

Abstract
An effective strategy for enzyme-enhanced electrochemical detection of human epidermal growth factor receptor 2 (HER2) is proposed for breast cancer diagnosis. This strategy utilizes a three-dimensional mesoporous covalent organic framework (COF), immobilized horseradish peroxidase (HRP), and a novel redox mediator, 4-acetamidophenol (APAP). The mesoporous structure, with encapsulation effect, and good biocompatibility of COF, makes the functionalized COF an efficient carrier for HRP immobilization (HRP-Ab-AuNPs@COF). It demonstrates superior catalytic activity, stability, and electrochemical performance compared to free HRP, thus making it an ideal probe for simultaneous target recognition and signal amplification. APAP is screened from four candidate phenolic compounds based on its high formal potential (0.32 V vs. Ag/AgCl), rapid electron transfer activity (kapp = 2.80 × 105 M− 1 s− 1), excellent solubility and stability. These properties prove significantly better than the conventional mediator hydroquinone (HQ), achieving a higher signal-to-background ratio. By integrating decorated multi-walled carbon nanotubes as substrate materials, the electrochemical aptasensor achieves a low HER2 detection limit (0.418 pg mL− 1) with high specificity. This method’s selectivity surpasses that of the HQ-mediated method by 59–73%. Moreover, the aptasensor can effectively distinguish breast cancer patients and healthy individuals, as well as patients at different stages of the disease with high accuracy (AUC = 0.928). This performance exceeds traditional biomarkers CEA and CA15-3. This work paves novel avenues for innovative applications of COF-immobilized enzymes and the novel mediator APAP in electrochemical biosensing, thus holding significant promise for individualized breast cancer diagnosis and treatment.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12951-024-03035-w.
Keywords: Covalent organic framework, Immobilized enzyme, Electrochemical aptasensing, 4-acetamidophenol, Breast cancer
Introduction
Breast cancer poses a significant global health threat to women, representing the leading cause of cancer-related deaths among them [1]. The World Health Organization (WHO) established the Global Breast Cancer Initiative in 2021 to combat this issue, aiming to decrease the global breast cancer mortality rate by 2.5% annually. In accordance, early detection and prompt diagnosis are crucial to achieving this objective [2]. Compared to current invasive pathological biopsy and radioactive imaging examination for breast cancer diagnosis, liquid biopsy, a non-invasive, non-radioactive, and simple approach, represents as a promising alternative [3, 4]. It primarily focuses on detecting biomarkers, particularly human epidermal growth factor receptor 2 (HER2, as one of the few potential biomarkers approved by the U.S. Food and Drug Administration [FDA] for breast cancer diagnosis and holds particular significance in this context) [5, 6]. Serum HER2 level is significantly correlated with the development and metastasis of breast cancer; however, detecting serum HER2 is challenging due to its extremely low concentration [7]. Therefore, developing a highly sensitive and specific HER2 assay is pivotal to meet the demand for clinical diagnosis.
Electrochemical biosensing has attracted significant attention for its simplicity, high sensitivity, and rapid response compared to traditional clinical laboratory methods [8]. Aptamer has similar or even higher specificity and affinity than antibodies and small molecules, however, it is easier to obtain with more stable and less costly properties. Moreover, aptamer binds the target molecules with smaller spatial resistance, which is considered more beneficial to the sensitivity of electrochemical biosensing compared with antibodies. Therefore, it is becoming increasingly popular for electrochemical biosensors to adopt aptamer as a recognition element. Numerous reaction strategies augment the effectiveness of electrochemical aptasensors; among which, enzymatic reaction strategies not only enhance the sensitivity, but the specificity of enzymes also ensures high selectivity [9, 10]. Thus, enzymatic reaction is a leading strategy, and enzyme-catalyzed electrochemical aptasensor is becoming increasingly compelling. Horseradish peroxidase (HRP) is a widely utilized labeled enzyme, known for its rapid catalysis, generating large quantities of electrochemically active products in the presence of H2O2 for quantitative analysis [11]. However, HRP exhibits the drawbacks common to natural enzymes, such as poor stability and stringent storage requirements [12]. Immobilized enzyme technology addresses these limitations by confining the enzyme to a specific region, promoting non-homogeneous catalysis, and thereby improving catalytic activity and stability [13]. Significantly, the immobilization carrier is a critical factor influencing immobilization efficiency and enzyme catalytic performance. Therefore, identifying an efficient and feasible carrier is paramount for successful enzyme immobilization.
Covalent organic frameworks (COFs) have emerged as a fascinating class of porous crystalline materials [14]. Constructed from lightweight organic building blocks connected by covalent bonds, COFs have found extensive applications in recent years in drug delivery, adsorption, separation, catalysis, and sensing [15–17]. Their adjustable structures, large surface areas, high porosity, and ease of modification render them particularly well-suited for enzyme immobilization. COFs effectively retain enzyme activity by offering sufficient space for immobilization while simultaneously preventing aggregation and promoting efficient substrate mass transfer [18]. These remarkable features position COFs as promising candidates for enhancing the performance of enzyme-catalyzed electrochemical aptasensors. However, the exploration of COF-based immobilized enzymes for electrochemical aptasensing remains in its early stages.
Redox mediators are vital to the performance of enzyme-catalyzed electrochemical aptasensors, directly affecting their sensitivity and accuracy. Hydroquinone (HQ) is a commonly utilized mediator for HRP-based enzymatic reactions [19]. It is catalytically oxidized to benzoquinone in the presence of hydrogen peroxide (H2O2). This benzoquinone is then electrochemically reduced, a process which can be effectively monitored to measure the reaction by measuring electron transfer. To achieve even better sensing performance, however, researchers are seeking more efficient redox mediators capable of even faster electron transfer between the enzyme and the electrode [20]. These sought-after mediators should ideally have a higher formal potential than HQ, allowing them to be rapidly oxidized by HRP in a reversible reaction. Besides, ideal candidates would exhibit high solubility and stability in the reaction environment, coupled with exceptional electron-mediating capacity. The discovery and implementation of such a novel redox mediator would represent a substantial advance in the field, paving the pathway for sensors with superior signal-to-background ratios and significantly lower detection limits.
Herein, we report an enzyme-enhanced electrochemical aptasensor based on COF-immobilized HRP and employing 4-acetamidophenol (APAP) as a novel mediator (Scheme 1). Functionalized COF acted as a nanocarrier for HRP immobilization to form the complexes (named HRP-Ab-AuNPs@COF) that displayed superior catalytic and electrochemical properties compared to free HRP. Representing a signaling probe, HRP-Ab-AuNPs@COF integrated target recognition with signal amplification, which were then combined with highly conductive, decorated multi-walled carbon nanotubes (MWCNTs) to construct the aptasensor. In an effort to optimize the system, several potential HRP mediators were screened: catechol (CAT), 4-aminophenol (p-AP), APAP, and 2-acetamidophenol (2AP). APAP demonstrated superior performance across all aspects and was chosen as a novel mediator in place of HQ, showcasing a high electrochemical signal-to-background ratio. When the target HER2 was present, HRP, in conjunction with H2O2, effectively catalyzed the oxidation of APAP. This oxidized form was then electrochemically reduced, generating an amplified electrical signal through the H2O2-APAP-HRP catalytic oxidation cycle, leading to the quantification of HER2. This novel electrochemical platform for HER2 detection had a low detection limit and high specificity. Furthermore, compared to detecting traditional biomarkers using conventional methods, the platform displayed greater accuracy and reliability in distinguishing between benign and malignant breast diseases, as well as breast cancer patients at different clinical stages. This work establishes a new platform for utilizing COF-immobilized enzymes and the novel mediator APAP in electrochemical biosensing, offering a powerful tool for early diagnosis and metastasis prediction of breast cancer.
Scheme. 1.

