

Abstract
PACIFIC‐PGx evaluated the feasibility of implementing pharmacogenetics (PGx) screening in Australia and the impact of DPYD/UGT1A1 genotype‐guided dosing on severe fluoropyrimidine (FP) and irinotecan‐related toxicities and hospitalizations, compared to historical controls. This prospective single arm trial enrolled patients starting FP/irinotecan for any cancer between 7 January 2021 and 25 February 2022 from four Australian hospitals (one metropolitan, three regional). During the accrual period, 462/487 (95%) consecutive patients screened for eligibility for DPYD and 50/109 (46%) for UGT1A1 were enrolled and genotyped (feasibility analysis), with 276/462 (60%) for DPYD and 30/50 (60%) for UGT1A1 received FP/irinotecan (safety analysis). DPYD genotyping identified 96% (n = 443/462) Wild‐Type, 4% (n = 19/462) Intermediate Metabolizers (50% dose reduction), and 0% Poor Metabolizers. UGT1A1 genotyping identified 52% (n = 26/50) Wild‐Type, 40% (n = 20/50) heterozygous, and 8% (n = 4/50) homozygous (30% dose reduction). Key demographics for the FP/irinotecan safety cohorts included: age range 23–89/34–74 years, male 56%/73%, Caucasian 83%/73%, lower gastrointestinal cancer 50%/57%. Genotype results were reported prior to cycle‐1 (96%), average 5–7 days from sample collection. PGx‐dosing for DPYD variant allele carriers reduced high‐grade toxicities compared to historic controls (7% vs. 39%; OR = 0.11, 95% CI 0.01–0.97, p = 0.024). High‐grade toxicities among Wild‐Type were similar (14% vs. 14%; OR = 0.99, 95% CI 0.64–1.54, p = 0.490). PGx‐dosing reduced FP‐related hospitalizations (−22%) and deaths (−3.7%) compared to controls. There were no high‐grade toxicities or hospitalizations for UGT1A1*28 homozygotes. PGx screening and prescribing were feasible in routine oncology care and improved patient outcomes. Findings may inform expanded PGx programs within cancer and other disease settings.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Pharmacogenetic (PGx) testing for DPYD and UGT1A1 variants, aimed at guiding fluoropyrimidine and irinotecan chemotherapy dosing, holds promise for reducing severe cancer‐treatment related toxicities and hospitalizations. Despite its utility, the potential implementation of PGx testing in routine oncology care, particularly in Australia, remains limited.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study explored the feasibility of implementing a PGx screening program in Australia and evaluated whether DPYD/UGT1A1 genotype‐guided dosing could reduce severe fluoropyrimidine and irinotecan‐related adverse events and hospitalizations, compared to standard body surface area dosing in historical controls.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
We found that PGx screening was feasible in both metropolitan and regional Australian healthcare settings. The program efficiently collected patient genetic samples and delivered results prior to planned treatment initiation, enabling genotype‐guided dosing. This approach reduced severe toxicities, hospitalizations, and deaths compared to historical controls.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The compelling feasibility and safety results from this study provided a foundation for scaled implementation of DPYD/UGT1A1 programs, and for expanding PGx testing within cancer care and across other therapeutic areas. Findings support broader adoption of personalized medicine approaches in routine care to improve patient outcomes.
INTRODUCTION
Evidence supporting the use of pharmacogenetic (PGx)‐guided dosing of fluoropyrimidines (FP) (i.e. 5‐fluorouracil (5‐FU) and capecitabine) dates back for over a decade. 1 Yet, clinical translation is limited globally, placing patients at risk of preventable life‐threatening toxicities. 1 FP is widely prescribed in curative and palliative treatments for many solid tumors. Nearly one‐third of patients experience severe treatment‐related toxicities resulting in death in ~1%. 2 The reduced capacity to metabolize FP (80%–90% by the dihydropyrimidine dehydrogenase (DPD) enzyme, coded by DPYD gene) can contribute to such events. 2 Partial and complete DPD enzyme deficiency is reported within predominantly Caucasian populations as ~5%/~0.1% respectively. 2
Irinotecan is also commonly prescribed in solid tumors, including pancreatic and colorectal cancers. 3 Neutropenia and diarrhea are the most common dose‐limiting toxicities that may lead to hospitalization, treatment interruptions, and delays. 4 , 5 Reduced capacity to metabolize the active metabolite of irinotecan, SN‐38 through glucuronidation by the UDP glucuronosyltransferase 1 family, polypeptide A1 (UGT1A1) enzyme, coded by the UGT1A1 gene, can contribute to such events. The plasma SN‐38 exposure is the main determinant of neutropenia, and SN‐38 intestinal accumulation via biliary excretion is associated with late‐onset diarrhea; both events can occur concurrently. 4 , 5
The Clinical Pharmacogenetics Implementation Consortium (CPIC) and the Dutch Pharmacogenetics Working Group (DPWG) have published recommendations for DPYD genotyping and DPWG for UGT1A1. 3 Recommendations for screening and dose adjustments based on DPYD status were recommended by the United States Food and Drug Administration (FDA) (in 2022 for capecitabine updated product label and in 2024 for 5‐FU), 6 European Medicines Agency (EMA) in 2019, 7 and Therapeutic Goods Administration (TGA) in 2022, 8 and the FDA has an equivalent recommendations for UGT1A1*28 (2005). 9 In 2024, the Association for Molecular Pathology (AMP) Pharmacogenomics (PGx) Working Group published guidance for standardizing PGx variant testing across clinical laboratories. 10 The two‐tier strategy considers the function impact of variant alleles, allele frequency across different ethnicities, availability of reference materials, and other technical factors. 10 Tier 1 specifies a minimum set of DPYD variant alleles for testing (c.1905+1G>A (rs3918290), c.1679T>G (rs55886062), c.1129‐5923C>G, c.1236G>A (rs75017182, rs56038477), c.557A>G (rs115232898), c.868A>G (rs146356975), c.2279C>T (r s112766203), c.2846A>T (rs67376798). Tier 2 includes an extended list of variants. 10
International landmark studies have demonstrated the feasibility and the cost effectiveness of pre‐treatment DPYD and UGT1A1 genotyping; however, clinical translation has not been widely adopted especially in the Australian healthcare system given that the feasibility as well as potential impact on efficacy outcomes remain unclear to clinicians. 2 , 4 , 11 , 12
The PACIFIC‐PGx trial 3 is the largest prospective multicenter clinical trial in Australia that evaluated the feasibility of implementing pre‐treatment DPYD and UGT1A1*28 genotyping, including a centralized model for testing and follow‐up of patients both at metropolitan and regional hospitals. We hypothesized that pre‐treatment PGx testing and genotype‐guided dose adjustments for FP and irinotecan would be feasible to implement within routine oncology care, and would reduce severe and life‐threatening toxicities, leading to fewer hospital admissions and reduced mortality compared to historical controls.
