considering two Expectation Maximization runs and eight generations since admixture. For the ancestry reference panel, we selected 982 individuals, including 250 Europeans, 250 East Asians, 250 Africans, and 232 individuals with predominant Indigenous American ancestry (Table S2). Europeans, Africans, and East Asian reference populations are part of the 1000 Genomes Project high coverage. Individuals with predominant Indigenous American ancestry include Indigenous Americans from the Peruvian Genome Project6,7 and individuals with predominant Indigenous American ancestry (above 99% of Indigenous American ancestry) from Guatemala (Table S2).
Distant genetic relatedness
IBD-community detection
For community detection, we calculated an IBD matrix by summing up all IBD segments with length within a specific range (>5cM, 5–9.3 or >9.3cM) across the genome for each pair of individuals and set all elements with values <12 cM to 0 in this matrix to reduce the density of non-zero elements in the matrix. The resulting symmetrical matrix was used as a weighted-adjacency matrix to build a bidirectional relatedness network. We used the infomap algorithm implemented with the python-igraph47 package to infer the community structure of the relatedness network. We kept individuals within the top 20 communities and with a degree ≥ 30 connections and used the Frutcherman Reingold layout87 for visualization purposes. Community enrichment in a given birth country is defined as the largest proportion of community labels for individuals born in the country. The number of communities enriched in a birth country is determined by counting the communities that have >1% enrichment in this country. Moreover, to explore other clustering strategies, we also performed community detection using the Louvain clustering algorithm via the find_partition function of the louvain-igraph package with the partition type of ModularityVertexPartition.88,89
IBD sharing among Latin American regions
To explore the recent relationship among Latin American regions, we focused on IBD segments greater than 21.4 cM. We calculated the IBD sharing at intra and interregional levels. For intraregional sharing, we summed the total amount of shared IBD and divided it by the number of pairs: N(N-1)/2, where N is the total number of individuals included for that region. For interregional sharing, we summed the total amount of shared IBD among individuals of populations 1 and 2 and divided it by N1xN2, where N1 and N2 are the total numbers of individuals included for populations 1 and 2 involved in the sharing, respectively.
Ancestry-specific IBD
Due to the multi-way admixed origin of Latin American populations, IBD (segments greater than 21.4 cM) and local ancestry analyses provide an opportunity to detect ancestry-specific signatures related to the similarity among individuals in a region (within-region analysis) or recent migration (across-region analysis) along the Americas.
We implemented a Python algorithm called GAfIS (“Getting Ancestry For IBD Segments”) that uses RFMIX outputs to identify local ancestry labels for an IBD segment shared by a pair of individuals under a certain probability threshold. As a probability threshold for local ancestry inferences in GAfIS, we set 90% for a genomic region being of the K ancestry. For this analysis, we included our processed IBD segments to reduce the proportion of false positives. Moreover, if an IBD segment contained several ancestries, we split the segment into segments corresponding to independent ancestries for each pair of individuals.
After ancestry identification of the IBD segments, we filter out ancestry specific-IBD segments based on the following criteria.
-
(1)
The ancestry profile of one of the individuals for the IBD region was unknown because the local ancestry probability was lower than 90%.
-
(2)
Both individuals have different ancestry labels of the IBD segment.
After those filters, we kept individuals with demographic information and calculated an ancestry-specific IBD score (asIBD score) within and across the 46 Latin American groups. Our asIBD score is defined in the following equations.
Within regions
| (Equation 1) |
Across regions:
| (Equation 2) |
Where:
anc K = African, European, or Indigenous American ancestries.
IBD anc K: The total amount of ancestry K IBD shared between a pair of individuals from regions i and j.
N region i: Total number of individuals from region i.
N region j: Total number of individuals from region j.
ɑ anc K region i: Global ancestry proportion for Ancestry K in region i.
ɑ anc K region j: Global ancestry proportion for Ancestry K in region j.
L: Total size of the genome that was included for IBD analysis.
In both equations, in the numerator, for a specific ancestry, we summed the total amount of IBD per ancestry for each pair of individuals from the same region (Equation 1) or between regions i and j (Equation 2). To control for sample size and ancestry proportions, for Equation 1, we divide the total amount of shared IBD by the product of the total number of combinations of individuals and the square of ancestry proportion. For Equation 2, we divide by the product of the sample size for each region and the product of the global ancestry proportion K for each region, respectively. To get a value relative to the total size of the genome, we included the genome size that was analyzed in the IBD inference in both equations. Finally, we removed IBD sharing signals that include less than 5 pairs. Codes and pipeline to estimate the asIBD score are available at: https://github.com/umb-oconnorgroup/GLAD_DemographicAnalysis.
Polygenic risk scores
Description of PRS cohorts
We utilized the following studies participating in GLAD.
-
(1)
Columbia University Study of Caribbean Hispanics and Late-Onset Alzheimer’s disease (phs000496).
-
(2)
Slim Initiative in Genomic Medicine for the Americas (SIGMA): Diabetes in Mexico Study (phs001388) eMERGE Network Phase III: HRC Imputed Array Data (phs001584).
-
(3)
Early Progression to Active Tuberculosis in Peruvians (phs002025).
-
(4)
EPIGEN-Brasil (Bambui, Pelotas, and SCAALA).
These studies all ascertained one or more of the following traits: height, body mass index (BMI), and/or type 2 diabetes (T2D). See Table S5 for a complete description of cohorts.