Schematic illustration of the proposed electrochemical aptasensor for HER2 detection in breast cancer. (A) Design and synthesis of the functionalized COF-immobolized HRP. (B) Schematic of the screening process for novel redox mediator. (C) Detection principle and clinical application of the aptasensor. Created with Figdraw
Experimental section
Reagents and materials
Acetonitrile, acetic acid, HQ, and p-AP, were purchased from Shanghai Macklin Biochemical Co., Ltd. MWCNTs were purchased from Nanjing XFNANO Materials Tech Co., Ltd. Chloroauric acid (HAuCl4), 1,3,5-tris(4-aminophenyl)benzene (TAPB), albumin from bovine serum (BSA), 2,5-dimethoxyterephaldehyde (DMTP), polyethylenimine (PEI), tris(2-carboxyethyl)phosphine hydrochloride (TCEP), APAP, 2AP, 3,3’,5,5’-tetramethylbenzidine (TMB), and 6-Mercapto-1-hexanol (MCH) were obtained from Bide Pharmatech Co., Ltd. Sodium borohydride (NaBH4) and CAT were from Shanghai Aladdin Biochemical Technology Co., Ltd. Anti-HER2 antibody (HER2 Ab) was obtained from Proteintech Group, Inc. Phosphate buffer saline (PBS, 0.1 M, pH 7.4) and ultrapure water were supplied by Nanjing SenBeiJia Biological Technology Co., Ltd. HAc-NaAc buffer solution (0.2 M, pH 4.0) was from Shanghai yuanye Bio-Technology Co., Ltd. H2O2 was purchased from Sigma-Aldrich. HRP was provided by Beijing Solarbio Science & Technology Co., Ltd. Thiol terminated HER2 aptamer (5’-SH-(CH2)6-AAC CGC CCA AAT CCC TAA GAG TCT GCA CTT GTC ATT TTG TAT ATG TAT TTG GTT TTT GGC TCT CAC AGA CAC ACT ACA CAC GCA CA-3’) [21] and tris-ethylenediaminetetraacetic acid (TE) buffer were supplied by Sangon Biotechnology Co., Ltd. Carcinoembryonic antigen (CEA) and carcinoma antigen 15 − 3 (CA15-3) were purchased from Beijing Key-Bio Biotech Co., Ltd. HER2 and PD-L1 was provided by Novoprotein Scientific Inc.
Synthesis of MWCNTs-PEI-AuNPs
The MWCNTs-PEI-AuNPs nanohybrids were synthesized according to the previous study with a slight modification [22]. Typically, 1 mg of MWCNTs and 25 mg of PEI were dispersed in 4 mL of ultrapure water, followed by sonicating for 30 min. Next, 1 mL of 10 mg mL− 1 HAuCl4 was added to the above solution and stirred for 2 h at 70 °C. After centrifugation and washing with ultrapure water, the MWCNTs-PEI-AuNPs nanohybrids were obtained by drying in vacuum at 55 °C overnight.
Synthesis of AuNPs@COF
The COF was synthesized via an uncomplicated solution-phase method. Briefly, 12 mg of DMTP and 12 mg of TAPB were dissolved into 24 mL of acetonitrile and sonicated for 1 min. Then, 0.6 mL of acetic acid was added under vigorous stirring and stirred for 24 h at room temperature. The precipitate was centrifuged and washed with ethanol, and then dried under vacuum at 55 °C for 12 h. The AuNPs@COF composites were prepared by in-situ growth of gold nanoparticles (AuNPs) on COF. Firstly, 10 mg of the as-prepared COF was suspended in 10 mL of ultrapure water, and sonicated for 10 min. Next, 2 mL of 1.5 mg mL− 1 HAuCl4 was added dropwise to the above solution under stirring. After stirring for 4 h at room temperature, 2 mL of 2 mg mL− 1 NaBH4 was added rapidly and stirred for another 2 h at room temperature. Afterwards, the precipitate was collected by centrifugation and washing with ultrapure water for 3 times. The obtained AuNPs@COF was dried overnight at 55 °C under vacuum.
Preparation of HRP-Ab-AuNPs@COF
To prepare HRP-Ab-AuNPs@COF, 10 µg HER2 Ab was added to 1 mL of 1 mg mL− 1 the above-obtained AuNPs@COF and stirred at 4 °C for 4 h. Subsequently, 1 mL of 1 mg mL− 1 HRP was added to the above solution and stirred at 4 °C overnight. Then, 1 mL of 1% BSA was added and further stirred at 4 °C for 2 h. The resultant precipitate was centrifuged and washed with PBS and resuspended in PBS for future use. The detailed experimental method for determining the immobilization efficiency of HRP was presented in the Supporting Information. All comparative experiments with free and immobilized enzymes utilized HRP quantities adjusted according to the measured immobilization efficiency.
Study on kinetics of enzyme-catalyzed reaction
The kinetics of the enzymatic reaction were evaluated in the presence of H2O2 utilizing TMB, a chromogenic substrate for HRP. A typical reaction procedure was conducted as follows: 4 µL of HRP or HRP-Ab-AuNPs@COF (13.125 µg mL− 1) and 1 µL of H2O2 (0.105, 0.21, 0.42, 0.84, 1.05 M) were introduced to 100 µL of TMB (1.68 mM) in HAc-NaAc buffer solution (0.2 M, pH 4.0). The final concentrations of HRP or HRP-Ab-AuNPs@COF, H2O2, and TMB were 0.5 µg mL− 1, 1 to 10 mM, and 1.6 mM, respectively. The absorbance change of the reaction system at 652 nm was monitored by a microplate reader at 30 s intervals for 6 min. The initial reaction velocity (V0) was determined utilizing the Beer-Lambert Law (Eqs. 1 and 2). Then, to quantify the enzymatic reaction kinetics, the maximum reaction velocity (Vmax) and the Michaelis-Menten constant (Km) were calculated employing the Michaelis-Menten equation (Eq. 3) and Lineweaver-Burk plot (Eq. 4) [23].
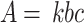 |
1 |
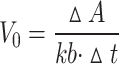 |
2 |
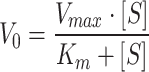 |
3 |
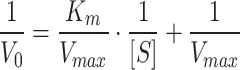 |
4 |
where k is the molar absorbance coefficient of oxTMB (39,000 M− 1 cm− 1), Δt is 60 s, ΔA is the change in absorbance from 0 to 60 s, and [S] is the H2O2 concentration.
Analysis of the catalytic stability of the enzyme
HRP or HRP-Ab-AuNPs@COF was prepared in solution to maintain a constant HRP concentration of 0.5 mg mL− 1 and stored at 4 or 37 °C for 7 or 15 days until use. The specific analysis procedure was carried out as follows: 4 µL of HRP or HRP-Ab-AuNPs@COF (13.125 µg mL− 1) prepared under different storage conditions and 1 µL of H2O2 (1.05 M) were added to 100 µL of TMB (1.68 mM) in HAc-NaAc buffer solution (0.2 M, pH 4.0). The final concentrations of HRP or HRP-Ab-AuNPs@COF, H2O2, and TMB were 0.5 µg mL− 1, 10 mM, and 1.6 mM, respectively. After a 5 min reaction period, the absorbance at 652 nm was measured utilizing a microplate reader.
