

Abstract
Objectives:
The objective of this study was to determine the effectiveness of WGS in identifying resistance genotypes of MDR Escherichia coli and whether these correlate with observed phenotypes.
Methods:
Seventy-six E. coli strains were isolated from farm cattle and measured for phenotypic resistance to 15 antimicrobials with the Sensititre® system. Isolates with resistance to at least four antimicrobials in three classes were selected for WGS using an Illumina MiSeq. Genotypic analysis was conducted with in-house Perl scripts using BLAST analysis to identify known genes and mutations associated with clinical resistance.
Results:
Over 30 resistance genes and a number of resistance mutations were identified among the E. coli isolates. Resistance genotypes correlated with 97.8% specificity and 99.6% sensitivity to the identified phenotypes. The majority of discordant results were attributable to the aminoglycoside streptomycin, whereas there was a perfect genotype–phenotype correlation for most antibiotic classes such as tetracyclines, quinolones and phenicols. WGS also revealed information about rare resistance mechanisms, such as structural mutations in chromosomal copies of ampC conferring third-generation cephalosporin resistance.
Conclusions:
WGS can provide comprehensive resistance genotypes and is capable of accurately predicting resistance phenotypes, making it a valuable tool for surveillance. Moreover, the data presented here showing the ability to accurately predict resistance suggest that WGS may be used as a screening tool in selecting anti-infective therapy, especially as costs drop and methods improve.
Introduction
Technological advancements and decreased sequencing costs may soon allow WGS to replace a number of traditional microbiology laboratory methods.1,2 While the operational processes for WGS are relatively undemanding, the management and analysis of these large datasets require specialized expertise and software tools. As a result, most clinical and diagnostic laboratories still rely on phenotypic measures to identify bacterial antibiotic resistance.3 Although in general phenotypic testing is reproducible, interlaboratory variability can be problematic.4,5 Thus genotypic methods, which rely on the identification of specific genes and mutations, may result in more clear-cut and consistent measures of resistance and thus play a useful role in resistance surveillance practices.
Although Escherichia coli is part of normal intestinal flora, several strains can cause a variety of enteric and extraintestinal infections, some of which can be life-threatening and require antimicrobial therapy. In addition, the rising antibiotic resistance of E. coli and other enteric pathogens is a critical public health issue worldwide.6 In E. coli, this problem is largely mediated by the acquisition of exogenous genes through transmissible plasmids, integrons and transposons.7,8 To better understand the origins, sources and spread of antimicrobial-resistant E. coli, it is important to catalogue and compare resistance genes in isolates from different sources. Further, correlating genotype and phenotype is a necessary aspect of uncovering novel resistance mechanisms and understanding the relative contribution of known resistance genes and their allelic variants. Therefore, we sought to correlate the phenotypic resistance patterns of E. coli with the genetic determinants that contribute to resistance.
Previous genotypic studies have used focused approaches with PCR tests or microarrays for detection of specific genes.9,10 These techniques can be useful to discover a number of features of isolates, including resistance and virulence determinants.11 Nevertheless, these methods only detect particular genes and are unable to uncover new or rare resistance mechanisms. Just as importantly, testing for specific genes for large numbers of isolates is more costly and laborious, as well as less informative, than what can be gleaned from WGS. Recently, several groups have used WGS to correlate resistance genotypes with phenotypes in various bacteria,12 including E. coli.4,13 These studies demonstrated the high sensitivity and specificity of the approach, but included large numbers of low-resistance isolates. To better evaluate the correlation of genotype and phenotype, we focused on MDR isolates and examined for the presence of hundreds of resistance genes and resistance-associated mutations.
Materials and methods
Bacterial strains, culture and MIC testing
MDR E. coli strains (n = 76) were selected from 2668 E. coli isolated from farm cattle in 2011. Samples were isolated as part of a pilot farm project in which caecal sample E. coli isolates were obtained from cattle sent for slaughter at facilities throughout the USA inspected by the US Department of Agriculture. Susceptibility testing was performed by broth microdilution using a Sensititre system (Trek Diagnostic Systems, Cleveland, OH, USA) according to standardized protocols, in which bacteria were incubated for 18 h at 35°C in antibiotic-containing plates (CMV2AGNF).14 CLSI interpretive criteria15 were used and the resistance breakpoints for each antibacterial agent were as follows: gentamicin, ≥16 mg/L; kanamycin, ≥64 mg/L; streptomycin, ≥64 mg/L; amoxicillin/clavulanic acid, ≥32/16 mg/L; ceftriaxone, ≥4 mg/L; cefoxitin, ≥32 mg/L; ceftiofur, ≥8 mg/L; trimethoprim/sulfamethoxazole, ≥4/76 mg/L; sulfisoxazole, ≥512 mg/L; azithromycin, ≥32 mg/L; ampicillin, ≥32 mg/L; chloramphenicol, ≥32 mg/L; ciprofloxacin, ≥4 mg/L; nalidixic acid, ≥32 mg/L; and tetracycline, ≥16 mg/L. Note that there are no CLSI interpretive criteria for streptomycin, azithromycin or the veterinary drug ceftiofur for E. coli, so interpretive criteria defined by the National Antimicrobial Resistance Monitoring System were used.14 Isolate-level susceptibility data are shown in Table S1 (available as Supplementary data at JAC Online).
