

Abstract
Adeno-associated virus (AAV) is a widely used vehicle for gene delivery, lending interest to developing methods for enhancing AAV transduction and transgene expression. Here, we profile the function of several topoisomerase poisons, which are small molecules that stabilize topoisomerase enzymatic intermediates, where topoisomerase enzymes are covalently bound at chromosomal DNA breaks. As previously observed, we found that the topoisomerase poisons camptothecin (CPT), doxorubicin (DOX), and etoposide (ETO) increased AAV transduction in cultured cell models. DOX and ETO, small molecules that specifically inhibit type II topoisomerases and so stabilize double-strand breaks, were found to boost integration of AAV DNA into the host cell chromosome. Analysis of integration site distributions showed that integration targeting was altered, so that integration in the presence of DOX or ETO was favored near actively transcribed regions. Locations of topoisomerase II binding sites were inferred from genomic data using a novel machine learning platform, and integration in the presence of DOX or ETO was found to be selectively favored near inferred topoisomerase II binding sites. These data help guide development of improved transduction protocols using these reagents and establish that DOX and ETO can control AAV integration targeting.
Keywords: AAV vectors, topoisomerase, doxorubicin, adriamycin, camptothecin, etoposide, AAV integration, machine learning, short-read sequencing, AAV gene therapy
Graphical abstract
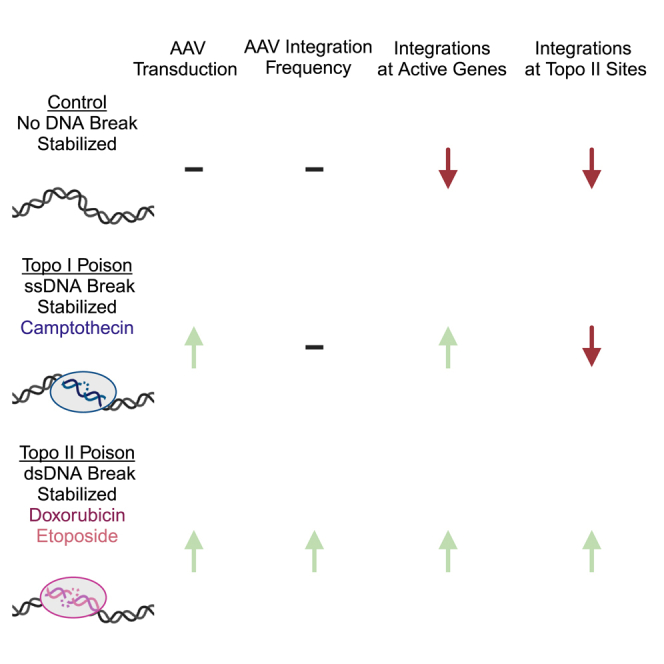
Co-delivery of type I and type II topoisomerase poisons is known to increase AAV transduction but impact on vector integration is unknown. Bushman and colleagues found that type II topoisomerase poison-treated cells have higher integration frequencies and more integration targeting to active genes and type II topoisomerase sites predicted by machine learning.
Introduction
Adeno-associated virus (AAV) vectors are a promising but sometimes inefficient transgene delivery system, motivating the development of methods to improve efficiency and reduce vector dose. Previously, small-molecule modulators of DNA repair pathways have been identified that can augment AAV transduction.1,2,3,4,5 However, the impact of modulators on AAV integration is understudied. Because integration of AAV can promote vector persistence in dividing cells,6 but may also increase risk of genotoxicity,7,8 it is important to understand the mechanisms and consequences in detail.
One category of small-molecule modulators is topoisomerase poisons. Topoisomerases relax supercoiling in DNA in a reaction involving cleaving DNA while remaining covalently associated at the broken DNA ends. This allows relaxation of super helical tension in DNA. The reaction cycle is completed with DNA rejoining and release of the topoisomerase protein.9 Topoisomerase I enzymes act via generating single-stranded DNA nicks, while topoisomerase II enzymes act by generating DNA double-strand breaks. Topoisomerase poisons stabilize topoisomerase complexes following DNA cleavage, thereby freezing the topoisomerase protein covalently bound to free DNA ends. Poisons of topoisomerase I enzymes such as camptothecin (CPT) stabilize single-strand DNA breaks, while poisons of topoisomerase II enzymes such as doxorubicin (DOX)/adriamycin or etoposide (ETO) stabilize double-strand DNA breaks10,11,12 Thus, CPT, ETO, and DOX induce DNA damage.13,14
These topoisomerase poisons have been reported to be enhancers of AAV transduction. CPT was reported to enhance AAV transduction in cultured human fibroblast cells and mouse cardiac tissue.15,16 DOX was reported to enhance AAV transduction17 in several different tissues and animal models.18,19 ETO was reported to increase AAV transduction in human fibroblast cells, primary human airway cells, human hepatocellular carcinomas, and in mouse and rat hepatocellular carcinomas.15,20 ETO was reported to be ineffective at increasing transduction in rabbit airways.21 Topoisomerase poisons have also been reported to increase AAV DNA synthesis.
ETO was also previously reported to increase AAV integration into cellular chromosomes. Assays tracked the stable incorporation of a selectable marker in cells following ETO treatment. Integration by this assay was also detectably increased by induction of DNA double-strand breaks by other means, including gamma-irradiation and cleavage with a targeted nuclease.22
Here, we evaluated the effects of topoisomerase poisons targeting topoisomerase I (CPT) and topoisomerase II (DOX and ETO) on AAV transduction systematically. We compared several cell models and demonstrated increased integration in the presence of DOX and ETO using qPCR. We quantified integration site distributions and compared results with locations of topoisomerase II binding sites inferred from a new machine learning model, revealing an association selectively in the presence of DOX or ETO. These results provide background and guidance for the use of topoisomerase poisons to optimize AAV transduction.
Results
Enhancement of AAV transduction by CPT, DOX, and ETO
Short-term tests of AAV transduction in the presence of the topoisomerase poisons CPT, DOX, and ETO have demonstrated an increase in vector transduction.15,16,20,21,23 These drugs and their derivatives are used clinically as chemotherapeutic agents to treat cancer, and so have been evaluated extensively for use in humans.24 We thus sought to better understand mechanisms and to optimize their use for increasing AAV transduction.
We carried out initial tests in a HeLa cell model. We surveyed the viability of HeLa cells in response to short-term exposure to CPT, DOX, and ETO and identified the highest dose of each compound that did not induce significant cell death (62.5 nM CPT, 50 nM DOX, and 3.13 μM ETO). To measure the influence of these compounds on transduction, cells were transduced with an ssAAV8 vector expressing a GFP reporter.
HeLa cells treated with all three drugs (Figure 1A) showed more GFP-positive cells by 84 h post transduction than were seen in untreated controls (p < 0.0001 for all three drugs, one-way ANOVA, corrected for multiple comparisons, n = 4). Transduction in both ETO- and DOX-treated cells were increased over the control by 48 h post transduction (p = 0.3792 for CPT, p = 0.0007 for DOX, and p = 0.0002 for ETO, n = 4), but CPT-treated cells did not show an increase until 60 h post transduction (p = 0.0492 for CPT, n = 4). These kinetics were reproducible in another experimental replicate (Figure S1).
Figure 1.

CPT, DOX, and ETO increase AAV transduction in HeLa cells
HeLa cells were treated overnight with either medium only (no drug control, green circles), 62.5 nM of CPT (blue squares), 50 nM DOX (maroon upward triangles), or 3.13 μM of ETO (pink downward triangles) before drug washout. Treated cells were transduced with AAV-GFP and subsequently cultured in the absence of topoisomerase poisons. GFP fluorescence was measured to track AAV gene expression. (A) Over the first 96 h post transduction, phase and green fluorescence live cell images were taken every 12 h using the Incucyte S3 and 25 images were taken per well. The Incucyte analysis software was used to quantify GFP-positive foci, with the reported count taking an average across the 25 images. Data displayed are the mean value and standard deviation of quadruplicate biological replicates (n = 4) per group. Highlighted time points (24, 36, 48, 60, 72, and 84 h) were analyzed using one-way ANOVA with Dunnett’s multiple comparison test of each drug against the control cells. (B and C) As cells were further passaged, harvested HeLa cells were stained with LIVE/DEAD dye, fixed and run on a flow cytometer to determine the percentage of cell population expressing GFP expression. Cells were analyzed at (B) 6 days post transduction and (C) 19 days post transduction. Data displayed are the percent GFP-positive values for each biological replicate with the mean displayed as a horizontal bar. (B) n = 4 and (C) n = 8. Data were analyzed using one-way ANOVA with Dunnett’s multiple comparison test of each drug against the control cells. In all graphs, significance is displayed as such (ns, no significance; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
After the initial drug treatment at the time of AAV transduction, HeLa cells were tracked over multiple passages in the absence of drug. HeLa cells double every ∼24 h. At each passage, we used flow cytometry to quantify the percentage of GFP-positive cells in each sample. In Figure 1B, all three drug conditions show higher percentage of GFP-positive cells at 6 days post transduction (p = 0.0086 for CPT, p = 0.0003 for DOX, and p = 0.0001 for ETO, one-way ANOVA, multiple comparisons, n = 4). The increase in the presence of the drugs was lost as cells were passaged further, possibly due to loss of episomal AAV vector DNA over time. By day 19, the GFP signal stabilized (Figure S2), with cells treated with the type II topoisomerase drugs DOX and ETO showing higher GFP signal compared with control cells. Cells treated with the type I topoisomerase poison CPT returned to control GFP expression levels by day 19 (Figure 1C, p = 0.7951 for CPT, p < 0.0001 for DOX, and p = 0.0062 for ETO, one-way ANOVA, corrected for multiple comparisons, n = 8). The data are consistent with increased frequencies of AAV vector DNA integration selectively in the presence of DOX and ETO.