Construction of the apatasensor and HER2 detection
Prior to the modification, GCE electrodes were polished with aluminum powder and ultrasonically cleaned with ethanol and ultrapure water. After drying under nitrogen flow, 10 µL of 1 mg mL− 1 MWCNTs-PEI-AuNPs was drop cast onto the electrode and dried at 37 °C for 2 h. Afterwards, 10 µL of 1 µM TCEP-activated HER2 aptamer was added dropwise to the electrode and incubated at 4 °C for 30 min. Followed by washing with TE buffer, 10 µL of 1 mM MCH was drop-casted onto the electrode and incubated at room temperature for 2 h for blocking non-specific binding sites. After rinsing with TE buffer, 10 µL of different concentrations of HER2 was added dropwise onto the electrode and maintained at 4 °C for 30 min, followed by washing with PBS. 10 µL of 10 µg mL− 1 HRP-Ab-AuNPs@COF was dropped onto the electrode and incubated at 4 °C for 30 min, followed by rinsing with PBS. Chronocoulometry was carried out at 0.0 V in PBS (0.1 M, pH 7.4) containing 2 mM APAP and 3 mM H2O2 under dark for 10 min.
Clinical samples detection
Serum samples from healthy individuals, patients diagnosed with benign breast disease, and patients diagnosed with breast cancer were collected at the Affiliated Taizhou People’s Hospital of Nanjing Medical University. This study received ethical approval from the Ethics Committee of Taizhou People’s Hospital under protocol KY2022-146-01. Analysis of both cell culture supernatants and clinical serum samples followed the procedure described previously (Construction of the apatasensor), where the aliquot of HER2 solution was replaced by an aliquot of the respective clinical sample for analysis.
Statistical analysis
Data were expressed as mean ± SD. Statistical analysis of the data was obtained utilizing GraphPad Prism 10.1.1 (GraphPad Software, USA) employing either the unpaired t test or one-way ANOVA. A p-value less than 0.05 was considered statistically significant.
Results and discussion
Characterization of MWCNTs-PEI-AuNPs nanohybrids
Transmission electron microscopy (TEM) was utilized to characterize the morphology of the MWCNTs-PEI-AuNPs nanohybrids (Synthesized diagram shown in Fig. S1A). As presented in Fig. S1B and S1C, the diameter of the MWCNTs (around 14 nm) increased after coating with PEI (around 18 nm). High-resolution transmission electron microscopy (HRTEM) images (Fig. S1D) clearly exhibited the interface between the sidewalls of the MWCNTs and the PEI. After HAuCl4 treatment, dark particles were observed attached to the nanotubes (Fig. S1E). HRTEM image in Fig. S1F indicated that AuNPs with a diameter of approximately 8 nm were uniformly dispersed on the sidewalls of the nanotubes, forming MWCNTs-PEI-AuNPs. The UV-vis spectra (Fig. S1G) exhibited an absorption peak near 520 nm after loading with AuNPs, which can be attributed to the absorption of the AuNPs [22]. These results confirmed the successful construction of MWCNTs-PEI-AuNPs nanohybrids. To analyze the electrochemical performance of the MWCNTs-PEI-AuNPs, Cyclic voltammetry (CV) responses of different modified electrodes were examined. Fig. S2 illustrated that, compared to the bare GCE, the peak currents sequentially increased after modifying with MWCNTs and MWCNTs-PEI-AuNPs. This indicated faster and more excellent electron transfer in the nanohybrids, suggesting their excellent electrical conductivity. Therefore, electrodes functionalized with MWCNTs-PEI-AuNPs could significantly accelerate electron transfer and act as anchoring substrates for biomolecules, such as aptamers, thereby enhancing the performance of electrochemical biosensors.
Solubility, stability, formal potential and kinetic parameters of candidate mediators
The electrochemical aptasensor was constructed based on HRP with H2O2 and a redox mediator, with HQ being the conventional mediator for this reaction system. To achieve higher analytical sensitivity and specificity, we sought a novel and efficient mediator for the following HER2 aptasensor construction. An effective redox mediator should exhibit good solubility and stability, high electron transfer activity, and a suitable formal potential relative to Ag/AgCl [24]. To select the optimal mediator, we comparatively evaluated the solubility, light stability, formal potential (vs. Ag/AgCl), and apparent rate constant (kapp) of the phenolic compounds, CAT, p-AP, APAP, and 2AP, in comparison with HQ. The basic properties of these five compounds were summarized in Table 1.
Table 1.
Comparative evaluation of the properties of the conventional and candidate mediators
| Mediators | Solubility (PBS, pH 7.4) |
Light stablility (PBS, pH 7.4) |
Formal Potential (V vs. Ag/AgCl, pH 7.4) |
Diffusion Coefficient (cm2 s-1) |
kapp (M-1 s-1) |
|---|---|---|---|---|---|
| HQ | Soluble with shake | Unstable | 0.14 | 8.98 × 10− 6 | 5.34 × 104 |
| CAT | Soluble with shake | Unstable | 0.25 | 9.50 × 10− 6 | 1.72 × 104 |
| p-AP | Soluble with long-time sonication | Unstable | 0.21 | 1.01 × 10− 5 | 1.02 × 105 |
| APAP | Soluble with shake | Stable | 0.32 | 7.60 × 10− 6 | 2.80 × 105 |
| 2AP | Soluble with sonication | Basically stable | 0.23 | 7.43 × 10− 6 | 2.84 × 105 |
HQ, CAT, and APAP exhibited greater solubility and dissolved by oscillation, whereas 2AP required sonication, and p-AP, with even lower solubility, dissolved only after prolonged sonication (Table 1). Therefore, the solubility of HQ, CAT, APAP, and 2AP can be considered sufficient. Ideally, the mediator should be highly resistant to light, so we dissolved the five compounds in PBS and left them at room temperature without light protection for 1 h. Fig. S3 demonstrated that APAP did not change color, demonstrating better light stability than the other compounds. 2AP remained largely undiscolored, indicating basic stability to light, while the light stability of HQ, CAT, and p-AP was poor.
To optimize electron transfer rates and signal amplification in this reaction system, the mediator’s formal potential should be carefully tuned. Ideally, it should be higher than the conventional mediator HQ’s formal potential, yet lower than that of HRP (0.7 V) [25]. We recorded CV curves (Fig. 1A) of the five mediators in PBS with a GCE electrode to study their electrochemical behaviors, and Fig. S4 demonstrated the electron transfer mechanisms of the five compounds. Table 1 listed the approximate formal potentials derived from the peak potentials in the CV curves. In PBS, the formal potentials were as follows: HQ (0.14 V), CAT (0.25 V), p-AP (0.21 V), APAP (0.32 V), and 2AP (0.23 V). Specifically, APAP possessed the most suitable formal potential for our electrochemical aptasensor. Upon modification of the GCE with MWCNTs-PEI-AuNPs, a significant enhancement in redox peak currents and a narrowing of peak spacing were observed for the mediators (Fig. 1B and S5). This observation highlighted the electrocatalytic activity of MWCNTs-PEI-AuNPs in mediating the redox reaction of the mediator (in the absence of an electrochemical reaction). This catalytic activity facilitated charge transfer and amplified current signals, consistent with the findings presented in Fig. S2. Importantly, even with the electrode modification, APAP maintained the highest formal potential among the five mediators. As depicted in Fig. 1B, the formal potential of APAP remained significantly higher than that of the conventional mediator HQ (approximately 0.18 V higher).
Fig. 1.