Genome sequencing and analysis
Genomic DNA was extracted with a DNeasy blood and tissue kit (Qiagen, Valencia, CA, USA) per the manufacturer’s instructions. DNA concentrations were measured using a Qubit fluorometer (Life Technologies, MD, USA) to determine DNA input from each isolate. WGS was performed using the MiSeq platform using v3 reagent kits with paired-end 2×300 bp reads (Illumina, San Diego, CA, USA). Libraries were prepared by following the Illumina Nextera XT sample preparation guide. Sequences for individual strains were demultiplexed by MiSeq Reporter version 2.5.1. Reads were trimmed by removing ambiguous nucleotides and those with Phred scores of <20. Assembly without scaffolding was performed de novo for each isolate with CLC Genomics Workbench version 7.5 (CLC bio, Aarhus, Denmark) using de Bruijn-based assembly with automatic word-size determination, discarding contigs of <200 bp. Contigs with low coverage (<10% of the average genome coverage) were also removed from final genome sequences. Genomes were annotated using the National Center for Biotechnology Information’s Prokaryotic Genome Automated Pipeline version 2.9.16 Among the 76 samples, there was a median of 154 contigs (range: 79–394) and 62-fold coverage (range: 34–114) per genome. Isolate-level sequencing data results are listed in Table S2. Whole-genome sequences and antibiogram data of the 76 E. coli isolates were deposited into GenBank under BioProject accession number PRJNA266657. Accession numbers for individual isolates are listed in Table S2.
Resistance genotype identification
Resistance genes were identified using Perl scripts to perform local BlastX with an in-house resistance gene database containing 2546 resistance genes and gene variants across all major antibiotic classes (Table S3; database version 1 October 2014). Hits were identified by having ≥85% amino acid identity and ≥50% sequence length to known resistance proteins. Hits of <100% identity and/or sequence length were analysed by additional manual BLAST analysis to identify the appropriate resistance genes.
For analysis of chromosomal structural gene mutations, Perl scripts were used to extract gyrA, gyrB, parC, parE and ampC genes, which were analysed for quinolone resistance-determining regions (QRDRs) or promoter mutations, as appropriate, with alignment by ClustalW in Mega version 6.06.17
Genotype was determined to match phenotype when a strain had phenotypic resistance in addition to known resistance genes or mutations or had phenotypic susceptibility in the absence of resistance genes or mutations. Intermediate phenotypes were counted as susceptible in this analysis. When mismatches occurred, antimicrobial susceptibility testing was repeated. If discrepancies still remained, then sequencing was reperformed using the same plate used for the repeated susceptibility testing. Any remaining discrepancies are shown at the isolate level in Table S4. CIs for genotype–phenotype correlations were calculated using OpenEpi version 3.03 with Fleiss quadratic continuity correction.18
Results
Antimicrobial susceptibility profiles
From cattle faecal samples, 76 E. coli isolates were selected that were resistant to at least four antimicrobials and three classes of antibiotics, determined by testing isolates for susceptibility to 15 antimicrobial compounds in nine classes (Figure 1). The most common resistances among these isolates were to sulfisoxazole (89.5%), tetracycline (88.2%), ampicillin (72.4%), chloramphenicol (72.4%) and streptomycin (68.4%). In contrast, there was less resistance to azithromycin (1.3%), gentamicin (3.9%) and ciprofloxacin (7.9%). Isolate-level resistance prevalence was in between these levels for the remaining seven antimicrobials tested.
Figure 1.