We next tested the ability of CPT, DOX, and ETO to enhance AAV transduction in primary IMR90 fibroblasts (Figure 2). Non-dividing fibroblasts are more responsive to AAV boosting by topoisomerase drugs and can tolerate higher topoisomerase poison concentrations than HeLa cells.15,25 IMR90 fibroblasts undergo cell-cycle arrest when contact inhibited and return to dividing state when no longer inhibited.26,27 IMR90 cells were grown to 100% confluency before treatment with CPT, DOX, ETO, and AAV transduction. Multiple doses of each drug were tested, supporting previous findings of higher drug tolerability in stationary fibroblast cells (Figures S3A–S3C). All three drugs increased transduction above controls by 96 h (0.1 μM CPT p < 0.0001, 3 μM DOX p < 0.0001, and 300 μM ETO p = 0.0007, one-way ANOVA, corrected for multiple comparisons, n = 4). Compared with HeLa cells, the IMR90 response to CPT was higher, which is consistent with previous studies in human fibroblasts.15 ETO-treated cells showed enhancement starting around 42 h post transduction, reaching significance at 60 h (300 μM ETO p < 0.0001, n = 4). CPT- and DOX-treated cells showed signal above the control GFP signal as early as 12 h post transduction, reaching significance at 18 h (0.3 μM CPT p = 0.0008, 3 μM DOX p < 0.0001, n = 4, one-way ANOVA, corrected for multiple comparisons) (Figure S3D). For unknown reasons, enhancement by DOX was greater than enhancement by ETO in this model (12- versus ∼8-fold), even when compared over multiple concentrations of each (Figures S3B and S3C).
Figure 2.
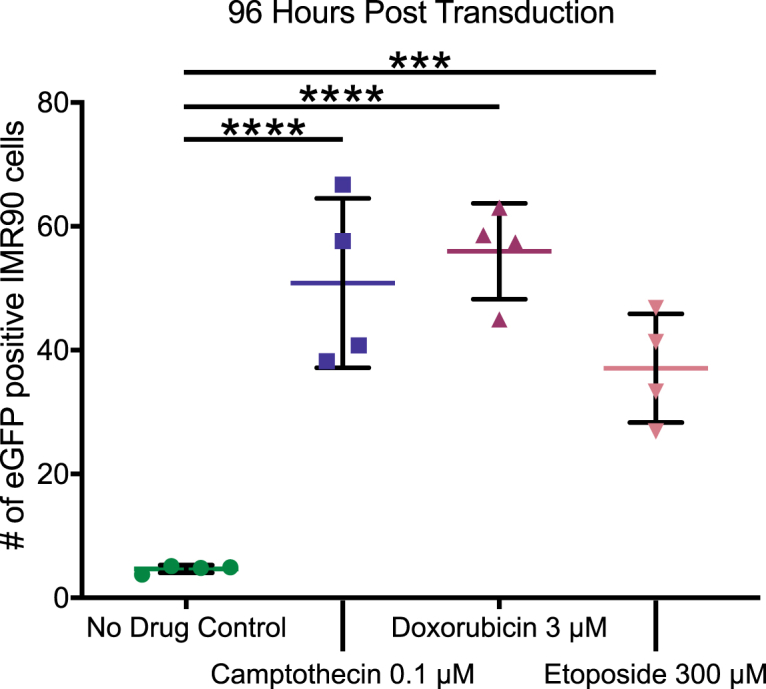
CPT, DOX, and ETO augment AAV transduction in IMR90 cells
Contact-inhibited IMR90 cells were treated overnight with either medium (no drug control, green circles), 0.1 μM CPT (blue squares), 300 μM DOX (maroon upward triangles), or 100 μM of ETO (pink downward triangles) before drug washout. Treated cells were transduced with AAV-GFP and cultured in the absence of topoisomerase drugs. Over the first 120 h post transduction, phase and green fluorescence live cell images were taken every 12 h using the Incucyte S3 and 25 images were taken per well. The Incucyte analysis software was used to quantify GFP-positive foci, with the reported count taking an average across the 25 images. Data displayed are the values for each biological replicate (n = 4) at 96 h post transduction with the mean displayed as a horizontal bar. Fold changes were calculated by comparing the means of each drug condition. Data were analyzed using one-way ANOVA with Dunnett’s multiple comparison test for each drug against the control cells. ns, no significance; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Increased AAV transduction is associated with an increase in vector copy number
We next evaluated whether the loss of GFP expression seen after AAV transduction of HeLa cells (Figures 1B and 1C) correlated with loss of the AAV vector DNA in cells (Figure 3), and whether higher expression was associated with higher copy number. We extracted gDNA from culture subsamples and quantified copy number using qPCR targeting the eGFP transgene. Copy numbers for all samples fell over time, consistent with dilution of unintegrated forms during cell division. At day 6 post transduction, CPT-treated cells had vector copy numbers similar to control cells, DOX-treated cells were 2.6-fold higher and ETO-treated cells were 1.5-fold higher than control cells (Figure 3A, p = 0.9976 for CPT, p = 0.0008 for DOX, and p = 0.2848 for ETO, one-way ANOVA, corrected for multiple comparisons, n = 4). By day 19, CPT-treated cells still had similar VCN to control, and DOX- and ETO-treated cells had higher vector copies than the control, 4- and 2-fold, respectively (Figure 3B, p = 0.4994 for CPT, p < 0.0001 for DOX, and p = 0.0117 for ETO, one-way ANOVA, corrected for multiple comparisons, n = 4).
Figure 3.

CPT, DOX, and ETO have different effects on AAV vector copy numbers in HeLa cells
HeLa cells were harvested during each passage, genomic DNA was extracted and subjected to vector copy number analysis using GFP-targeted primers with three technical replicates per sample. Vector copy number was quantified from cells harvested at (A) 6 days post transduction and (B) 19 days post transduction. Data displayed are the values for each biological replicate (n = 4) with the mean displayed as a horizontal bar. Medium only (no drug control, green circles), 62.5 nM of CPT (blue squares), 50 nM DOX (maroon upward triangles), or 3.13 μM of ETO (pink downward triangles). Fold changes were calculated by comparing the means of each drug condition. Data were analyzed using one-way ANOVA with Dunnett’s multiple comparison test of each drug against the control cells. ns, no significance; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Assessing integration frequency and targeting using ligation-mediated PCR
The elevated AAV vector copy numbers in DOX- and ETO-treated cells even after ∼19 passages indicated that some of the AAV genomes were likely integrated into the host cell genome. If true, this raised the question of how treatment with the topoisomerase drugs affected frequency of integration and selection of integration target sites.
We thus investigated integration frequency and targeting by characterizing AAV-host junctions using ligation-mediated PCR, Illumina paired-end DNA sequencing, and alignment to the human genome.28,29,30,31,32,33,34,35 We analyzed integration sites from the control, CPT-, DOX-, and ETO-treated cells 6 days after transduction (n = 4 replicates per condition). Counts of recovered integration sites showed that CPT-treated cells yielded similar numbers of integration sites as the control cells, but cells treated with both DOX and ETO showed an increase of approximately 2.4- to 2.8-fold over controls (Figure 4A, p = 0.0588 for CPT, p < 0.0001 for DOX, and p < 0.0001 for ETO, one-way ANOVA, corrected for multiple comparisons, n = 4). No single-cell clones were notably expanded in any condition (Figure S4).
Figure 4.