Evaluation of the candidate redox mediators. (A) CV responses obtained using GCE electrodes in 0.1 M PBS (pH 7.4) containing 2 mM different mediators. Inset: CV response of 2AP. (B) CV responses obtained using MWCNTs-PEI-AuNPs modified- or bare GCE electrodes in 0.1 M PBS (pH 7.4) containing 2 mM HQ or APAP. (C) Chronocoulometric curves obtained using GCE electrodes at 0.0 V in 0.1 M PBS (pH 7.4) containing 2 mM APAP, PBS containing 2 mM APAP and 3 mM H2O2, PBS containing 2 mM APAP and 40 µg mL-1 HRP, and PBS containing 2 mM APAP, 3 mM H2O2, and 40 µg mL-1 HRP. (D) Charge values at 100 s from chronocoulograms using GCE electrodes in 0.1 M PBS (pH 7.4) containing 2 mM mediators and 3 mM H2O2, and PBS containing 2 mM mediators, 3 mM H2O2, and 40 µg mL-1 HRP
To verify the viability of the candidate compounds as mediators in the electrochemical-enzymatic redox cycle, bare electrodes were subjected to chronocoulometry after incubation in various solutions. As depicted in Fig. 1C, the charge values exhibited a marginal increase when either H2O2 or HRP was added to the APAP solution, implying the direct reaction between APAP and H2O2 or HRP was sluggish. However, the chronocoulogram exhibited a significantly steeper slope when both H2O2 and HRP were introduced to the APAP solution. This indicated the occurrence of a rapid and highly efficient electrochemical-enzymatic redox cycle, facilitating electron transfer. This observation aligned with the phenomenon observed in HQ (Fig. S6A). Likewise, similar results were obtained for CAT, p-AP, and 2AP (Fig. S6B-D), suggesting their feasibility as candidate mediators in the electrochemical-enzymatic redox cycle of HRP.
Then, we evaluated the electron transfer kinetics of the five compounds. This assessment utilized kapp calculated utilizing Eq. 5 for the HRP-mediated redox system [26].
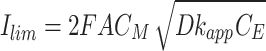 |
5 |
where Ilim, F, A, CM, D, and CE represent the background-corrected limit current, Faradaic constant, electrode surface area, mediator concentration, mediator diffusion coefficient, and HRP concentration, respectively. Values for Ilim were obtained from the chronoamperograms depicted in Fig. S7. Diffusion coefficients for each of the five compounds were determined by measuring limit currents. This measurement employed an ultramicroelectrode through linear scanning voltammetry (Fig. S8) and was calculated according to Eq. 6 [26].
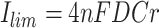 |
6 |
where n is the number of transferred electrons, C is the mediator concentration, and r is the radius of the ultramicroelectrode. Calculated diffusion coefficients, as well as kapp, for the five compounds in PBS, were presented in Table 1. The kapp values for both APAP and 2AP were similar, at approximately 2.8 × 105 M− 1 s− 1. These values were higher than those of the other compounds, especially HQ, revealing their potential to be utilized as highly efficient electron mediators for enzymatic reactions.
To effectively utilize the mediator in the following electrochemical analysis, achieving a low detection limit and high selectivity required minimizing the electrochemical signal in the absence of HRP, while maximizing signal enhancement upon HRP introduction. Therefore, we evaluated the signal-to-background (S/B) ratios of the different mediators by measuring their charges at 100 s. Among the five mediators evaluated, APAP demonstrated the highest S/B ratio (Fig. 1D), suggesting that APAP was a superior mediator for sensitive and specific electrochemical analysis compared to the conventional mediator, HQ. These results collectively pointed to APAP as the optimal redox mediator for this electrochemical-enzymatic reaction system, leading to its deployment in the following electrochemical aptasensor studies.
Design, synthesis and characterization of HRP-Ab-AuNPs@COF composites
AuNPs@COF composites were synthesized through a facile solution-phase method utilizing TAPB and DMTP as precursors. The spherical COF, acting as a growth template for the AuNPs, was prepared through an aldimine condensation reaction. Then, antibodies were immobilized onto the composite via Au-NH2 bonding, while HRP was integrated in the COF pores through host-guest interactions (Scheme 1 A) [27]. TEM was employed to analyze the morphology of the synthesized materials. As depicted in Fig. 2A, the COF exhibited a uniform spherical morphology with a rough surface. Following the in-situ growth of AuNPs on the COF, uniformly dispersed dark particles were observed on its surface, with no evidence of aggregation or free AuNPs (Fig. 2B). This observation implicated that the COF structure facilitated the growth of AuNPs. Energy dispersive X-ray spectroscopy (EDS) elemental mapping confirmed the presence of C, N, O, and Au elements, all essential for the successful synthesis of AuNPs@COF. In addition, the mapping analysis verified the uniform distribution of Au on the COF surface (Fig. 2C-I).
Fig. 2.

TEM images of (A) COF and (B) AuNPs@COF. (C-H) EDS-elemental mapping images of AuNPs@COF. (I) EDS spectrum of AuNPs@COF
The Fourier transform infrared spectra (FT-IR) were presented in Fig. 3A. The absorption peaks at 3430, 3346, and 3208 cm− 1 for TAPB arose from N-H vibrations [18]. The absorption peak at 2759 cm− 1 for DMTP was attributed to C-H vibrations in the aldehyde group, while the peak at 1676 cm− 1 corresponded to C = O bonding. The synthesized COF exhibited characteristic peaks at 1618, 1465, and 1288 cm− 1, assigned to C = N, CH3, and C-O, respectively [28]. These were accompanied by a weakening of the N-H stretching vibration peaks of TAPB and the C = O stretching vibration peak of DMTP, offering strong evidence for successful aldimine condensation. Upon the introduction of AuNPs to the COF, the positions of the characteristic absorption peaks remained largely unchanged, indicating that the AuNPs did not significantly affect the COF structure. However, a slight shift was observed for the peak at 3372 cm− 1 to 3366 cm− 1, potentially due to interactions between [AuCl4]− and NH3+. The XRD pattern of AuNPs@COF, depicted in Fig. 3B, indicated additional diffraction peaks at 38.08°, 44.28°, and 64.47° compared to COF. These peaks corresponded to the (111), (200), and (220) reflective surfaces of Au, respectively [7]. X-ray photoelectron spectroscopy (XPS) (Fig. S9) confirmed the presence of C, N, O, and Au elements in AuNPs@COF, consistent with the EDS results (Fig. 2). In the N 1s spectrum (Fig. 3C), two fitted peaks were observed, corresponding to C = N and residual -NH2, further supporting the successful synthesis of COF. The Au 4f spectrum displayed two peaks at 87.38 and 83.68 eV, representing the Au(0) 4f5/2 and 4f7/2, respectively (Fig. 3D) [29]. Specifically, the binding energy of N 1s in AuNPs@COF was lower compared to COF, suggesting a strong interaction between AuNPs and -NH2. Considering the results holistically, the successful synthesis of AuNPs@COF could be confirmed.
Fig. 3.

Synthesis and characterization of AuNPs@COF. (A) FT-IR spectra of TAPB, DMTP, COF, and AuNPs@COF. (B) XRD patterns of COF and AuNPs@COF. (C) XPS spectra of N 1s in COF and AuNPs@COF. (D) XPS spectra of Au 4f in AuNPs@COF. (E) Nitrogen adsorption-desorption isotherms, (F) pore size distribution, and (G) TGA curves of COF and AuNPs@COF
Nitrogen suction-desorption experiments were employed to analyze the pore characteristics of AuNPs@COF. As depicted in Fig. 3E, COF displayed type IV isotherms at 77 K, indicating mesoporous properties. The Brunauer-Emmett-Teller (BET) specific surface area of COF decreased from 246.1 m2 g− 1 to 35.6 m2 g− 1 after the introduction of AuNPs, while the mesoporous structure was retained. NLDFT analysis (Fig. 3F) indicated that the pore volumes of COF before and after loading with AuNPs were 0.425 cm3 g− 1 and 0.144 cm3 g− 1, respectively. This reduction in BET specific surface area and pore volume suggested that the AuNPs were likely located either inside or on the surface of the COF. In addition, thermogravimetric analysis (TGA) indicated good thermal stability for both COF and AuNPs@COF up to 400 °C (Fig. 3G). The abundance of open mesopores in the synthesized AuNPs@COF not only facilitated the loading of antibodies and enzymes but also enhanced charge transfer and molecular diffusion during the catalytic process.
