

Abstract
α,β-unsaturated carbonyl compounds have extensive applications in various fields, such as organic, inorganic, analytical, and biological. In the modern era, they offer excellent pharmacological application prospects and find widespread use in the pharmaceutical industry. The current study revealed the synthesis and characterization of a novel 3-(2-bromo-5-fluorophenyl)-1-(thiophen-2-yl) prop-2-en-1-one (CY3). In vitro their antimicrobial (Pseudomonas aeruginosa, Klebsiella pneumonia, Escherichia coli, Staphylococcus aureus, and Acinetobacter baumannii), antifungal ( Candida parapsilosis, Candida tropicalis, and Candida albicans), cytotoxicity (VERO and Hep-G2 cells), in silico, and molecular docking analysis were also performed. The in-silico analysis evaluated the drug-likeness properties of the compound CY3 using various filtering rules, including Lipinski’s, Ghose filter, Veber, Egan, Muegge, and Medicinal Chemistry alerts such as Pan Assay Interference Structures (PAINS), Brenk, and Lead-likeness. Then, molecular docking studies performed using the AutoDock (AD4), Vina, and iGEMDOCK tools to determine the mechanism by which the CY3 compound interact with the bacterial strains. Here, five different receptors were selected, such as DNA gyrase, glucose 6-phosphate synthase (GlmS), dihydrofolate reductase (DHFR), dehydrosqualene synthase (DHSS), and undecaprenyl pyrophosphate synthase (UDPPS), for molecular docking analysis. The CY3 compound showed a good binding affinity with the two target proteins, DHFR and DHSS, respectively, with maximum binding energies of about − 7.07 and − 7.05 kcal/mol. The synthesized CY3 compound exhibited moderate antibacterial activity with a MIC value > 100 µg/mL against all five bacterial strains and moderate antifungal activity with a MIC value > 50 µg/mL against all three fungal strains. Drug-likeness analyses also support their favourable bioavailability.
Keywords: Synthesis, Antibacterial, Antifungal, Drug-likeness, Molecular docking, ADME calculation
Subject terms: Analytical chemistry, Chemical biology, Cheminformatics, Computational biology and bioinformatics
Introduction
α, β-unsaturated carbonyl compounds play a key role in both heterocyclic chemistry and medicinal chemistry. Promising potential exists for α,β-unsaturated carbonyl compounds as antifungal and antibacterial agents1. By interfering with vital cellular functions or structures, these compounds’ structural motifs render them effective against fungi that cause infections2. α,β-unsaturated carbonyl compounds have been recognized as versatile organic skeletons participating in various organocatalytic reactions for the formation of five- and six-membered saturated nitrogen-containing heterocycles3. They are also a precursor to many flavonoids. The biological activities of the chalcones are diverse4. Few skeletons can assert a broad spectrum of pharmacological actions, including antioxidant, cytotoxicity, anti-tumor, anti-inflammatory, anti-plasmodial, and immunosuppressive properties5. The conjugated double bonds that are usually present next to a carbonyl group in α β-unsaturated carbonyl compounds provide reactivity and biological activity6. Their ability to form covalent adducts with thiol-containing biomolecules7, like cysteine residues in proteins, allows them to function as Michael acceptors8. Fungal growth inhibition or cell death can result from interfere with important enzymes and cellular functions9. The percentage of Staphylococcus aureus that is resistant to common antibiotics like methicillin, oxacillin, or naproxenicillin is currently over 50% in intensive care units in the United States10. This number is still rising. Vancomycin was reportedly the last medication to treat Staphylococcus aureus that was resistant to several drugs11. The agents would then permeate and insert itself into bacterial membrane to exert its lethal disruptive effect12. Synthesized chalcones containing piperazine or 2,5-dichlorothiophene and some of the compounds to show good antibacterial activity13. They are believed to be the most reactive substructures of molecules, either naturally occurring or produced artificially14. These groups’ reactivity is influenced by a wide range of pharmacological activities15. The most important and well-studied α,β-unsaturated carbonyl-based compounds are the naturally occurring compounds chalcone (3), zerumbone (2), and curcumin (1), as well as their analogues and derivatives16. Some of the biological activities associated with them include, antimicrobial17, antioxidant18, anti-inflamatory19, antitubucular20, anticancer21, cardioprotective, antidiabetic22, antiviral23, anti-ageing24, antiallergic25, and hepatoprotective26 (Fig. 1).
Fig. 1.
Different medicinal activity of α,β-unsaturated carbonyl compounds.
Recent studies has shown that chalcone derivatives can exhibit significant antimicrobial activity by targeting different bacterial enzymes such as DNA gyrase, glucose 6-phosphate synthase, dihydrofolate reductase, dehydrosqualene synthase, and undecaprenyl pyrophosphate synthase27–30. The WHO regularly monitors and evaluates the safety and efficacy of antimicrobial agents, including those derived from natural sources like α,β-unsaturated carbonyl compounds31.
This study looked at the antibacterial activity of the α,β-unsaturated carbonyl compound ((3-(2-bromo-5-fluorophenyl)-1-(thiophen-2-yl) prop-2-en-1-one) by using several computational chemistry tools, such as ADMET (absorption, distribution, metabolism, excretion, and toxicity) and molecular docking analysis. In this study, utilized the AutoDock (AD4), Vina, and iGEMDOCK tools to predict the best fit orientation of the compound that binds to a specific protein target. This helped us figure out the drug’s activity and affinity. This will enable us to establish the mechanism by which the α,β-unsaturated carbonyl prevents bacterial growth. DNA gyrase, glucose 6-phosphate synthase (GlmS), dihydrofolate reductase (DHFR), dehydrosqualene synthase (DHSS), and undecaprenyl pyrophosphate synthase (UDPPS) with their Protein Data Bank (PDB) IDs are 1KZN, 1MOQ, 2W9S, 2ZCO, and 4H8E were used to evaluate action mechanism of the synthesized compound.
Methods
All chemical reagents and solvents were obtained from commercial companies and did not further purify them during use. The nuclear magnetic resonance (NMR) analysis was carried out using a Bruker Avance 400 MHz Ultrashield™ spectrometer for 1H and 13C-NMR. About 15 mg of the sample was dissolved in CDCl3 and DMSO-d632. The ES-MS spectra were recorded on an ion trap LCQ Advantage Max mass spectrometer (Thermo Electron Corporation)33. The melting point apparatus (Stuart SMP10) was used to determine the melting points of the synthesized compounds34. Fourier transform infrared spectroscopy (FTIR) analysis was carried out using PerkinElmer Nicolet 6700 FTIR spectrometer with attenuated total reflection in the frequency range of 150–750 cm−135,36.
General synthesis
A simple synthesis process was developed, as outlined in Scheme 1, to produce the desired anti-microbial active agent. Initially, equivalent amounts of 1-(thiophen-2-yl) ethan-1-one (0.01 mol) and 2-bromo-5-fluorobenzaldehyde (0.01 mol) were dissolved in 25 mL of ethanol. Then, slowly add sodium hydroxide solution (0.02 mol) and stir the mixture for 24 h37. TLC monitored the reaction’s completion. Upon completion, slowly pour the mixture into 400 mL of cold water, stirring constantly, and add 10% HCl until it reaches a pH of 7. Then, it was filtered and washed with distilled water to obtain pure 3-(2-bromo-5-fluorophenyl)-1-(thiophen-2-yl) prop-2-en-1-one (CY3) compound in quantitative yield38.