Effects of CPT, DOX, and ETO on AAV integration frequency in HeLa cells
HeLa DNA harvested 6 days post vector transduction was subject to integration site sequencing. (A) Unique integration sites recovered from each biological replicate, quantified using the AAVengeR pipeline. Integration sites were filtered to exclude sites supported by fewer than three reads. (B) Abundance measures were used to estimate the minimum population size of integrations in each biological replicate (Chao1). Data displayed are the values for each biological replicate (n = 4) with the mean displayed as a horizontal bar. Medium only (no drug control, green circles), 62.5 nM of CPT (blue squares), 50 nM DOX (maroon upward triangles), or 3.13 μM of ETO (pink downward triangles). Fold changes were calculated by comparing the means of each drug condition. Data were analyzed using one-way ANOVA with Dunnett’s multiple comparison test of each drug against the control cells. ns, no significance; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Each ligation-mediated PCR assay captures only a portion of the integrated vectors present in a sample.36 Minimum population sizes of cell clones harboring AAV can be estimated using the Chao1 method,37,38 which models a minimum population size based on the abundance of recovered subsamples. Chao1 estimates for each sample type supported the conclusions that CPT-treated cells have similar integration frequencies as control cells, and that DOX- and ETO-treated cells have higher integration frequencies, approximately 1.5- to 1.7-fold over controls (Figure 4B, p = 0.9331 for CPT, p = 0.0018 for DOX, and p = 0.0055 for ETO, one-way ANOVA, corrected for multiple comparisons, n = 4).
Integration targeting in the presence of CPT, DOX, and ETO
We next investigated whether the topoisomerase drug treatments altered the selection of AAV integration target sites. Previous studies of AAV integration target site selection have shown profiles similar to randomly selected positions, sometimes with modest favoring of transcription units (TUs) and associated features such as CpG islands and transcription start sites (TSSs).28,39,40,41,42,43 Integration site selection may influence AAV transgene expression or genotoxicity,6,7,44,45 and so was studied here in detail.
We first compared the frequency of integration within TUs in the presence of the three drugs or in untreated controls (Figure 5A). Integration in the presence of all three drugs was increased in TUs compared with the control. Previous work has suggested that topoisomerase binding sites in the human chromosomes are enriched near TUs,46,47,48,49,50 consistent with our results. The favoring of integration in TUs was stronger in DOX- and ETO-treated cells than in CPT-treated cells (p = 0.0102 for CPT, p <0.0001 for DOX, and p = 0.0001 for ETO, one-way ANOVA, corrected for multiple comparisons, n = 4).
Figure 5.

CPT-, DOX-, and ETO-treated HeLa cells have different patterns of AAV integration relative to genomic features
(A) The percentage of integration sites in transcription units was calculated for each biological replicate. Data displayed are the values for each biological replicate (n = 4) with the mean displayed as a horizontal bar. Medium only (no drug control, green circles), 62.5 nM of CPT (blue squares), 50 nM DOX (maroon upward triangles), or 3.13 μM of ETO (pink downward triangles). Data were analyzed using one-way ANOVA with Dunnett’s multiple comparison test of each drug against the control cells. ns, no significance; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. (B and C) Distribution of the AAV vector integration sites in the human genome relative to random controls (three times the number of sites as each experimental measure). Tracks are grouped by (B) genomic features or (C) histone markers/chromatin features. All biological replicates are collapsed by treatment condition, and associations were calculated versus random distributions using the ROC area method.35 Values of the ROC were scaled between 0.4 (negatively associated, (B) blue or (C) aqua) and 0.6 (positively associated, (B) yellow or (C) red). Significance was calculated by the ROC method (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
We next assessed integration site favoring near TUs and associated features (Figure 5B), and sites of histone modifications (Figure 5C), comparing with a distribution of randomly selected genomic locations over multiple genomic window sizes. The strength of each association was displayed by the color on the heatmap (Figures 5B and 5C), and the significance of the association is marked with asterisks (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Integration in the no drug control showed a slight disfavoring of integration in TUs, integration in the presence of CPT was close to random, and integration in the presence of DOX and ETO favored integration in TUs. Integration in the presence of DOX and ETO was also favored in or near TU-associated features including CpG islands and TSSs (Figure 5B). For many of the histone post-translational modifications associated with transcriptional activity, integration distributions were weakly negatively associated in the untreated control and were positively associated in the samples treated with CPT, DOX, and ETO. An exception was the heterochromatic mark H3K27me3, where associations were negative for both the untreated control and drug-treated cells (Figure 5C).51,52,53,54
Thus, over a variety of measures, integration in the presence of the topoisomerase poisons was more favored in regions associated with active transcription, and the effect was typically stronger for DOX and ETO.
Integration targeting near inferred topoisomerase II DNA binding sites
Next, we investigated whether the increased integration near TUs occurring in the DOX and ETO cells was associated with integration near topoisomerase binding sites. Previous work has identified topoisomerase II binding sites in human cells by ChIP-seq. Following this, a machine learning-based model was developed to annotate topoisomerase II binding sites based on experimentally mapped binding sites for CTCF, RAD21, and DNAseI cleavage sites.55 We generated such a model based on HeLa-S3 CTCF, RAD21, and DNAseI tracks from ENCODE.56,57,58
We then assessed integration site distributions in our samples near inferred topoisomerase II binding sites compared with a random distribution of integrations (Figure 6). We found that integration near inferred topoisomerase II binding sites was selectively favored in the presence of ETO and DOX, but modestly disfavored in the presence of CPT or in the mock-treated controls. Favoring was seen over multiple window sizes used for comparison (Figure 6A). A comparison of percentage of integration sites associated with the inferred topoisomerase II binding sites between the experimental measures and 1,000 random distribution also shows favoring by the topoisomerase II drug (Figure 6B, CPT 30.0%, DOX 32.4%, ETO 34.3%). We thus conclude that treatment with ETO and DOX favors AAV integration in or near topoisomerase II binding sites.
Figure 6.

AAV favors integration near inferred topoisomerase IIB binding sites selectively in the presence of the topoisomerase II poisons DOX and ETO
TOP2B binding sites were predicted by machine learning methods55 for HeLa cells. (A) Distribution of the AAV vector integration sites in the human genome relative to random controls (three times the number of sites as each experimental measure). TOP2B binding sites were predicted by machine learning. All biological replicates are collapsed by treatment condition, and associations were calculated using the ROC area method.35 Values of the ROC were scaled between 0.4 (negatively associated, green) and 0.6 (positively associated, purple). Significance was calculated by the ROC method (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). (B) Percentage of integration sites within 10,000 bp windows of inferred TOP2B binding sites. Random simulations are depicted in black and were generated from 1 million bootstraps of 1,000 randomly selected integration sites. Percent of sites in TOP2B tracks are presented as colored lines. No drug control (green), 62.5 nM of CPT (blue), 50 nM DOX (maroon), or 3.13 μM of ETO (pink).
Vector rearrangement is not detectably altered in the presence of CPT, ETO, or DOX
Lastly, we asked whether treatment with the topoisomerase drugs might affect transduction via a different mechanism, specifically by modulating the frequency of AAV vector DNA rearrangements. We and others have reported previously that AAV genomes are commonly rearranged after transduction in cells, and integrated genomes may be particularly affected.40,41,42,43,59,60,61,62,63,64,65 In addition, DNA damage responses may influence rearrangement, and all three topoisomerase drugs are known to induce DNA damage.13,14 We used the integration site sequence data to quantify rearrangement frequency at the inverted terminal repeat (ITR)-genomic junction and to ask whether AAV DNA rearrangement character or frequency was different in the presence of CPT, ETO, or DOX.
For this, we analyzed the AAV DNA recovered from ligation-mediated PCR reactions for break points within the AAV DNA prior to reaching junctions with cellular DNA (Figure 7). Each integration site was scored for whether there were additional AAV DNA fragments between the ITR and host DNA, and how many different AAV DNA segments were identifiable. The percentage of rearranged DNAs was scored for each condition. Each condition was tested in four independent biological replicates. We found that the percentage of rearranged DNAs was not significantly different across drug conditions (Figure 7A, p = 0.9986 for CPT, p = 0.9247 for DOX, and p = 0.6906 for ETO, n = 4). Maps of ITR remnant structures (Figure 7B) also did not show large variations in rearrangement frequencies. For all conditions, breakpoint hotspots were seen near ITR positions 71 and 95, which correspond to the tips of the folded DNA “dumbbells” in the ITRs (annotated by the dashed lines). These locations have been reported previously to be hotspots for ITR breakage and genomic junctions.62,66 Thus, we conclude that available data do not support the hypothesis that differences in rearrangement frequencies in the presence of CPT, DOX, or ETO mediated the observed differences in transduction frequencies.
Figure 7.