Finally, to verify the successful synthesis of HRP-Ab-AuNPs@COF, we employed CV to appraise electrodes modified with various components. These electrodes were immersed in PBS solution containing both APAP and H2O2. As shown in Fig. S10, the bare electrode, along with electrodes modified with AuNPs@COF and Ab-AuNPs@COF, exhibited only capacitive currents. In contrast, the electrode modified with HRP-Ab-AuNPs@COF displayed significant cathodic currents. This observation pointed to the presence of enzymatic redox reactions, facilitating electron transfer, and thus confirming the successful synthesis of HRP-Ab-AuNPs@COF.
Catalytic and electrochemical performance of HRP-Ab-AuNPs@COF
Leveraging the catalytic properties of HRP, the interaction with H2O2 leads to the formation of the [HRP + H2O2] complex. This complexation, accordingly, facilitates the oxidation of a range of hydrogen donors, with TMB representing a suitable candidate, significantly exhibiting significant absorption at 652 nm upon oxidation (ox-TMB) [23]. To evaluate the catalytic activity of both free HRP and HRP-Ab-AuNPs@COF in the presence of H2O2, the TMB colorimetric method was employed. It should be noted that immobilized HRP quantities were normalized to equivalent free HRP amounts utilizing the immobilization efficiency (see more discussion in Table S1). Our initial analyses focused on steady-state catalytic kinetics utilizing a mixture of free or immobilized HRP with TMB, while varying the concentrations of H2O2 (Fig. S11). The V0 of HRP, determined at different H2O2 concentrations, was then calculated through the Beer-Lambert Law, facilitating a comprehensive evaluation of catalytic activity. Michaelis-Menten curves were plotted, representing the relationship between H2O2 concentration and corresponding V0 values (Fig. 4A and C). In addition, Lineweaver-Burk plots were constructed utilizing linear double inverse transformations (Fig. 4B and D). The kinetic parameters Vmax and Km were then calculated to offer a quantitative assessment of catalytic capacity. Specifically, our results demonstrated that the Km value (2.69 mM) of HRP-Ab-AuNPs@COF was lower compared with that of free HRP, indicating a significant increase in catalytic efficiency. This was primarily attributed to the mesoporous structure and domain-limiting effect of COF. These properties allowed for coupling between internal diffusion and the catalytic reaction, effectively eliminating diffusion barriers. Accordingly, this increased the concentration of local catalytic sites and mediators, finally enhancing substrate mass transfer efficiency. Meanwhile, the host-guest interaction between HRP and COF scaffold resulted in a partial reduction in the maximum reaction rate, as evidenced by the lower Vmax by the immobilized enzyme. However, for certain immobilized enzymes, a slight increase in the initial reaction rate was observed, which we hypothesized to be a consequence of the substrate channeling effect, i.e., the overall catalytic turnover rate of the immobilized enzyme was higher than that of the randomly distributed free enzyme in solution [30]. Additionally, the synergistic effect introduced by the Au particles attached in the COF pores, potentially exhibiting enzyme-mimicking activity, may have acted as a synergistic booster, thereby enhancing the initial reaction rate to a certain extent [31, 32].
Fig. 4.
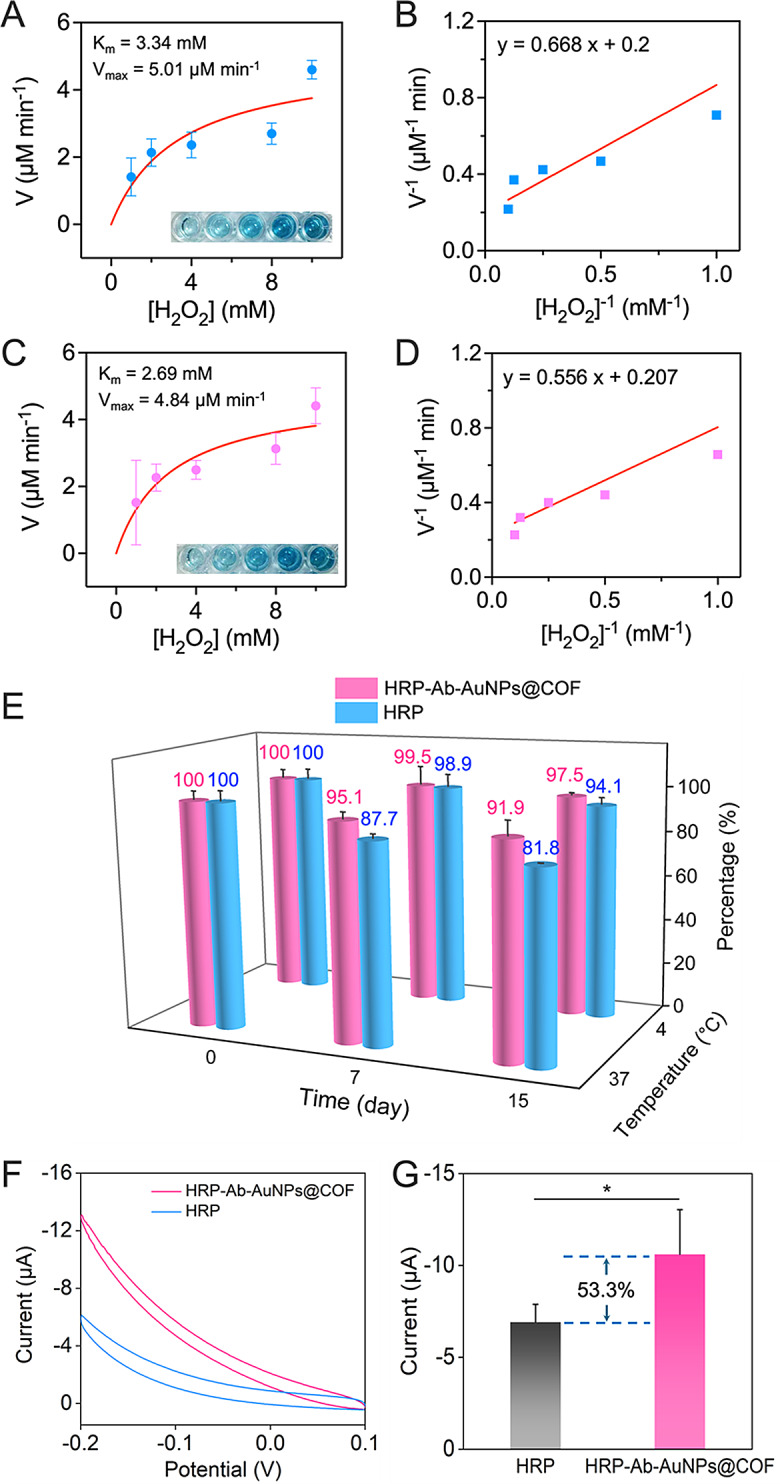
Comparative analysis of the catalytic and electrochemical performance between free HRP and HRP-Ab-AuNPs@COF. Michaelis-Menten fitting curves of (A) free HRP and (C) HRP-Ab-AuNPs@COF. Lineweaver-Burk plots obtained from Michaelis-Menten fitting curves of (B) free HRP and (D) HRP-Ab-AuNPs@COF. (E) Stability test of free HRP and HRP-Ab-AuNPs@COF under different storage conditions. (F) CV responses and (G) histogram for the current values of free HRP and HRP-Ab-AuNPs@COF in 0.1 M PBS (pH 7.4) containing 2 mM APAP and 3 mM H2O2. *p < 0.05 by unpaired t test
The catalytic stability of HRP-Ab-AuNPs@COF was evaluated utilizing a colorimetric TMB assay. As depicted in Fig. 4E, HRP-Ab-AuNPs@COF retained 97.5% of its catalytic activity after 15 days of storage at 4 °C, marginally surpassing free HRP (94.1%). Additionally, HRP-Ab-AuNPs@COF exhibited remarkable stability at 37 °C, retaining 91.9% of its catalytic activity after 15 days. In contrast, free HRP displayed a significant decline in activity under identical conditions, with its activity dropping below 90% after only 7 days (compared to 95.1% for HRP-Ab-AuNPs@COF) and declining to a mere 81.8% after 15 days. This superior catalytic stability of HRP-Ab-AuNPs@COF could be attributed to the protective encapsulation and anchoring interactions offered by the COF structure.
