

Abstract
The growth and development of the skeleton is regulated by bone morphogenetic proteins of which several are linked to genetic skeletal disorders. So far, no human skeletal malformations have been associated with variants in BMP5. Here, we report a patient with biallelic loss of function variants in BMP5 and a syndromic phenotype including skeletal dysostosis, dysmorphic features, hypermobility, laryngo‐tracheo‐bronchomalacia and atrioventricular septal defect. We discuss the phenotype in relation to the known tissue‐specific expression of Bmp5 and similar morphological abnormalities previously reported in experimental animal models. Our findings suggest a new association between BMP5 variants and a range of developmental anomalies, involving ears, heart and skeleton, thereby increasing understanding of BMP5's role in human development.
Keywords: BMP5, cardiac malformation, rare disease, skeletal dysostosis, skeletal dysplasia
We report a patient with biallelic loss of function variants in BMP5 and a syndromic phenotype including ischio‐pubic‐patellar dysostosis, dysmorphism, hypermobility, laryngo‐tracheo‐bronchomalacia, and atrioventricular septal defect. Our findings suggest a new association between BMP5 variants and a range of developmental anomalies, thereby increasing understanding of BMP5's role in human development.
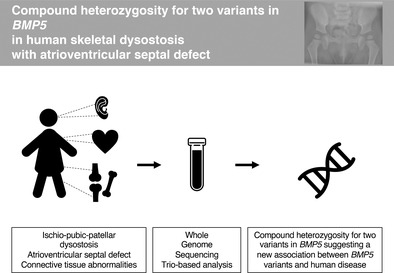
1. INTRODUCTION
Bone Morphogenetic Proteins (BMPs) belong to the transforming growth factor‐beta (TGF‐beta) superfamily and are essential for development and growth. 1 , 2 , 3 , 4 BMP5 acts in the extracellular matrix, where it is released, cleaved, and dimerized to obtain the final active protein that is encoded only by exons 5, 6, and 7. Only a few BMP‐related genetic skeletal disorders have been reported, for example, those related to BMP1, BMP2, BMPR1B, and BMPER. 5 In addition, pathogenic variants in several other genes involved in the BMP‐signaling pathway give rise to genetic skeletal disorders through their effect on ligands, receptors (ACVR1), mediators (SMAD4) and inhibitors (NOG). 5 , 6 To our knowledge, no genetic skeletal disease has been linked to BMP5 in humans. 5 , 7 However, studies in mice have shown that Bmp5 has multiple roles in skeletal, cartilage, ear and heart development. 8 , 9 , 10 This report presents a child, compound heterozygous for two variants in BMP5, and a complex syndromic phenotype involving skeletal dysostosis, dysmorphic features, hypermobility, laryngo‐tracheo‐bronchomalacia, and atrioventricular septal defect (AVSD).
2. METHODS
2.1. Ethics
This study was approved by the Regional Ethical Review Board Karolinska University Hospital and Karolinska Institutet (protocol numbers 2014/983‐31/1, 2012/2106‐31/4, 2018/2207‐32, 2021‐05360). Written informed consent was obtained.
2.2. Genetic analyses
Genomic DNA was extracted from blood from the patient and her parents, and buccal swabs from her unaffected siblings.
Genome sequencing was performed as a singleton to identify structural and single nucleotide variants (SNVs) in all known skeletal dysplasia genes using a gene panel according to PanelApp 11 and the Nosology of Genetic Skeletal disorders. 5 Furthermore, we performed family‐trio analysis for all known human genes. Induction of BMP5 expression in primary fibroblasts and RNA sequencing were performed as previously described. 12 Further details are provided in S1.
3. RESULTS
3.1. Clinical data
The patient is a 7‐year‐old girl and the second child of healthy non‐consanguineous Danish parents. The pregnancy was conceived naturally. At gestational age (GA) 13 + 0, routine ultrasound showed an increased nuchal translucency (3.9 mm, ref. <3.5 mm); chorionic villus sampling was normal (Elucigene QF‐PCR, Agilent 180 K). Fetal ultrasound scans at GA 16 + 2 and 20 + 5 were normal. Birth was uncomplicated at GA 39 + 3 (birth weight 3716 g [+1 standard deviation (SD)], birth length 50 cm [median], head circumference 36 cm [+1SD] 13 ). At day 12, she was admitted to hospital due to dyspnea. Echocardiography showed an AVSD that was surgically repaired at age 6 weeks. Surgery was complicated by third‐degree AV block necessitating pacemaker. At 6 months, she was admitted to hospital due to an upper airway infection. An echocardiography showed dyssynchronous left ventricle and reduced ejection fraction. Treatment with anti‐asthmatic and diuretic medication was successful. At 1 year and 5 months, she had tachypnea and sparse weight gain and was diagnosed with severe left AV valve insufficiency requiring surgery.