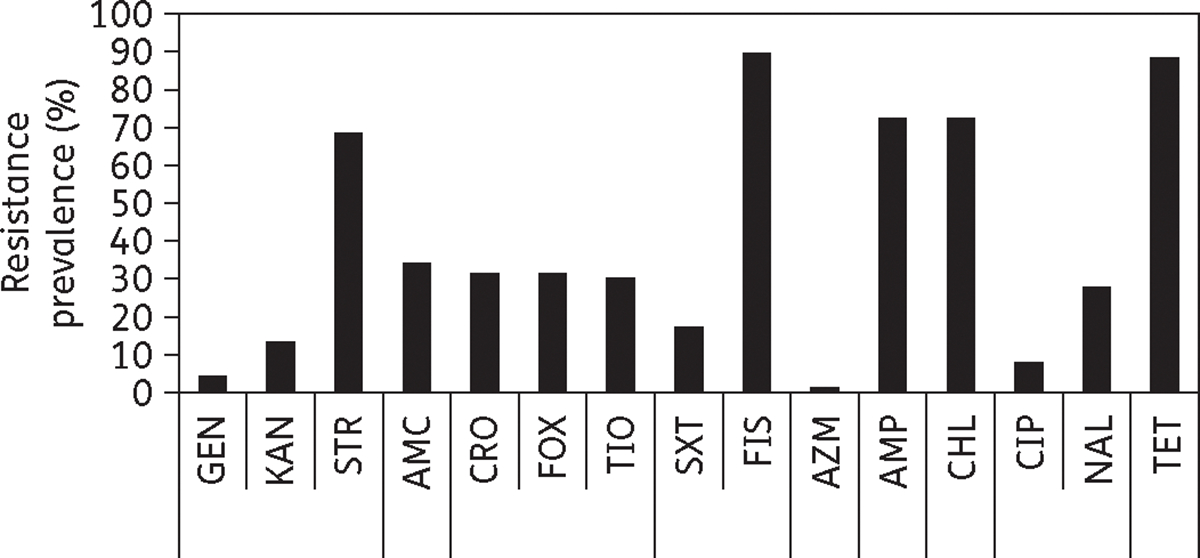
Isolate-level prevalence of resistance among E. coli isolates. The percentage of resistant isolates is depicted for each antibiotic tested. Resistance is grouped by classes, separated by lines, in the order of: aminoglycosides, β-lactam/β-lactam inhibitors, cephems, folate synthesis inhibitors, macrolides, penicillins, phenicols, quinolones and tetracyclines. GEN, gentamicin; KAN, kanamycin; STR, streptomycin; AMC, amoxicillin/clavulanic acid; CRO, ceftriaxone; FOX, cefoxitin; TIO, ceftiofur; SXT, trimethoprim/sulfamethoxazole; FIS, sulfisoxazole; AZM, azithromycin; AMP, ampicillin; CHL, chloramphenicol; CIP, ciprofloxacin; NAL, nalidixic acid; TET, tetracycline.
Among these MDR isolates, 55.3% had resistance to at least five classes of antibiotics and 17.1% had resistance to at least seven of the nine classes tested. WGS-based genotypic analysis was performed to correlate resistance genotypes and phenotypes. For this analysis, Perl scripts were used to perform local BlastX to query genome sequences for genes similar to those in our in-house resistance gene database. A complete list of each strain and its resistance phenotype and genotype is shown in Table S4, with MIC values from susceptibility testing in Table S1.
Aminoglycosides
Streptomycin resistance genes were present in 84.2% of isolates. The most frequently identified resistance genes were strA [aph(3′)-Ib] and strB [aph(6′)-Id], which were always present together in the same strains (75% of all strains). Also common were aadA genes, present in 21.1% of all strains (Table 1). Although most strains had strA/strB and/or aadA genes, only 81.3% containing at least one resistance gene had phenotypic resistance to streptomycin. Resistance was more common among strains with strA/strB genes as 51/57 (89.5%) were resistant. In contrast, only 10/16 (62.5%) strains with at least one aadA gene were resistant, including just 1/7 (14.3%) that did not have strA/strB genes as well. All of the strA/strB and aadA genes that did not confer phenotypic resistance had intact coding regions and promoters, including some with 100% identity to genes present in isolates with observed resistance.
Table 1.
List of resistance genes and their prevalence
| Resistance gene | Antibiotic class | Resistance phenotype | Prevalence (%) |
|---|---|---|---|
|
| |||
| strA [aph(3’)-Ib] | aminoglycosides | STR | 75.0 |
| strB [aph(6’)-Id] | aminoglycosides | STR | 75.0 |
| aadAl | aminoglycosides | STR | 9.2 |
| aadA2 | aminoglycosides | STR | 9.2 |
| aadA5 | aminoglycosides | STR | 2.6 |
| aadA7 | aminoglycosides | STR | 1.3 |
| aadA24 | aminoglycosides | STR | 1.3 |
| aph(3’)-Ia | aminoglycosides | KAN | 14.5 |
| aac(3’)-VI | aminoglycosides | GEN | 2.6 |
| aac(3’)-IId | aminoglycosides | GEN | 1.3 |
| bla TEM-1 | β-lactams | AMP | 43.4 |
| bla OXA-1 | β-lactams | AMP | 1.3 |
| bla CMY-2 | β-lactams | AMC, AMP, CRO, FOX, TIO | 30.3 |
| ampC (−42 T→C) | β-lactams | AMC, AMP, FOX | 5.3 |
| sul1 | folate synthesis inhibitors | FIS | 17.1 |
| sul2 | folate synthesis inhibitors | FIS | 78.9 |
| sul3 | folate synthesis inhibitors | FIS | 2.6 |
| dfrA1 | folate synthesis inhibitors | SXT | 5.3 |
| dfrA5 | folate synthesis inhibitors | SXT | 1.3 |
| dfrA12 | folate synthesis inhibitors | SXT | 9.2 |
| dfrA17 | folate synthesis inhibitors | SXT | 2.6 |
| mphA | macrolides | AZM | 1.3 |
| floR | phenicols | CHL | 68.4 |
| cmlA | phenicols | CHL | 2.6 |
| catA1 | phenicols | CHL | 3.9 |
| catB3 | phenicols | CHL | 1.3 |
| qnrB2 a | quinolones | NAL, CIP | 1.3 |
| qnrB6 a | quinolones | NAL, CIP | 1.3 |
| qnrS2 a | quinolones | NAL, CIP | 1.3 |
| gyrA mutations | quinolones | NAL, CIP | 27.6 |
| parC mutations | quinolones | NAL, CIP | 10.5 |
| parE mutations | quinolones | NAL, CIP | 3.9 |
| tet(A) | tetracyclines | TET | 71.1 |
| tet(B) | tetracyclines | TET | 22.4 |
| tet(C) | tetracyclines | TET | 21.1 |
| tet(D) | tetracyclines | TET | 19.7 |
| tet(M) | tetracyclines | TET | 9.2 |
STR, streptomycin; KAN, kanamycin; GEN, gentamicin; AMP, ampicillin; AMC, amoxicillin/clavulanic acid; CRO, ceftriaxone; FOX, cefoxitin; TIO, ceftiofur; FIS, sulfisoxazole; SXT, trimethoprim/sulfamethoxazole; AZM, azithromycin; CHL, chloramphenicol; NAL, nalidixic acid; CIP, ciprofloxacin; TET, tetracycline.