To verify the advantages of HRP-Ab-AuNPs@COF, we evaluated the electrochemical properties of both the novel structure and free HRP. Figure 4F demonstrated the CV curves of both free HRP and HRP-Ab-AuNPs@COF in PBS containing APAP and H2O2. Specifically, the current of HRP-Ab-AuNPs@COF was 53.3% higher than that of free HRP (Fig. 4G). This confirmed that the HRP-Ab-AuNPs@COF structure exhibited significantly stronger catalytic performance and signal amplification. These results suggested that HRP-Ab-AuNPs@COF could be utilized as an efficient catalyst for future studies.
Fabrication and optimization of the aptasensor
An electrochemical aptasensor was developed for sensitive and specific detection of HER2. The sensor’s electrode surface was modified with a layer of MWCNTs-PEI-AuNPs, which enhanced electron transfer and offered a scaffold for aptamer immobilization. Upon introduction of HER2, the aptamer selectively bound to the target, which was then captured by the HER2 Ab conjugated to COF-based composites (Fig. 5A). This complexation then facilitated an enzymatic reaction: HRP immobilized on the COF catalyzed the oxidation of APAP in the presence of H2O2. The oxidized APAP was then reduced electrochemically, creating an electrochemical-enzymatic redox cycle that amplified the signal for detection (Scheme 1 C). Specifically, the binding sites of the aptamer and the antibody against HER2 were evaluated utilizing surface plasmon resonance (SPR) assay, which demonstrated that they two targeted different HER2 binding sites and were suitable for the construction of the aptasensor (see Fig. S12 for more discussion).
Fig. 5.

Establishment and analytical performance of the aptasensor. (A) Schematic description of the HER2 aptasensor assembly. (M-P-A refers to MWCNTs-PEI-AuNPs, H-A-A@C refers to HRP-Ab-AuNPs@COF). (B) CV responses of (a) bare GCE, (b) MWCNTs-PEI-AuNPs/GCE, (c) HER2 aptamer/MWCNTs-PEI-AuNPs/GCE, (d) MCH/HER2 aptamer/MWCNTs-PEI-AuNPs/GCE, (e) HER2/MCH/HER2 aptamer/MWCNTs-PEI-AuNPs/GCE, (f) HRP-Ab-AuNPs@COF/HER2/MCH/HER2 aptamer/MWCNTs-PEI-AuNPs/GCE in 0.1 M PBS (pH 7.4) containing 2 mM K3Fe(CN)6. (C) Chronocoulometric curves and (D) calibration plot of the presented aptasensor with different concentrations of HER2 in 0.1 M PBS (pH 7.4) containing 2 mM APAP and 3 mM H2O2. (E) Stability of the as-fabricated aptasensor for 1 ng mL− 1 HER2 after a storage of 2, 7, and 15 days. (F) Charge values of the as-fabricated aptasensor for HER2 and interfering substances with a concentration of 1 ng mL− 1. ***p < 0.001 and ns, not significant by one-way ANOVA. (G) Comparison of charge values of the APAP-mediated system and the HQ-mediated system for PD-L1, CEA, CA15-3, and HER2
CV was employed to study the stepwise assembly of the aptasensor. As depicted in Fig. 5B, the bare GCE exhibited a well-defined [Fe(CN)6]3− redox peak with a peak-to-peak separation of less than 80 mV (curve a), indicating facile electron transfer kinetics. The incorporation of MWCNTs-PEI-AuNPs, known for their excellent electron transfer properties, led to a significant increase in peak current at the functionalized electrode (curve b). The successive change with the aptamer, MCH, HER2, and HRP-Ab-AuNPs@COF resulted in a progressive decrease in peak currents. This observation was attributed to the hindered penetration of the redox probe and impeded electron transfer caused by the presence of non-electroactive proteins and molecules. These findings confirmed the successful assembly of the aptasensor.
To achieve optimal signal output from the aptasensor, we conducted a series of optimization experiments, utilizing chronocoulometry to monitor the effects of varying construction conditions. Recognizing the direct effect of aptamer concentration on detection effectiveness, we first optimized this parameter. As illustrated in Fig. S13A, the absolute charge value recorded at 100 s exhibited an increasing trend with increasing aptamer concentrations, finally reaching a plateau at 1 µM. This saturation point indicated complete binding of the aptamer to the MWCNTs-PEI-AuNPs substrate, establishing 1 µM as the optimal aptamer concentration for the following experiments. Then, we explored the significant effect of incubation temperature on detection sensitivity. We selected two representative incubation temperatures, 4 °C and 37 °C, for this analysis. As depicted in Fig. S13B, both HER2 and HRP-AuNPs@COF yielded maximum absolute charge values when incubated at 4 °C. Therefore, we designated 4 °C as the optimal incubation temperature for both HER2 and HRP-Ab-AuNPs@COF. Finally, we evaluated the critical role of incubation time in optimizing the detection signal. Our findings demonstrated that an incubation time of 0.5 h consistently yielded the highest absolute charge values for the aptamer, HER2, and HRP-Ab-AuNPs@COF (Fig. S13C). Based on these results, we established 0.5 h as the optimal incubation time for the aptamer, HER2, and HRP-Ab-AuNPs@COF.
Analytical performance of the HER2 aptasensor
Under optimal experimental conditions, the developed aptasensor, utilizing chronocoulometry with APAP as the redox mediator, successfully detected HER2. As expected, the absolute charge exhibited a positive correlation with increasing HER2 concentrations (Fig. 5C). A linear relationship was observed between the absolute charge at 100 s and the logarithm of HER2 concentration over a range of 0.5 pg mL− 1 to 100 ng mL− 1 (Fig. 5D). This relationship was defined by the linear regression equation Q (µC) = − 47.766 lg [HER2 (ng mL− 1)] − 208.13, with a correlation coefficient of 0.993. In addition, the limit of detection (LOD) was determined to be 0.418 pg mL− 1 (S/N = 3). This aptasensor demonstrated superior analytical performance compared to numerous previously reported biosensors designed for HER2 detection (Table S2). To evaluate stability, the constructed aptasensors were stored at 4 °C for 2, 7, and 15 days. Specifically, the electrochemical signal responses remained at 97.638%, 94.025%, and 93.150% of the initial response, respectively (Fig. 5E), indicating satisfactory stability. The coefficient of variation (RSD) of charge, representing the determination of 1 ng mL− 1 of HER2 utilizing five independently fabricated aptasensors, was 3.275% (Fig. S14). This result confirmed the good reproducibility of the aptasensing platform.
To determine the selectivity of the newly developed electrochemical analytical method, 1 ng mL− 1 PD-L1, CEA, CA15-3, and mixtures of these potential interferents with HER2 were individually evaluated alongside HER2 under identical experimental conditions. Figure 5F illustrated that the absolute charges of the interferents at 100 s were significantly lower than that of HER2. However, the absolute charges of the mixture were similar to that of HER2, indicating excellent aptasensor selectivity. This was likely attributable to the efficient enzymatic reaction and the high affinity and specific recognition of HER2 by the aptamer or antibody. For comparison, an aptasensor utilizing HQ as the electron mediator was also constructed (schematic demonstrated in Fig. S15). As depicted in Fig. 5G, the APAP-mediated aptasensor detected only 59–73% of the interfering agent signals compared to the HQ-mediated counterpart. This result clearly demonstrated that APAP exhibited superior selectivity and immunity to interferents compared to HQ, the most commonly utilized mediator. To evaluate the accuracy of this method, recovery experiments were performed utilizing the developed aptasensor for various concentrations of HER2 in healthy human serum. The analytical recoveries ranged from 91.104 to 102.925% with an RSD in 7.531% (Table S3), confirming the aptasensor’s accuracy and reliability. The high sensing performance was attributed to the highly efficient electron mediator APAP and the high catalytic performance of COF-immobilized HRP.