METHODS
Study design
PACIFIC‐PGx was a prospective, multi‐site, single arm trial at four Australian hospitals: a metropolitan tertiary specialist cancer center (primary site leading the centralized model of care), and three regional (satellite) centers. 3 Single arm design was implemented given the lack of equipoise for randomization. 3 High‐grade toxicities (Grade ≥ 3) were compared to standard dosing (historic control) for FP. 13 The trial was conducted under the Networked Teletrial Model, allowing regional patients access to the trial via their local hospital. 3 , 14
Patient demographics and treatment
Adults age ≥18 with any cancer diagnosis and planned first exposure to FP or irinotecan chemotherapy as a single agent or in combination with other anti‐cancer therapy were eligible for enrolment (refer to published protocol for expanded details). 3 Consecutively presenting patients were screened against the study eligibility criteria with all eligible patients included in feasibility analysis, and those that received standard clinical dose FP/irinotecan included in safety analysis (Figure 1a,b). The safety analysis was limited to standard clinical dosing (i.e., excluding dose reductions for clinical reasons unrelated to PGx), to improve confidence that any differences in toxicity event rates would be likely attributable to PGx‐guided dosing, without the confounding influence of dose reductions due to other clinical or patient‐related factors.
FIGURE 1.
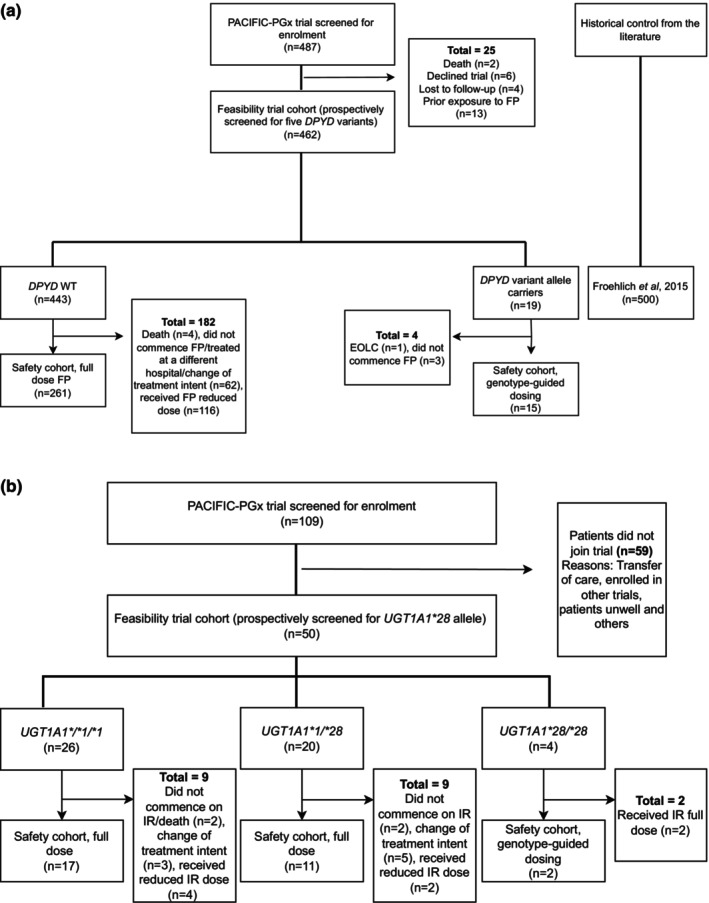
(a) Trial profile: fluoropyrimidine patients. (b) Trial profile: irinotecan patients. (a) Historical control cohort from Froehlich et al. 2015. 13 Out of 25 patients excluded, 11 patients may have had UGT1A1*28 tested if irinotecan treatment was confirmed. EOLC, end of life care; FP, fluoropyrimidines; WT, wild‐type. (b) IR, Irinotecan.
Historical control data for toxicity events was obtained from previously published studies, with all studies from the 2015 Meulendijks meta‐analysis formally assessed for similarity to the PACIFIC‐PGx trial cohort (Table S1). 15 The selection of an appropriate historic control cohort (Froehlich 2015) 13 was based on similarity of PGx screening alleles matched [except one additional variant (c.557A>G, (rs115232898)) added to our panel relevant to African populations], 16 cancer diagnoses, treatment types, and duration of follow‐up (2 cycles). Multiple comparator cohorts for hospitalization events were selected (White 2023, 17 Toffoli 2019, 18 Lunenberg 2018 19 ) each with similarities/differences to the PACIFIC‐PGx cohort (Table S2). The trial was registered with the Australian New Zealand Clinical Trials Registry (ANZCTR12621000251820). Investigators obtained informed consent from each participant or each participant's guardian and approved by the Peter MacCallum Cancer Centre Human Research Ethics Committee (HREC/66681/PMCC‐2020).
Study procedures
Patients underwent screening for five DPYD variants recommended by CPIC and the AMP (tier 1) 10 : c.1905+1G>A (rs3918290), c.1679T>G (rs55886062), c.2846A>T (rs67376798), c.1236G>A (rs56038477) c.557A>G (rs115232898) and UGT1A1*28 (rs8175347) genotyping as per DPWG recommendations. Blood or cheek swab sample was collected prior to commencement of FP and/or irinotecan. Prospectively planned exploratory analyses with whole exome sequencing, including DPYD variant alleles included in AMP Tier 2 10 and UGT1A1*6, have been conducted on stored samples and will be reported as a separate discovery publication.
Following referral by treating oncologists across four sites, centralized PGx testing was conducted at an accredited laboratory, and clinical follow‐up was coordinated by a pharmacist at the primary metropolitan site with PGx expertise. Patients were followed for acute toxicities and health resource utilization until completion of cycle‐2. 3 A subset of DPYD intermediate metabolizer (IM) patients were followed up for an additional two cycles, to evaluate the effect of gradual FP dose increase on toxicity and hospitalizations. Patients were included in the final analysis if they were: (1) DPYD wild‐type (WT)—normal DPD enzyme activity for tested variants with activity score of 2, receiving full FP dose 16 ; or (2) DPYD IM—reduced DPD enzyme activity with activity score of 1 or 1.5, receiving 50% FP dose reduction per the CPIC guidelines 16 ; or (3); DPYD poor metabolizers (PM)—complete DPD enzyme deficiency with activity score of 0 or 0.5 avoiding FP exposure per the CPIC guidelines 16 ; and/or (4) UGT1A1 WT (*1/*1) or heterozygous (*1/*28) receiving full irinotecan dose; or (5) UGT1A1*28 homozygous (*28/*28) receiving 30% irinotecan dose reduction per DPWG guidelines. 5 Variant allele carriers received PGx‐guided dose reductions without further changes to supportive care. Details on DNA extraction and genotyping assays are summarized in Supplementary Material 1 in Appendix S1.
Primary and secondary endpoints
The primary endpoint was feasibility of implementing a PGx screening program. Secondary endpoints included any grade and high‐grade toxicities (Grade ≥ 3) according to the Common Terminology Criteria for Adverse Events (CTCAE) version 5.0, the number and type of unplanned hospital admissions, and treatment‐related deaths. High‐grade toxicities for DPYD variant allele carriers receiving PGx‐guided reduced dosing (PACIFIC‐PGx) were compared with (1) DPYD variant allele carriers receiving standard dosing (historic control) to show impact of PGx and (2) DPYD WT patients receiving FP standard dose (PACIFIC‐PGx) to assess toxicity rates with the assumption that genotyping‐guided dosing should deliver equivalent drug exposures with similar rates of high‐grade toxicity. High‐grade toxicities for UGT1A1 WT (*1/*1) and heterozygous (*1/*28) patients receiving full dose irinotecan were compared to homozygous (*28/*28) patients receiving reduced irinotecan dose. Endpoints were further described in the published study protocol. 3
Statistical analysis
There was no formal sample size calculation for this feasibility study (primary endpoint) with pragmatic inclusion of eligible participants consecutively enrolled over a 13‐month period (January 2021–February 2022). 3 A post hoc power analysis was conducted for the secondary endpoint of high‐grade toxicity with PGx versus body surface area (BSA) dosing, based on the observed event rates and sample sizes, yielding 65% power to detect a statistically significant difference at a 0.05 significance level and 78% power at a 0.10 significance level.