Only confers reduced susceptibility.
E. coli isolates were also tested for susceptibility to other aminoglycosides, gentamicin and kanamycin. Resistance to each was uncommon, with just 13.2% resistant to kanamycin and 3.9% resistant to gentamicin. Each isolate with kanamycin resistance possessed an aph(3′)-Ia gene, as did one isolate without phenotypic resistance that possessed a truncated gene. All gentamicin-resistant isolates had aac genes, which were not encoded by any isolates lacking resistance.
β-Lactams
We detected the presence of three main β-lactamases that contributed to resistance phenotypes in our isolates: blaTEM-1, blaOXA-1 and blaCMY-2. TEM-1 confers ampicillin resistance and did so for each strain that had the gene (43.4%; Table 1). This was also the case for the one isolate that expressed OXA-1. In contrast, CMY-2, expressed by 30.3% of strains, confers expanded resistance to potentiated β-lactams as well as third-generation cephalosporins such as ceftiofur and ceftriaxone. Each isolate except one with this gene demonstrated resistance to ampicillin, amoxicillin/clavulanic acid, cefoxitin, ceftiofur and ceftriaxone. The one isolate that differed was not resistant to the veterinary cephalosporin ceftiofur, instead having an intermediate phenotype (MIC 4 mg/L).
In addition to these well-characterized resistance genes, overexpression of chromosomal ampC β-lactamase also confers clinical resistance.19 We identified four isolates with −42 thymine-to-cytosine transitions in their promoters, which is known to cause increased ampC expression (Table 1).19 Three of the isolates had β-lactam resistance in the absence of other resistance genes (N35912PS, N36834PS and N33633PS), while the other isolate (N33552PS) carried blaCMY-2, which masked any potential resistance conferred by the ampC mutation. Overproduction of AmpC is predicted to confer resistance to ampicillin, amoxicillin/clavulanic acid and cefoxitin.20 However, two of the three isolates had only intermediate susceptibility to cefoxitin (MIC 16 mg/L). One of these (N36834PS) had additional unanticipated resistance to ceftiofur and ceftriaxone (Table S4). This is likely due to an S287R amino acid substitution, as other substitutions of this residue have been shown to confer expanded β-lactam resistance.21
Folate synthesis inhibitors
Resistance to sulfisoxazole was common (89.5%), with each resistant isolate encoding dihydropteroate synthase sul genes.22 Each sul gene conferred the appropriate resistance and there was no resistance shown by any isolates lacking these genes. In addition, trimethoprim/sulfamethoxazole resistance was found in 17.1% of strains, with dihydrofolate reductase (dfrA) genes being responsible in each instance. There was no unexpected resistance, but one isolate had a truncated dfrA1 gene that resulted in its antibiotic susceptibility.
Macrolides
In our panel of MDR E. coli, there was only one isolate with resistance to the macrolide azithromycin, mediated by an mphA gene, which encodes a macrolide phosphotransferase.23 No additional isolates had any macrolide resistance genes.
Phenicols
Chloramphenicol resistance was found in 72.4% of the MDR E. coli. Resistance was predominantly mediated by the floR gene, present in 94.5% of resistant isolates. However, a minority of strains were also found to have other resistance genes, including cmlA, catA1 and catB3. Each isolate with at least one resistance gene demonstrated chloramphenicol resistance, while all isolates lacking resistance genes were susceptible to chloramphenicol.