CPT, DOX, and ETO do not alter AAV ITR-genome junction rearrangement in HeLa cells
(A) Percentage of integration sites with at least one rearrangement in the ITR remnant were calculated per biological replicate. Medium only (no drug control, green circles), 62.5 nM of CPT (blue squares), 50 nM DOX (maroon upward triangles), or 3.13 μM of ETO (pink downward triangles). The data displayed are the value for each biological replicate (n = 4) with the mean displayed as a horizontal bar. Data were analyzed using one-way ANOVA with Dunnett’s multiple comparison test for each drug against the control cells. ns, no significance; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. (B) Composite visualization of ITR remnants of all biological replicates in each drug condition. Integration sites are mapped by the terminal ITR position of the ITR remnant (x axis) and sites sharing the same terminal position were summed (y axis). Sites with no rearrangements were colored green and sites containing rearrangements were colored according to the number of breaks (1, light green; 2, yellow; 3, orange; 4, red; ≥5, maroon). The dashed lines indicate the tips of the ITR dumbbells.
Discussion
Here, we investigate the mechanism of action of topoisomerase poisons in enhancing AAV transduction. Several previous reports have investigated the effects of these drugs, revealing increased AAV transduction as measured by AAV gene expression.13,15,16,20,21,23,67 Preliminary mechanistic studies have proposed a mechanism of increased second-strand synthesis due to topoisomerase poison treatment16,20; however, other studies using self-complimentary vectors that bypass second-strand synthesis also showed increases, and so support additional mechanisms of action.13,68 Previous studies of the topoisomerase II poison ETO supported an increase in AAV integration as measured by transduction of an antibiotic resistance marker.22
Here, we report increased AAV transduction in transformed and primary cells in the presence of CPT, ETO, and DOX. We found that these drugs behaved differently early after transduction, with drug-dependent and cell line-dependent kinetic variations, although CPT, ETO, and DOX all boosted in our assays. Using the HeLa cell model, we show that AAV transduction is increased in all three drugs, yet ETO and DOX achieve boosting at an earlier time point. As topoisomerase II drugs stabilize double-stranded DNA breaks, there may be an impact on the type of damage on early transduction. However, this is still speculative, and these kinetics were not seen in IMR90 cells, indicating a potential effect of cell type.
The two poisons show different enhancement at longer times after transduction. For all conditions, the percent transduction dropped with time, likely reflecting the dilution of episomal AAV genomes with cell growth. Transduced cells showed higher transduction frequencies at late times in the presence of DOX and ETO, consistent with higher integration frequencies. These results are also consistent with expected differences in the cellular responses to damage from type I (ssDNA breaks) and type II (dsDNA breaks) topoisomerase poisons. We compared integration site distributions generated in the presence of CPT, ETO, and DOX to untreated control transductions, and showed that integration targeting was measurably different in the presence of all three of the topoisomerase drugs, with greater favoring of integration in TUs and associated features.
We took advantage of novel machine learning-based methods to identify type II topoisomerase binding sites and compared them with AAV integration site distributions generated in the presence of the three drugs. Previous work has shown that experimentally mapped topoisomerase II binding sites identified by ChIP-seq correlate with several other forms of genomic annotation that are more widely available.55 These include open sites mapped by DNAse I cleavage, and binding sites for the proteins CTCF and RAD21, which together with topoisomerase IIB are found at topology-associated domain (TAD) boundaries.69,70 These three types of annotation were available for HeLa cells, thereby making possible the machine learning-based annotation of likely topoisomerase II binding sites. Integration was selectively favored at these predicted sites in the presence of the topoisomerase II drugs ETO or DOX, but not in the presence of the topoisomerase I poison CPT or controls. These data thus support a model in which ETO and DOX stabilize dsDNA breaks generated by stalled topoisomerase II catalytic intermediates, and these breaks are then able to host AAV integration. The fact that cells treated with ETO and DOX showed preferential integration near topoisomerase II sites, but not cells treated with CPT or mock treated, also helps validate the results from the machine learning approach.
These data imply that integration by AAV in the presence of ETO and DOX is favored at TAD boundaries. TAD boundaries are commonly shared between cell types69 and define the higher-order looping structure in chromosomes. In published work, ETO-induced double-stranded breaks were reported to co-localize to sites of CTCF occupancy in B and T cells, neurons, and embryonic fibroblasts, and many of the break locations were conserved across cell types.55,71 These data suggest that the integration preferences reported here for HeLa cells may often be shared with other cell types, although further work will be useful on this point.
While increased integration at stalled topoisomerase intermediates largely explains our results, we note that other mechanisms may also be involved in the enhancement of AAV transduction. Vector gene expression was elevated for CPT-, ETO-, and DOX-treated cells at early times (up to 6 days post transduction) suggesting that increased integration may not have been solely responsible, especially as CPT-treated cells did not demonstrate increased integration relative to transgene expression. It is possible that the topoisomerase drugs induced cellular DNA damage responses that increased vector DNA synthesis. An alternative model, that CPT, ETO, and DOX altered the frequency of rearrangement of AAV vector DNA, was not supported by the data.
Our study has several limitations. While CPT, DOX, and ETO target topoisomerases, these drugs may induce damage and enhance AAV transduction by other mechanisms. One such example is that DOX has effects on the proteasome and vector nuclear transport.23,68,72 We only studied two human cells lines; others have shown increased transduction with CPT, DOX, and ETO in murine, rat, and other animal models. More extensive testing in preclinical animal models would be valuable. For example, a previous report demonstrated increased second-strand synthesis in CPT-treated mouse hearts 2 weeks post transduction, yet our methods did not detect an impact of CPT on vector copy number.16 Machine learning was effective in identifying topoisomerase II binding sites, but more direct experimental work could clarify binding sites more fully and specify local bound proteins at integration sites. For translation, although CPT, ETO, and DOX are used clinically to treat cancer, they are toxic, and it is unknown whether low doses could be used safely in human subjects to increase AAV transduction and improve stability with integration.
If sufficiently safe, augmenting AAV vector transduction with CPT, ETO, and DOX could be valuable in clinical applications. In one implementation, Spirovant Sciences has begun recruitment for a phase 1/2 trial which will assess an inhaled combination of AAV and DOX.73 Augmenting AAV integration with topoisomerase poisons also could be beneficial for long-term gene therapy outcomes. For example, in any tissue where dividing cells are targeted, stability of transgene production could potentially be increased with integration. Of course, the toxicity of CPT, ETO, and DOX, and possible genotoxicity of integration, would need to be carefully monitored, but clinical application may be justified in treatment of severe disorders lacking effective alternative therapies.
Materials and methods
Cell culture
HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS) and 1% penicillin-streptomycin. IMR90 cells were cultured in minimum essential medium (MEM) supplemented with 10% HI-FBS and 1% penicillin-streptomycin.
AAV vector preparation
AAV8-GFP was produced from AAV trans, cis, and helper plasmids procured from Addgene, pAAV-GFP74 was a gift from John T. Gray (Addgene, Watertown, MA, plasmid no. 32395; http://n2t.net/addgene:32395; Addgene, 32395). pAAV2/8 (Addgene, plasmid no. 112864; http://n2t.net/addgene:112864; Addgene, 112864) and pAdDeltaF6 (Addgene, plasmid no. 112867; http://n2t.net/addgene:112867; Addgene, 112867) were a gift from James M. Wilson. Plasmids were validated through whole-plasmid sequencing, which was performed by Plasmidsaurus using Oxford Nanopore technology with custom analysis and pLannotate annotation. AAV-GFP vectors were prepared as described below.
The AAV8-GFP vectors were generated by triple transfection of 293T cells. Cells were plated in DMEM 10% HI-FBS medium 24 h before transfection. AAV trans plasmid (pAAV-GFP), AAV cis plasmid (pAAV2/8), and helper plasmid (pAdDeltaF6) were complexed with PEI in serum free Opti-MEM medium. The AAV genome in pAAV-GFP produces a single-stranded AAV with a CMV enhancer, a CMV promoter, a β-globin intron, EGFP transgene, and a β-globin poly(A) sequence.
Plasmid-PEI mixtures were spiked into the medium of plated 293T cells and allowed to incubate for 24 h before basal medium was replaced with lower serum medium (DMEM, 5% HI-FBS, 1% penicillin-streptomycin). Cells were grown in lower serum medium for 48 h. After 48 h, medium was collected, cells were washed, harvested, and subjected to freeze-thaw cycles for lysis.
Lysed cell supernatant was mixed with cell medium and subjected to chloroform extraction and overnight polyethylene glycol precipitation. Preparations were centrifuged and resuspended in DPBS containing calcium and potassium. They were then DNaseI and RNaseI treated in the presence of MgCl2 before a second chloroform extraction. Final vector stocks were stored at 4°C. During transduction, AAV8-GFP was prepared to desired concentration with basal cell culture medium and added dropwise to cells.
Chemical treatments
HeLa cells were plated in quadruplicate 6 h before treatment in basal medium (DMEM, 10% HI-FBS, 1% penicillin-streptomycin) at 80% confluency (3.5 × 105 cells/mL in a 24-well plate). IMR90 cells were plated in quadruplicate 6 h before treatment in basal medium (MEM, 10% HI-FBS, 1% penicillin-streptomycin) at 100% confluency to induce contact inhibition (4 × 105cells/mL). ETO, CPT, and DOX stock solutions were prepared in dimethyl sulfoxide and stored at −20°C. Solutions were thawed and diluted in basal DMEM medium immediately before addition to cells. HeLa cells were incubated with chemicals for 16–18 h and IMR90 cells were incubated with chemicals for 24 h. Both cell types were washed twice with respective basal medium. Vector was diluted in basal medium immediately before use and added to cells after washing. Cultures were maintained in basal medium without chemicals.