Scheme 1.

Synthesis of 3-(2-bromo-5-fluorophenyl)-1-(thiophen-2-yl) prop-2-en-1-one (CY3).
DFT calculations
DFT helped evaluate electron transport and electronic properties39. In this study, DFT performed by using Gaussian 9W and Gauss View v6.040. To optimise molecular structure, the DFT/B3LYP/6-31G(d,p) method is used in gaseous and solvent phases. The polarised continuum model (PCM) is used to compute the energies and intensities of the lowest-energy spin-permitted electronic excitations in solvent and vacuum phases using the B3LYP method41,42. Frontier molecular orbitals (FMOs), like the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), provide valuable information about the molecule’s electron density clouds43. Electrophilic reactions favour the non-bonding molecular orbital (NBMO), which is also called the HOMO. On the other hand, nucleophilic reactions favour the LUMO44. The fundamental quantum chemical parameters (QCPs) measure the reactivity of chemical species. Therefore, biological activity is more effective with a lower EGAP. Thus, a lower EGAP value suggests stronger chemical species reactivity and improved electron donation.
In-vitro antibacterial activity
Preparation of bacterial culture for the experiment
A loop of bacteria cultured on blood agar plates was moved to 10 mL of 3% w/v tryptic soy broth (TSB) and allowed to grow for a duration of 12–18 h. The test bacteria were then grown to the mid-logarithmic phase by adding 200 µL of the overnight culture to fresh 10 mL of 3% w/v tryptic soy broth (TSB). To get 1–2 × 109 CFU/mL of the bacterial suspension, the pellet was diluted in 10 mMTris, pH 7.4, and the bacteria were centrifuged at 10,000 RPM for 10 min. The supernatant was then discarded45.
Minimum inhibitory concentration
Antimicrobial activities of the compound was screened against Staphylococcus aureus (ATCC 29213), Escherichia coli (ATCC 25922), Klebsiela pneumoniae (ATCC 27736), Pseudomonas aeruginosa (ATCC 27853) and Acinetobacter baumannii (ATCC 1605). The synthesized compound was prepared as stock solutions in dimethyl sulfoxide (DMSO) at a concentration of 5 mg/mL, which is comparable to Colistin, levofloxacin and vancomycin. After being grown in 3% TSB for the entire night until they reached the mid-logarithmic phase, the bacteria were centrifuged, two times washed in 10 mMTris at pH 7.4, and then resuspended in 2X MHB broth (Becton Dickinson, USA) to obtain 1 × 105 CFU/mL. The peptide concentration was serially diluted (from 100 to 0.78 µg/mL) in a 96-well microtiter plate (made of polypropylene, Costar Corp., Cat. No. 3790). Followed, 50 µL of 2X MHB broth containing 1 × 105 CFU/mL of bacteria was added to each corresponding well. The plate was then incubated at 37 °C for 16 h. The Resazurin method was used to visually inspect the plates to determine the minimum inhibitory concentration (MIC), and the value that was observed to be greater than the MIC in > 100. Assay and MIC determination were carried out in triplicate46.
In vitro antifungal activity
In vitro antifungal activity of compound was tested against fungi; Candida parapsilosis (ATCC -22019), Candida tropicalis (ATCC-750), and Candida albicans (ATCC-90028) The Standard Broth Micro dilution method was used to conduct the susceptibility testing in accordance with the guidelines set forth by the Clinical and Laboratory Standard Institute (CLSI) using RPMI 1640 Medium buffered with MOPS [3-(N-morpholino propanesulphonic acid] for fungal cultures in 96 well plates employing a standard broth microdilution methodology in line with CLSI47. The highest concentration that was tested was 50 µg/mL, and each test well had an inoculum load that ranged from 1 to 5 × 103 cells. The plates were incubated for yeasts for 24–48 h, 72–96 h for mycelial fungi at 35 ± 1 °C and read visually for determination of MIC48.
Cytotoxicity
The synthesized CY3 compound was evaluated for cytotoxicity profiles using the MTT assay on the VERO cell line and Hep-G2. VERO and Hep-G2 cells were cultured in complete RPMI media supplemented with HEPES, 0.2% sodium bicarbonate, 0.2% d-glucose, 10% FBS, fungizone (0.25 mg/L), and gentamycin (50 mg/L) at 37 °C in a humidified CO2 incubator. Hep-G2 cells and VERO cells (10*4/well) were seeded in a 96-well plate, and cells were treated with different dilution of compound. Podophyllotoxin (P4405, Sigma) was used as the positive control. The drug was diluted for 72 h, and then 25 µL of MTT dye (M2128, Sigma) (stock 5 mg/mL) was added to each well. The plates were then put in a CO2 incubator for 2 h. The supernatant was removed carefully without disturbing the cells, and 150 µL of DMSO was added to each well to dissolve the purple precipitate. Absorbance was recorded at 540 nm using an ELISA plate reader, and the data was analysed to determine the 50% cytotoxic concentration (CC50)49.
Computational analysis
Molecular descriptors (MDs)
A chemical agent’s MDs affect its physiochemical properties including molecular weight (MW), rotatable bonds (RB), hydrogen bond donors (OHNH), hydrogen bond acceptor sites (ON), topological polar surface area (TPSA), and partition coefficient (log P). MW, RB, and TPSA also clarify the drug-likeness of a molecule. These are well-known MDs, and an increase in MW, TPSA, and RB values implies a decrease in their cell penetration power. Today, a variety of MDs rules are available to identify the drug-likeness of a chemical agent, such as the Ghose filter and Lipinski’s rule of five (Ro5). All the MD’s rules in Drug Design and Development (DDD) are highly effective in reducing miss-lead. Here, the MDs were calculated using ChemDraw 15.1 and Molinspiration50 (Fig. 2).
Fig. 2.

3D structures of 3-(2-bromo-5-fluorophenyl)-1-(thiophen-2-yl) prop-2-en-1-one (CY3). (a) CPK model; (b) 3D surface model; (c) ball-stick model.
Bioactivity score (BAS) calculation
Molinspiration v2018.10 facilitates the computation of BAS, a statistic that gauges the overall drug-likeness of a chemical entity. The general BAS rule suggests that a stronger reflected value increases the likelihood of the chemical agent under investigation exhibiting biological activity50.
ADMET evaluation
In this study, utilised the SwissADME and admetSAR tools to evaluate the ADMET properties of a CY3 compound. These data bases are frequently uses to conduct structural-based searches to identify ADMET characteristics51,52.
Bioavailability radar (BAR)
The use of bioavailability radar (BAR) or spider plots is highly beneficial in assessing the drug-likeness of a CY3 compound. The pink zone corresponds to six physicochemical properties: lipophilicity, size, polarity, solubility, flexibility, and saturation. The pink zone was the optimal area for achieving a compound’s oral bioavailability51.
Brain or IntestinaL EstimateD (BOILED-Egg)
Recently, researchers introduced the BOILED-Egg model to measure a chemical substance’s ability to cross the blood–brain barrier and enter the digestive system. This model is quite useful for evaluating the drug-likeness of a proposed compound. Lipophilicity (WLOGP) and TPSA are two crucial determinants of a molecular system’s bioavailability. To generate a BOILED-Egg plot, the SwissADME server uses WLOGP and TPSA data51,53.