Quinolones
We found that 27.6% of isolates were resistant to nalidixic acid, with 7.9% being resistant to ciprofloxacin. Isolates with a single mutation in the QRDR of gyrA typically have nalidixic acid resistance, with two mutations being required for ciprofloxacin resistance.24 These genotypes correlated well in our study, as there were 21 isolates with gyrA mutations (S83L, S83F, D87G and D87N amino acid changes), all of which were resistant to nalidixic acid (Table S4). Six isolates had multiple gyrA mutations, with each isolate being resistant to both nalidixic acid and ciprofloxacin. Only strains with multiple mutations had ciprofloxacin resistance, consistent with our genotypic prediction. Eight of the isolates with gyrA mutations also had mutations in parC, while three had mutations in parE (Table S4). Mutations of each have also been associated with increased quinolone resistance, but no isolates had parC or parE mutations in the absence of gyrA mutations.25 None of the E. coli was found to contain relevant gyrB mutations, which can also alter susceptibility to quinolones.26
In addition to mutation of gyrase and topoisomerase genes, three isolates also contained qnr genes that can confer quinolone resistance.27 Individual isolates had qnrB2, qnrB6 or qnrS2 genes, yet each was still clinically susceptible to both nalidixic acid and ciprofloxacin. This is consistent with previous reports that these qnr genes do not confer actual resistance, but instead elevated MIC levels of both antibiotics.28–30 All three isolates encoding qnr genes had elevated ciprofloxacin and nalidixic acid MICs of ≥0.25 and 8 mg/L, respectively (Table S1). Although this still resulted in these isolates being classified as susceptible to each drug, the only other isolates with MICs at least that high had at least one gyrA QRDR mutation. This confirms the capability of these genes to confer decreased quinolone susceptibility.
Tetracyclines
Tetracycline resistance was widespread among our isolates, with 88.2% of isolates demonstrating resistance. The most prevalent resistance gene was tet(A) (in 80.6% of tetracycline-resistant strains), although tet(B)-, tet(C)- and tet(D)-mediated resistance was also common (Table 1). The tet(M) gene was found in several isolates, each of which also had tet(A). Genotype was an excellent predictor of phenotype, as each isolate that had at least one resistance gene was resistant to tetracycline. Furthermore, there were no isolates lacking tet genes that had resistance.
Resistance elements
In addition to the identification of resistance determinants, WGS can also provide information about elements that contain multiple resistance genes, such as integrons or resistance islands. For example, we found that 35 isolates (46.1%) had the resistance genes tet(A), strA/strB and sul2 together on the same contig. Of these 35, 11 also had a floR gene present, 8 had a blaTEM-1 gene and 8 had both (Figure 2). These numbers are likely underestimates, since only isolates with these genes on individual contigs were included and many additional isolates had these genes on multiple contigs.
Figure 2.
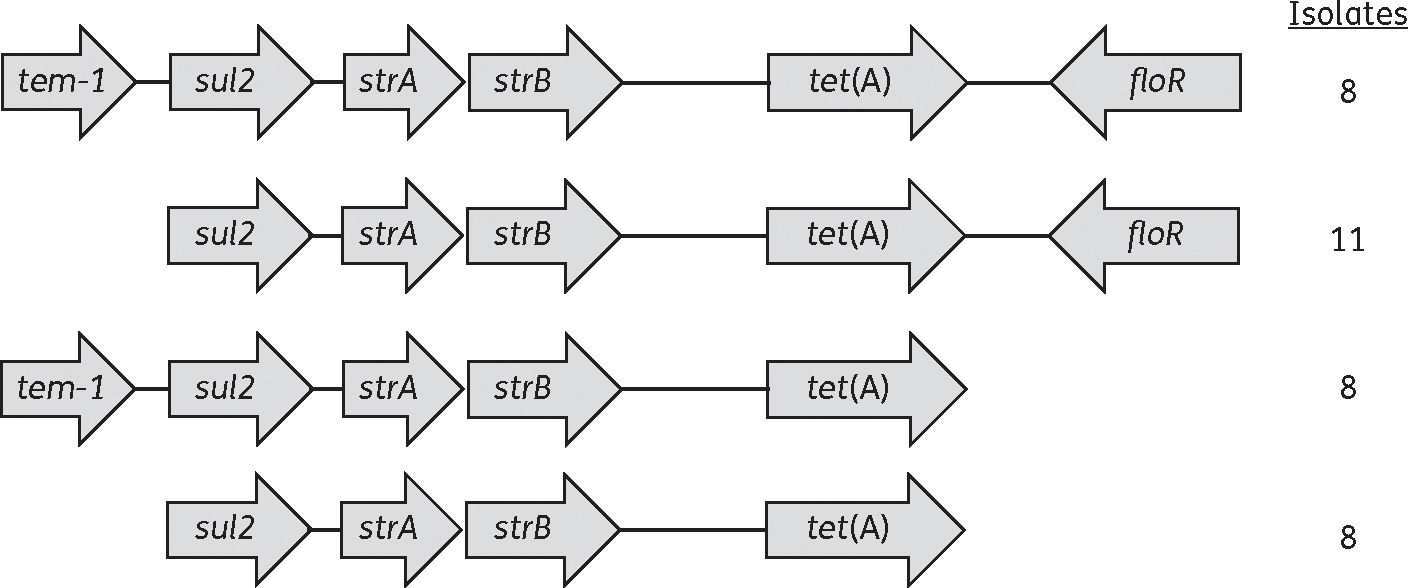
Depiction of resistance elements containing contiguous antibiotic resistance genes. The values indicate the number of isolates with each resistance element.