Clinical application of the HER2 aptasensor
To explore the clinical viability of a newly developed enzyme-enhanced electrochemical aptasensor, we utilized it to analyze HER2 levels in the culture supernatants of several cell lines, including the human mammary epithelial line MCF-10 A and the human breast cancer cell lines MDA-MB-231, MCF-7, and ZR-75-1. Our analysis indicated that the breast cancer cells possessed a higher absolute charge compared to normal mammary epithelial cells (Fig. S16). This suggested that breast cancer cells may release more HER2 than their normal counterparts, thus illustrating the practical utility of the aptasensor.
Subsequently, the aptasensor was employed to analyze serum samples from patients with benign and malignant breast diseases. Patient information was available in Tables S4 and S5. As presented in Fig. 6A, we measured the charge of clinical samples from healthy individuals, patients with benign breast disease (BBD), and patients with breast cancer (BC). By analysis, HER2 levels were found to be significantly higher in BC patients compared to both BBD patients and healthy individuals (Fig. 6B). Then, we compared HER2 levels in BC patients at different clinical stages (Fig. 6C; patient information was available in Tables S6 and S7). We determined that samples from metastatic BC patients displayed significantly higher absolute charge than those from non-metastatic BC patients (Fig. 6D). Importantly, the varying intensities of electrochemical signals observed in samples from BC patients were largely consistent with BC progression, suggesting that HER2 may be a valuable biomarker for the clinical diagnosis of BC. Utilizing receiver operating characteristic (ROC) curve analysis, an optimal diagnostic cut-off value of -220.90 µC for HER2 was established (Fig. 6E). The aptasensor demonstrated high diagnostic accuracy for breast cancer (area under the curve, AUC = 0.928), with good accuracy (84.44%) and high positive (PPV) and negative (NPV) predictive values (Table-Fig. 6E, PPV = 90.91%, NPV = 78.26%). Specifically, the AUC value and accuracy of HER2 detection utilizing the developed aptasensor were significantly higher than those achieved with conventional biomarkers, such as CEA (AUC = 0.702, see Fig. S17A for specific information) and CA15-3 (AUC = 0.807, see Fig. S17B for specific information), as measured by clinical immunoassay analysis. These results indicated that, compared to the use of classical biomarkers in standard methods, this aptasensor more effectively differentiated between BC patients and healthy individuals, as well as between BC patients at different clinical stages, significantly improving the accuracy of breast cancer diagnosis. Thus, this high-performance aptasensor had strong potential for clinical application.
Fig. 6.

Application of the aptasensor in clinical serum samples. (A) HER2 levels in clinical serum samples derived from healthy individuals (n = 10), BBD patients (n = 10), and BC patients (n = 10) measured using the aptasensor. (C) HER2 levels in clinical serum samples derived from non-metastatic (n = 8) and metastatic (n = 7) BC patients measured using the aptasensor. (B and D) Box plots reflected the statistical results of the determined HER2 levels shown in (A and C), respectively. *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired t test. (E) ROC curve and diagnostic testing evaluation elucidated the value of this method for breast cancer diagnosis
Conclusion
In summary, we demonstrated a novel COF-immobilized enzyme-enhanced electrochemical aptasensor for the ultrasensitive and ultraspecific detection of HER2 employing APAP as mediator, enabling the accurate diagnosis of breast cancer. Selected from several candidates (CAT, p-AP, APAP, and 2AP), APAP represented the optimal choice, replacing the conventional mediator HQ in HRP-catalyzed aptasensors. APAP exhibited high formal potential (0.32 V) and excellent stability. Importantly, HRP-based enzymatic reaction involving APAP possessed high kapp value (2.80 × 105 M− 1 s− 1), indicating strong electron exchange properties to facilitate superior signal amplification and enhance signal-to-background ratio. The COF’s abundant, open mesoporous structure effectively immobilized HRP, offering spatially separated and catalytically active sites. This efficient immobilization, as demonstrated through enzyme-catalyzed kinetic analysis, accelerated the overall reaction rate by minimizing mass transfer time and maximizing mass transfer efficiency. In addition, the biocompatible COF, with its encapsulation properties, functioned as a protective shell, ensuring a stable environment for the enzyme-catalyzed reaction. Exhibiting remarkable catalytic activity and stability, HRP-Ab-AuNPs@COF acted as an efficient catalyst, underpinning its exceptional electrochemical performance. This performance was further augmented when combined with functionalized MWCNTs composites, known for their excellent electrical conductivity. This combination enabled the ultrasensitive detection of HER2, achieving a detection limit of 0.418 pg mL− 1. Compared to methods relying on HQ as the mediator, this method demonstrated a significant improvement in selectivity, ranging from 59 to 73%. Remarkably, this method accurately differentiated between breast cancer patients and healthy individuals, and further distinguished between breast cancer patients at different stages with high accuracy (AUC = 0.928), outperforming the current classical methods focused on traditional biomarkers such as CEA and CA15-3. This work holds immense promise in guiding individualized diagnosis and facilitating effective monitoring of breast cancer progression.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are very grateful to Le Wang from CNOOC Taizhou Petrochemical Co., Ltd for his kind help in this study.
Author contributions
ZY made conceptualization, performed the laboratory correlative experiments, and drafted the manuscript. CSY and MJ made contributions to methodology and data curation. ZXB and SXC collected the clinical samples and validation. ZCL revised the manuscript.
Funding
The High-level Innovation and Entrepreneurship Talent Introduction Plan of Jiangsu Province (JSSCBS20211599), Taizhou 311 High-level Talent Cultivation Project to ZY, and the NSFC Cultivation Project of Nanjing Medical University (TZKY20220103).
Data availability
All data generated or analyzed during this study are included in the article and supplementary material.
Declarations
Ethics approval and consent to participate
The human studies were reviewed and approved by the Ethics Committee of Taizhou People’s Hospital under protocol KY2022-146-01.
Consent for publication
All authors agree to be published.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yue Zhang, Email: yue_zhang@aa.seu.edu.cn.
Chenglin Zhou, Email: 18762340015@njmu.edu.cn.