Molecular docking
Molecular docking analysis can predict the binding interaction between a ligand and a receptor protein. The study explores, the ligand binds to the active pocket of the receptor.
Most favourable target prediction
Computational approaches are highly beneficial for finding the optimal medication target. At first, using computational techniques to screen and find suitable targets, as well as develop an alternate target for a conventional chemical structure, is beneficial. Here, employed the online Prediction of Activity Spectra for Substances (PASS) tool to determine the most favourable target for the CY3 compound (https://www.way2drug.com/passonline/) (Table 1).
Table 1.
Prediction of activity spectra for substances (PASS) analysis of CY3 compound.
| Probability to be active (Pa) | Probability to be inactive (Pi) | Activity |
|---|---|---|
| 0.571 | 0.082 | Antineurotic |
| 0.447 | 0.004 | Lipoxygenase inhibitor |
| 0.387 | 0.006 | Orexin receptor 1 antagonist |
| 0.374 | 0.005 | Cyclooxygenase inhibitor |
| 0.370 | 0.004 | 5-Lipoxygenase inhibitor |
| 0.320 | 0.073 | Antifungal |
| 0.257 | 0.079 | Glycerol-3-phosphate dehydrogenase inhibitor |
| 0.206 | 0.085 | Antihelmintic |
| 0.187 | 0.066 | Aldose reductase substrate |
| 0.183 | 0.065 | Antimycoplasmal |
| 0.226 | 0.119 | Antipsoriatic |
| 0.243 | 0.140 | Antiviral (Influenza) |
Target protein retrieval
It was found that DNA gyrase, GlmS, DHFR, DHSS, and UDPPS would be the best proteins to study the CY3 compound antibacterial properties. The crystal structures of the proteins were obtained from Protein Data Bank (PDB) with the IDs of 1KZN, 1MOQ, 2W9S, 2ZCO, and 4H8E (Fig. 3)27–29.
Fig. 3.

Target proteins exhibit 3D ribbon and surface structures. (a) DNA gyrase; (b) glucose 6-phosphate synthase (GlmS); (c) dihydrofolate reductase (DHFR); (d) dehydrosqualene synthase (DHSS); and (e) undecaprenyl pyrophosphate synthase (UDPPS).
Pocket analysis and refinement
The active pocket analysis is anticipated to be performed using POCASA1.1 (http://g6altair.sci.hokudai.ac.jp/g6/service/pocasa/) and CASTp v3.0 (http://sts.bioe.uic.edu/castp/). The results indicate that DHFR and DHSS comprise the top five pockets. The most favourable pocket information includes area (SA & MS) and volume (SA & MS), with values of 441.6333, 766.3973, 305.925, and 1105.858 Å3 for DHFR, and 783.893, 1242.083, 658.970, and 2042.566 Å3 for DHSS. Similarly, pocket mouth information includes area (SA & MS) and volume (SA & MS), with values of 89.092, 240.36, 98.314, and 124.70 Å3 for DHFR, and 96.325, 228.15, 85.977, and 103.57 Å3 for DHSS (Table 2). The study also improved the structures of proteins and ligand. To achieve this, they first obtained the crystal structure of the target protein from the Protein Data Bank (PDB) and subsequently employed UCSF Chimaera v1.14 to remove unnecessary hetatom, water, ligands, and other chains. Furthermore, used the YASARA minimisation server (http://www.yasara.org/minimizationserver.htm) to optimise the energy of the target protein’s 3D structures. UCSF Chimaera v1.14 software was also used to visualize the pocket where the substrate found. MGL Tools 1.5.6 used to add Kollman charge, polar hydrogen bonds, and margin non-polar hydrogen bonds before molecular docking analysis (Fig. 4).
Table 2.
Sequence ID, pocket amino acid residues (atom) of the target proteins (Dihydrofolate reductase (DHFR) and Dehydrosqualene synthase (DHSS)) selected for multiple grids molecular docking.
| Target | Area (SA) | Volume (SA) | Pocket amino acid with sequence ID (atom) |
|---|---|---|---|
| Dihydrofolate reductase (DHFR) | 441.633 | 305.925 | ILE6-C; ILE6-O; ILE6-CG1; ILE6-CG2; VAL7-CA; VAL7-C; VAL7-O; VAL7-CG2;ALA8-N; ALA8-O; ALA8-CB; ILE15-O; ILE15-CB; ILE15-CG1; ILE15-CD1;GLY16-CA; TYR17-N; ASN19-N; ASN19-CA; ASN19-C; ASN19-O; ASN19-OD1; GLN20-N;GLN20-CA; GLN20-C; GLN20-O; LEU21-N; LEU21-CG; LEU21-CD1; LEU21-CD2; TRP23-NE1;ASP28-OD1; ASP28-OD2; LEU29-CD1; LEU29-CD2; HIS31-CD2; HIS31-NE2; ILE32-CG1; ILE32-CD1; ILE32-CD1; ALA44-CB; RG45-N; ARG45-CB; ARG45-NE; ARG45-NH2; LYS46-N; LYS46-CB; LYS46-CG; LYS46-CD; LYS46-CE; THR47-N; THR47-CA; THR47-OG1; THR47-CG2; SER50-CB; SER50-OG; ILE51-CG1; ILE51-CG2; ILE51-CD1; LYS53-CE; LYS53-NZ; LEU55-CG; LEU55-CD2; LEU63-O; LEU63-CG; LEU63-CD1; LEU63-CD2; THR64-CA; THR64-C; THR64-O; THR64-OG1; ASN65-N; ASN65-CB; ASN65-CG; ASN65-OD1; ASN65-ND2; GLN66-CG; GLN66-NE2; ASN78-O; PHE93-O; PHE93-CD1; PHE93-CD2; PHE93-CE1; PHE93-CE2; PHE93-CZ; GLY94-CA; GLY95-N; GLY95-CA; GLN96-N; GLN96-CB; THR97-N; THR97-CB; THR97-OG1; THR97-CG2; TYR99-CE1; TYR99-OH; TYR110-C; TYR110-O; ILE111-C; THR112-N; THR112-OG1; ASP121-OD1; THR122-CB; THR122-OG1; THR122-CG2 |
| 46.977 | 23.196 | TYR127-O; TYR127-CD2; TYR127-CE2; THR128-CA; THR128-C; THR128-O; PHE129-N; PHE129-CA; PHE129-CD1; PHE129-CD2; PHE129-CE1; PHE129-CE2; PHE129-CZ; TRP132-O; TRP132-CB; GLU133-CA; VAL134-CG2; LEU153-CD2; LEU155-CD1; LEU155-CD2; ARG158-NH2 | |