Ancestry proportions, relationship inference, principal components, and imputation
Within each cohort, PCs were calculated using PC-Air to utilize as covariates. Related individuals were resolved to the 3rd degree using a kinship matrix generated in Identity-by-descent and relatedness analyses section. Genotyped data from each cohort were separately merged with the 1000 Genomes Project (1KGP).40 Global ancestry proportions were estimated using ADMIXTURE,41 a K of 5, and 20 replicates. For PRS estimation, imputed variants were filtered for a minimum imputation Rsq of 0.9 and a MAF of 0.01. Both imputed and genotyped data were down-sampled to Hapmap Phase 3 variants as required by PRS-CS53 and we kept the same variants for all cohorts. Phenotype data was harmonized across cohorts, though all analyses were conducted on a per-cohort basis.
GWAS summary statistics
Genome-wide association statistics were obtained from the GWAS Catalog,90 Biobank Japan31 (BBJ), and UK Biobank29 (UKBB). African-ancestry GWAS summary statistics were combined using a random-effects meta-analysis using the GAP package in R to improve the sample size. See Table S6 for a description of the summary statistics used for this study.
Heritability estimation
Per-cohort additive heritability for each trait was estimated using GCTA,81 adjusting for sex, age, age,2 and PCs 1–10. For each set of GWAS summary statistics, heritability was estimated using LD score regression,91 using the appropriate 1KGP super-population for calculating LD scores.
Polygenic risk score calculation
Pruning/Thresholding PRS: We used PRS calculated with PRSice-252 as the representative pruning and thresholding (P + T) method. For P + T, we trained the r2 parameters (r2 thresholds of 0.2, 0.4, 0.6, and 0.8), window size (+/− 250 kb, 500kb, 750kb, 1000 kb), and p-value thresholds (iterated by PRSice-2) in one cohort (eMERGE) and validated the parameters in the other cohorts.
Bayesian Mixture PRS: We used PRS estimated with PRS-CS53 as the baseline Bayesian mixture method. For PRS-CS, we trained the phi (ϕ) parameter (phi = 1e−06, 1e−04, 1-e02, and 1e+00) in one cohort (eMERGE, as this cohort included information for all tested traits) via a small grid search and validated it in the other cohorts. In addition, we also evaluated the fully Bayesian pseudo-validation method (phi = auto) for obtaining phi.
Multi-ancestry PRS using PRS-CSx: We leveraged PRS-CSx54 to compute a multi-ancestry PRS, which simultaneously fits multiple sets of GWAS summary statistics while modeling population-specific LD, resulting in more accurate posterior effect sizes for any relatively underpowered GWAS. PRS-CSx outputs a PRS corresponding to each GWAS population and an inverse variance meta-analysis of the posterior effect sizes. We trained the best linear combination of each single-population PRS in one cohort using the mixing weights method proposed by Márquez-Luna et al.92,93 (Equations 3 and 4) with validation in other cohorts. Prior to combining, each PRS is scaled (mean 0, standard deviation 1). In addition, we also evaluated weighting PRS by ancestry proportions (Equation 5), weighting by ancestry proportions after collapsing East Asian and Indigenous American ancestries (Equation 6), and regressing on ancestry proportions prior to model fitting. We compared these linear combinations to the PRS generated from the inverse-variance meta-analysis of PRS-CSx posterior effect sizes.
| (Equation 3) |
| (Equation 4) |
| (Equation 5) |
| (Equation 6) |
where α, α1, α2, and α3 represent mixing weights, , , and represent a PRS calculated using African, European, and East Asian ancestry GWAS, respectively, for individual i. , , , and represent the East Asian, African, European, and Indigenous American ancestry proportions for individual i.
For BMI, height, and T2D, GWAS summary statistics from East Asian, European, and African populations are publicly available (see Table S6). In addition, we were able to train the full range of parameters thanks to multiple independent Latin American cohorts containing data for these traits. We first compared pruning and thresholding (P + T), PRS-CS, and PRS-CSx models. We then evaluated PRS-CSx based multi-ancestry models, comparing linear combinations (the best-performing linear combination model for each cohort) and inverse-variance meta-analyses of PRS-CSx posterior effects. These multi-ancestry models were derived from East Asian and European GWAS (referred to as SUM2 and META2) or derived from East Asian, European, and African GWAS (referred to as SUM3 and META3). Finally, we compared these multi-ancestry models against the best single ancestry PRS (EUR2 and EUR3 estimated using PRS-CSx).
PRS model evaluation
All models were evaluated using the 10-fold cross-validation framework outlined by Pain et al..94 In this approach, the primary metric is the Pearson correlation between the predicted and true values with a standard error of , where r is the Pearson correlation and n is the sample size. Correlations were compared using the two-sided William’s test implemented in the psych R package that accounts for the non-independence of the model predictions. R2 was calculated as the square of Pearson’s r; partial R2 was estimated by subtracting the R2 of the base model (only covariates) from R2 of the full model (covariates and PRS). In general, the base model included age, age,2 sex, and PCs 1–10 except for cohorts with a categorical age variable (eMERGE for T2D). In the Pelotas cohort, as a birth-year cohort, age and age2 were not included as all subjects were the same age. We tested the association of mean ancestry proportions of the cohorts with model performance using linear regression, adjusting for the GWAS trait (R2∼ scaled ancestry proportion + trait).