Discussion
Here, we report on the utility of WGS for accurately predicting antibiotic resistance. Overall, >30 resistance genes were identified from 76 MDR E. coli isolates, along with a number of resistance-associated mutations (Table 1 and Table S4). In addition, the resultant resistance genotypes correlated with 99.6% sensitivity (95% CI 98%–100%) and 97.8% specificity (95% CI 96%–99%) to resistance phenotypes (Table 2). Although our analysis only included MDR isolates, this demonstrates the robustness of WGS in predicting resistance phenotypes. Overall, among 1140 phenotypic resistance tests, there were only 17 discrepancies between genotype and phenotype, with each remaining after phenotypes were retested (Table 3).
Table 2.
Evaluation of genotypic analysis for the prediction of resistance phenotypes
| Antibiotic | Phenotype: susceptible |
Phenotype: resistant |
||||
|---|---|---|---|---|---|---|
| genotype: resistant | genotype: susceptible | genotype: resistant | genotype: susceptible | Sensitivity (%) | Specificity (%) | |
|
| ||||||
| GEN | 0 | 73 | 3 | 0 | 100 | 100 |
| KAN | 0 | 66 | 10 | 0 | 100 | 100 |
| STR | 12 | 12 | 52 | 0 | 100 | 50 |
| AMC | 0 | 50 | 26 | 0 | 100 | 100 |
| CRO | 0 | 52 | 23 | 1 | 95.8 | 100 |
| FOX | 2 | 50 | 24 | 0 | 100 | 96.1 |
| TIO | 1 | 52 | 22 | 1 | 95.6 | 98.1 |
| SXT | 0 | 63 | 13 | 0 | 100 | 100 |
| FIS | 0 | 8 | 68 | 0 | 100 | 100 |
| AZM | 0 | 75 | 1 | 0 | 100 | 100 |
| AMP | 0 | 21 | 55 | 0 | 100 | 100 |
| CHL | 0 | 21 | 55 | 0 | 100 | 100 |
| CIP | 0 | 70 | 6 | 0 | 100 | 100 |
| NAL | 0 | 55 | 21 | 0 | 100 | 100 |
| TET | 0 | 9 | 67 | 0 | 100 | 100 |
| Overall | 15 | 677 | 446 | 2 | 99.6 | 97.8 |
GEN, gentamicin; KAN, kanamycin; STR, streptomycin; AMC, amoxicillin/clavulanic acid; CRO, ceftriaxone; FOX, cefoxitin; TIO, ceftiofur; SXT, trimethoprim/ sulfamethoxazole; FIS, sulfisoxazole; AZM, azithromycin; AMP, ampicillin; CHL, chloramphenicol; CIP, ciprofloxacin; NAL, nalidixic acid; TET, tetracycline.
Table 3.
List of resistance genotype-phenotype discrepancies
| Strain(s) | Drug(s) | Gene(s) | Predicted genotype | Phenotype | Explanation |
|---|---|---|---|---|---|
|
| |||||
| N33707PS | TIO | bla CMY-2 | R | I | unknown; intermediate susceptibility |
| N35912PS, N36834PS | FOX | ampC | R | I | unknown; intermediate susceptibility |
| N36834PS | CRO, TIO | ampC | S | R | ampC coding mutations |
| Six strains | STR | aadA genes | R | S | unknown; high resistance threshold |
| Six strains | STR | strA+strB | R | S | unknown; high resistance threshold |
TIO, ceftiofur; FOX, cefoxitin; CRO, ceftriaxone; STR, streptomycin; S, susceptible; I, intermediate; R, resistant.
Among the discordant results, 12 (70.6%) were from isolates with streptomycin resistance genes that lacked phenotypic resistance. There is no CLSI-defined streptomycin breakpoint for E. coli, although resistance is often demarcated by an MIC of ≥64 mg/L.31 In our dataset, a vast majority of strA/strB-encoding isolates did have streptomycin MICs ≥64 mg/L, although this was not true of strains with aadA genes. This confirms the previous results of Sunde and Norstrom,32 who demonstrated that strA/strB genes confer higher resistance than aadA genes. Although a reduction in the MIC cut-off may result in better genotype–phenotype correlation, even optimized streptomycin breakpoints can result in substantial discrepancies with genotypic data.33 Thus, the presence of these genes could instead be used as an indicator of resistance potential and may present a reasonable alternative to conventional phenotypic testing.
Besides streptomycin, some minor discrepancies were observed for β-lactam antibiotics (Table 3). One strain carried blaCMY-2 and exhibited an intermediate phenotype for ceftiofur (MIC 4 mg/L). Similarly, two strains with ampC promoter mutations displayed intermediate phenotypes to cefoxitin (MIC 16 mg/L). The only two resistance phenotypes not predicted by genotype were from strain N36834PS, which had unexpected ampC-mediated resistance to ceftiofur and ceftriaxone. Although not previously described, the S287R substitution of N36834PS AmpC likely resulted in its resistance to the third-generation cephalosporins. This was confirmed by the fact that the S287R amino acid change is the only difference between it and AmpC from N33633PS, which does not have resistance to these cephems.