| 28.382 | 13.156 | ASN27-CA; ASN27-CB; ASN27-CG; ASN27-OD1; ASN27-ND2; LYS30-CE; LYS30-NZ; GLN141-O; GLN141-B; GLN141-CG; ASP143-OD2; ASN146-ND2 | |
| 30.209 | 10.27 | ILE103-O; ILE103-CG1; ILE103-CG2; ILE103-CD1; ASP104-CA; ASP104-OD1; PRO126-O; TYR127-CA; TYR127-CB; ASP131-CB; ASP131-CG; ASP131-OD2; TRP132-NE1; TRP132-CZ2; ARG157-NH1 | |
| 18.895 | 6.73 | LYS33-CD; LYS33-CE; LYS33-NZ; THR37-OG1; THR37-CG2; PRO56-CB; PRO56-CG; ASN57-CB; ASN57-ND2; ARG58-NH2 | |
| Dehydrosqualene synthase (DHSS) | 783.893 | 658.97 |
MET21-SD; LYS23-O; HIS24-C; HIS24-O; HIS24-CD2; HIS24-CE1; HIS24-NE2; SER25-CA; SER25-CB; SER25-OG; LYS26-N; LYS26-CB; LYS26-CG; LYS26-NZ; SER27-N; SER27-CB; SER27-OG; PHE28-CB; PHE28-CG; PHE28-CD1; PHE28-CD2; PHE28-CE1; PHE28-CE2; PHE28-CZ; PHE32-CD2; PHE32-CE2; PHE32-CZ; VAL43-CG1; TYR47-CA; TYR47-O; TYR47-CD1; TYR47-CD2; TYR47-CE2; TYR47-CZ; TYR47-OH; CYS500-CB; ARG51-D; ARG51-N1; ARG51-NH; ASP54-CA; ASP54-O; ASP54-CB; ASP54-CG; ASP54-OD1; ASP54-OD2; ASP55-CA; ASP55-C; ASP55-O; ASP55-OD1; ILE57-N; ILE57-CB; ILE57-CG1; ILE57-CG2; ASP58-N; ASP58-CB; ASP58-CG; ASP58-D1; ASP58-OD2; THR116-CG2; VAL117-CA; VAL117-O; VAL117-CG1; VAL117-CG2; ASP120-CB; ASP120-CG; ASP120-OD1; ASP120-OD2; GLN121-N; GLN121-CA; GLN121-CG; TYR135-CE1; TYR135-E2; TYR135-OH; VAL139-O; VAL139-CB; VAL139-CG1; VAL139-CG2; ALA140-CA; ALA140-O; ALA140-CB; VAL143-CB; VAL143-CG1; VAL143-CG2; GLY144-N; GLY144-CA; GLY144-O; LEU147-CB; LEU147-CD1; LEU151-CD1; ALA163-CA; ALA163-O; ALA163-CB; LEU166-CB; LEU166-CG; LEU166-CD2; GLY167-N; GLY167-CA; GLY167-O; LEU170-CB; LEU170-CD1; GLN171-N; GLN171-CA; GLN171-OE1; GLN171-NE2; ASN174-C; ASN174-O; ASN174-CB; ASN174-CG; ASN174-OD1; ASN174-ND2; ILE175-CA; ILE175-CG1; ARG177-CD; ARG177-NH1; ARG177-NH2; ASP178-CB; ASP178-CG; ASP178-OD2; GLU181-CG; GLU181-CD; GLU181-OE1; ASP182-CA; ASP182-OD1; ASP182-OD2; ASN185-O; ASN185-CB; ASN185-CG; ASN185-ND2; GLU186-CB; GLU186-OE2; ARG187-CB; ARG187-CG; ARG187-NE; ARG187-CZ; ARG187-NH1; ARG187-NH2; TYR189-CE2; TYR189-CZ; TYR189-OH; PHE239-CZ; ILE247-CG1; ILE247-CD1; ALA250-CB; TYR254-CE2; TYR254-OH; ILE257-CG1; ILE257-CD1; ARG271-CD; ARG271-NH1; ARG271-NH2; VAL272-O; PHE273-CA; PHE273-CB; VAL274-N; VAL274-CB; LYS279-CE; LYS279-NZ |
| 120.764 | 64.167 | PHE14-CE1; PHE14-CE2; PHE14-CZ; ASP38-CA; ASP38-C; ASP38-O; ASP38-CB; ASP38-CG; ASP38-OD1; ASP38-OD2; GLN39-CA; GLN39-CG; GLN39-CD; GLN39-OE1; GLN39-NE2; LYS41-CB; LYS41-0CD; LYS41-E; LYS41-NZ; ALA41-N; ALA42-CA; ALA42-CB; HIS98-CD2; HIS98-NE2; VAL99-CA; VAL99-CG1; VAL99-CG2; HIS102-CB; HIS102-CG; HIS102-ND1; HIS102-CD2; LYS103-CG; LYS103-CD; LYS103-CE; LYS103-NZ; PRO149-O; ILE150-CA; ILE150-O; ILE150-CG2; ILE150-CD1; SER152-N; SER152-C; SER1520-O; ASP153-CA; ASP153-OD1; SER240-CB; SER240-OG; GLU242-OE1 | |
| 68.819 | 51.524 | THR116-O; THR116-OG1; THR116-G2; LYS119-CB; LYS119-CD; LYS119-CE; LYS119-NZ; ASP120-N; ASP120-CA; ASP120-CB; ASP120-OD2;PHE123-CD1; PHE126-CD1; PHE126-CE1; PHE126-CZ; GLU131-CA; GLU131-O; GLU131-CB; GLU131-CG; GLU131-OE1; GLY134-C; GLY134-O; TYR135-N; TYR135-CA; TYR135-CB; TYR135-CD2; TYR135-CE2; GLY138-CA | |
| 72.789 | 15.734 | LEU176-O; ARG177-CA; ARG177-O; ASP178-CA; ASP178-OD1; VAL179-N; VAL179-CB; VAL179-CG2; GLY180-N; GLY180-CA; GLU181-N; GLU181-CB; GLU181-OE2; TYR214-OH; TRP218-CD1; TRP218-NE1; VAL261-CA; VAL261-CG1; VAL261-CG2; ALA264-CB; TYR266-CA; TYR266-C; TYR266-O; TYR266-CB; THR267-N; THR267-O; THR267-CG2; LEU268-CA; LEU268-C; LEU268-O; LEU268-CG; LEU268-CD1; GLU270-N; GLU270-C; GLU270-O; ARG271-N; ARG271-CA; ARG271-CB; VAL272-N; VAL272-CG1; VAL272-CG2 | |
| 30.712 | 13.298 | LYS22-CG; LYS22-CE; LYS26-CA; LYS26-O; LYS26-CB; LYS26-CD; SER29-CB; TYR30-N; TYR30-CA; TYR30-CB; TYR30-CD1; LYS276-CG; LYS276-CD; LYS276-CE; LYS279-CD; LYS279-CE; LYS279-NZ |
Fig. 4.

Target proteins exhibit cartoon structure with the top five pockets highlighted with different colures, the priority order of pockets is as red > blue > green > purple > organ and their amino acid sequence. (a) Dihydrofolate reductase (DHFR) and (b) Dehydrosqualene synthase (DHSS).
Ligand preparation
In this study, ChemDraw 15.1 was used to prepare the 2D structure of CY3 compound, and Chem3D 15.1 used to prepare their 3D structure. The 3D structure of CY3 compound was optimized by using Avogadro v1.2.0 with the help of the mark method of the force field (MMFF94). Similarly, the SMILES and PDB files of the ligand was prepared by using Biovia DSV 20.1.0. By using MGL Tools 1.5.6, added rotatable bonds and the Gasteiger charge to CY3 compound. Here, 2ʹ-hydroxy-4ʹ-methoxychalcone (HMOC) and trimethoprim (TOP) were taken as reference molecules. Likewise, fluconazole, levofloxacin, and podophylotoxin were also selected as standard drugs. The crystalline structures of reference and standards were downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov).