Matching
Both the baseline bipartite matching51 algorithm and the nearest neighbor simulated annealing matching algorithm operate on a principal components space composed of the first 50 components computed using 246,799 LD-pruned SNPs from GLADdb. The external-user-provided query is also embedded into the PCA space with a saved transformation matrix, and pairwise distances are computed using a variance-weighted Minkowski distance metric. Once a suitable matching set has been found, we return summary statistics to the external user, including alternate allele frequency, genotype counts, and haplotype ancestry counts by segment.
The baseline algorithm is outlined in Algorithm 1 and consists of iteratively applying scikit-learn’s95 bipartite matching implementation until enough controls have been found.
Algorithm 1. Greedy Control Match.
Input: - number of matches, - distance metric, - embedding matrix of the query, - embedding matrix of the database, ( is the number of query genotypes, is the number of dataset genotypes, and is the embedding dimension).
Output: satisfying
while do
if then
else
while do
end while
end if
end while
return
Given a desired control cohort size m and hyperparameters α, β, γ, and n, the nearest neighbor simulated annealing matching algorithm, outlined in Algorithm 2, proceeds as follows. The computed pairwise distances between the query and GLADdb PCA embeddings are used to find the α nearest neighbors of each query genome from the potential controls, which we then merge into a candidate set. We sample m controls from the candidate set and do so β times to generate β control cohorts. We use the genomic control λ, calculated between a control cohort and the query, to evaluate the β control cohorts. The λ values are then used to select the optimal starting control cohort, and a function of their standard deviation is used to initialize our simulated annealing temperature.
Algorithm 2. Simulated Annealing Control Match.
Input: - number of matches, - distance metric, - evaluation metric, - embedding matrix of the query, - embedding matrix of the database, - genotype tensor of the query, - genotype tensor of the database, - number of simulated annealing iterations, - number of nearest neighbors to consider, - number of starting configurations to choose from, - number of individuals per iteration to swap, ( is the number of SNPs, is the number of query genotypes, is the number of dataset genotypes, and is the embedding dimension).
Output: satisfying
▹ Define set of candidate matches with nearest neighbors
for do
For do
end for
end for.
▹ Select best starting match set from several random trials
for do
while do
end while.
end for.
▹ Run simulated annealing
for do
for do
end for
if then
if then
end if
end if
end for.
To evaluate the performance of our algorithm compared to the bipartite matching (PCAmatch), we performed an empirical test using several cohorts with a different ancestral background (Table 1). We identified controls using our algorithm and PCAmatch. We created a dummy binary phenotype that defined query individuals as cases and matched individuals as controls. Then, a pseudo-GWAS was performed with the dummy phenotype, and finally, we estimated the genomic inflation parameter and compared which method provided the lowest.
GLADdb
We developed an online portal where investigators both 1) find controls and 2) interact with and visualize GLAD cohorts. In the first use case, investigators can provide summary statistics from their cases and we match and provide summary statistics as controls. As GLADdb samples were ascertained for various phenotypes, options are provided so that samples with known phenotypes are removed from consideration (e.g., in a Parkinson’s Disease (PD) case study any cases with PD are not included as potential controls) as well as the option to remove ADMIXTURE-defined individuals. No individual-level genotype data is communicated in either direction. For the second use case, we provide a visualization portal wherein users view GLAD samples and cohorts in a variety of embedding spaces (PCA, UMAP). The visualizations are built with the Plotly library, enabling in-browser interaction, zooming, and filtering. The control matching page enables filtering by self-identified ethnicity, PHS numbers, and some phenotypic traits. The external user is asked to prepare and anonymize their data using a Dockerfile provided at github.com/umb-oconnorgroup/gladprep.
Figure S20 contains screenshots from the portal. The online portal is hosted on virtual machines, with a separate computer cluster handling the computation required by the matching service.
Published: October 31, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2024.100692.
Contributor Information
Victor Borda, Email: vicbp1@gmail.com.
Timothy D. O’Connor, Email: timothydoconnor@gmail.com.
Supplemental information
Ancestry proportions were inferred using supervised ADMIXTURE. Latin American groups were organized at an administrative division level (i.e., country, state or city when information is available). IA, EUR, EAS and AFR stand for the Average proportion of Indigenous American, European, East Asian, and African ancestries, respectively, related to STAR Methods.
Interregional sharing with less than 5 pairs were removed, related to Figure 4.