Patient demographics and study outcomes were summarized using appropriate descriptive statistics. Comparison of patient characteristics between groups with and without high‐grade toxicities was conducted using the chi‐squared test or Fisher exact test for categorical variables, and Mann–Whitney U test for continuous variables. Odds ratios (OR) were calculated to assess differences in high‐grade toxicity rates between the tested cohorts, as specified under secondary endpoints. A one‐tailed test was performed to evaluate the significance of the differences in event rates, given the study's aim to determine whether PGx‐dosing could reduce toxicity, with no clinical rationale for increased toxicity due to lower dosing. A two‐tailed test was performed for robustness. Multivariable analysis was conducted using Cox Proportional Hazards models (events from cycle 1 day 1) and logistic regression to describe the relationship between toxicity and predictors (age, sex, cancer type, and treatment intent). Additional predictors (i.e., ethnicity, ECOG, BSA, cancer status, prior cancer treatment, and all treatment) were tested for the same relationship. Median time to event and IQR were reported, 95% confidence intervals were calculated, and two‐sided p‐values of <0.05 were considered statistically significant. In terms of the hospitalizations, we have utilized two‐by‐two‐tables for statistical analysis and Mid‐P exact to report the p‐values. All other statistical analyses were performed using R version 4.3.0 in RStudio version 2022.12.0.353. 20
Ethics statement
The study was approved by the Peter MacCallum Cancer Centre Human Research Ethics Committee (HREC/66681/PMCC‐2020). The study was performed in accordance with the Declaration of Helsinki.
RESULTS
Patient demographics and treatment characteristics
During the 13‐month accrual period (January 2021–February 2022), 462/487 (95%) patients screened for trial eligibility for DPYD and 50/109 (46%) for UGT1A1 were enrolled and underwent genotype testing (feasibility analysis), with 276/462 (60%) for DPYD genotyping and 30/50 (60%) for UGT1A1 receiving FP or irinotecan (safety analysis). Enrolment by genotype/treatment and reasons for exclusion are included in the trial profiles for FP patients (Figure 1a) and irinotecan patients (Figure 1b).
Patient demographics and treatment characteristics were reported for 276 patients in the FP safety cohort (Tables 1 and 2) and 30 patients in the irinotecan safety cohort (Tables S3 and S4). Most patients were males [56% (154/276) for FP and 73% (22/30) for irinotecan] and of Caucasian ethnicity [83% (228/276) for FP, and 73% (22/30) for irinotecan]. Ages ranged from 23 to 89 years for FP and 34–74 years for irinotecan. Most patients had a diagnosis of lower gastrointestinal cancer [50% (138/276) FP and 57% (17/30) irinotecan], and metastatic disease [55% (152/276) FP and 77% (23/30) irinotecan]. Most FP patients (59%) were treated with a 5‐FU‐based chemotherapy combination (Table 2). All but one patient treated with irinotecan received multi‐agent combination chemotherapy (Table S4). Historic control groups for FP are described in Tables 1 and 2.
TABLE 1.
Demographics of treated patients with fluoropyrimidines (PACIFIC‐PGx trial vs. historical control).
| Characteristic | DPYD WT full dose | DPYD variant allele carriers genotype‐guided dosing | Total | Historical cohort |
|---|---|---|---|---|
| (n = 261) | (n = 15) | (n = 276) | (Froehlich et al. 2015 13 ) | |
| (Total = 500) | ||||
| n (%) | n (%) | n (%) | n (%) | |
| Sex | ||||
| Male | 149 (57%) | 5 (33%) | 154 (56) | 301 (60) |
| Female | 112 (43%) | 10 (67%) | 122 (44) | 199 (40) |
| Race/Ethnicity | ||||
| Caucasian | 215 (82) | 13 (87) | 228 (83) | 493 (99) |
| Indigenous | 3 (1) | 0 (0) | 3 (1) | NR |
| Asian | 30 (11) | 2 (13) a | 32 (12) | |
| African | 3 (1) | 0 (0) | 3 (1) | |
| Middle Eastern | 8 (3) | 0 (0) | 8 (3) | |
| Others b | 2 (1) | 0 (0) | 2 (1) | |
| Age, years | ||||
| Median | 61 | 57 | 61 | 63 |
| Range | [23–89] | [33–81] | [23–89] | [18–99] |
| ECOG | ||||
| 0–1 | 236 (90) | 12 (80) | 248 (90) | NR |
| 2–3 | 16 (6) | 2 (13) | 18 (7) | |
| NR | 9 (3) | 1 (7) | 10 (4) | |
| BSA | ||||
| Median | 1.85 | 1.84 | 1.85 | NR |
| Range | [1.29–2.76] | [1.47–2.44] | NA | NR |
| Cancer status | ||||
| New | 202 (77) | 12 (80) | 214 (78) | NR |
| Recurrent | 59 (23) | 3 (20) | 62 (22) | |
| Disease status | ||||
| Local | 51 (20) | 6 (40) | 57 (21) | NR |
| Locally advanced | 66 (25%) | 1 (7) | 67 (24) | |
| Metastatic | 144 (55) | 8 (53) | 152 (55) | |
| Chemotherapy intent | ||||
| Curative | 118 (45) | 10 (67) | 128 (46) | NR |
| Palliative | 143 (55) | 5 (33) | 148 (54) | |
| Type of cancer | ||||
| Upper GI | 85 (33) | 4 (27) | 89 (32) | 143 (29) |
| Lower GI | 131 (50) | 7 (47) | 138 (50) | 275 (55) |
| Breast | 24 (9) | 4 (27) | 28 (10) | 26 (5) |
| H&N | 16 (6) | 0 (0) | 16 (6) | 31 (6) |
| Other c | 5 (2) | 0 (0) | 5 (2) | 25 (5) |
Note: Definitions: Full dose is classified as equal or greater than 94.5%. If 5‐fluorouracil (5‐FU) infusion and 5‐FU bolus were part of the chemotherapy regimen, 5‐FU infusion full dose was applied, otherwise 5FU bolus full dose was applied (i.e., QUASAR 5‐FU).
Abbreviations: ECOG, Eastern Coorperative Oncology Group; BSA, Body Surface Area; GI, gastrointestinal; H&N, Head and Neck; NR, not reported; WT, wild‐type.
South Asians (n = 1 Indian carrying c.1236G>A, n = 1 Sir Lankan carrying c.1905+1G>A).
Others include mixed ethnicities such as Indian/English/Ashkenazi Jew, English/Filipino.
Other [Gynecological (n = 1)/Urogenital (n = 1)/cancer of unknown primary (n = 3)].
TABLE 2.