Until age 2.5 years, the girl suffered from several wheezing episodes and airway infections, but spirometry and bronchodilatator response were normal. A trachea‐bronchoscopy at 1 year and 11 months, revealed moderate laryngo‐tracheo‐bronchomalacia, a stalked omega‐shaped epiglottis, and a short right main bronchus. She is hypomimic with no cranial nerve palsies, has malar hypoplasia, short palpebral fissures, short nose, low nasal bridge, anteverted nares, long philtrum, small ears with abnormally folded antihelix (Figure 1), redundant but not hyperextensible skin, asymmetrical pectus carinatum, and thoracolumbar scoliosis. She has severe hypermobility of shoulders (100° rotation), elbows (20° hyperextension), knees (30° hyperextension), and ankles. She has a minor conductive hearing loss due to serous otitis media, mild muscular hypotonia and delay of gross motor development (independent walk at 25 months). Her cognitive development is normal but below the mean. She is doing well in a regular school program (further information in Supplementary Material).
FIGURE 1.
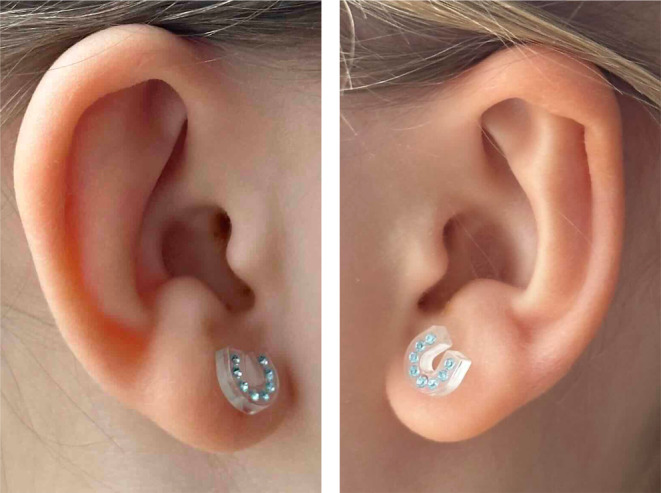
Photographs of ears (7 years, 6 months). Note small, dysplastic ears.
Skeletal survey showed mild thoracolumbar scoliosis, four sacral segments, absent ossification of the inferior pubic rami, the adjacent part of the pubic body and the ischial rami, and suspicion of patellar aplasia. Sonography showed absence of patellar cartilage anlagen bilaterally and of the cartilage anlagen of the inferior pubic and the ischial rami. MRI showed four sacral segments and anterior angulation of the coccyx (Figure 2).
FIGURE 2.
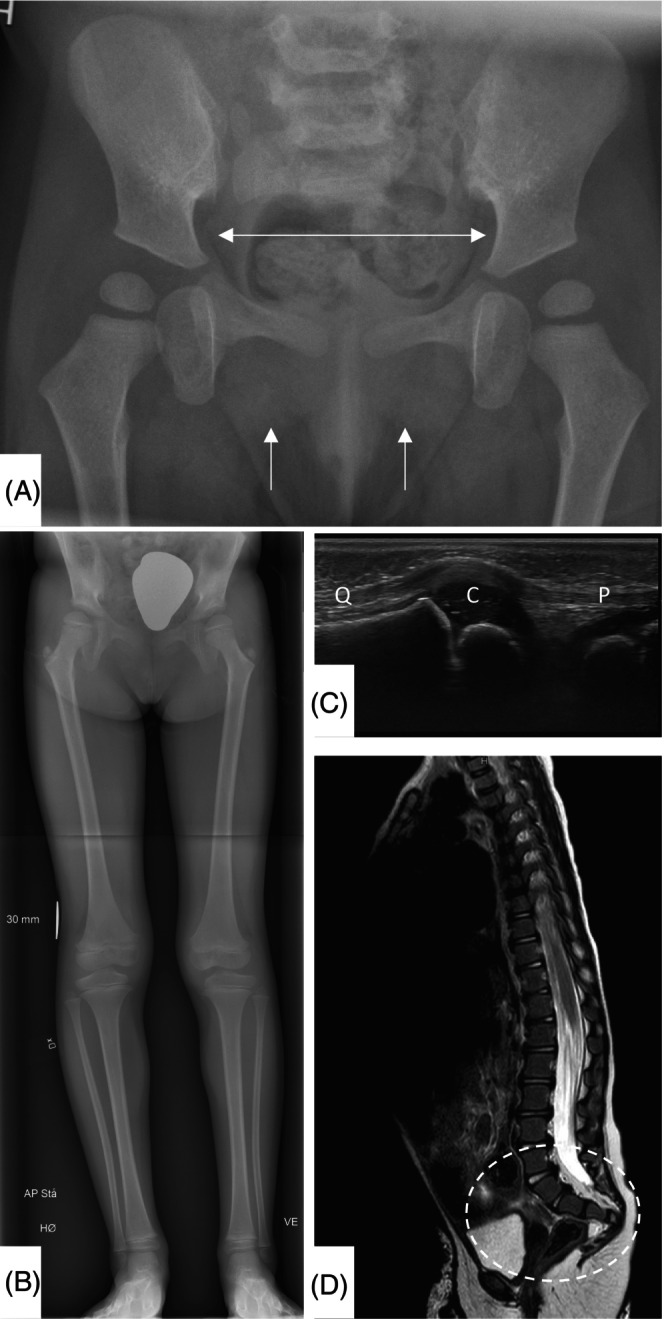
(A) Pelvic radiograph (1 year, 8 months). Note absent ossification of the inferior pubic rami, the adjacent part of the pubic body and the ischial rami. The central part of the pelvis is broad, with horizontal superior pubic rami. (B) Frontal standing radiograph of legs (6 years, 0 months). Mild anisomelia. Pelvic findings as described above. Bilateral coxa valga. Ossification centers in the greater trochanter small for age. (C) Sonography left knee (1 year, 10 months). Absence of patellar cartilage. Quadriceps tendon (Q), patellar ligament (P), cartilaginous epiphysis distal femur (C). (D) Sagittal T2 weighted spine MRI (2 years, 2 months). Horizontal sacrum, and anterior angulation of the coccyx.