References
- 1.Manichaikul A., Palmas W., Rodriguez C.J., Peralta C.A., Divers J., Guo X., Chen W.-M., Wong Q., Williams K., Kerr K.F., et al. Population Structure of Hispanics in the United States: The Multi-Ethnic Study of Atherosclerosis. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Plecher H. Statista; 2019. Latin America - Statistics & Facts.https://www.statista.com/topics/3287/latin-america/ [Google Scholar]
- 3.Noe-Bustamante L., Hugo Lopez M., Manuel Krogstad J. Pew Res. Cent. US Hisp. Popul. Surpassed 60 Million 2019 Growth Has Slowed. 2020. U.S. Hispanic population surpassed 60 million in 2019, but growth has slowed.https://www.pewresearch.org/fact-tank/2020/07/07/u-s-hispanic-population-surpassed-60-million-in-2019-but-growth-has-slowed/ [Google Scholar]
- 4.Mills M.C., Rahal C. The GWAS Diversity Monitor tracks diversity by disease in real time. Nat. Genet. 2020;52:242–243. doi: 10.1038/s41588-020-0580-y. [DOI] [PubMed] [Google Scholar]
- 5.Taliun D., Harris D.N., Kessler M.D., Carlson J., Szpiech Z.A., Torres R., Taliun S.A.G., Corvelo A., Gogarten S.M., Kang H.M., et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris D.N., Song W., Shetty A.C., Levano K.S., Cáceres O., Padilla C., Borda V., Tarazona D., Trujillo O., Sanchez C., et al. Evolutionary genomic dynamics of Peruvians before, during, and after the Inca Empire. Proc. Natl. Acad. Sci. USA. 2018;115:E6526–E6535. doi: 10.1073/pnas.1720798115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borda V., Alvim I., Mendes M., Silva-Carvalho C., Soares-Souza G.B., Leal T.P., Furlan V., Scliar M.O., Zamudio R., Zolini C., et al. The genetic structure and adaptation of Andean highlanders and Amazonians are influenced by the interplay between geography and culture. Proc. Natl. Acad. Sci. USA. 2020;117:32557–32565. doi: 10.1073/pnas.2013773117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loesch D.P., Horimoto A.R.V.R., Heilbron K., Sarihan E.I., Inca-Martinez M., Mason E., Cornejo-Olivas M., Torres L., Mazzetti P., Cosentino C., et al. Characterizing the Genetic Architecture of Parkinson’s Disease in Latinos. Ann. Neurol. 2021;90:353–365. doi: 10.1002/ana.26153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.SIGMA Type 2 Diabetes Consortium. Estrada K., Aukrust I., Bjørkhaug L., Burtt N.P., Mercader J.M., García-Ortiz H., Huerta-Chagoya A., Moreno-Macías H., Walford G., et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA. 2014;311:2305–2314. doi: 10.1001/jama.2014.6511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pino-Yanes M., Gignoux C.R., Galanter J.M., Levin A.M., Campbell C.D., Eng C., Huntsman S., Nishimura K.K., Gourraud P.-A., Mohajeri K., et al. Genome-wide association study and admixture mapping reveal new loci associated with total IgE levels in Latinos. J. Allergy Clin. Immunol. 2015;135:1502–1510. doi: 10.1016/j.jaci.2014.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moreno-Estrada A., Gignoux C.R., Fernández-López J.C., Zakharia F., Sikora M., Contreras A.V., Acuña-Alonzo V., Sandoval K., Eng C., Romero-Hidalgo S., et al. The genetics of Mexico recapitulates Native American substructure and affects biomedical traits. Science. 2014;344:1280–1285. doi: 10.1126/science.1251688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moreno-Estrada A., Gravel S., Zakharia F., McCauley J.L., Byrnes J.K., Gignoux C.R., Ortiz-Tello P.A., Martínez R.J., Hedges D.J., Morris R.W., et al. Reconstructing the Population Genetic History of the Caribbean. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kehdy F.S.G., Gouveia M.H., Machado M., Magalhães W.C.S., Horimoto A.R., Horta B.L., Moreira R.G., Leal T.P., Scliar M.O., Soares-Souza G.B., et al. Origin and dynamics of admixture in Brazilians and its effect on the pattern of deleterious mutations. Proc. Natl. Acad. Sci. USA. 2015;112:8696–8701. doi: 10.1073/pnas.1504447112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adhikari K., Mendoza-Revilla J., Sohail A., Fuentes-Guajardo M., Lampert J., Chacón-Duque J.C., Hurtado M., Villegas V., Granja V., Acuña-Alonzo V., et al. A GWAS in Latin Americans highlights the convergent evolution of lighter skin pigmentation in Eurasia. Nat. Commun. 2019;10:358. doi: 10.1038/s41467-018-08147-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonfante B., Faux P., Navarro N., Mendoza-Revilla J., Dubied M., Montillot C., Wentworth E., Poloni L., Varón-González C., Jones P., et al. A GWAS in Latin Americans identifies novel face shape loci, implicating VPS13B and a Denisovan introgressed region in facial variation. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abc6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chacón-Duque J.-C., Adhikari K., Fuentes-Guajardo M., Mendoza-Revilla J., Acuña-Alonzo V., Barquera R., Quinto-Sánchez M., Gómez-Valdés J., Everardo Martínez P., Villamil-Ramírez H., et al. Latin Americans show wide-spread Converso ancestry and imprint of local Native ancestry on physical appearance. Nat. Commun. 2018;9:5388. doi: 10.1038/s41467-018-07748-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naslavsky M.S., Scliar M.O., Yamamoto G.L., Wang J.Y.T., Zverinova S., Karp T., Nunes K., Ceroni J.R.M., de Carvalho D.L., da Silva Simões C.E., et al. Whole-genome sequencing of 1,171 elderly admixed individuals from Brazil. Nat. Commun. 