GFP expression
Early GFP expression post transduction was measured using the Incucyte live-cell imaging and analysis system (Incucyte S3 two-color Green/Red, software v.2019 RevB, Sartorius). Fluorescent and phase images of live cells were obtained every 6 h for the first 6 days post transduction and GFP-positive cells were quantified using adherent cell-by-cell scanning and Incucyte Basic Analysis methods; note that this method does not yield a count of the untransduced cells.
After 6 days, HeLa cells were passaged twice a week upon reaching confluency and a subset of cells was flash frozen in PBS for DNA extraction. Once a week, an additional subset of the HeLa cells was harvested for flow cytometry analysis. Cells were washed in PBS and stained with LIVE/DEAD Aqua (Invitrogen) stain before fixation with BD Cytofix. Cell samples were measured on a BD LSR II (BD Biosciences) and analyzed using FlowJo v.10.9.0 software. GFP BrightComp beads and ArC Amine reactive beads (Thermo Fisher Scientific) with and without LIVE/DEAD Aqua staining were used to calculate compensation. An FSC-A/SSC-A plot was used to gate on epithelial cells and an FSC-A/FSC-H plot was used to gate on single cells. The cell viability stain LIVE/DEAD Aqua was used to distinguish live vs. dead cells using an FSC-H/Violet 515/20-A plot. An FSC-H/Blue 515/20-A plot was used for GFP fluorescence and untransduced HeLa cells were used to gate on GFP+ cells (Figure S5); both GFP+ and GFP− cells were quantified.
Vector copy number
DNA was extracted from frozen pellets using the QIAGEN DNeasy Blood and Tissue kit and resuspended in AE buffer. DNA mass was quantified using the Quant-iT PicoGreen dsDNA assay kit and read on a Synergy LX plate reader (BioTek). Quantitative PCR (qPCR) was carried out on a QuantStudio 5 (Thermo Fisher Scientific) with the TaqMan Fast Universal PCR Master mix (Applied Biosystems). GFP-targeted qPCR primers were as follows (also in Table S3).
Forward primer: 5′-GAACCGCATCFAFCTGAA-3ʹ.
Reverse primer 5′-TGCTTGTCGGCCATGATATAG-3ʹ.
Probe 5′-56-FAM/ATCGACTTC/ZEN/AAGGAGGACGGCAAC/3ʹ.
qPCR reactions were prepared in 10 μL reaction volume with 300 μM forward primer, 300 μM reverse primer, 0.0825 μM probe, 1× TaqMan Fast Universal PCR with 2 μL of gDNA. Thermocycling parameters were as follows: initial denaturation at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 3 s and elongation at 60°C for 30 s. The standard curve was created with pAAV-GFP plasmid and diluted to an appropriate range for primer efficiency to estimate vector copies per reaction. Genome copies per reaction were calculated from DNA mass in 2 μL using a conversion factor of 7 pg per genome copy of human DNA. Vector copies per cell were calculated as a ratio of vector copies to genome copies per reaction. All reactions were carried out with four biological replicates per drug condition and three technical replicates per biological replicate.
Integration site sequencing
Integration analysis was performed as described previously34 using ligation-mediated PCR and the sonic abundance method. A positive control was freshly prepared by spiking an equimolar amount of six plasmids each with variable length ITRs fused to different segments of the yeast genome into human gDNA. All samples and positive control were sheared to a size of ∼1,000 bp with a Covaris M220 ultrasonicator with the following conditions (peak power 50 W, duty factor 2%, 200 cycles for 95 s). After sonication, samples were subjected to bead purification with AMPureXP beads (Beckman Coulter) and a water-only negative control was added. Samples were subjected to end repair and dA tailing with the NEBNext Ultra II End Repair/dA-Tailing Module. Adaptors with unique sample-specific linkers were ligated to repaired DNA using the NEBNext Ultra II Ligation Module. Adaptor-ligated gDNA was then subjected to nested PCRs under stringent conditions to enrich for integration sites. Nested PCR reactions with primers landing near the inner edge of ITR sequences and primers specific to the linker selectively amplified genomic fragments containing ITR/DNA junction sequences (Table S3). All PCRs were carried out in quadruplicate to account for PCR jackpotting with unique barcodes for each PCR replicate.
The first PCR reaction (PCR1) was performed with a total volume of 25 μL per reaction, 300 nM PCR1 linker primer, 300 nM ITR primer 1, 1× Clontech Advantage 2 PCR Buffer (Takara Bio), 200 μM dNTPs, and 1× Clontech Advantage 2 Polymerase Mix (Takara Bio). PCR1 thermocycling parameters were as follows: initial denaturation for 1 min at 95°C, 5 linear amplification cycles of 95°C for 30 s, 80°C for 30 s (annealing), and 72°C for 1 min 30 s (extension), followed by 20 exponential amplification cycles of 95°C for 30 s, 80°C for 30 s (annealing), and 70°C for 1 min 30 s (extension), followed by a final extension at 72°C for 4 min and an infinite hold at 4°C. Two microliters of PCR1 product was diluted into the second PCR reaction (PCR2), keeping samples in quadruplicate, with a total volume of 25 μL per reaction, 300 nM PCR2 linker primer, 300 nM ITR primer 2, 1× Clontech Advantage 2 PCR Buffer (Takara Bio), 200 μM dNTPs, and 1× Clontech Advantage 2 Polymerase Mix (Takara Bio). PCR2 thermocycling parameters were as follows: 1 min initial denaturation at 95°C, 5 linear amplification cycles of 95°C for 30 s, 80°C for 30 s (annealing), and 72°C for 1 min 30 s (extension), followed by 15 exponential amplification cycles of 95°C for 30 s, 80°C for 30 s (annealing), at 70°C for 1 min 30 s (extension), followed by a final extension at 72°C for 4 min and an infinite hold at 4°C.
Technical replicates were pooled at the sample level, purified with AMPureXP beads post-PCR and quantified using KAPA SYBR qPCR (KapaBiosciences) run on a QuantStudio 5 (Thermo Fisher Scientific). The normalized pooled library was quantified with another KAPA SYBR qPCR and Tapestation (Agilent) to determine library concentration. The library was denatured and diluted to 10 pM with a 40% PhiX v.3 (Illumina) spike-in due to low diversity in common linker regions. Illumina R1, I1, and R2 primers were spiked with custom sequencing primers (Table S3). The library was sequenced with an Illumina MiSeq 600 cycle kit using an R1/R2 split of 250–350 cycles due to R2 reads harboring the complex ITR regions.
Integration site analysis
AAV genome integrations and ITR remnant structures were determined using the AAVengeR software pipeline (https://github.com/helixscript/AAVengeR) v.2.1.0. Integration positions were identified by aligning paired-end short-read sequences from sheared genomic fragments containing the internal edge of ITR sequences to the human genome. Reads were aligned with the BLAT aligner software and reads that aligned near one another with opposite orientations were used to define genomic fragments where one end of fragments contained the genomic juncture sequences and the other contained sonically sheared genomic DNA sequences. Integration positions, defined as the transition between ITR remnant sequences and genomic DNA were defined for each fragment and fragment boundaries were standardized to correct for PCR, sequencing, and alignment errors. Genomic fragments were grouped by standardized integration positions and the number of unique sonic break positions associated with each integration position was used as a proxy for clonal abundance (SonicAbundance method34,75). Alignments between recovered ITR remnant sequences and the vector-plasmid were used to determine ITR remnant rearrangements (Figure 7).
Due to the potential presence of episomal DNA and subsequent PCR recombination, integration site data were filtered to require three or more reads supporting an integration site, as determined by previous optimization with integration-negative controls (purified AAV vector DNA spiked into purified genomes, data not shown). The numbers of unique integration sites recovered from each sample were counted. SonicAbundance cell estimates were used to infer minimum population size with Chao1. Transcription unit annotations used by AAVengeR (Figure 5A) were downloaded from the UCSC genome browser76 (http://genome.ucsc.edu/, hg38 RefSeq Curated, 2019).77,78,79 The percentage of sites in TUs was calculated over the unique sites recovered for each sample.