3.2. Genetic findings
Chromosomal microarray and clinical exome trio analyses were normal. Genome sequencing trio analysis identified compound heterozygosity for two variants in BMP5 (NM_021073.4): c.88_89del, p.(Gly30Argfs*11) (paternal) and c.1104+2del, p.(?) (maternal). Splice prediction for the c.1104+2del variant indicates loss of donor splice site (SpliceAI delta score 0.98 and Pangolin delta score 0.84). 14 , 15 Her two healthy siblings were heterozygous for the maternal c.1104+2del variant.
3.3. Confirmation of abnormal splicing on the maternally inherited allele
Alternative splicing of BMP5 in maternal fibroblasts resulted in multiple annotated isoforms with a single aberrant transcript due to skipping of exon 5 (NM_021073.4:r.1028_1104del) (Figure S1A). The resulting frameshift was predicted to introduce a premature termination codon early in the penultimate exon (NP_066551.1:p.(Asp343Glyfs*15)) expected to trigger nonsense‐mediated decay (NMD). Sanger sequencing and RT‐PCR using primers in exon 5 and the 3′UTR region for two on genomic sequence heterozygous 3′UTR downstream variants indicated complete loss of activity at the donor splice site of exon 5 in the allele containing the c.1104+2del variant (Figure S1B). BMP5 is a highly evolutionarily conserved protein in different species as well as among paralogues (Figure S2A,B). In addition, exon 5, that is deleted in the maternal allele, encodes the conserved cysteine 353 of the TGF‐β‐like domain (Figure S2C,D) that is important for the proper folding required for receptor binding. In addition, in silico analysis of the in BMP5 signaling pathway proteins showed interactions with BMPER, TBX4 and PITX1 (Figure S3); genes associated with dysostoses involving pubic and ischiatic bones, sacrum and patella.
4. DISCUSSION
We report a patient with biallelic loss of function variants in BMP5 presenting with a rare phenotype including skeletal dysostosis, dysmorphic features, hypermobility, laryngo‐, tracheo‐bronchomalacia and AVSD. Searching for similar patients in GeneMatcher and our collaborative networks did not lead to identification of other affected individuals, indicating that this diagnosis is ultrarare.