Although our results show a high degree of correlation between resistance genotypes and phenotypes for most antibiotic classes, this study does have some drawbacks. For example, the use of whole-genome shotgun sequencing results in fragmentary genomes with sequences assembled into contigs. Although this did not appear to hinder our analysis, it complicates the identification of resistance plasmids and cassettes. In addition, strains with multiple resistance genes for a single antimicrobial may demonstrate phenotypic resistance, but it is unclear which gene(s) confer this resistance. Furthermore, genotypic prediction of resistance relies on curated databases of known resistance determinants, so currently unknown resistance mechanisms would not be identified by this approach. Since any resistance gene database is necessarily incomplete, this means that errors cannot always be avoided.
In general, WGS accurately predicted the vast majority of resistance phenotypes from MDR E. coli strains. Additional studies based on this work may focus on utilizing genotypic methods to predict resistance profiles in the absence of phenotypic information. Subsequent phenotypic susceptibility testing can then be used to validate the approach.
Although phenotypic testing is the current norm for determining antibiotic resistance, WGS can provide additional useful information. This is because WGS detected resistance mechanisms such as qnr genes that result in elevated MICs without meeting clinical resistance thresholds, providing information that may be of use in patient treatment. Since resistance genes are either present or absent and do not have breakpoints, this makes WGS a more unbiased and consistent method for determining at least the genotypic potential for antibiotic resistance. In many cases, phenotypic testing is still faster and cheaper than WGS-based genotypic analysis. However, this is changing and once WGS data are obtained, hundreds or thousands of resistance genes can be simultaneously identified in an automated process. This simplifies the detection of rare resistance genes for phenotypes that may not be tested in a cost-effective manner and supports the use of WGS-based genotypic analysis in resistance surveillance programmes. WGS can also be used to identify the relatedness of bacterial isolates and therefore support the investigation of bacterial source attribution. Thus, WGS could supplement existing scientific and clinical techniques while gaining new insight into resistance patterns, virulence genes and emerging outbreaks.
Overall, we used WGS to successfully identify the resistance genotypes for 76 MDR E. coli strains. These genotypes correlated well with resistance phenotypes, demonstrating the potential of WGS-based techniques to replace phenotypic indicators of resistance. The vast amount of data gained by WGS also potentiates its use for a number of additional research and clinical applications.
Supplementary Material
Acknowledgements
We would like to thank Maureen Davidson for critical review of the manuscript prior to submission.
Funding
This work was supported by the US Food and Drug Administration with internal funds as part of routine work. This project was also supported in part by an appointment to the Research Participation Program at the Center for Veterinary Medicine administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the US Department of Energy and US Food and Drug Administration.
Footnotes
Transparency declarations
G. H. L. has provided scientific advice to various pharmaceutical companies that market antimicrobial drugs for administration to animals. He has on occasion billed for his service. G. H. L. has also received honoraria for service on advisory boards and presentations from pharmaceutical companies. All other authors: none to declare.
Disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the official policy of the Department of Health and Human Services, the US Food and Drug Administration or the US Government. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture or Food and Drug Administration.
Supplementary data
Tables S1 to S4 are available as Supplementary data at JAC Online (http://jac.oxfordjournals.org/).
References
- 1.Aarestrup FM, Brown EW, Detter C et al. Integrating genome-based informatics to modernize global disease monitoring, information sharing, and response. Emerg Infect Dis 2012; 18: e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Didelot X, Bowden R, Wilson DJ et al. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet 2012; 13: 601–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dudley MN, Ambrose PG, Bhavnani SM et al. Background and rationale for revised Clinical and Laboratory Standards Institute interpretive criteria (breakpoints) for Enterobacteriaceae and Pseudomonas aeruginosa: I. Cephalosporins and aztreonam. Clin Infect Dis 2013; 56: 1301–9. [DOI] [PubMed] [Google Scholar]
- 4.Stoesser N, Batty EM, Eyre DW et al. Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. J Antimicrob Chemother 2013; 68: 2234–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Threlfall EJ, Fisher IS, Ward LR et al. Harmonization of antibiotic susceptibility testing for Salmonella: results of a study by 18 national reference laboratories within the European Union-funded Enter-net group. Microb Drug Resist 1999; 5: 195–200. [DOI] [PubMed] [Google Scholar]
- 6.Paterson DL. Resistance in Gram-negative bacteria: Enterobacteriaceae. Am J Med 2006; 119 Suppl 1: S20–8; discussion S62–70. [DOI] [PubMed] [Google Scholar]
- 7.Fluit AC, Schmitz FJ. Class 1 integrons, gene cassettes, mobility, and epidemiology. Eur J Clin Microbiol Infect Dis 1999; 18: 761–70. [DOI] [PubMed] [Google Scholar]