AD4
The AD4 tool is used to investigate the optimal ligand conformation that binds to the target protein. The current study used AutoDock 4.0 and ADT v4.2.6 software to perform molecular docking. It used the Genetic Algorithm (GA) and Lamarckian genetic algorithm (LGA). Docking parameters, including population size, number of generations, crossover rate, and mutation rate, were generally set to their default values of 150, 27,000, 0.8, and 0.02, respectively. It was observed that in most studies, these default settings yield satisfactory results. The AD4 tool’s results include binding energies and inhibition constants. AutoDock uses an empirical free energy scoring function (Intermolecular energies include Van der Waals interactions (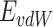 ) and electrostatic interactions (
) and electrostatic interactions ( ); desolvation energy (
); desolvation energy (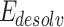 ); hydrogen bonding energy (
); hydrogen bonding energy (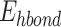 ); and thermal free energy (
); and thermal free energy ( )) to estimate ligands’ binding energy to their target proteins. Here, Eq. (1) shows the total binding energy and their formula as:
)) to estimate ligands’ binding energy to their target proteins. Here, Eq. (1) shows the total binding energy and their formula as:
 |
1 |
Vina
Vina version 1.1.2 conducts the analysis of the ligand-receptor interaction profile. This algorithm systematically explores the degrees of freedom within the system and calculates the interaction profile based on the minimum energy required. Vina is more accurate than AD4 in docking estimation because it uses the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm and an empirical scoring algorithm. Vina commonly sets its exhaustion, mode number, and energy range to their default values of 8, 9, and 3 kcal/mol, respectively, and adjusts the search space based on the requirements. Higher values of exhaustion increase the search time, but they also improve the accuracy of the results. A scoring function that is a weighted sum of different atomic interactions, like steric interactions ( ), hydrophobic interactions (
), hydrophobic interactions ( ), hydrogen bonding (
), hydrogen bonding ( ), and rotational entropy (
), and rotational entropy (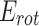 ), is used by Vina to figure out the ligand interaction with the target proteins. Here, Eq. (2) shows the total binding energy and their formula as:
), is used by Vina to figure out the ligand interaction with the target proteins. Here, Eq. (2) shows the total binding energy and their formula as:
 |
2 |
iGEMDOCK
To assess the effectiveness of virtual screening, iGEMDOCK v2.1 was utilized. This software tool automates post-analysis, screening, and combinational docking processes. The docking analysis necessitates a pdb file and generates results in the form of a fitness score. iGEMDOCK uses the genetic algorithm (GA) and docking parameters, which include factors such as population size, number of generations, number of solutions, preparation of the binding site, and post-screening analysis. For the current analysis, specific parameters were set, including 800 individuals, 80 generations, and a population comprised of 10 solutions. The ligand was docked with the binding site, utilising a precise docking function to facilitate interaction. In iGEMDOCK, the scoring function (Van der Waals interactions ( ), electrostatic interactions (
), electrostatic interactions ( ), hydrogen bonding (
), hydrogen bonding ( ), and pharmacological interactions (
), and pharmacological interactions (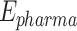 )) combines energy-based terms to predict the binding affinity of ligands to their target proteins. Here, Eq. (3) shows the total binding energy and their formula as:
)) combines energy-based terms to predict the binding affinity of ligands to their target proteins. Here, Eq. (3) shows the total binding energy and their formula as:
 |
3 |
Results
Chemistry
The synthesis of CY3 compound was optimized by exploring different reaction conditions, catalysts, and purification methods to improve the yield, selectivity, and efficiency of the reaction. Finally, compound (3) was successfully synthesized via Claisen-Schmidt condensation reaction between respective aldehyde and methyl ketone using sodium hydroxide as base in the presence of ethanol at room temperature54. The reaction proceeded smoothly and almost completed in 24 h. CY3 compound was obtained in good yield (73%), after purification with water followed by n-hexane. The synthesized compound was characterized using NMR, ES-MS, and FTIR spectral techniques55 (S. Figures 1-6).
HOMO‑LUMO analysis
To find out about the chemical stability and electronic properties of CY3, used the DFT/B3LYP/6-31G(d,p) method to figure out the EHOMO, ELUMO, and their EGAP values, respectively. The HOMO and the LUMO are essential parameters for facilitating charge transfer between orbitals during chemical processes. Figure 5 displays the computed values of EHOMO, ELUMO, and their EGAP. In this figure, red indicates the presence of a negative charge, while green indicates a positive charge. The HOMO–LUMO band gap values for CY3 in the gas, water, methanol, and ethanol phases are 4.2793, 4.1863, 4.1893, and 4.1909 eV, respectively. Notably, the energy gap varies by approximately 0.09 eV as the molecule transitions from the gaseous phase to the solvent phase. A smaller HOMO–LUMO energy gap enhances electron mobility and reactivity. Compared to the gas phase, the molecule in the water phase demonstrates increased softness and decreased hardness, resulting in higher reactivity. Molecules with low hardness are generally more susceptible to chemical reactions and exhibit significant electron transfers, such as nucleophilic attacks.
Fig. 5.

Atomic orbital composition of the HOMO, LUMO, and their EGAP obtained from TD-DFT/B3LYP/6-31G(d,p) level at gas and solvent phase.
In vitro antibacterial activity
The synthesized CY3 compound was evaluated against five bacterial strains. The results have been tabulated in Table 3. It could be seen that the synthesized CY3 compound show inhibitory effect up to > 100 μg/mL concentration.
Table 3.
Antibacterial activity of synthesized CY3 compound.
| MIC (μg/mL) | |||||
|---|---|---|---|---|---|
| Entry | S. aureus (ATCC29213) | E. coli (ATCC25922) | K. pneumoniae (ATCC27736) | P. aeruginosa (ATCC27853) | A. baumannii (ATCC1605) |
| CY3 | > 100 | > 100 | > 100 | > 100 | > 100 |
| Colistin | > 100 | 6.25 | 3.12 | 3.12 | 3.12 |
| Levofloxacin | 0.39 | 0.39 | 0.39 | 0.39 | 3.12 |
| Vancomycin | 0.39 | > 100 | > 100 | > 100 | 50 |
In vitro antifungal activity
The synthesized CY3 compound was subjected to in vitro antifungal assay against three strains of fungus, namely Candida albicans, Candida parapsilosis and Candida tropicalis, utilizing the Standard Broth Micro dilution method CLSI guidelines47. The obtained results were then compared, in terms of MIC (50 µg/mL), with those of the standard drug (fluconazole) Table 4.
Table 4.
Antifungal activity of synthesized CY3 compound.
| Entry | MIC (µg/mL) | ||
|---|---|---|---|
| Candida albicans (ATCC 90028) | Candida parapsilosis (ATCC 22019) | Candida tropicalis (ATCC 750) | |
| CY3 | > 50 | > 50 | > 50 |
| Fluconazole | 0.25 | 2 | 1 |
Cytotoxicity
The synthesized CY3 compound was testing cytotoxicity against two cell lines vero and HepG2 cell lines, using MTT assay. The obtained results were then compared, in terms of cytotoxic concentration of two concentration vero cell line CC50 (200 µM) and HepG2 cell lines CC50 200 µM), with those of the standard drugs (Podophylotoxin) Table 5.