The variant inherited from the father (c.88_89del) leads to a premature stop codon and is predicted to lead to nonsense mediated decay. The maternal variant (c.1104+2del) leads to abnormal transcript due to skipping of exon 5 that encodes the TGF‐β‐like domain. This is also predicted to trigger NMD, but if the abnormal RNA transcript results in synthesis of a truncated protein, it will lack the functionally important TGF‐β‐like domain. This domain is involved in BMP5 interaction with bone morphogenetic protein receptors BMPR1A and BMPR2 to initiate the signaling cascade. 4
In mice, loss of function mutations in this locus reduce outer ear growth due to disrupted formation and activity of the surface perichondrium around the ear. 8 Monozygotic twins with unilateral ear hypoplasia were reported to be heterozygous for variants in BMP2 (p.Ser111Ile) and BMP5 (c.833‐4C>G). However, segregation or cDNA sequencing were not performed, 10 and the pathogenicity of those variants remains unclear. Previous studies of chicken embryonic heart development showed BMP5 expression in the endoderm underlying the precardiac mesoderm, the myocardium of the atrioventricular canal and outflow tract regions. 9 These are all sites where the valvuloseptal endocardial cushion tissue and the primordia of the valves and septa of the adult heart are induced. 16 , 17 These findings suggest that BMP5 is essential for the development of myocardial and endocardial cushion tissue. 9
BMP5 expression is confined to specific parts of the skeleton and cartilage in mice and is tightly regulated by different enhancers. 18 , 19 The skeletal abnormalities observed in our patient, including dysostosis of ischial, pubic and sacral bones, and bilateral patellar aplasia, are similar to those seen in the known human syndromes diaphanospondylodysostosis, BMPER‐related, mandibular‐pelvic patellar syndrome, PITX1‐related and ischiocoxopodopatellar syndrome, TBX4‐related, as well as in campomelic dysplasia, SOX9‐related. 7 The BMP5 locus controls development of several bones, including processes on vertebrae, xiphoid, the fibula, the number of ribs along vertebra, and the volume of the thoracic cavity. 18 In addition, several individuals with BMPER‐related conditions, 20 , 21 as well as our patient, show laryngo‐tracheo‐bronchomalacia, indicating the importance of the BMP‐signaling pathway for cartilage maturation in the respiratory tract. Previous in vitro studies showed that BMP5 modifies chondrogenesis. 22 Considering the multiple malformations in our patient, and the molecular interactions of BMP5 with TBX4, PITX1, LMX1B and BMPER pathways, (Figure S3), it is plausible that the loss of function variants in BMP5 may cause our patient's ischio‐pubic‐patellar dysostosis and cardiac malformation. However, this study includes a single patient with an ultrarare condition. Further exploration including characterization of additional patients and studies of the disease mechanisms behind BMP5‐related conditions are necessary to confirm and expand the findings of this study.
FUNDING INFORMATION
Financial support from Stiftelsen Sällskapet Barnavård to (A.H.), Stiftelsen Promobilia to (G.G., H.L.), Stiftelsen Frimurare to (A.N., G.G., H.L.), Swedish Research Council to (A.N., G.G.), Region Stockholm to (A.N., G.G., H.L.), and Karolinska Institutet to (A.H., G.G., S.P.).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
Data S1. Supporting Information.
Figure S1. (A) RNA‐Seq of fibroblasts from the mother, heterozygous for the splice donor site variant c.1104+2del in BMP5 (NM_021073.4) and a wildtype control after CRISPR activation to induce expression of BMP5 and cycloheximide treatment to inhibit nonsense‐mediated decay. The count of supportive reads is shown at each splice junction and colored to enable comparison between the mother and the wildtype control. Only junctions supported by a minimum of five spliced reads are shown. (B) RT‐PCR and Sanger sequencing from exon 5 to exon 7 confirmed complete loss of natural splice donor site activity resulting in mono‐allelic expression of exon 5 as demonstrated by apparent hemizygosity for two variants in the untranslated region of exon 7, which were known to be in trans with the variant from genome sequencing.