References
- 1.Nishimura T, Kakiuchi N, Yoshida K, Sakurai T, Kataoka TR, Kondoh E, Chigusa Y, Kawai M, Sawada M, Inoue T, et al. Evolutionary histories of breast cancer and related clones. Nature. 2023;620:607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, Jemal A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229–63. [DOI] [PubMed] [Google Scholar]
- 3.Loibl S, Poortmans P, Morrow M, Denkert C, Curigliano G. Breast cancer. Lancet. 2021;397:1750–69. [DOI] [PubMed] [Google Scholar]
- 4.Hashimoto K, Ochiya T, Shimomura A. Liquid biopsy using non-coding RNAs and extracellular vesicles for breast cancer management. Breast Cancer. 2024. [DOI] [PubMed]
- 5.Freitas M, Nouws HPA, Keating E, Delerue-Matos C. High-performance electrochemical immunomagnetic assay for breast cancer analysis. Sens Actuat B: Chem. 2020;308:127667–76. [Google Scholar]
- 6.Chen Z, Yang L, Yang Z, Wang Z, He W, Zhang W. Ultrasonic-responsive piezoelectric stimulation enhances sonodynamic therapy for HER2-positive breast cancer. J Nanobiotechnol. 2024;22:369–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bai L, Gu C, Liu J, Gai P, Li F. Photofuel cell-based self-powered biosensor for HER2 detection by integration of plasmonic-metal/conjugated molecule hybrids and electrochemical sandwich structure. Biosens Bioelectron. 2023;220:114850–6. [DOI] [PubMed] [Google Scholar]
- 8.Wu Y, Arroyo-Curras N. Nucleic acid-based Electrochemical Sensors Facilitate the study of DNA binding by platinum (II)-based Antineoplastics. Angew Chem Int Ed Engl. 2024;63:e202312402–202312410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang S, Liu Y, Zhu A, Tian Y. Vivo Electrochemical biosensors: recent advances in Molecular Design, Electrode materials, and Electrochemical devices. Anal Chem. 2023;95:388–406. [DOI] [PubMed] [Google Scholar]
- 10.Kadam US, Trinh KH, Kumar V, Lee KW, Cho Y, Can MT, Lee H, Kim Y, Kim S, Kang J, et al. Identification and structural analysis of novel malathion-specific DNA aptameric sensors designed for food testing. Biomaterials. 2022;287:121617–30. [DOI] [PubMed] [Google Scholar]
- 11.Chu GB, Li WY, Han XX, Sun HH, Han Y, Zhi GY, Zhang DH. Co-immobilization of GOD & HRP on Y-Shaped DNA Scaffold and the regulation of Inter-enzyme Distance. Small. 2023;19:e2301413–2301422. [DOI] [PubMed] [Google Scholar]
- 12.Alvarado-Ramirez L, Machorro-Garcia G, Lopez-Legarrea A, Trejo-Ayala D, Rostro-Alanis MJ, Sanchez-Sanchez M, Blanco RM, Rodriguez-Rodriguez J, Parra-Saldivar R. Metal-organic frameworks for enzyme immobilization and nanozymes: a laccase-focused review. Biotechnol Adv. 2024;70:108299–312. [DOI] [PubMed] [Google Scholar]
- 13.Jabeen R, Ali N, Tajwar MA, Liu Y, Luo D, Li D, Qi L. Encapsulation of an enzyme-immobilized smart polymer membrane in a metal-organic framework for enhancement of catalytic performance. J Mater Chem B. 2024;12:3996–4003. [DOI] [PubMed] [Google Scholar]
- 14.Zhang W, Chen L, Dai S, Zhao C, Ma C, Wei L, Zhu M, Chong SY, Yang H, Liu L, et al. Reconstructed covalent organic frameworks. Nature. 2022;604:72–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akhzari S, Raissi H, Ghahari A. Architectural design of 2D covalent organic frameworks (COFs) for pharmaceutical pollutant removal. npj Clean Water. 2024;7:31–45. [Google Scholar]
- 16.Zhou T, Deng Y, Qu X, Wang L, Xie H, Xu Y, Sun L, Yang J, Li G. Preparation of Well-Constructed and Metal-Modified Covalent Organic Framework nanoparticles for Biosensing Design with Cascade Catalytic Capability. Anal Chem. 2023;95:18814–20. [DOI] [PubMed] [Google Scholar]
- 17.Shi Y, Xu R, Wang S, Zheng J, Zhu F, Hu Q, Huang J, Ouyang G. Fluorinated-Squaramide Covalent Organic frameworks for high-performance and interference-free extraction of Synthetic cannabinoids. Adv Sci. 2023;10:e2302925–2302933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiong Z, Sun B, Zou H, Wang R, Fang Q, Zhang Z, Qiu S. Amorphous-to-Crystalline Transformation: General Synthesis of Hollow Structured Covalent Organic frameworks with High Crystallinity. J Am Chem Soc. 2022;144:6583–93. [DOI] [PubMed] [Google Scholar]
- 19.Quinchia J, Blazquez-Garcia M, Torrente-Rodriguez RM, Ruiz-Valdepenas Montiel V, Serafin V, Rejas-Gonzalez R, Montero-Calle A, Orozco J, Pingarron JM, Barderas R, Campuzano S. Disposable electrochemical immunoplatform to shed light on the role of the multifunctional glycoprotein TIM-1 in cancer cells invasion. Talanta. 2024;267:125155–64. [DOI] [PubMed] [Google Scholar]
- 20.Nandhakumar P, Lee W, Nam S, Bhatia A, Seo J, Kim G, Lee NS, Yoon YH, Joo JM, Yang H. Di(Thioether Sulfonate)-Substituted quinolinedione as a rapidly dissoluble and stable Electron mediator and its application in sensitive biosensors. Adv Healthc Mater. 2022;11:e2101819–2101827. [DOI] [PubMed] [Google Scholar]
- 21.Arya SK, Zhurauski P, Jolly P, Batistuti MR, Mulato M, Estrela P. Capacitive aptasensor based on interdigitated electrode for breast cancer detection in undiluted human serum. Biosens Bioelectron. 2018;102:106–12. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Chen S, Ma J, Zhou X, Sun X, Jing H, Lin M, Zhou C. Enzyme-catalyzed electrochemical aptasensor for ultrasensitive detection of soluble PD-L1 in breast cancer based on decorated covalent organic frameworks and carbon nanotubes. Anal Chim Acta. 2023;1282:341927–35. [DOI] [PubMed] [Google Scholar]
- 23.Cheng Y, Xia YD, Sun YQ, Wang Y, Yin XB. Three-in-one Nanozyme Composite for Augmented Cascade Catalytic Tumor Therapy. Adv Mater. 2024;36:e2308033–2308046. [DOI] [PubMed] [Google Scholar]
- 24.Morshed J, Nakagawa R, Hossain MM, Nishina Y, Tsujimura S. Disposable electrochemical glucose sensor based on water-soluble quinone-based mediators with flavin adenine dinucleotide-dependent glucose dehydrogenase. Biosens Bioelectron. 2021;189:113357–62. [DOI] [PubMed] [Google Scholar]
- 25.Hayashi Y, Yamazaki I. The oxidation-reduction potentials of compound I/compound II and compound II/ferric couples of horseradish peroxidases A2 and C. J Biol Chem. 1979;254:9101–6. [PubMed] [Google Scholar]
- 26.Ogino Y, Takagi K, Kano K, Ikeda T. Reactions between diaphorase and quinone compounds in bioelectrocatalytic redox reactions of NADH and NAD + 1. J Electroanal Chem. 1995;396:517–24. [Google Scholar]
- 27.Chen Y, Lu W, Schroder M, Yang S. Analysis and refinement of host-guest interactions in Metal-Organic frameworks. Acc Chem Res. 2023;56:2569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang L, Jiang Y, Liu F, Li S, Wu J, Zhao S, Ye F. Three-in-one covalent organic framework nanozyme: Self-reporting, self-correcting and light-responsive for fluorescence sensing 3-nitrotyrosine. Biosens Bioelectron. 2023;237:115542–9. [DOI] [PubMed] [Google Scholar]
- 29.Feng S, Yan M, Xue Y, Huang J, Yang X. Electrochemical Immunosensor for Cardiac Troponin I Detection based on covalent Organic Framework and enzyme-catalyzed Signal amplification. Anal Chem. 2021;93:13572–9. [DOI] [PubMed] [Google Scholar]
- 30.Li P, Chen Q, Wang TC, Vermeulen NA, Mehdi BL, Dohnalkova A, Browning ND, Shen D, Anderson R, Gómez-Gualdrón DA, et al. Hierarchically Engineered Mesoporous Metal-Organic frameworks toward cell-free immobilized enzyme systems. Chem. 2018;4:1022–34. [Google Scholar]
- 31.Bretschneider JC, Reismann M, von Plessen G, Simon U. Photothermal Control of the activity of HRP-Functionalized gold nanoparticles. Small. 2009;5:2549–53. [DOI] [PubMed] [Google Scholar]
- 32.Samui A, Happy, Sahu SK. Integration of α-amylase into covalent organic framework for highly efficient biocatalyst. Micropor Mesopor Mat. 2020;291:109700–8. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in the article and supplementary material.