2022;13:1004. doi: 10.1038/s41467-022-28648-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franceschini N., Carty C.L., Lu Y., Tao R., Sung Y.J., Manichaikul A., Haessler J., Fornage M., Schwander K., Zubair N., et al. Variant Discovery and Fine Mapping of Genetic Loci Associated with Blood Pressure Traits in Hispanics and African Americans. PLoS One. 2016;11 doi: 10.1371/journal.pone.0164132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sofer T., Baier L.J., Browning S.R., Thornton T.A., Talavera G.A., Wassertheil-Smoller S., Daviglus M.L., Hanson R., Kobes S., Cooper R.S., et al. Admixture mapping in the Hispanic Community Health Study/Study of Latinos reveals regions of genetic associations with blood pressure traits. PLoS One. 2017;12 doi: 10.1371/journal.pone.0188400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi Q., Stilp A.M., Sofer T., Moon J.-Y., Hidalgo B., Szpiro A.A., Wang T., Ng M.C.Y., Guo X., et al. MEta-analysis of type 2 DIabetes in African Americans MEDIA Consortium Genetics of Type 2 Diabetes in U.S. Hispanic/Latino Individuals: Results From the Hispanic Community Health Study/Study of Latinos (HCHS/SOL) Diabetes. 2017;66:1419–1425. doi: 10.2337/db16-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graff M., Emery L.S., Justice A.E., Parra E., Below J.E., Palmer N.D., Gao C., Duan Q., Valladares-Salgado A., Cruz M., et al. Genetic architecture of lipid traits in the Hispanic community health study/study of Latinos. Lipids Health Dis. 2017;16:200. doi: 10.1186/s12944-017-0591-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saccone N.L., Emery L.S., Sofer T., Gogarten S.M., Becker D.M., Bottinger E.P., Chen L.-S., Culverhouse R.C., Duan W., Hancock D.B., et al. Genome-Wide Association Study of Heavy Smoking and Daily/Nondaily Smoking in the Hispanic Community Health Study/Study of Latinos (HCHS/SOL) Nicotine Tob. Res. 2018;20:448–457. doi: 10.1093/ntr/ntx107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Justice A.E., Young K., Gogarten S.M., Sofer T., Graff M., Love S.A.M., Wang Y., Klimentidis Y.C., Cruz M., Guo X., et al. Genome-wide association study of body fat distribution traits in Hispanics/Latinos from the HCHS/SOL. Hum. Mol. Genet. 2021;30:2190–2204. doi: 10.1093/hmg/ddab166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerr K.F., Avery C.L., Lin H.J., Raffield L.M., Zhang Q.S., Browning B.L., Browning S.R., Conomos M.P., Gogarten S.M., Laurie C.C., et al. Genome-wide association study of heart rate and its variability in Hispanic/Latino cohorts. Heart Rhythm. 2017;14:1675–1684. doi: 10.1016/j.hrthm.2017.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cade B.E., Chen H., Stilp A.M., Gleason K.J., Sofer T., Ancoli-Israel S., Arens R., Bell G.I., Below J.E., Bjonnes A.C., et al. Genetic Associations with Obstructive Sleep Apnea Traits in Hispanic/Latino Americans. Am. J. Respir. Crit. Care Med. 2016;194:886–897. doi: 10.1164/rccm.201512-2431OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sofer T., Emery L., Jain D., Ellis A.M., Laurie C.C., Allison M.A., Lee J., Kurniansyah N., Kerr K.F., González H.M., et al. Variants Associated with the Ankle Brachial Index Differ by Hispanic/Latino Ethnic Group: a genome-wide association study in the Hispanic Community Health Study/Study of Latinos. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-47928-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernández-Rhodes L., Graff M., Buchanan V.L., Justice A.E., Highland H.M., Guo X., Zhu W., Chen H.-H., Young K.L., Adhikari K., et al. Ancestral diversity improves discovery and fine-mapping of genetic loci for anthropometric traits—The Hispanic/Latino Anthropometry Consortium. Hum. Genet. Genomics Adv. 2022;3 doi: 10.1016/j.xhgg.2022.100099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ziyatdinov A., Torres J., Alegre-Díaz J., Backman J., Mbatchou J., Turner M., Gaynor S.M., Joseph T., Zou Y., Liu D., et al. Genotyping, sequencing and analysis of 140,000 adults from Mexico City. Nature. 2023;622:784–793. doi: 10.1038/s41586-023-06595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sudlow C., Gallacher J., Allen N., Beral V., Burton P., Danesh J., Downey P., Elliott P., Green J., Landray M., et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLoS Med. 2015;12 doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wall J.D., Stawiski E.W., Ratan A., Kim H.L., Kim C., Gupta R., Suryamohan K., Gusareva E.S., Purbojati R.W., Bhangale T., et al. The GenomeAsia 100K Project enables genetic discoveries across Asia. Nature. 2019;576:106–111. doi: 10.1038/s41586-019-1793-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagai A., Hirata M., Kamatani Y., Muto K., Matsuda K., Kiyohara Y., Ninomiya T., Tamakoshi A., Yamagata Z., Mushiroda T., et al. Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol. 2017;27:S2–S8. doi: 10.1016/j.je.