Table 5.
Cytotoxicity of synthesized CY3 compound against vero cell line and Hep-G2 cell line.
| Entry | CC50 (µM) | |
|---|---|---|
| VERO cell line | HepG2 | |
| CY3 | > 200 | > 200 |
| Podophylotoxin | 6.2 | 6.2 |
Computational screening
Molecular descriptors (MDs)
Molecular weight (MW), molar refractivity (MR), number of rotatable bonds (nRB), hydrogen bond donor (nOHNH), hydrogen bond acceptor (nON), fraction Csp3 (fCsp3), and topological polar surface area (TPSA) are some of MDs parameters. MDs are generally used to narrow down the library molecules. SA serves as a scale for assessing molecular synthesis based on its level of complexity. It provides details regarding the complexity and viability of the molecule’s synthesis56–58. The percent absorption also aids in estimating the oral availability of CY3 compound. Both the number of atoms and the number of aromatic atoms play an important role. Cells’ ability to transport and use the molecular system heavily depends on their molecular weight (MW). Studies have shown that molecules in the 350–400 Dalton (500) range are best for use in drug design. ON and OHNH are important parameters that may play a role in biological systems’ interactions50. Rotatable bonds (RB) play a significant role in molecular flexibility, impacting a molecular system’s oral bioavailability. Molecular refractivity (MR) is used to evaluate the interaction profile within the target-receptor complex. The logP (octanol/water partition coefficient) is employed to determine the fragment-based support and correction factor. Consequently, several filtering standards have been established, such as lead likeness, Ro5, Veber, and Ghose. The synthesized CY3 compound demonstrates favourable oral bioavailability, which meets filtering measures. Both standard drugs and CY3 compound exhibit excellent oral bioavailability and absorption, as observed (Tables 6, 7) (S. Figures 7, 8).
Table 6.
Molecular descriptors (MDs) of the synthesized CY3 compound.
| Molecular descriptors | CY3 |
|---|---|
| miLogP (octanol/water partition coefficient) | 4.43 |
| Topological polar surface area (TPSA, Å2) | 45.31 |
| Number of atoms (nAtoms) | 17 |
| Number of aromatic atoms | 11 |
| Molecular weight (MW) | 311.18 |
| Number of hydrogen bond acceptor (nON) | 1 |
| Number of hydrogen bond donor (nOHNH) | 0 |
| Number of rotatable bonds (nRB) | 3 |
| Volume | 215.38 |
| Synthetic accessibility (SA) | 2.79 |
| Molar refractivity (MF) | 71.78 |
| Bioavailability score | 0.55 |
| Percentage absorption (109 − [0.345 × topological polar surface area]) | 93.37 |
| Synthetic accessibility | 2.79 |
Table 7.
Lipinski, Muegge, Ghose, Veber, and lead-likeness screening rules were used to compare the bioavailability and drug-likeness of the synthesized CY3 compound.
| Filtering rules | Number of violations |
|---|---|
| Drug-likeness violations | |
| Lipinski’s | 0 |
| Ghose filter | 0 |
| Veber | 0 |
| Egan | 0 |
| Muegge | 0 |
| Medicinal chemistry alerts | |
| Pan Assay Interference Structures (PAINS) | 0 |
| Brenk | 1 |
| Leadlikeness | 1 |
Bioactivity Score (BAS) Analysis
The study found that the synthesized CY3 compound shows good BAS values for ion channel modulator, kinase inhibitor, nuclear receptor ligand, enzyme inhibitors, GPCR ligands, and protease inhibitors. The synthesized CY3 compound has been shown negative values against all targets. Thus, they are now ready for the next step of evaluation (Table 8) (S. Figure 9).
Table 8.
Parameters of BAS analysis of the synthesized CY3 compound.
| Parameters | CY3 |
|---|---|
| GPCR ligand | − 0.63 |
| Ion channel modulator | − 0.66 |
| Kinase inhibitor | − 0.82 |
| Nuclear receptor ligand | − 0.88 |
| Protease inhibitor | − 0.87 |
| Enzyme Inhibitor | − 0.40 |
ADMET analysis
To assess the chemical agent’s oral bioavailability, the ADMET potentials are important considerations. To qualify as a drug, a chemical must meet the ADMET requirements. Clinical studies will only be permitted if ADMET properties are evaluated. The study observed the oral bioavailability of the synthesized CY3 compound. In this study, observed the blood–brain barrier (BBB) permeability, Caco-2 permeability, and human intestinal absorption (HIA). Also, evaluated synthesized CY3 compound BBB penetration and found that it crosses the BBB. CY3 compound has also shown a favourable amount of HIA. Moreover, CY3 compound showed minimal Caco-2 cell permeability. The obtained results also revealed a high HIA percent in all the CY3 compound. The synthesized CY3 compound showed any toxicity or carcinogenicity in the Ames mutagenicity test. Therefore, further research and analysis are performed for CY3 compound (Table 9) (S. Figures 8, 10).
Table 9.
Shows the relative ADMET profiles of the synthesized CY3 compound.
| Parameters | Probability |
|---|---|
| Absorption | |
| Human intestinal absorption (HIA+) | 1.0000 |
| Bloo-Brain Barrier (BBB+) | 0.9917 |
| Caco-2 permeability (Caco2+) | 0.6621 |
| Renal Organic Cation Transporter | 0.8353 |
| P-glycoprotein substrate | 0.7615 |
| P-glycoprotein inhibitor | 0.7730 |
| Distribution | |
| Subcellular localization (Mitochondria) | 0.6441 |
| Metabolism | |
| CYP450 2C9 Substrate (Non-substrate) | 0.7416 |
| CYP450 2D6 Substrate (Non-substrate) | 0.8664 |
| CYP450 3A4 Substrate (Non-substrate) | 0.7385 |
| CYP450 1A2 Inhibitor (Inhibitor) | 0.8132 |
| CYP450 2C9 Inhibitor (Inhibitor) | 0.6243 |
| CYP450 2D6 Inhibitor (Non-inhibitor) | 0.8819 |
| CYP450 2C19 Inhibitor (Inhibitor) | 0.8278 |
| CYP450 3A4 Inhibitor (Non-inhibitor) | 0.7911 |
| CYP Inhibitory Promiscuity (High CYP Inhibitory Promiscuity) | 0.9183 |
| Excretion | |
| Human Ether-a-go-go-Related Gene Inhibition (weak inhibitor) | 0.9674 |
| Biodegradation (not ready biodegradable) | 0.9856 |
| Toxicity | |
| AMES Toxicity (non-AMES toxic) | 0.6447 |
| Carcinogens (non-carcinogens) | 0.7716 |
Bioactivity Radar (BAR) and Brain Or IntestinaL EstimateD (BOILED-Egg) analysis
Based on its various molecular properties, the BAR model provides a concise visual representation of a drug’s characteristics. The pink zone in the BAR indicates the range of properties that are optimal for oral bioavailability, including polarity, size, solubility, flexibility, and lipophilicity. The BAR prediction showed that the synthesized CY3 compound fell within the desirable pink zone. Furthermore, the BOILED-Egg model used to assess CY3 compound permeability and absorption across the blood–brain barrier and gastrointestinal tract. The BOILED-Egg model suggests that the white area represents the likelihood of gastrointestinal absorption, while the yellow yolk indicates the ability to cross the blood–brain barrier. Here, found that the CY3 compound to be well-absorbed through the gastrointestinal system and capable of crossing the blood–brain barrier, making it a promising biologically active candidate (Fig. 6).