Figure S2. (A) Conservation analysis of BMP5 between different species (from top to bottom: mouse, rat, rhesus macaque, chimpanzee, human, cat, sheep, cow, chicken, zebrafish). Amino sequence corresponding to exon 5 is highlighted in magenta. (B) Conservation analysis of BMP5, BMP2, and BMP4. Exon 5 is highlighted in magenta. Cysteine 353 is marked with an arrow. (C) AlphaFold model of BMP5 colored by confidence score (pLDDT), from blue (very high confidence) to orange (very low). (D) Superposition of the AlphaFold model of BMP5 (protein residues after‐cleavage) in white and x‐ray experimental structure of BMP2 (PDB code 3BMP) in yellow, indicating structural similarities in the paralogues. Structure corresponding to exon 5 is shown in magenta. The arrow points to Cys353 that is making a disulfide bond with Cys419.
Figure S3. Analysis of the molecular interactors using bioinformatics resource STRING https://string-db.org/. Please note the interactions of BMP5 with LMX1B, BMPER, TBX4, and PITX1, the encoding genes of which have been associated with conditions with the similar pelvic and patellar abnormalities as seen in our patient 7 : Nail‐patella syndrome, LMX1B‐related; Diaphanospondylodysostosis, BMPER‐related; Mandibulor‐pelvic patellar syndrome, PITX1‐related; Ischiocoxopodopatellar syndrome, TBX4‐related.
ACKNOWLEDGMENTS
The authors warmly thank the family for participating and collaborating in this study.
Gregersen PA, Hammarsjö A, Graversen L, et al. Compound heterozygosity for two variants in BMP5 in human skeletal dysostosis with atrioventricular septal defect. Clinical Genetics. 2025;107(1):78‐82. doi: 10.1111/cge.14616
DATA AVAILABILITY STATEMENT
The identified variants are submitted to ClinVar database (accession numbers SCV005038946 and SCV005038947). Other data are available from the authors upon reasonable request.
REFERENCES
- 1. Kawabata M, Imamura T, Miyazono K. Signal transduction by bone morphogenetic proteins. Cytokine Growth Factor Rev. 1998;9(1):49‐61. [DOI] [PubMed] [Google Scholar]
- 2. Urist MR. Bone: formation by autoinduction. Science. 1965;150(3698):893‐899. [DOI] [PubMed] [Google Scholar]
- 3. Wang RN, Green J, Wang Z, et al. Bone morphogenetic protein (BMP) signaling in development and human diseases. Genes Dis. 2014;1(1):87‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu M, Wu S, Chen W, Li YP. The roles and regulatory mechanisms of TGF‐β and BMP signaling in bone and cartilage development, homeostasis and disease. Cell Res. 2024;34(2):101‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Unger S, Ferreira CR, Mortier GR, et al. Nosology of genetic skeletal disorders: 2023 revision. Am J Med Genet A. 2023;191(5):1164‐1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Costantini A, Guasto A, Cormier‐Daire V. TGF‐β and BMP signaling pathways in skeletal dysplasia with short and tall stature. Annu Rev Genomics Hum Genet. 2023;24:225‐253. [DOI] [PubMed] [Google Scholar]
- 7. OMIM . Online Mendelian Inheritance in Man, O., John Hopkins University. 2024.
- 8. Kingsley DM, Bland AE, Grubber JM, et al. The mouse short ear skeletal morphogenesis locus is associated with defects in a bone morphogenetic member of the TGF beta superfamily. Cell. 1992;71(3):399‐410. [DOI] [PubMed] [Google Scholar]
- 9. Yamagishi T, Nakajima Y, Nishimatsu SI, Nohno T, Ando K, Nakamura H. Expression of bone morphogenetic protein‐5 gene during chick heart development: possible roles in valvuloseptal endocardial cushion formation. Anat Rec. 2001;264(4):313‐316. [DOI] [PubMed] [Google Scholar]
- 10. Liu W, Wang Q, Guo Y, Lin L, Yang Q, Jiang H. Whole‐genome sequencing identifies two novel rare mutations in BMP5 and BMP2 in monozygotic twins with microtia. J Craniofac Surg. 2022;33(2):e212‐e217. [DOI] [PubMed] [Google Scholar]
- 11. Martin AR, Williams E, Foulger RE, et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet. 2019;51(11):1560‐1565. [DOI] [PubMed] [Google Scholar]
- 12. Terkelsen T, Mikkelsen NS, Bak EN, et al. CRISPR activation to characterize splice‐altering variants in easily accessible cells. Am J Hum Genet. 2024;111(2):309‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tinggaard J, Aksglaede L, Sørensen K, et al. The 2014 Danish references from birth to 20 years for height, weight and body mass index. Acta Paediatr. 2014;103(2):214‐224. [DOI] [PubMed] [Google Scholar]