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gouveia M.H., Borda V., Leal T.P., Moreira R.G., Bergen A.W., Kehdy F.S.G., Alvim I., Aquino M.M., Araujo G.S., Araujo N.M., et al. Origins, Admixture Dynamics, and Homogenization of the African Gene Pool in the Americas. Mol. Biol. Evol. 2020;37:1647–1656. doi: 10.1093/molbev/msaa033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luisi P., García A., Berros J.M., Motti J.M.B., Demarchi D.A., Alfaro E., Aquilano E., Argüelles C., Avena S., Bailliet G., et al. Fine-scale genomic analyses of admixed individuals reveal unrecognized genetic ancestry components in Argentina. PLoS One. 2020;15 doi: 10.1371/journal.pone.0233808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagar S.D., Conley A.B., Chande A.T., Rishishwar L., Sharma S., Mariño-Ramírez L., Aguinaga-Romero G., González-Andrade F., Jordan I.K. Genetic ancestry and ethnic identity in Ecuador. HGG Adv. 2021;2 doi: 10.1016/j.xhgg.2021.100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Artomov M., Loboda A.A., Artyomov M.N., Daly M.J. Public platform with 39,472 exome control samples enables association studies without genotype sharing. Nat. Genet. 2024;56:327–335. doi: 10.1038/s41588-023-01637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown D.W., Myers T.A., Machiela M.J. PCAmatchR: a flexible R package for optimal case–control matching using weighted principal components. Bioinformatics. 2021;37:1178–1181. doi: 10.1093/bioinformatics/btaa784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Machiela M.J., Grünewald T.G.P., Surdez D., Reynaud S., Mirabeau O., Karlins E., Rubio R.A., Zaidi S., Grossetete-Lalami S., Ballet S., et al. Genome-wide association study identifies multiple new loci associated with Ewing sarcoma susceptibility. Nat. Commun. 2018;9:3184. doi: 10.1038/s41467-018-05537-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin A.R., Gignoux C.R., Walters R.K., Wojcik G.L., Neale B.M., Gravel S., Daly M.J., Bustamante C.D., Kenny E.E. Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. Am. J. Hum. Genet. 2017;100:635–649. doi: 10.1016/j.ajhg.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin A.R., Kanai M., Kamatani Y., Okada Y., Neale B.M., Daly M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019;51:584–591. doi: 10.1038/s41588-019-0379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Byrska-Bishop M., Evani U.S., Zhao X., Basile A.O., Abel H.J., Regier A.A., Corvelo A., Clarke W.E., Musunuri R., Nagulapalli K., et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell. 2022;185:3426–3440.e19. doi: 10.1016/j.cell.2022.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alexander D.H., Novembre J., Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Das S., Forer L., Schönherr S., Sidore C., Locke A.E., Kwong A., Vrieze S.I., Chew E.Y., Levy S., McGue M., et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Browning S.R., Browning B.L., Daviglus M.L., Durazo-Arvizu R.A., Schneiderman N., Kaplan R.C., Laurie C.C. Ancestry-specific recent effective population size in the Americas. PLoS Genet. 2018;14 doi: 10.1371/journal.pgen.1007385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mooney J.A., Huber C.D., Service S., Sul J.H., Marsden C.D., Zhang Z., Sabatti C., Ruiz-Linares A., Bedoya G., Freimer N., et al. Understanding the Hidden Complexity of Latin American Population Isolates. Am. J. Hum. Genet. 2018;103:707–726. doi: 10.1016/j.ajhg.2018.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ongaro L., Scliar M.O., Flores R., Raveane A., Marnetto D., Sarno S., Gnecchi-Ruscone G.A., Alarcón-Riquelme M.E., Patin E., Wangkumhang P., et al. The Genomic Impact of European Colonization of the Americas. Curr. Biol. 2019;29:3974–3986.e4. doi: 10.1016/j.cub.2019.09.076. [DOI] [PubMed] [Google Scholar]
- 46.Rosvall M., Bergstrom C.T. Maps of random walks on complex networks reveal community structure. Proc. Natl. Acad. Sci. USA. 2008;105:1118–1123. doi: 10.1073/pnas.0706851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Csardi G., Nepusz T. The igraph software package for complex network research. InterJournal Complex Systems. 2006;1695 [Google Scholar]
- 48.Baharian S., Barakatt M., Gignoux C.R., Shringarpure S., Errington J., Blot W.J., Bustamante C.D., Kenny E.E., Williams S.M., Aldrich M.C., Gravel S. The Great Migration and African-American Genomic Diversity. PLoS Genet. 2016;12 doi: 10.1371/journal.pgen.1006059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Devlin B., Roeder K. Genomic Control for Association Studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341X.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 50.Dadd T., Weale M.E., Lewis C.M. A critical evaluation of genomic control methods for genetic association studies. Genet. Epidemiol. 2009;33:290–298. doi: 10.1002/gepi.20379. [DOI] [PubMed] [Google Scholar]
- 51.Munkres J. Algorithms for the Assignment and Transportation Problems. J. Soc. Ind. Appl. Math. 1957;5:32–38. doi: 10.1137/0105003. [DOI] [Google Scholar]