Fluoropyrimidine regimen characteristics of treated patients (PACIFIC‐PGx trial vs. historical control).
| Characteristic | PACIFIC‐PGx | Historical cohorts (Froehlich et al. 2015 13 ) | ||
|---|---|---|---|---|
| DPYD WT full dose | DPYD variant allele carriers genotype‐guided dosing | Total | Historical cohort | |
| (n = 261) | (n = 15) | (n = 276) | (Total = 500) | |
| n (%) | n (%) | n (%) | n (%) | |
| Prior anticancer treatment | ||||
| Previously treated with anticancer treatment a | 113 (43) | 7 (47) | 120 (43) | NR |
| No prior treatment | 148 (57) | 8 (53) | 156 (57) | NR |
| Type of treatment regimen | ||||
| Total 5‐FU regimens | 181 (69) | 10 (67) | 191 (69) | 394 (79) |
| 5‐FU monotherapy (+/− LV) | 3 (1) | 0 (0) | 3 (1) | 100 (20) |
| 5‐FU‐based combination chemotherapy plus RT | 25 (10) | 1 (7) | 26 (9) | NR |
| 5‐FU‐based combination chemotherapy plus other anti‐cancer treatments b | 153 (59) | 9 (60) | 162 (59) | 294 (59) |
| Total CAP regimens | 80 (31) | 5 (33) | 85 (31) | 109 (22) |
| CAP monotherapy | 28 (11) | 3 (20) | 31 (11) | 40 (8) |
| CAP‐based combination chemotherapy plus RT | 31 (12) | 0 (0) | 31 (11) | NR |
| CAP‐based combination chemotherapy plus other anti‐cancer treatments a | 21 (8) | 2 (13) | 23 (8) | 69 (14) |
| DPYD variants | ||||
| Total | NA | 15 | 15 (5) | 31 c (6) |
| c.1905+1G>A | 0 (0) | 4 (27) | 4 (1) | 4 (1) |
| c.1679T>G | 0 (0) | 0 (0) | 0 (0) | 2 (0) |
| c.2846A>T | 0 (0) | 3 (20) | 3 (1) | 3 (1) |
| c.1236G>A | 0 (0) | 8 (53) | 8 (3) | 22 d (4) |
| c.557A>G | 0 (0) | 0 (0) | 0 (0) | NR |
Note: Full dose is classified as equal or greater than 94.5%. If 5FU infusion and 5FU bolus were part of the chemotherapy regimen, 5FU infusion full dose was applied, otherwise 5FU bolus full dose was applied (i.e., QUASAR 5FU).
Abbreviations: 5‐FU, 5‐fluorouracil; RT, radiotherapy; CAP, capecitabine; LV, leucovorin; NA, not applicable; NR, not reported; WT, wild‐type.
Chemotherapy/RT/PRRT/surgery/immunotherapy/target therapy/other.
Targeted therapy, immunotherapy etc.
DPYD variant: c.1601G>A – tested as part of historical cohort but it is not applicable to the PACIFIC trial, therefore excluded from the analysis.
Does not include extra two patients who were homozygous or compound heterozygous.
DPYD genotyping (n = 462) identified 96% (n = 443/462) WT, 4% (n = 19/462) IM, and 0% PM (Figure 1a). UGT1A1 genotyping (n = 50) identified 52% (n = 26/50) WT (UGT1A1*1/*1), 40% (n = 20/50) heterozygous (UGT1A1*1/*28), and 8% (n = 4/50) homozygous (UGT1A1*28/*28) (Figure 1b). All treated patients received protocol specified PGx‐guided dosing reductions.
Historical control for fluoropyrimidines: patient demographics and treatment characteristics
The historic control cohort included 500 patients treated with standard dose FP‐based chemotherapy. 2 , 13 Of these patients, 31 patients were DPYD variant allele carriers (c.1679T>G c.1236G>A/HapB3 c.1601G>A) (Table 2). Similar to the PACIFIC‐PGx cohort, the majority of patients were males, 60% (301/500) with age ranging from 18 to 99 years and of Caucasian ethnicity 99% (493/500) (Table 1). Colorectal cancers were the most common 55% (275/500), and most patients were treated with 5‐FU‐based combination chemotherapy plus anti‐cancer treatment 59% (294/500) (Tables 1 and 2).
Trial feasibility outcomes
Most patients screened were eligible and enrolled in the trial for DPYD genotyping [96% (331/345) at the primary site and 92% (131/142) at the regional sites] (feasibility cohort). Similarly, high enrolment rates were observed for irinotecan at regional sites (100%, 26/26) but not the primary site (29%, 24/83). Overall, 96% of gene test results were analyzed and reported prior to cycle 1 for both DPYD and UGT1A1 [primary site 99% (340/345) and regional sites 91% (123/135)] (Table 3). Teleheatlh consultations were utilized to conduct program visits by the PGx pharmacist (including consent, education, remote DNA patient self‐sampling and post cycle 1 follow‐up) for nearly half of the cohort with 100% of regional and 28% of metropolitan patients having at least their first visit via telehealth. All cheek swab samples collected gave conclusive assay results without requiring subsequent blood sampling. The average time from sample collection to reporting of results was five and seven days for DPYD and UGT1A1 respectively. Detailed trial feasibility outcomes are presented in Table 3.
TABLE 3.
PACIFIC‐PGx trial feasibility outcomes.
| Primary site (PS) | Satellite sites (SS) | PS and SS | |||
|---|---|---|---|---|---|
| n (%) | Dominator | n (%) | Dominator | Total n (%) | |
|
Feasibility cohort |
331 (96) | 345 | 131 (92) | 142 | 462 (95) |
|
Feasibility cohort |
24 (29) | 83 | 26 (100) | 26 | 50 (46) |
| Safety cohort (DPYD genotyping) | 176 (53) | 331 | 100 (76) | 131 | 276 (60) |
| Safety cohort (UGT1A1 genotyping) | 17 (71) | 24 | 15 (58) | 26 | 32 (64) |
| First visit by telehealth for both DPYD/UGT1A1 | 98 (28) | 345 | 135 (100) | 135 | 233 (49) |
| Cheek swab samples collected for both DPYD/UGT1A1 | 25 (7) | 345 | 135 (100) | 135 | 160 (33) |
| Inconclusive assay results from the cheek swab for both DPYD/UGT1A1 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) |
| Gene test results reported prior to cycle one (DPYD/UGT1A1) | 340 (99) | 345 | 123 (91) | 135 | 463 (96) |
| Average days to results (DPYD) c | 5 | NA | 5 | NA | NA |
| Average days to results (UGT1A1) c | 7 | NA | 7 | NA | NA |
| Guidelines implemented by clinicians (DPYD IM) | 7 (88) | 8 | 8 (100) | 8 | 15 (94) d |
| Guidelines implemented by clinicians (UGT1A1*28 PM) | 2 (100) | 2 | 0 (0) | 2 | 2 (50) e |
Abbreviations: IM, intermediate metabolizer; PM, poor metabolizer.
Proportionate of patients that were genotyped.
A subset of patients invited to submit an additional gene sample for research purposes (whole exome sequencing, results to be reported separately), 94% provided samples. Some patients were tested for both DPYD and UGT1A1.
Average days to results for DPYD and UGT1A1 were calculated based on the feasibility cohort.