Integration distribution heatmaps were generated using the hotROCs package (https://github.com/BushmanLab/hotROCs). Detailed methods for obtaining receiver operator characteristic (ROC) curve areas and calculating p values have been reported previously.35 The random distribution datasets were produced with three times the number of sites as the experimental datasets. ROC values of each association are presented by color scales and p values are represented as asterisks (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). The percentage of integration sites for each sample, feature, and window combination was calculated by taking the number of integration sites at each combination, dividing by the total number of integration sites, and multiplying by 100. To generate the random distribution of integration site percentages in each feature, the above process was performed for 1 million bootstraps of 1,000 randomly selected integration sites. To evaluate each feature, we downloaded the call sets from the ENCODE portal56,57,58 (https://www.encodeproject.org/) with the following identifiers: ENCFF162RSB (H3K4me1), ENCFF429OQI (H3K4me2), ENCFF903JDG (H3K4me3), ENCFF021PYM (H3K9ac), ENCFF831XSS (H3K27ac), ENCFF584RYA (H3K27me3), ENCFF406DAM (H4K20me1), ENCFF582MED (H3K36me3), ENCFF238XWI (H3K79me2), ENCFF094MFL (H2AFZ). Transcription unit annotations in Figure 5B were downloaded from the UCSC genome browser76 (http://genome.ucsc.edu/, hg38 RefSeq Curated, 2024).77,78,79 CpG island annotations were downloaded from the UCSC genome browser76 (http://genome.ucsc.edu) hg38 track.80
TOP2B binding sites were calculated using machine learning as described previously55 using HeLa-S3 input tracks CTCF, RAD21, and DNAseI hypersensitivity from ENCODE. We downloaded the call sets from the ENCODE portal56,57,58 (https://www.encodeproject.org/) with the following identifiers: ENCFF855ZMQ (DNase-seq), ENCFF035SOZ (CTCF), ENCFF155UQU (RAD21), ENCFF089KHK (CTCF input), and ENCFF232RQF (RAD21 input). Predicted TOP2B sites were filtered to only include regions with a prediction score >0.8.
Statistics
GFP and vector copy number data are either presented as the mean of four biological replicates ± standard deviation or with four biological replicates as individual data points with a bar representing the mean. GraphPad Prism v.10 was used to display data and calculate statistical significance. Comparison among the four groups was made using one-way ANOVA with correction for multiple comparisons, comparing each drug condition to the no drug control.
Integration site data are presented per biological replicate, with each as a composite of four technical replicates. Number of sites, percent in Tus, and percent rearrangement was plotted in GraphPad Prism v.10. Comparison among the four groups was made using one-way ANOVA with correction for multiple comparisons, comparing each drug condition to the no drug control.
Integration site distributions are presented by treatment condition, condensing the four biological replicates. Statistical significance of the area under the curve (ROC) was as described previously,35 and p values are represented as asterisks (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, determined with Welch’s t test).
Data and code availability
All DNA sequence information generated in this study is available at the NCBI SRA PRJNA1131384; samples are annotated in Table S1. The AAVengeR pipeline is available at https://github.com/helixscript/AAVengeR. The code used to train machine learning models and generate genome wide predictions is available at https://gitlab.com/mgarciat/genome-wide-prediction-of-topoisomerase-iibeta-binding.
Acknowledgments
We are grateful to members of the Bushman and Sabatino laboratories for help and suggestions. This work was supported in part by National Institutes of Health grant (P30AI045008, U01AI125051, R01CA241762, R01HL142791, R01HL160748, U19AI149680, and T32 in Virology T32-AI-007324). We thank the Viral Molecular High Density Sequencing Core at the University of Pennsylvania (RRID: SCR_022433) for support of integration site analysis. We are grateful to the University of Pennsylvania Department of Microbiology for use of shared resources and the Incucyte S3 machine. The flow cytometry data for this manuscript were generated in the Penn Cytomics and Cell Sorting Shared Resource Laboratory at the University of Pennsylvania and is partially supported by the Abramson Cancer Center NCI Grant (P30 016520). The research identifier number is RRID: SCR_022376. We would like to thank Zahidul Alam at the University of Pennsylvania and Marie Seleme at the Children’s Hospital of Pennsylvania for their training in running flow cytometry on epithelial cells. Plasmids were validated through whole-plasmid sequencing, which was performed by Plasmidsaurus using Oxford Nanopore technology with custom analysis and pLannotate annotation.
Author contributions
A.K. designed and performed experiments, analyzed data, and wrote the manuscript. Y.H., J.K.E., A.G.M., S.A.Z., and T.L. performed experiments, analyzed data, and revised the manuscript. A.M.R. contributed to experimental design, data organization, data analysis, and revised the manuscript. P.M.M.-G. designed the machine learning model and revised the manuscript. D.E.S. conceptualized experiments and revised the manuscript. F.D.B. designed and conceptualized experiments, provided resources and training, and wrote the manuscript.
Declaration of interests
D.E.S. is a consultant for Poseida Therapeutics and Biomarin Pharmaceuticals. D.E.S. receives licensing royalties from Spark Therapeutics. F.D.B. is a founder of Biocept and has intellectual property licensed to Novartis.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101364.
Supplemental information
References
- 1.Schwartz R.A., Palacios J.A., Cassell G.D., Adam S., Giacca M., Weitzman M.D. The Mre11/Rad50/Nbs1 complex limits adeno-associated virus transduction and replication. J. Virol. 2007;81:12936–12945. doi: 10.1128/JVI.01523-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi V.W., McCarty D.M., Samulski R.J. Host Cell DNA Repair Pathways in Adeno-Associated Viral Genome Processing. J. Virol. 2006;80:10346–10356. doi: 10.1128/jvi.00841-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maurer A.C., Weitzman M.D. Adeno-Associated Virus Genome Interactions Important for Vector Production and Transduction. Hum. Gene Ther. 2020;31:499–511. doi: 10.1089/hum.2020.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lentz T.B., Samulski R.J. Insight into the Mechanism of Inhibition of Adeno-Associated Virus by the Mre11/Rad50/Nbs1 Complex. J. Virol. 2015;89:181–194. doi: 10.1128/jvi.01990-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang C.C., Xiao X., Zhu X., Ansardi D.C., Epstein N.D., Frey M.R., Matera A.G., Samulski R.J. Cellular recombination pathways and viral terminal repeat hairpin structures are sufficient for adeno-associated virus integration in vivo and in vitro. J. Virol. 1997;71:9231–9247. doi: 10.1128/jvi.71.12.9231-9247.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greig J.A., Martins K.M., Breton C., Lamontagne R.J., Zhu Y., He Z., White J., Zhu J.X., Chichester J.A., Zheng Q., et al. Integrated vector genomes may contribute to long-term expression in primate liver after AAV administration. Nat. Biotechnol. 2024;42:1232–1242. doi: 10.1038/s41587-023-01974-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandler R.J., LaFave M.C., Varshney G.K., Trivedi N.S., Carrillo-Carrasco N., Senac J.S., Wu W., Hoffmann V., Elkahloun A.G., Burgess S.M., Venditti C.P. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Invest. 2015;125:870–880. doi: 10.1172/jci79213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kasimsetty A., Sabatino D.E. Integration and the risk of liver cancer-Is there a real risk? J. Viral Hepat. 2024;31:26–34. doi: 10.1111/jvh.13915. [DOI] [PubMed] [Google Scholar]
- 9.Berger J.M., Gamblin S.J., Harrison S.C., Wang J.C. Structure and mechanism of DNA topoisomerase II. Nature. 1996;379:225–232. doi: 10.1038/379225a0. [DOI] [PubMed] [Google Scholar]
- 10.Fan M., Chen G., Sun B., Wu J., Li N., Sarker S.D., Nahar L., Guo M. Screening for natural inhibitors of human topoisomerases from medicinal plants with bio-affinity ultrafiltration and LC-MS. Phytochemistry Rev. 2020;19:1231–1261. doi: 10.1007/s11101-019-09635-x. [DOI] [Google Scholar]