- 52.Choi S.W., O’Reilly P.F. PRSice-2: Polygenic Risk Score software for biobank-scale data. GigaScience. 2019;8:giz082. doi: 10.1093/gigascience/giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ge T., Chen C.-Y., Ni Y., Feng Y.-C.A., Smoller J.W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 2019;10:1776. doi: 10.1038/s41467-019-09718-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruan Y., Lin Y.-F., Feng Y.-C.A., Chen C.-Y., Lam M., Guo Z., Martin A.R., Stanley Global Asia Initiatives. He L., Sawa A., et al. Improving polygenic prediction in ancestrally diverse populations. Nat. Genet. 2022;54:573–580. doi: 10.1038/s41588-022-01054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fonseca L., Sena B.F., Crossley N., Lopez-Jaramillo C., Koenen K., Freimer N.B., Bressan R.A., Belangero S.I., Santoro M.L., Gadelha A. Diversity matters: opportunities in the study of the genetics of psychotic disorders in low- and middle-income countries in Latin America. Br. J. Psychiatry. 2021;43:631–637. doi: 10.1590/1516-4446-2020-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Durand J., Massey D.S. New World Orders: Continuities and Changes in Latin American Migration. Ann. Am. Acad. Polit. Soc. Sci. 2010;630:20–52. doi: 10.1177/0002716210368102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fleisher B.M. Some Economic Aspects of Puerto Rican Migration to the United States. Rev. Econ. Stat. 1963;45:245. doi: 10.2307/1923894. [DOI] [Google Scholar]
- 58.Meléndez Vélez E. The Ohio State University Press; 2017. Sponsored Migration: The State and Puerto Rican Postwar Migration to the United States. [Google Scholar]
- 59.Mintz S.W. PUERTO RICAN EMIGRATION: A THREEFOLD COMPARISON. Soc. Econ. Stud. 1955;4:311–325. [Google Scholar]
- 60.Souza B.C. Trabajo y Tristeza - “Work and Sorrow”: the Puerto Ricans of Hawaii 1900 to 1902. Hawaii. Jew Hist. 1984;18:156–173. [Google Scholar]
- 61.Amaral E.F. In: The Encyclopedia of Global Human Migration. Ness I., editor. Wiley; 2013. Brazil: internal migration. [DOI] [Google Scholar]
- 62.Bastian M. Media and Accountability in Latin America Studies in International, Transnational and Global Communications. Springer Fachmedien Wiesbaden; 2019. Brazil, Argentina, Uruguay: Historical and political background; pp. 15–62. [DOI] [Google Scholar]
- 63.Elizaincín A., Behares L.E., Barrios G. 1987. Nos falemo brasilero: dialectos portugueses en Uruguay (Editorial Amesur) [Google Scholar]
- 64.Sirugo G., Williams S.M., Tishkoff S.A. The Missing Diversity in Human Genetic Studies. Cell. 2019;177:26–31. doi: 10.1016/j.cell.2019.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y., Tsuo K., Kanai M., Neale B.M., Martin A.R. Challenges and Opportunities for Developing More Generalizable Polygenic Risk Scores. Annu. Rev. Biomed. Data Sci. 2022;5:293–320. doi: 10.1146/annurev-biodatasci-111721-074830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Márquez-Luna C., Gazal S., Loh P.-R., Kim S.S., Furlotte N., Auton A., 23andMe Research Team, Price A.L. Incorporating functional priors improves polygenic prediction accuracy in UK Biobank and 23andMe data sets. Nat. Commun. 2021;12:6052. doi: 10.1038/s41467-021-25171-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weissbrod O., Kanai M., Shi H., Gazal S., Peyrot W.J., Khera A.V., Okada Y., Biobank Japan Project. Martin A.R., Finucane H.K., Price A.L. Leveraging fine-mapping and multipopulation training data to improve cross-population polygenic risk scores. Nat. Genet. 2022;54:450–458. doi: 10.1038/s41588-022-01036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tian P., Chan T.H., Wang Y.-F., Yang W., Yin G., Zhang Y.D. Multiethnic polygenic risk prediction in diverse populations through transfer learning. Front. Genet. 2022;13 doi: 10.3389/fgene.2022.906965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Castro e Silva M.A., Ferraz T., Couto-Silva C.M., Lemes R.B., Nunes K., Comas D., Hünemeier T. Population Histories and Genomic Diversity of South American Natives. Mol. Biol. Evol. 2022;39 doi: 10.1093/molbev/msab339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luo Y., Suliman S., Asgari S., Amariuta T., Baglaenko Y., Martínez-Bonet M., Ishigaki K., Gutierrez-Arcelus M., Calderon R., Lecca L., et al. Early progression to active tuberculosis is a highly heritable trait driven by 3q23 in Peruvians. Nat. Commun. 2019;10:3765. doi: 10.1038/s41467-019-11664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Picard toolkit Broad Inst. GitHub Repos. 2019 [Google Scholar]
- 72.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Delaneau O., Zagury J.-F., Robinson M.R., Marchini J.L., Dermitzakis E.T. Accurate, scalable and integrative haplotype estimation. Nat. Commun. 2019;10:5436. doi: 10.1038/s41467-019-13225-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou Y., Browning S.R., Browning B.L. A Fast and Simple Method for Detecting Identity-by-Descent Segments in Large-Scale Data. Am. J. Hum. Genet. 2020;106:426–437. doi: 10.1016/j.ajhg.2020.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou Y., Browning S.R., Browning B.L. IBDkin: fast estimation of kinship coefficients from identity by descent segments. Bioinformatics. 2020;36:4519–4520. doi: 10.1093/bioinformatics/btaa569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leal T.P., Furlan V.C., Gouveia M.H., Saraiva Duarte J.M., Fonseca P.A., Tou R., Scliar M.d.O., Araujo G.S.d., Costa L.F., Zolini C., et al. NAToRA, a relatedness-pruning method to minimize the loss of dataset size in genetic and omics analyses. Comput. Struct. Biotechnol. J. 2022;20:1821–1828. doi: 10.1016/j.csbj.2022.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zheng X., Levine D., Shen J., Gogarten S.M., Laurie C., Weir B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Conomos M.P., Miller M.B., Thornton T.A. Robust Inference of Population Structure for Ancestry Prediction and Correction of Stratification in the Presence of Relatedness. Genet. Epidemiol. 