Overall 94% (15/16) of patients had dose recommendations implemented by their treating clinician for DPYD intermediate metabolizers; the single outlier did have a dose reduction but to 66.7% instead of the recommended 50%. Therefore, this patient was excluded from the analysis.
Overall 50% (2/4) were homozygous for UGT1A1*28 and received the recommended PGx‐guided dose reduction; the two outliers required urgent treatment with the decision made by the clinician to proceed without UGT1A1 results. Therefore, these patients were excluded from the analysis.
High‐grade toxicities between PACIFIC‐PGx trial and historical control
For the secondary outcome of high‐grade toxicity rates among DPYD variant allele carriers, we report 7% (1/15) in the PACIFIC‐PGx group with PGx‐guided dosing versus 39% (12/31) in the historic control group receiving standard dosing 13 (OR 0.11, 95% CI 0.01–0.97, p = 0.024) (Figure 2, Table S5). Rates of high‐grade toxicities among WT patients were similar (14% vs. 14%; OR 0.99, 95% CI 0.64–1.54, p = 0.490) (Figure 2, Table S5). The OR for high‐grade toxicity for DPYD variants versus DPYD WT in PACIFIC‐PGx was 0.45 (95% CI 0.06–3.50, p = 0.485), compared to 3.93 (95% CI 1.82–8.47, p = 0.001) in the historic control. 13 Using a one‐tailed test (assumption that PGx‐guided dose reductions would reduce toxicity), there was a significant difference between the OR of the two studies (OR 0.11, 95% CI 0–0.72, p = 0.026) (Figure 2). However, this difference was no longer significant when the two‐tailed test was applied (OR 0.11, 95% CI 0.01–1.02, p = 0.053).
FIGURE 2.
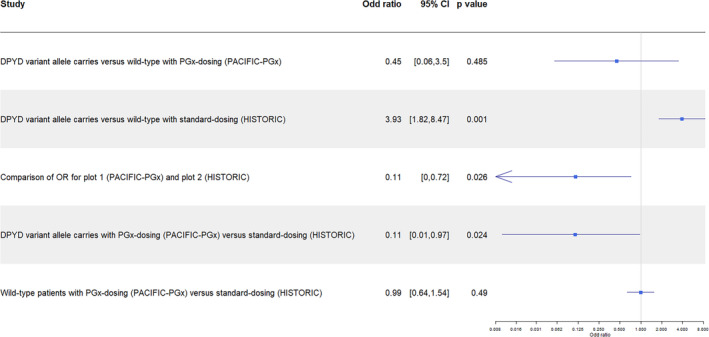
Odd ratio (OR) for high‐grade toxicity between DPYD variant allele carriers and wild‐type patients, and between PGx‐dosing (PACIFIC‐PGx) and standard dosing (HISTORIC control). Historical control cohort from Froehlich et al. 2015. 13 Cl, confidence interval; OR, odds ratio; PGx, pharmacogenetics.
PACIFIC‐PGx: fluoropyrimidine related toxicities
High‐grade toxicities were observed in 13% (37/276) of patients (Table 4) whilst 82% (225/276) had grade 1/2 toxicities (Table S6). The median time to first grade 3/4 toxicity event was 14 days ([IQR 10, 32], p < 0.001) (Table S7). In multivariable time‐to‐event analyses, gender predicted high‐grade toxicities (Figure 3) with males less likely to develop high‐grade toxicities than females (HR 0.45, 95% CI [0.22–0.92], p = 0.03). In logistic regression, cancer type also predicted high‐grade toxicity: patients with a non‐GI solid cancers (OR = 3.828, 95% CI 1.473–10.149, p = 0.006) and upper GI cancers (OR = 2.381, 95% CI 0.968–6.002, p = 0.060) were more likely to experience high grade toxicities compared to lower‐GI cancer (Figure S3). Univariate analyses (Table S7) and Forest Plots for expanded multivariable analysis of all grade events (Figure S1) and high‐grade events (Figure S2) are provided.
TABLE 4.
Grade 3/4 toxicities for PACIFIC‐PGx trial patients treated with fluoropyrimidines.
| Toxicities | DPYD WT full dose | DPYD variant allele carriers genotype‐guided dosing | Overall DPYD population |
|---|---|---|---|
| (n = 261) | (n = 15) | (n = 276) | |
| n (%) | n (%) | n (%) | |
| Grade 3/4 toxicity at either timepoint (assessment 1 or assessment 2) | 36 (14) | 1 (7) | 37 (13) |
| Total Grade 3/4 toxicity at both timepoints (assessment 1 and assessment 2) | 52 | 2 | 54 |
| Assessment 1: Post C1–PreC2 toxicities | |||
| n= | 260 | 15 | 275 |
| G3/4 overall toxicities (at least one toxicity event per patient) | 20 (8) | 0 | 20 (7) |
| Total G3/4 toxicity events | 30 | 0 (0) | 30 |
| Diarrhea | 9 (3) | 0 (0) | 9 (3) |
| Nausea | 1 (0) | 0 (0) | 1 (0) |
| Vomiting | 1 (0) | 0 (0) | 1 (0) |
| Mucositis | 4 (2) | 0 (0) | 4 (1) |
| Fever | 1 (0) | 0 (0) | 1 (0) |
| UTI resulting in fall and ED admission | 1 (0) | 0 (0) | 1 (0) |
| Hepatitis | 1 (0) | 0 (0) | 1 (0) |
| Community acquired pneumonia | 2 (1) | 0 (0) | 1 (0) |
| Neutrophil count decreased | 3 (1) | 0 (0) | 3 (1) |
| Febrile Neutropenia | 1 (0) | 0 (0) | 1 (0) |
| Platelet count decreased | 2 (1) | 0 (0) | 2 (1) |
| Anemia | 2 (1) | 0 (0) | 2 (1) |
| White blood cell count decreased | 2 (1) | 0 (0) | 2 (1) |
| Assessment 2: PreC3 visit toxicities | |||
| n= | 240 | 15 | 225 |
| G3/4 overall toxicities (at least one toxicity event per patient) | 23 (10) | 1 (7) | 24 (9) |
| Total G3/4 toxicity events | 32 | 2 | 34 |
| Diarrhea | 9 (4) | 1 (0) | 10 (4) |
| Mucositis | 2 (1) | 0 (0) | 2 (1) |
| Fever | 1 (0) | 0 (0) | 1 (0) |
| Colitis | 1 (0) | 0 (0) | 1 (0) |
| Neutrophil count decreased | 7 (3) | 0 (0) | 7 (3) |
| Neutropenic sepsis | 0 (0) | 1 (0) | 1 (0) |
| Febrile neutropenia | 2 (1) | 0 (0) | 2 (1) |
| Platelet count decreased | 2 (1) | 0 (0) | 2 (1) |
| Anemia | 2 (1) | 0 (0) | 2 (1) |
| White blood cell count decreased | 6 (3) | 0 (0) | 6 (2) |
Abbreviations: G, grade; UTI, urinary tract infection; WT, wild‐type.
FIGURE 3.