- 8.Schultsz C, Geerlings S. Plasmid-mediated resistance in Enterobacteriaceae: changing landscape and implications for therapy. Drugs 2012; 72: 1–16. [DOI] [PubMed] [Google Scholar]
- 9.Hu X, Xu B, Yang Y et al. A high throughput multiplex PCR assay for simultaneous detection of seven aminoglycoside-resistance genes in Enterobacteriaceae. BMC Microbiol 2013; 13: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis MA, Besser TE, Orfe LH et al. Genotypic-phenotypic discrepancies between antibiotic resistance characteristics of Escherichia coli isolates from calves in management settings with high and low antibiotic use. Appl Environ Microbiol 2011; 77: 3293–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bugarel M, Granier SA, Weill FX et al. A multiplex real-time PCR assay targeting virulence and resistance genes in Salmonella enterica serotype Typhimurium. BMC Microbiol 2011; 11: 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gordon NC, Price JR, Cole K et al. Prediction of Staphylococcus aureus antimicrobial resistance by whole-genome sequencing. J Clin Microbiol 2014; 52: 1182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zankari E, Hasman H, Kaas RS et al. Genotyping using whole-genome sequencing is a realistic alternative to surveillance based on phenotypic antimicrobial susceptibility testing. J Antimicrob Chemother 2013; 68: 771–7. [DOI] [PubMed] [Google Scholar]
- 14.FDA. National Antimicrobial Resistance Monitoring System—Enteric Bacteria (NARMS): 2011 Executive Report. Rockville, MD: US Department of Health and Human Services, Food and Drug Administration, 2013. [Google Scholar]
- 15.Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing: Twenty-fifth Informational Supplement M100-S25. CLSI, Wayne, PA, USA, 2015. [Google Scholar]
- 16.Angiuoli SV, Gussman A, Klimke W et al. Toward an online repository of standard operating procedures (SOPs) for (meta)genomic annotation. OMICS 2008; 12: 137–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamura K, Stecher G, Peterson D et al. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 2013; 30: 2725–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sullivan KM, Dean A, Soe MM. OpenEpi: a web-based epidemiologic and statistical calculator for public health. Public Health Rep 2009; 124: 471–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caroff N, Espaze E, Gautreau D et al. Analysis of the effects of −42 and −32 ampC promoter mutations in clinical isolates of Escherichia coli hyperproducing ampC. J Antimicrob Chemother 2000; 45: 783–8. [DOI] [PubMed] [Google Scholar]
- 20.Mammeri H, Nazic H, Naas T et al. AmpC β-lactamase in an Escherichia coli clinical isolate confers resistance to expanded-spectrum cephalosporins. Antimicrob Agents Chemother 2004; 48: 4050–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mammeri H, Poirel L, Fortineau N et al. Naturally occurring extended-spectrum cephalosporinases in Escherichia coli. Antimicrob Agents Chemother 2006; 50: 2573–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antunes P, Machado J, Sousa JC et al. Dissemination of sulfonamide resistance genes (sul1, sul2, and sul3) in Portuguese Salmonella enterica strains and relation with integrons. Antimicrob Agents Chemother 2005; 49: 836–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howie RL, Folster JP, Bowen A et al. Reduced azithromycin susceptibility in Shigella sonnei, United States. Microb Drug Resist 2010; 16: 245–8. [DOI] [PubMed] [Google Scholar]
- 24.Saenz Y, Zarazaga M, Brinas L et al. Mutations in gyrA and parC genes in nalidixic acid-resistant Escherichia coli strains from food products, humans and animals. J Antimicrob Chemother 2003; 51: 1001–5. [DOI] [PubMed] [Google Scholar]
- 25.Khodursky AB, Zechiedrich EL, Cozzarelli NR. Topoisomerase IV is a target of quinolones in Escherichia coli. Proc Natl Acad Sci USA 1995; 92: 11801–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida H, Bogaki M, Nakamura M et al. Quinolone resistance-determining region in the DNA gyrase gyrB gene of Escherichia coli. Antimicrob Agents Chemother 1991; 35: 1647–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cattoir V, Poirel L, Rotimi V et al. Multiplex PCR for detection of plasmid-mediated quinolone resistance qnr genes in ESBL-producing enterobacterial isolates. J Antimicrob Chemother 2007; 60: 394–7. [DOI] [PubMed] [Google Scholar]
- 28.Marti E, Balcazar JL. Multidrug resistance-encoding plasmid from Aeromonas sp. strain P2G1. Clin Microbiol Infect 2012; 18: E366–8. [DOI] [PubMed] [Google Scholar]
- 29.Gutierrez B, Herrera-Leon S, Escudero JA et al. Novel genetic environment of qnrB2 associated with TEM-1 and SHV-12 on pB1004, an IncHI2 plasmid, in Salmonella Bredeney BB1047 from Spain. J Antimicrob Chemother 2009; 64: 1334–6. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz E, Saenz Y, Zarazaga M et al. qnr, aac(6′)-Ib-cr and qepA genes in Escherichia coli and Klebsiella spp.: genetic environments and plasmid and chromosomal location. J Antimicrob Chemother 2012; 67: 886–97. [DOI] [PubMed] [Google Scholar]
- 31.Tadesse DA, Zhao S, Tong E et al. Antimicrobial drug resistance in Escherichia coli from humans and food animals, United States, 1950–2002. Emerg Infect Dis 2012; 18: 741–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sunde M, Norstrom M. The genetic background for streptomycin resistance in Escherichia coli influences the distribution of MICs. J Antimicrob Chemother 2005; 56: 87–90. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Migura L, Sunde M, Karlsmose S et al. Establishing streptomycin epidemiological cut-off values for Salmonella and Escherichia coli. Microb Drug Resist 2012; 18: 88–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.