2015;39:276–293. doi: 10.1002/gepi.21896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maples B.K., Gravel S., Kenny E.E., Bustamante C.D. RFMix: A Discriminative Modeling Approach for Rapid and Robust Local-Ancestry Inference. Am. J. Hum. Genet. 2013;93:278–288. doi: 10.1016/j.ajhg.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang J., Lee S.H., Goddard M.E., Visscher P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wiesner G.L., Kulchak Rahm A., Appelbaum P., Aufox S., Bland S.T., Blout C.L., Christensen K.D., Chung W.K., Clayton E.W., Green R.C., et al. Returning Results in the Genomic Era: Initial Experiences of the eMERGE Network. J. Personalized Med. 2020;10:30. doi: 10.3390/jpm10020030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hu Y., Graff M., Haessler J., Buyske S., Bien S.A., Tao R., Highland H.M., Nishimura K.K., Zubair N., Lu Y., et al. Minority-centric meta-analyses of blood lipid levels identify novel loci in the Population Architecture using Genomics and Epidemiology (PAGE) study. PLoS Genet. 2020;16 doi: 10.1371/journal.pgen.1008684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guo B., Takala-Harrison S., O’Connor T.D. Benchmarking and Optimization of Methods for the Detection of Identity-By-Descent in High-Recombining Plasmodium falciparum Genomes. bioRxiv. 2024 doi: 10.1101/2024.05.04.592538. Preprint at. [DOI] [Google Scholar]
- 85.Browning B.L., Browning S.R. Improving the Accuracy and Efficiency of Identity-by-Descent Detection in Population Data. Genetics. 2013;194:459–471. doi: 10.1534/genetics.113.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McInnes L., Healy J., Melville J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv. 2018 doi: 10.48550/ARXIV.1802.03426. Preprint at. [DOI] [Google Scholar]
- 87.Fruchterman T.M.J., Reingold E.M. Graph drawing by force-directed placement. Software Pract. Ex. 1991;21:1129–1164. doi: 10.1002/spe.4380211102. [DOI] [Google Scholar]
- 88.Blondel V.D., Guillaume J.-L., Lambiotte R., Lefebvre E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008;2008 doi: 10.1088/1742-5468/2008/10/P10008. [DOI] [Google Scholar]
- 89.Shemirani R., Belbin G.M., Burghardt K., Lerman K., Avery C.L., Kenny E.E., Gignoux C.R., Ambite J.L. Biocomputing 2023 (WORLD SCIENTIFIC) 2022. Selecting Clustering Algorithms for Identity-By-Descent Mapping; pp. 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.MacArthur J., Bowler E., Cerezo M., Gil L., Hall P., Hastings E., Junkins H., McMahon A., Milano A., Morales J., et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog) Nucleic Acids Res. 2017;45:D896–D901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bulik-Sullivan B.K., Loh P.R., Finucane H.K., Ripke S., Yang J., Schizophrenia Working Group of the Psychiatric Genomics Consortium. Patterson N., Daly M.J., Price A.L., Neale B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Márquez-Luna C., Loh P.-R., South Asian Type 2 Diabetes SAT2D Consortium, and SIGMA Type 2 Diabetes Consortium. Price A.L. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol. 2017;41:811–823. doi: 10.1002/gepi.22083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bitarello B.D., Mathieson I. Polygenic Scores for Height in Admixed Populations. G3 (Bethesda) 2020;10:4027–4036. doi: 10.1534/g3.120.401658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pain O., Glanville K.P., Hagenaars S.P., Selzam S., Fürtjes A.E., Gaspar H.A., Coleman J.R.I., Rimfeld K., Breen G., Plomin R., et al. Evaluation of polygenic prediction methodology within a reference-standardized framework. PLoS Genet. 2021;17 doi: 10.1371/journal.pgen.1009021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Müller A., Nothman J., Louppe G., et al. Scikit-learn: Machine Learning in Python. arXiv. 2012 doi: 10.48550/ARXIV.1201.0490. Preprint at. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ancestry proportions were inferred using supervised ADMIXTURE. Latin American groups were organized at an administrative division level (i.e., country, state or city when information is available). IA, EUR, EAS and AFR stand for the Average proportion of Indigenous American, European, East Asian, and African ancestries, respectively, related to STAR Methods.
Interregional sharing with less than 5 pairs were removed, related to Figure 4.
Data Availability Statement
No data were generated for this study. The code utilized for this study is publicly available on GitHub at the following:
-
•
IBD analysis: https://github.com/umb-oconnorgroup/ibdtools. All original code has been deposited at Zenodo and is publicly available at https://doi.org/10.5281/zenodo.13851024 as of the date of publication.
-
•
ROH and asIBD: https://github.com/umb-oconnorgroup/GLAD_DemographicAnalysis. All original code has been deposited at Zenodo and is publicly available at https://doi.org/10.5281/zenodo.13850990 as of the date of publication.
-
•
PRS estimation and evaluation: https://github.com/dloesch/PRSHelpDesk. All original code has been deposited at Zenodo and is publicly available at https://doi.org/10.5281/zenodo.13851588 as of the date of publication.
-
•
GLADdb: https://github.com/umb-oconnorgroup/gladprep and https://github.com/umb-oconnorgroup/gladdb. All original code has been deposited at Zenodo and is publicly available at https://doi.org/10.5281/zenodo.13851635 as of the date of publication.