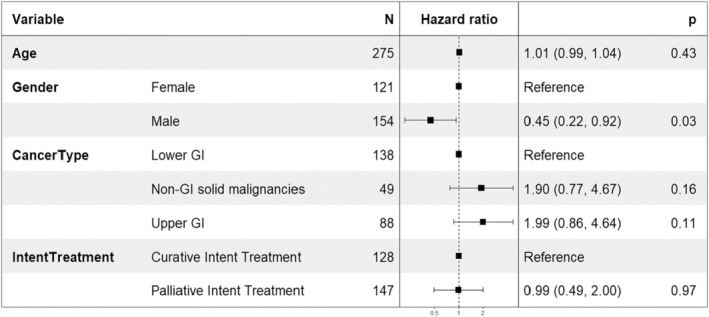
Forest plot of the multivariable analysis for high grade toxicity (grade 3 or 4) using COX Proportional Hazards Models for patients treated with fluoropyrimidines. GI, gastrointestinal.
Of the 15 patients identified as DPYD variant allele carriers, one (7%) experienced high‐grade toxicities (grade ≥3 diarrhea and neutropenic sepsis) at cycle 2 day 10. This patient was c.1905+1G>A variant allele carrier and received capecitabine alone (50% dose at cycle 1 and 2) for metastatic breast cancer. The patient was hospitalized and received uridine triacetate (antidote for FP toxicity), with the capecitabine subsequently discontinued.
In the capecitabine group, two patients (DPYD normal metabolizers) were admitted to the intensive care unit (ICU) and resulting in higher average ICU hours of 216 (range 168–264) in comparison to one 5‐FU patient with 18 h (range 18–18) (Table S8). Of the two patients that were admitted to ICU with capecitabine related toxicities, one received bevacizumab/CAPOX (capecitabine 94% of the dose) for metastatic colon cancer, admitted to emergency department (ED) with grade 4 colitis at cycle 1 day 13, followed by ICU admission (168 h) and ward admission (12 days). The other patient received capecitabine (94.7%) and trastuzumab for metastatic breast cancer and presented to ED with diarrhea complicated with ileus at cycle 1 day 17, requiring ICU admission (264 h) and ward admission (21 days) (Table S8).
Among DPYD variant allele carriers (6/15) followed up for a additional 2 cycles, no high‐grade toxicities or hospitalizations were observed in the context of proposed dose escalation. All six patients received 50% of FP dose at cycles 1 and 2. Only one clinician increased the FP dose to 100% for one patient carrying c.1236G>A variant (1/6), the other five patients continued to receive reduced dose of FP (ranged from 57.4% to 75%).
Hospitalizations and death between PACIFIC‐PGx trial and historical control
PGx dosing for DPYD variant allele carriers in PACIFIC‐PGx trial reduced hospitalization for G3/4 treatment‐related toxicities [7% (1/15)] compared to historical controls [White 2023: 56% (9/16), RR 0.12 (95% CI 0.02–0.83, p = 0.002), 17 Toffoli 2019: 30% (11/37), RR 0.22 (95% CI 0.03–1.59, p = 0.040), 18 Lunenburg 2018 18% (6/34), RR 0.38 (95% CI 0.05–2.87, p = 0.180)]. 19 Pooled hospitalizations: [29% (26/89), RR 0.22 (95% CI 0.03–1.56, p = 0.031)]. The comparison versus White 2023 17 remained statistically significant when the two‐tailed test was applied (p = 0.004). Median duration of hospitalization for variant allele carriers with PGx‐guided dosing in PACIFIC‐PGx was 5.5 days (range 1–10), comparable to the Lunenberg PGx‐dosing cohort (4 days, range 2–5), and shorter than the Lunenberg standard‐dose cohort (23 days, range 6–36). 19 The WT cohort, as part of the PACIFIC‐PGx trial, showed similar average hours in terms of ED utilization (12 h vs. 11 h) and ward days (6.9 days vs. 6.5 days) between capecitabine and 5‐FU treatments, Table S8. There were zero deaths reported in the genotype‐guided dosing compared to 3.7% in historical controls when tested for the following variants c.1679T>G, c.1905+1G>A, and/or c.2846A>T. 21 Full details on hospitalizations are presented in Table S8.
Irinotecan‐related toxicities and hospitalizations
There were no high‐grade toxicities or hospitalizations reported for UGT1A1*28 homozygotes, in comparison to 6% (1/17) and 18% (2/11) in WT and UGT1A1*28 heterozygotes (for both toxicities and hospitalizations) (Tables S9 and S10). Grade 1/2 toxicity events for irinotecan are summarized in Table S11.
DISCUSSION
This is the largest prospective multicenter clinical trial in Australia that evaluated the feasibility of implementing a PGx screening program for DPYD and UGT1A1 into routine oncology care. We demonstrated that PGx screening was feasible in both metropolitan and regional healthcare settings, with testing efficiently performed and reported prior to planned treatment initiation, enabling genotype‐guided dosing that reduced severe toxicities, hospitalizations, and deaths.
Our results showed that dose individualization improved the safety of FP with the rate of high‐grade toxicities reduced from 39% in the historical control to 7% in the genotype‐guided dosing cohort (p = 0.024). 13 Similarly, Deenan 2016 reported reduction in rates of Grade ≥3 toxicities from 73% in the historical control to 28% in the genotype‐guided cohort for DPYD*2A. 12 While PGx‐guided dosing has been associated with potential ‘under‐dosing’, 19 we found no difference in high‐grade toxicities between DPYD variant allele carriers receiving reduced dose compared to WT patients receiving full dose. This is consistent with findings from a large United States (US) study (n = 757) also reporting no difference in high‐grade toxicity rates between DPYD carriers receiving reduced dose versus WT receiving full dose (31% vs. 30%). 22
Our study demonstrated a reduction in FP‐related hospitalizations compared to historical controls. 17 , 18 , 19 Although only one comparison (White 2023) maintained statistical significance with a two‐tailed test, likely due to the small sample size. This cohort was the most comparable to ours, as it involved an Australian population with similar treatment regimens, dosing strategies, and healthcare systems. 17 In White 2023, 56% of DPYD variant allele carriers receiving standard FP dosing required hospitalization, compared to just 7% in our genotype‐guided dosing cohort p = 0.004. 17 In contrast, Lunenberg (2018) reported comparable hospitalization rates between DPYD variant allele carriers receiving reduced and full FP doses, but observed significantly shorter hospital stays for those receiving reduced doses, consistent with our findings. 19 Notably, there were no FP‐induced deaths in our cohort, compared to a 3.7% mortality rate in historical controls. 21 Similarly, Deenen 2016 showed an absolute risk reduction in drug‐induced mortality from 10% in the historical full‐dose group to 0% in their genotype‐guided cohort for DPYD*2A carriers. 12
Higher‐grade toxicities were observed more frequently among patients with upper GI and non‐GI cancers compared to lower GI cancers. This may reflect the former group being a more heavily pre‐treated population, presenting with a higher tumor burden and receiving greater intensity chemotherapy. However, the lack of statistical significance suggests this may be a chance finding of our study. Females were at a greater risk of developing high‐grade FP toxicities, consistent with international studies, 23 , 24 possibly due to reduced 5‐FU clearance in females than males. 24 , 25
Our study is the largest to explore the prevalence of the five common DPYD variants among Australian multicultural ethnicities, adding to the recently reported single center study (n = 104) by White (2023). 17 In addition to standard DPYD variants, we tested for c.557A>G variant, predominant in African populations 16 ; however, we did not observe this variant due to the smaller than expected number of patients with African ethnicity (n = 3). Most patients recruited were of Caucasian ethnicity with 4% of patients genotyped as DPYD IM, consistent with studies previously published in European countries. 26 , 27
In our study, patients who were homozygous for UGT1A1*28 did not experience high‐grade toxicities or hospitalizations. Given the variable uptake of genotype‐guided dosing recommendations, this may be a chance finding in our small irinotecan cohort. Reduction in high‐grade toxicities was demonstrated by Hulshof 2022, 4 comparing UGT1A1 genotype‐guided dosing to historical controls, with a 17.5% reduction in febrile neutropenia (6.5% vs. 24%). 4 The study also reported that there was adequate SN38 exposure in the UGT1A1*28 homozygous receiving dose reduction in comparison to UGT1A1 standard irinotecan dosing (WT/heterozygous) and hence concluded that a 30% dose reduction was optimal in UGT1A1*28 homozygous. 4 UGT1A1*6 is more common in Asian population 4 ; however, due to the lack of DPWG dosing guidelines at the time when this trial was initiated (Jan‐2021), it was not tested in our study. However, it will be tested as part of whole exome sequencing on stored samples, and it will be reported in a separate publication.