- 14. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(3):535‐548.e24. [DOI] [PubMed] [Google Scholar]
- 15. Zeng T, Li YI. Predicting RNA splicing from DNA sequence using Pangolin. Genome Biol. 2022;23(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Markwald RR, Mjaatvedt CH, Krug EL, Sinning AR. Inductive interactions in heart development. Role of cardiac adherons in cushion tissue formation. Ann N Y Acad Sci. 1990;588:13‐25. [DOI] [PubMed] [Google Scholar]
- 17. Wenink AC, Gittenberger‐de Groot AC. The role of atrioventricular endocardial cushions in the septation of the heart. Int J Cardiol. 1985;8(1):25‐44. [DOI] [PubMed] [Google Scholar]
- 18. Guenther C, Pantalena‐Filho L, Kingsley DM. Shaping skeletal growth by modular regulatory elements in the Bmp5 gene. PLoS Genet. 2008;4(12):e1000308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. King JA, Marker PC, Seung KJ, Kingsley DM. BMP5 and the molecular, skeletal, and soft‐tissue alterations in short ear mice. Dev Biol. 1994;166(1):112‐122. [DOI] [PubMed] [Google Scholar]
- 20. Kuchinskaya E, Grigelioniene G, Hammarsjö A, et al. Extending the phenotype of BMPER‐related skeletal dysplasias to ischiospinal dysostosis. Orphanet J Rare Dis. 2016;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Braun F, Gangfuß A, Stöbe P, et al. Expansion of the mutational spectrum of BMPER leading to diaphanospondylodysostosis and description of the associated disease process. Mol Genet Genomic Med. 2021;9(12):e1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Snelling SJ, Hulley PA, Loughlin J. BMP5 activates multiple signaling pathways and promotes chondrogenic differentiation in the ATDC5 growth plate model. Growth Factors. 2010;28(4):268‐279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.
Figure S1. (A) RNA‐Seq of fibroblasts from the mother, heterozygous for the splice donor site variant c.1104+2del in BMP5 (NM_021073.4) and a wildtype control after CRISPR activation to induce expression of BMP5 and cycloheximide treatment to inhibit nonsense‐mediated decay. The count of supportive reads is shown at each splice junction and colored to enable comparison between the mother and the wildtype control. Only junctions supported by a minimum of five spliced reads are shown. (B) RT‐PCR and Sanger sequencing from exon 5 to exon 7 confirmed complete loss of natural splice donor site activity resulting in mono‐allelic expression of exon 5 as demonstrated by apparent hemizygosity for two variants in the untranslated region of exon 7, which were known to be in trans with the variant from genome sequencing.
Figure S2. (A) Conservation analysis of BMP5 between different species (from top to bottom: mouse, rat, rhesus macaque, chimpanzee, human, cat, sheep, cow, chicken, zebrafish). Amino sequence corresponding to exon 5 is highlighted in magenta. (B) Conservation analysis of BMP5, BMP2, and BMP4. Exon 5 is highlighted in magenta. Cysteine 353 is marked with an arrow. (C) AlphaFold model of BMP5 colored by confidence score (pLDDT), from blue (very high confidence) to orange (very low). (D) Superposition of the AlphaFold model of BMP5 (protein residues after‐cleavage) in white and x‐ray experimental structure of BMP2 (PDB code 3BMP) in yellow, indicating structural similarities in the paralogues. Structure corresponding to exon 5 is shown in magenta. The arrow points to Cys353 that is making a disulfide bond with Cys419.
Figure S3. Analysis of the molecular interactors using bioinformatics resource STRING https://string-db.org/. Please note the interactions of BMP5 with LMX1B, BMPER, TBX4, and PITX1, the encoding genes of which have been associated with conditions with the similar pelvic and patellar abnormalities as seen in our patient 7 : Nail‐patella syndrome, LMX1B‐related; Diaphanospondylodysostosis, BMPER‐related; Mandibulor‐pelvic patellar syndrome, PITX1‐related; Ischiocoxopodopatellar syndrome, TBX4‐related.
Data Availability Statement
The identified variants are submitted to ClinVar database (accession numbers SCV005038946 and SCV005038947). Other data are available from the authors upon reasonable request.