- 11.McClendon A.K., Osheroff N. DNA topoisomerase II, genotoxicity, and cancer. Mutat. Res. 2007;623:83–97. doi: 10.1016/j.mrfmmm.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pommier Y. DNA Topoisomerase I Inhibitors: Chemistry, Biology, and Interfacial Inhibition. Chem. Rev. 2009;109:2894–2902. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gómez-Moreno A., San Sebastian E., Moya J., Gomollón-Zueco P., Isola S., Vales A., Gonzalez-Aseguinolaza G., Unzu C., Garaigorta U. Topoisomerase Inhibitors Increase Episomal DNA Expression by Inducing the Integration of Episomal DNA in Hepatic Cells. Pharmaceutics. 2023;15:2459. doi: 10.3390/pharmaceutics15102459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang T., Shi S., Shi Y., Jiang P., Hu G., Ye Q., Shi Z., Yu K., Wang C., Fan G., et al. Chemical-induced phase transition and global conformational reorganization of chromatin. Nat. Commun. 2023;14:5556. doi: 10.1038/s41467-023-41340-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russell D.W., Alexander I.E., Miller A.D. DNA-synthesis and topoisomerase inhibitors increase transduction by adenoassociated virus vectors. Proc. Natl. Acad. Sci. USA. 1995;92:5719–5723. doi: 10.1073/pnas.92.12.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prasad K.M.R., Xu Y., Yang Z., Toufektsian M.C., Berr S.S., French B.A. Topoisomerase inhibition accelerates gene expression after adeno-associated virus-mediated gene transfer to the mammalian heart. Mol. Ther. 2007;15:764–771. doi: 10.1038/sj.mt.6300071. [DOI] [PubMed] [Google Scholar]
- 17.Yan Z., Zak R., Zhang Y., Ding W., Godwin S., Munson K., Peluso R., Engelhardt J.F. Distinct classes of proteasome-modulating agents cooperatively augment recombinant adeno-associated virus type 2 and type 5-mediated transduction from the apical surfaces of human airway epithelia. J. Virol. 2004;78:2863–2874. doi: 10.1128/jvi.78.6.2863-2874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang T., Hu J., Ding W., Wang X. Doxorubicin augments rAAV-2 transduction in rat neuronal cells. Neurochem. Int. 2009;55:521–528. doi: 10.1016/j.neuint.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Gong H., Yuan N., Shen Z., Tang C., Shipp S., Qian L., Lu Y., Andolina I.M., Zhang S., Wu J., et al. Transduction catalysis: Doxorubicin amplifies rAAV-mediated gene expression in the cortex of higher-order vertebrates. iScience. 2021;24 doi: 10.1016/j.isci.2021.102685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng D., Qian C., Sun Y., Barajas M.A., Prieto J. Transduction of hepatocellular carcinoma (HCC) using recombinant adeno-associated virus (rAAV):: in vitro and in vivo effects of genotoxic agents. J. Hepatol. 2000;32:975–985. doi: 10.1016/s0168-8278(00)80102-6. [DOI] [PubMed] [Google Scholar]
- 21.Halbert C.L., Standaert T.A., Aitken M.L., Alexander I.E., Russell D.W., Miller A.D. Transduction by adeno-associated virus vectors in the rabbit airway: Efficiency, persistence, and readministration. J. Virol. 1997;71:5932–5941. doi: 10.1128/jvi.71.8.5932-5941.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller D.G., Petek L.M., Russell D.W. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 2004;36:767–773. doi: 10.1038/ng1380. [DOI] [PubMed] [Google Scholar]
- 23.Nicolson S.C., Li C., Hirsch M.L., Setola V., Samulski R.J. Identification and Validation of Small Molecules That Enhance Recombinant Adeno-associated Virus Transduction following High-Throughput Screens. J. Virol. 2016;90:7019–7031. doi: 10.1128/jvi.02953-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delgado J.L., Hsieh C.M., Chan N.L., Hiasa H. Topoisomerases as anticancer targets. Biochem. J. 2018;475:373–398. doi: 10.1042/bcj20160583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bushman F. Targeting Retroviral Integration. Science. 1995;267:1443–1444. doi: 10.1126/science.7878462. [DOI] [PubMed] [Google Scholar]
- 26.Nichols W.W., Murphy D.G., Cristofalo V.J., Toji L.H., Greene A.E., Dwight S.A. Characterization of a new human diploid cell strain, IMR-90. Science. 1977;196:60–63. doi: 10.1126/science.841339. [DOI] [PubMed] [Google Scholar]
- 27.Ciuffi A., Mitchell R.S., Hoffmann C., Leipzig J., Shinn P., Ecker J.R., Bushman F.D. Integration site selection by and growth-arrested HIV-Based vectors in dividing IMR-90 lung fibroblasts. Mol. Ther. 2006;13:366–373. doi: 10.1016/j.ymthe.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 28.Berry C., Hannenhalli S., Leipzig J., Bushman F.D. Selection of target sites for mobile DNA integration in the human genome. PLoS Comput. Biol. 2006;2:e157. doi: 10.1371/journal.pcbi.0020157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berry C.C., Gillet N.A., Melamed A., Gormley N., Bangham C.R.M., Bushman F.D. Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics. 2012;28:755–762. doi: 10.1093/bioinformatics/bts004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schroder A.R.W., Shinn P., Chen H.M., Berry C., Ecker J.R., Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 31.Mitchell R.S., Beitzel B.F., Schroder A.R.W., Shinn P., Chen H., Berry C.C., Ecker J.R., Bushman F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004;2:E234. doi: 10.1371/journal.pbio.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang G.P., Ciuffi A., Leipzig J., Berry C.C., Bushman F.D. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007;17:1186–1194. doi: 10.1101/gr.6286907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang G.P., Berry C.C., Malani N., Leboulch P., Fischer A., Hacein-Bey-Abina S., Cavazzana-Calvo M., Bushman F.D. Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial. Blood. 2010;115:4356–4366. doi: 10.1182/blood-2009-12-257352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sherman E., Nobles C., Berry C.C., Six E., Wu Y., Dryga A., Malani N., Male F., Reddy S., Bailey A., et al. INSPIIRED: A Pipeline for Quantitative Analysis of Sites of New DNA Integration in Cellular Genomes. Mol. Ther. Methods Clin. Dev. 2017;4:39–49. doi: 10.1016/j.omtm.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berry C.C., Nobles C., Six E., Wu Y., Malani N., Sherman E., Dryga A., Everett J.K., Male F., Bailey A., et al. INSPIIRED: Quantification and Visualization Tools for Analyzing Integration Site Distributions. Mol. Ther. Methods Clin. Dev. 2017;4:17–26. doi: 10.1016/j.omtm.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bushman F.D., Cantu A., Everett J., Sabatino D., Berry C. Challenges in estimating numbers of vectors integrated in gene-modified cells using DNA sequence information. Mol. Ther. 2021;29:3328–3331. doi: 10.1016/j.ymthe.2021.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chao A., Chazdon R.L., Colwell R.K., Shen T.J. Abundance-based similarity indices and their estimation when there are unseen species in samples. Biometrics. 2006;62:361–371. doi: 10.1111/j.1541-0420.2005.00489.x. [DOI] [PubMed] [Google Scholar]
- 38.Chiu C.H., Chao A. Estimating and comparing microbial diversity in the presence of sequencing errors. PeerJ. 2016;4 doi: 10.7717/peerj.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller D.G., Trobridge G.D., Petek L.M., Jacobs M.A., Kaul R., Russell D.W. Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. J. Virol. 2005;79:11434–11442. doi: 10.1128/jvi.79.17.11434-11442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakai H., Montini E., Fuess S., Storm T.A., Grompe M., Kay M.A. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat. Genet. 2003;34:297–302. doi: 10.1038/ng1179. [DOI] [PubMed] [Google Scholar]
- 41.Zhong L., Malani N., Li M., Brady T., Xie J., Bell P., Li S., Jones H., Wilson J.M., Flotte T.R., et al. Recombinant Adeno-Associated Virus Integration Sites in Murine Liver After Ornithine Transcarbamylase Gene Correction. Hum. Gene Ther. 2013;24:520–525. doi: 10.1089/hum.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen G.N., Everett J.K., Kafle S., Roche A.M., Raymond H.E., Leiby J., Wood C., Assenmacher C.A., Merricks E.P., Long C.T., et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021;39:47–55. doi: 10.1038/s41587-020-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martins K.M., Breton C., Zheng Q., Zhang Z., Latshaw C., Greig J.A., Wilson J.M. Prevalent and Disseminated Recombinant and Wild-Type Adeno-Associated Virus Integration in Macaques and Humans. Hum. Gene Ther. 2023;34:1081–1094. doi: 10.1089/hum.2023.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Donsante A., Miller D.G., Li Y., Vogler C., Brunt E.M., Russell D.W., Sands M.S. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317:477. doi: 10.1126/science.1142658. [DOI] [PubMed] [Google Scholar]
- 45.Donsante A., Vogler C., Muzyczka N., Crawford J.M., Barker J., Flotte T., Campbell-Thompson M., Daly T., Sands M.S. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Ther. 2001;8:1343–1346. doi: 10.1038/sj.gt.3301541. [DOI] [PubMed] [Google Scholar]