Pre‐treatment DPYD and UGT1A1 genotyping was conducted at metropolitan and regional hospitals, as part of daily practice showing the feasibility of implementation in a variety of Australian healthcare settings. Importantly, there was no delay in starting treatment (96% of results analyzed and reported prior to cycle‐1, within 5–7 days). A study conducted by Nguyen 2024 from the US has demonstrated similar outcomes to our study showing that 90% of patients received their results before starting FP treatment, with median turn‐around time of 6 days. 22 A US study by Tracksdorf 2024 reported ~90% of sites having DPYD results within 10 days prior to FP commencement. 28 This should provide assurance to the 76% clinicians identified in a national survey conducted recently by our group that turn‐around times for PGx testing were a barrier to implementation within health services in Australia. 29
An important feature of our study was the pharmacist‐led centralized tele‐trial model, allowing regional patients to access PGx testing service not otherwise available at their local hospital and incorporated genotype‐guided dosing changes into clinical verification of chemotherapy orders. The centralized model in which all regional patients were consulted via telehealth and cheek swab kits utilized to enable at home DNA sampling, proved feasible/acceptable. The PGx screening program was successfully implemented and continues post‐trial as part of routine care at two out of four hospitals. One of the top barriers to implementing PGx testing in Australia is the lack of financial reimbursement for DPYD genotyping, as it is not currently government funded. Recent surveys of Australian clinicians identified the cost of the service as the primary obstacle to implementing PGx screening programs. 29 , 30 To address this challenge, our group is preparing a cost‐effectiveness analysis for PGx testing in Australian healthcare settings to provide evidence supporting broader implementation. Similarly, a recent U.S. survey also identified cost as a key barrier to PGx implementation 28 despite Medicare reimbursement for DPYD testing in 40 out of 50 states based on the local coverage for PGx testing assigned by CPIC level A or B, and additional coverage from some private insurers. 31 Some institutions also offset testing costs through research or institutional funding. 28
This study has several limitations, particularly regarding the use of historical control cohorts. For hospitalization comparisons, follow‐up duration varied from 2 to 12 cycles, with longer follow‐up likely leading to more hospitalizations, though acknowledging acute FP‐related hospitalizations most common during initial treatment cycles. Differences in cohort populations and treatment regimens were more pronounced for some hospitalization controls, such as the Toffoli 2019 cohort, 18 which was limited to colorectal cancer, and the Lunenburg 2018 cohort, 19 which included only patients receiving FP with radiotherapy—a combination associated with fewer and different toxicities compared to multi‐agent chemotherapy regimens. These differences are clearly described to aid in the interpretation of results. Importantly, one hospitalization control (White 2023) 17 and one toxicity control (Froehlich 2015) 13 were largely comparable to our study. Additionally, while detailed demographic and treatment information from the historical controls was limited, the key factors required for comparison—such as PGx screening alleles, cancer diagnoses, treatment types, and follow‐up duration—were sufficiently similar. The consistency in toxicity rates among WT patients receiving standard BSA dosing further supports cohort comparability regarding treatment‐related toxicities. To enhance comparability, our toxicity control cohort was derived from a single historical study, which limited sample size and power but helped reduce bias from using controls with differing patient and treatment characteristics. Lastly, given that most patients in this study were of Caucasian ethnicity, further research is needed to explore the frequency and clinical relevance of DPYD variants in other ethnic populations, including the impact of UGT1A1*6 and other variants with CPIC dosing recommendations.
Implementation of a coordinated PGx screening program in Australian health care settings was feasible within routine oncology care. PACIFIC‐PGx has established the potential of a new model of care associated with reduced high‐grade toxicities, hospitalizations, and deaths than historical controls.
AUTHOR CONTRIBUTIONS
S.G., M.M., and M.A. designed the research, performed the research, analyzed the data, and wrote manuscript. M.J. and S.S. analyzed research and wrote manuscript. S.L., B.L., I.C., M.I., M.F., S.H., C.G., C.U., M.W., R.C., J.H.M., and J.T. wrote the manuscript.
ACKNOWLEDGMENTS
Open access publishing facilitated by The University of Melbourne, as part of the Wiley ‐ The University of Melbourne agreement via the Council of Australian University Librarians.
FUNDING INFORMATION
This work was supported by the International Society of Oncology Pharmacy Practitioners (ISOPP) under ISOPP Research Grant Award.
CONFLICT OF INTEREST STATEMENT
The authors declared no competing interests for this work.
Supporting information
Appendix S1
Glewis S, Lingaratnam S, Lee B, et al. Pharmacogenetic‐guided dosing for fluoropyrimidine (DPYD) and irinotecan (UGT1A1*28) chemotherapies for patients with cancer (PACIFIC‐PGx): A multicenter clinical trial. Clin Transl Sci. 2024;17:e70083. doi: 10.1111/cts.70083
Marliese Alexander and Michael Michael equal contributions.
Trial registration: ANZCTR registration number 12621000251820.