Fig. 6.

(a) Bioactivity Radar (BAR) and (b) Brain Or IntestinaL EstimateD (BOILED-Egg) model of CY3 compound.
Blind docking analysis
The blind-docking studies is performed to identify the most favorable targets to interact with the synthesized CY3 compound. Several proteins as targets, including DNA gyrase, GlmS, DHFR, DHSS, and UDPPS were used to find the most favorable target. Here, AD4 docking tool with the default docking parameters were used to perform blind docking analysis and found that DHFR and DHSS had the lowest binding energies. The decreasing order of targets based on the binding energy is as follows: DHFR > DHSS > DNA gyrase > GlmS > UDPPS. The synthesized CY3 compound exhibits a proximity of less than 5.0 Å within the deep active region of the receptor protein. The blind docking study revealed potential interactions between the CY3 compound and the target proteins (DHFR and DHSS) (Table 10). The synthesized CY3 compound is also subjected for multiple grids docking assessments to determine the most favorable binding pocket.
Table 10.
Binding energy of blind docking analysis of synthesized CY3 compound against DNA gyrase, glucose 6-phosphate synthase (GlmS), dihydrofolate reductase (DHFR), dehydrosqualene synthase (DHSS), and undecaprenyl pyrophosphate synthase (UDPPS).
| Ligands | Target | AD4 | |
|---|---|---|---|
| Binding energies (Kcal/mol) | Inhibition constant (μM) | ||
| CY3 | DNA gyrase | − 5.84 | 51.98 |
| GlmS | − 4.79 | 306.35 | |
| DHFR | − 6.95 | 8.02 | |
| DHSS | − 6.78 | 10.76 | |
| UDPPS | − 4.63 | 406.4 | |
Significant values are in bold.
Multi-Grid docking (MGD) analysis
The blind docking analysis shows that the CY3 compound can work as an antibacterial due to its interaction with target proteins (DHFR and DHSS). Here, MGD identifies the grid and pocket where the CY3 compound exhibits the strongest interactions or binding potentials with the targets. We performed the MGD using the widely used docking software (AD4), which offers multiple docking configurations. Here, MGD was conducted using a population size of 270, a maximum of 30,100,000 evaluations, 301,000 generations and other default parameters. MGD used different dimensions and coordinates to cover all the amino acid residues in the active pocket. The active pocket analysis was performed by utilising POCASA1.1 and CASTp v3.0, respectively. To enhance the accuracy and reliability of docking simulations in identifying active binding sites, it is essential to concentrate on the primary region where physiological ligands and standard drugs interact naturally. This study found that Grid IV of DHFR and DHSS was the most favourable pocket for binding interaction and inhibition constant analysis. The grid file had x, y, and z dimensions of 22 × 3 Å for both DHFR and DHSS, and grid centres of 9.992, 0.852, and 45.362 Å for DHFR, and 55.514, 12.682, and 51.969 Å for DHSS. For CY3 compound to bind to the amino acid residues of DHFR and DHSS, it needs − 7.07 and − 7.05 kcal/mol of binding energy and 6.60 and 6.78 µM of inhibition constant (Table 11).
Table 11.
Selected blind and target grids were used for binding interaction analysis of CY3 compound against dihydrofolate reductase (DHFR), dehydrosqualene synthase (DHSS).
| Target | Mode | Grid size (Å) | Grid coordinate (Å) | Inhibition constant (μM) | Binding energies (Kcal/mol) | ||||
|---|---|---|---|---|---|---|---|---|---|
| x | y | z | x | y | z | ||||
| DHFR | I | 48 | 48 | 48 | 9.982 | 0.879 | 45.320 | 8.02 | − 6.95 |
| II | 26 | 26 | 26 | 7.002 | 0.852 | 42.064 | 10.07 | − 6.82 | |
| III | 26 | 26 | 26 | 9.995 | 0.858 | 45.367 | 6.69 | − 7.06 | |
| IV | 22 | 22 | 22 | 9.992 | 0.852 | 45.362 | 6.60 | − 7.07 | |
| DHSS | I | 58 | 58 | 58 | 55.510 | 12.689 | 51.961 | 10.76 | − 6.78 |
| II | 28 | 28 | 28 | 55.518 | 12.680 | 51.963 | 9.86 | − 6.83 | |
| III | 24 | 24 | 24 | 55.514 | 12.682 | 51.969 | 7.86 | − 6.96 | |
| IV | 22 | 22 | 22 | 55.514 | 12.682 | 51.969 | 6.78 | − 7.05 | |
Significant values are in bold.
Target docking analysis
In vitro antibacterial and molecular docking analyses showed that the CY3 compound could have antibacterial activity due to its interaction with both DHFR and DHSS target proteins (Table 12, Figs. 7, 8). This study conducted target docking analyses using the established tools AD4, iGEMDOCK, and Vina to achieve more accurate docking results. To enhance the precision of the outcomes, the docking parameters were configured as follows: for AD4, a population size of 270, with a maximum of 30,100,000 evaluations and 301,000 generations, alongside other default parameters; for iGEMDOCK, 800 individuals, 80 generations, and a population of 10 solutions; and for Vina, an exhaustiveness of 64, mode number of 09, and an energy range of 3 kcal/mol. During target docking, CY3 compound demonstrated superior binding potentials and interactions with DHFR and DHSS, utilizing the most prominent grid. However, CY3 compound stood out due to its lower binding energies compared to the reference molecules for DHFR and DHSS. In this study, we selected fluconazole, levofloxacin, and podophylotoxin as standard drugs. The CY3 compound interacts with the target proteins deep in the active site, showing closer contacts (< 5.0 Å). We found that CY3 compound interacted with ALA8 and GLN96 through hydrogen bonds. It also interacted with ILE6, VAL7, ILE15, ASN19, ILE32, THR47, SER50, GLY95, TYR99, and THR122 ~ 3.20 Å and through vdW interactions. These interactions were all against DHFR. The π-π interaction with LEU21 and PHE93, respectively, was also found against DHFR. Similarly, CY3 compound made vdW interaction with PHE32, CYS50, ARG51, GLY144, LEU151, ALA163, GLY167, GLN171, and ASN174. Additionally, it established antagonistic interaction with PHE28, TYR47, VAL143, LEU147, LEU166, and LEU170. With a binding energy of − 7.07 kcal/mol and an inhibition constant of 6.60 µM, the CY3 compound binds to DHFR. Similarly, it binds to DHSS with a binding energy of − 7.05 kcal/mol and an inhibition constant of 6.78 µM. In addition to being a standard drug, the CY3 compound exhibits antibacterial activity because it contains the dual inhibitors DHFR and DHSS (Table 12).
Table 12.