- 46.Yang F., Kemp C.J., Henikoff S. Anthracyclines induce double-strand DNA breaks at active gene promoters. Mutat. Res. 2015;773:9–15. doi: 10.1016/j.mrfmmm.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baranello L., Kouzine F., Wojtowicz D., Cui K., Przytycka T.M., Zhao K., Levens D. DNA Break Mapping Reveals Topoisomerase II Activity Genome-Wide. Int. J. Mol. Sci. 2014;15:13111–13122. doi: 10.3390/ijms150713111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uusküla-Reimand L., Hou H.Y., Samavarchi-Tehrani P., Rudan M.V., Liang M.G., Medina-Rivera A., Mohammed H., Schmidt D., Schwalie P., Young E.J., et al. Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016;17:182. doi: 10.1186/s13059-016-1043-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manville C.M., Smith K., Sondka Z., Rance H., Cockell S., Cowell I.G., Lee K.C., Morris N.J., Padget K., Jackson G.H., Austin C.A. Genome-wide ChIP-seq analysis of human TOP2B occupancy in MCF7 breast cancer epithelial cells. Biol. Open. 2015;4:1436–1447. doi: 10.1242/bio.014308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitchell R., Chiang C.Y., Berry C., Bushman F. Global analysis of cellular transcription following infection with an HIV-based vector. Mol. Ther. 2003;8:674–687. doi: 10.1016/s1525-0016(03)00215-6. [DOI] [PubMed] [Google Scholar]
- 51.Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 52.Hosogane M., Funayama R., Shirota M., Nakayama K. Lack of Transcription Triggers H3K27me3 Accumulation in the Gene Body. Cell Rep. 2016;16:696–706. doi: 10.1016/j.celrep.2016.06.034. [DOI] [PubMed] [Google Scholar]
- 53.Kundu S., Ji F., Sunwoo H., Jain G., Lee J.T., Sadreyev R.I., Dekker J., Kingston R.E. Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation (vol 65, pg 432, 2017) Mol. Cell. 2018;71:191. doi: 10.1016/j.molcel.2018.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morey L., Helin K. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 2010;35:323–332. doi: 10.1016/j.tibs.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 55.Martínez-García P.M., García-Torres M., Divina F., Terrón-Bautista J., Delgado-Sainz I., Gómez-Vela F., Cortés-Ledesma F. Genome-wide prediction of topoisomerase IIβ binding by architectural factors and chromatin accessibility. PLoS Comput. Biol. 2021;17 doi: 10.1371/journal.pcbi.1007814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hitz B.C., Jin-Wook L., Jolanki O., Kagda M.S., Graham K., Sud P., Gabdank I., Strattan J.S., Sloan C.A., Dreszer T., et al. The ENCODE Uniform Analysis Pipelines. bioRxiv. 2023 doi: 10.1101/2023.04.04.535623. Preprint at. [DOI] [Google Scholar]
- 57.Luo Y., Hitz B.C., Gabdank I., Hilton J.A., Kagda M.S., Lam B., Myers Z., Sud P., Jou J., Lin K., et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 2020;48:D882–D889. doi: 10.1093/nar/gkz1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R., et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nowrouzi A., Penaud-Budloo M., Kaeppel C., Appelt U., Le Guiner C., Moullier P., von Kalle C., Snyder R.O., Schmidt M. Integration frequency and intermolecular recombination of rAAV vectors in non-human primate skeletal muscle and liver. Mol. Ther. 2012;20:1177–1186. doi: 10.1038/mt.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tai P.W.L., Xie J., Fong K., Seetin M., Heiner C., Su Q., Weiand M., Wilmot D., Zapp M.L., Gao G. Adeno-associated Virus Genome Population Sequencing Achieves Full Vector Genome Resolution and Reveals Human-Vector Chimeras. Mol. Ther. Methods Clin. Dev. 2018;9:130–141. doi: 10.1016/j.omtm.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun X., Lu Y., Bish L.T., Calcedo R., Wilson J.M., Gao G. Molecular analysis of vector genome structures after liver transduction by conventional and self-complementary adeno-associated viral serotype vectors in murine and nonhuman primate models. Hum. Gene Ther. 2010;21:750–761. doi: 10.1089/hum.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calabria A., Cipriani C., Spinozzi G., Rudilosso L., Esposito S., Benedicenti F., Albertini A., Pouzolles M., Luoni M., Giannelli S., et al. Intrathymic AAV delivery results in therapeutic site-specific integration at TCR loci in mice. Blood. 2023;141:2316–2329. doi: 10.1182/blood.2022017378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dalwadi D.A., Calabria A., Tiyaboonchai A., Posey J., Naugler W.E., Montini E., Grompe M. AAV integration in human hepatocytes. Mol. Ther. 2021;29:2898–2909. doi: 10.1016/j.ymthe.2021.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakai H., Iwaki Y., Kay M.A., Couto L.B. Isolation of recombinant adeno-associated virus vector-cellular DNA junctions from mouse liver. J. Virol. 1999;73:5438–5447. doi: 10.1128/jvi.73.7.5438-5447.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Deyle D.R., Russell D.W. Adeno-associated virus vector integration. Curr. Opin. Mol. Therapeut. 2009;11:442–447. [PMC free article] [PubMed] [Google Scholar]
- 66.Inagaki K., Ma C., Storm T.A., Kay M.A., Nakai H. The role of DNA-PKcs and artemis in opening viral DNA hairpin termini in various tissues in mice. J. Virol. 2007;81:11304–11321. doi: 10.1128/jvi.01225-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kanazawa T., Mizukami H., Nishino H., Okada T., Hanazono Y., Kume A., Kitamura K., Ichimura K., Ozawa K. Topoisomerase inhibitors enhance the cytocidal effect of AAV-HSVtk/ganciclovir on head and neck cancer cells. Int. J. Oncol. 2004;25:729–735. [PubMed] [Google Scholar]
- 68.Yan Z.Y., Zak R., Zhang Y.L., Ding W., Godwin S., Munson K., Peluso R., Engelhardt J.F. Distinct classes of proteasome-modulating agents cooperatively augment recombinant adeno-associated virus type 2 and type 5-mediated transduction from the apical surface of human airway epithelia. Mol. Ther. 2004;9:S287–S288. doi: 10.1128/JVI.78.6.2863-2874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Canela A., Maman Y., Jung S., Wong N., Callen E., Day A., Kieffer-Kwon K.R., Pekowska A., Zhang H., Rao S.S.P., et al. Genome Organization Drives Chromosome Fragility. Cell. 2017;170:507–521.e18. doi: 10.1016/j.cell.2017.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McArthur E., Capra J.A. Topologically associating domain boundaries that are stable across diverse cell types are evolutionarily constrained and enriched for heritability. Am. J. Hum. Genet. 2021;108:269–283. doi: 10.1016/j.ajhg.2021.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Canela A., Maman Y., Huang S.Y.N., Wutz G., Tang W., Zagnoli-Vieira G., Callen E., Wong N., Day A., Peters J.M., et al. Topoisomerase II-Induced Chromosome Breakage and Translocation Is Determined by Chromosome Architecture and Transcriptional Activity. Mol. Cell. 2019;75:252–266.e8. doi: 10.1016/j.molcel.2019.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jennings K., Miyamae T., Traister R., Marinov A., Katakura S., Sowders D., Trapnell B., Wilson J.M., Gao G., Hirsch R. Proteasome inhibition enhances AAV-mediated transgene expression in human synoviocytes in vitro and in vivo. Mol. Ther. 2005;11:600–607. doi: 10.1016/j.ymthe.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 73.Spirovant Sciences, Inc. A Single Ascending Dose, Phase 1/2 Trial to Evaluate the Safety, Pharmacokinetics, and Pharmacodynamics of SP-101 via Nebulizer for the Treatment of Cystic Fibrosis (CF) 2024. https://clinicaltrials.gov/study/NCT06526923
- 74.Gray J.T., Zolotukhin S. Design and construction of functional AAV vectors. Methods Mol. Biol. 2011;807:25–46. doi: 10.1007/978-1-61779-370-7_2. [DOI] [PubMed] [Google Scholar]
- 75.Berry C.C., Gillet N.A., Melamed A., Gormley N., Bangham C.R.M., Bushman F.D. Estimating Abundances of Retroviral Insertion Sites from DNA Fragment Length Data. Bioinformatics. 2012;28:755–762. doi: 10.1093/bioinformatics/bts004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nassar L.R., Barber G.P., Benet-Pagès A., Casper J., Clawson H., Diekhans M., Fischer C., Gonzalez J.N., Hinrichs A.S., Lee B.T., et al. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 2023;51:D1188–D1195. doi: 10.1093/nar/gkac1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brister J.R., Ako-adjei D., Bao Y., Blinkova O. NCBI Viral Genomes Resource. Nucleic Acids Res. 2015;43:D571–D577. doi: 10.1093/nar/gku1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.O'Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D., et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–D745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tatusova T., DiCuccio M., Badretdin A., Chetvernin V., Nawrocki E.P., Zaslavsky L., Lomsadze A., Pruitt K.D., Borodovsky M., Ostell J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–6624. doi: 10.1093/nar/gkw569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gardinergarden M., Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All DNA sequence information generated in this study is available at the NCBI SRA PRJNA1131384; samples are annotated in Table S1. The AAVengeR pipeline is available at https://github.com/helixscript/AAVengeR. The code used to train machine learning models and generate genome wide predictions is available at https://gitlab.com/mgarciat/genome-wide-prediction-of-topoisomerase-iibeta-binding.