REFERENCES
- 1. Meyer UA. Pharmacogenetics – five decades of therapeutic lessons from genetic diversity. Nat Rev Genet. 2004;5(9):669‐676. [DOI] [PubMed] [Google Scholar]
- 2. Henricks LM, Lunenburg CATC, de Man FM, et al. DPYD genotype‐guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 2018;19(11):1459‐1467. [DOI] [PubMed] [Google Scholar]
- 3. Glewis S, Alexander M, Lingaratnam S, et al. Pharmacogenomics guided dosing for fluoropyrimidine and irinotecan chemotherapies for patients with cancer (PACIFIC‐PGx): study protocol of a multicentre clinical trial. Acta Oncol. 2022;61(9):1136‐1139. [DOI] [PubMed] [Google Scholar]
- 4. Hulshof EC, De With M, De Man FM, et al. UGT1A1 genotype‐guided dosing of irinotecan: a prospective safety and cost analysis in poor metaboliser patients. Eur J Cancer. 2022;162:148‐157. [DOI] [PubMed] [Google Scholar]
- 5. Karas S, Innocenti F. All you need to know about UGT1A1 genetic testing for patients treated with irinotecan: a practitioner‐friendly guide. JCO Oncol Pract. 2022;18(4):270‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. U.S. Food and Drug Administration . United States: FDA [updated 2024; cited 2024 Nov 23]. Available from: https://www.fda.gov/drugs/resources‐information‐approved‐drugs/fda‐approves‐safety‐labeling‐changes‐regarding‐dpd‐deficiency‐fluorouracil‐injection‐products
- 7. European Society for Medical Oncology . EMA starts review on screening patients before treatment with fluorouracil, capecitabine, tegafur and flucytosine. Ginevra Lugano (USA): ESMO; 2019 Mar 21. Accessed July 28, 2021. https://www.esmo.org/oncology‐news/EMA‐Starts‐Review‐on‐Screening‐Patients‐Before‐Treatment‐with‐Fluorouracil‐Capecitabine‐Tegafur‐and‐Flucytosine
- 8. Australian Government Department of Health and Aged Care Therapeutic Good Administrations . Fluorouracil and capecitabine ‐ DPD deficiency [Internet]. Australia: TGA 2022 [Updated 2022, cited 2022 September 14]. Availiable from: https://www.tga.gov.au/news/safety‐updates/fluorouracil‐and‐capecitabine‐dpd‐deficiency
- 9. Perera MA, Innocenti F, Ratain MJ. Pharmacogenetic testing for uridine diphosphate glucuronosyltransferase 1A1 polymorphisms: are we there yet? Pharmacotherapy. 2008;28:755‐768. doi: 10.1592/phco.28.6.755 [DOI] [PubMed] [Google Scholar]
- 10. Pratt VM, Cavallari LH, Fulmer ML, et al. DPYD genotyping recommendations. J Mol Diagn. 2024;26:851‐863. [DOI] [PubMed] [Google Scholar]
- 11. Brooks GA, Tapp S, Daly AT, Busam JA, Tosteson ANA. Cost‐effectiveness of DPYD genotyping prior to fluoropyrimidine‐based adjuvant chemotherapy for colon cancer. Clin Colorectal Cancer. 2022;21(3):e189‐e195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deenen MJ, Meulendijks D, Cats A, et al. Upfront genotyping of DPYD * 2A to individualize fluoropyrimidine therapy: a safety and cost analysis. J Clin Oncol. 2016;34(3):227‐234. [DOI] [PubMed] [Google Scholar]
- 13. Froehlich TK, Amstutz U, Aebi S, Joerger M, Largiadèr CR. Clinical importance of risk variants in the dihydropyrimidine dehydrogenase gene for the prediction of early‐onset fluoropyrimidine toxicity. Int J Cancer. 2015;136(3):730‐739. [DOI] [PubMed] [Google Scholar]
- 14. Collins IM, Burbury K, Underhill CR. Teletrials: implementation of a new paradigm for clinical trials. Med J Aust. 2020;213(6):263‐265.e1. [DOI] [PubMed] [Google Scholar]
- 15. Meulendijks D, Henricks LM, Sonke GS, et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine‐associated toxicity: a systematic review and meta‐analysis of individual patient data. Lancet Oncol. 2015;16(16):1639‐1650. [DOI] [PubMed] [Google Scholar]
- 16. Amstutz U, Henricks LM, Offer SM, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther. 2018;103(2):210‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. White C, Paul C, Liet E, et al. Feasibility of DPYD Genotyping in Australian Cancer Patients. 2023. Accessed September 10, 2024. https://www.researchsquare.com/article/rs‐2838793/v1
- 18. Toffoli G, Innocenti F, Polesel J, et al. The genotype for dpyd risk variants in patients with colorectal cancer and the related toxicity management costs in clinical practice. Clin Pharmacol Ther. 2019;105(4):994‐1002. [DOI] [PubMed] [Google Scholar]
- 19. Lunenburg CATC, Henricks LM, Dreussi E, et al. Standard fluoropyrimidine dosages in chemoradiation therapy result in an increased risk of severe toxicity in DPYD variant allele carriers. Eur J Cancer. 2018;104:210‐218. [DOI] [PubMed] [Google Scholar]
- 20. R Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2023. https://www.R‐project.org/ [Google Scholar]
- 21. Sharma BB, Rai K, Blunt H, Zhao W, Tosteson TD, Brooks GA. Pathogenic DPYD variants and treatment‐related mortality in patients receiving fluoropyrimidine chemotherapy: a systematic review and meta‐analysis. Oncologist. 2021;26(12):1008‐1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nguyen DG, Morris SA, Hamilton A, et al. Real‐world impact of an in‐house dihydropyrimidine dehydrogenase (DPYD) genotype test on fluoropyrimidine dosing, toxicities, and hospitalizations at a multisite cancer center. JCO Precis Oncol. 2024;8:e2300623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chansky K, Benedetti J, Macdonald JS. Differences in toxicity between men and women treated with 5‐fluorouracil therapy for colorectal carcinoma. Cancer. 2005;103(6):1165‐1171. [DOI] [PubMed] [Google Scholar]
- 24. Schwab M, Zanger UM, Marx C, et al. Role of genetic and nongenetic factors for fluorouracil treatment‐related severe toxicity: a prospective clinical trial by the German 5‐FU Toxicity Study Group. J Clin Oncol. 26(13):2131‐2138. doi: 10.1200/JCO.2006.10.4182 [DOI] [PubMed] [Google Scholar]
- 25. Amstutz U, Froehlich TK, Largiadèr CR. Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5‐fluorouracil toxicity. Pharmacogenomics. 2011;12(9):1321‐1336. [DOI] [PubMed] [Google Scholar]
- 26. Varughese LA, Lau‐Min KS, Cambareri C, et al. DPYD and UGT1A1 pharmacogenetic testing in patients with gastrointestinal malignancies: an overview of the evidence and considerations for clinical implementation. Pharmacotherapy. 2020;40(11):1108‐1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miarons M, Manzaneque Gordón A, Riera P, et al. Allelic frequency of DPYD genetic variants in patients with cancer in Spain: the PhotoDPYD study. Oncologist. 2023;28(5):e304‐e308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tracksdorf T, Smith DM, Pearse S, et al. Strategies for DPYD testing prior to fluoropyrimidine chemotherapy in the US. Support Care Cancer. 2024;32(8):497. doi: 10.1007/s00520-024-08674-1 [DOI] [PubMed] [Google Scholar]
- 29. Glewis S, Lingaratnam S, Krishnasamy M, et al. Pharmacogenetics testing (DPYD and UGT1A1) for fluoropyrimidine and irinotecan in routine clinical care: perspectives of medical oncologists and oncology pharmacists. J Oncol Pharm Pract. 2023;30:30‐37. [DOI] [PubMed] [Google Scholar]
- 30. Glewis S, Krishnasamy M, Lingaratnam S, et al. Patient and healthcare professional acceptability of pharmacogenetic screening for DPYD and UGT1A1: a cross sectional survey. Clin Transl Sci. 2023;16(12):2700‐2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koo K, Pasternak AL, Henry NL, Sahai V, Hertz DL. Survey of US medical oncologists' practices and beliefs regarding DPYD testing before fluoropyrimidine chemotherapy. JCO Oncol Pract. 2022;18(6):e958‐e965. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1