Binding energy of target docking analysis of CY3 compound against dihydrofolate reductase (DHFR), and dehydrosqualene synthase (DHSS).
| Target | Binding energies (Kcal/mol) | Most interacting amino acids | |||||
|---|---|---|---|---|---|---|---|
| AD4 | iGEM | Vina | vdW | H bonds | π–π interaction | ||
| CY3 | DHFR | − 7.07 | − 124.73 | − 7.8 | ILE6, VAL7, ILE15, ASN19, ILE32, THR47, SER50, GLY95, TYR99, THR122 | ALA8 3.18, GLN96 | LEU21, PHE93 |
| HMOC | − 6.40 | − 117.87 | − 8.7 | VLA7, ILE15, TYR17, ASN19, GLN20, GLN96, THR122 | ALA8, TYR99 | GLY16, LEU21, ILE32, PHE93 | |
| TOP | − 6.19 | − 110.66 | − 7.8 | GLY16, GLN20, ILE32, THR47 | ALA8, ILE15, ASN19, ASP28, SER50, PHE93, THR122 | VAL7, LEU21 | |
| Fluconazole | − 4.70 | − 86.52 | − 9.1 | ILE51, GLY94, GLY95 | ALA8, ASN19, THR47, TYR99, THR122, | VAL7, LEU21, PHE93 | |
| Levofloxacin | − 5.71 | − 97.49 | − 9.0 | GLY16, ASN19, GLN20, LEU29, ILE32, LEU55 | ILE15, THR47, SER50, PHE93, THR122 | LEU21, ILE51 | |
| Podophylotoxin | − 7.30 | − 129.16 | − 9.1 | VLA7, TRP23, ILE32, SER50, GLY95, GLN96, TYR99, THR122 | ALA8 | LIE15, LEU21, LEU29, ILE51, LEU55, PHE93 | |
| CY3 | DHSS | − 7.05 | − 118.79 | − 8.3 | PHE32, CYS50, ARG51, GLY144, LEU151, ALA163, GLY167, GLN171, ASN174 | – | PHE28, TYR47, VAL143, LEU147, LEU166, LEU170 |
| HMOC | − 6.23 | − 99.94 | − 8.2 | PHE32, ASP54, VAL139, GLY144, LEU151, GLY167, LEU170, GLN171, ASN174 | TYR47, ARG51 | CYS50, ALA140, VAL143, LEU147 | |
| TOP | − 5.08 | − 92.89 | − 7.5 | ARG51, GLY144, ALA163, GLN171, TYR254 | GLY167, ASN174 | PHE28, PHE32, TYR47, ALA140, VAL143, LEU147, LEU166, LEU170 | |
| Fluconazole | − 5.01 | − 78.20 | − 7.4 | PHE28, ASP54, VAL139, GLY144, LEU147, GLN171, ASN174 | HIS24, TYR47, ARG51 | CYS50, ALA140, VAL143, LEU170 | |
| Levofloxacin | − 4.05 | − 73.43 | − 7.2 | TYR47, TYR135, VAL139, GLN171, ASP178, ARG187, TYR189, ARG271 | ASP120, ASN174, ARG177, TYR254 | HIS24, ARG51 | |
| Podophylotoxin | − 4.19 | − 75.27 | − 7.6 | TYR47, ASP55, TYR135, GLN171, ARG177, ASP178 | ARG51, ASP54 | HIS24, PHE28, VAL117, VAL139 | |
Significant values are in bold.
AD4 AutoDock, iGEM iGEMDOCK, Vina AutoDock Vina, DOXO Doxorubicin, DACA Dacarbazine, THIO Thioridazine.
Fig. 7.

Molecular docking interaction of CY3 compound with the dihydrofolate reductase (DHFR). (a) CY3 ball-stick model showing interaction; (b) CY3 ball-stick model surrounded by surface to show the desirable site of H-bond donor and acceptor; (c) 2D diagram showing the types of contacts formed between DHFR and the CY3; (d) 2D diagram LigPlot + showing the types of contacts formed between DHFR and the CY3.
Fig. 8.

Molecular docking interaction of CY3 compound with the dehydrosqualene synthase (DHSS). (a) CY3 ball-stick model showing interaction; (b) CY3 ball-stick model surrounded by surface to show the desirable site of H-bond donor and acceptor; (c) 2D diagram showing the types of contacts formed between DHSS and the CY3; (d) 2D diagram LigPlot + showing the types of contacts formed between DHSS and the CY3.
Discussion
Preparation of 3-(2-bromo-5-fluorophenyl)-1-(thiophen-2-yl) prop-2-en-1-one (CY3)
Firstly, we dissolved equimolar amounts of a substituted ketone and a substituted aldehyde in ethanol. We carefully incorporated the sodium hydroxide solution into the mixture and diligently stirred it for a duration of 24 h. Monitoring the completion of the reaction was a priority for the TLC. After finishing, we carefully poured the mixture into 400 mL of cold water, stirring continuously, and added 10% HCl until we reached a neutral pH. After filtering and thoroughly washing the precipitate with distilled water, obtain pure CY3 compound in excellent yields.
Characterization: 3-(2-bromo-5-fluorophenyl)-1-(thiophen-2-yl) prop-2-en-1-one (CY3)
Pale yellow solid, yield 73%, mp 149–150 °C, IR (KBr) νmax: 2921 (C–H aromatic), 1654 (C=O α,β-unsaturated), 1466 (–C=C– aromatic) cm−1; 1H-NMR (400 MHz, CDCl3) δH: 6.61–6.58 (1H, dd, J = 7.9, 7.6 Hz), 7.08 (1H, s), 7.15 (1H, s), 7.50–7.46 (1H, dd, J = 8.6, 8.7 Hz), 7.59 (1H, s, J = 4.8), 7.63 (1H, s, J = 4.8), 7.71 (1H, s), 7.91 (1H, s); 13C-NMR (100 MHz, CDCl3) δc: 115.6, 118.8, 128.2 (d, JC-F = 14 Hz), 132.2, 132.6, 134.2 (d, JC-F = 17 Hz), 134.4 (d, JC-F = 8 Hz), 134.8, 143.8, 144.0, 145.8, 162.9 (d, JC-F = 46 Hz), 190.5; ES-MS (m/z): 311[M]+, calculated for C13H8BrFOS.
Antibacterial activity
α,β-unsaturated carbonyl compound influences diverse biological activities. This study the synthesized CY3 compound was tested for its antibiotic potential. The CY3 compound was evaluated against five bacterial strains (S. aureus, E. coli, K. pneumoniae, P. aeruginosa, and A. baumannii). It could be seen that the CY3 compound show inhibitory effects up to > 100 μg/mL concentration. Thus, the CY3 compound demonstrated antimicrobial activity in vitro against the strains of Gram-positive bacteria (S. aureus) and Gram-negative bacteria (E. coli, K. pneumoniae, P. aeruginosa, and A. baumannii). It was effective as the standard antibiotics including Colistin, Levofloxacin, and Vancomycin against all pathogens tested (S. aureus, E. coli, K. pneumoniae, P. aeruginosa, and A. baumannii) with up to > 100 μg/mL concentration.
Antifungal activity
This study also evaluated the antifungal potential of the synthesized CY3 compound. The synthesized CY3 compound tested against three fungal strains (Candida albicans, Candida parapsilosis, and Candida tropicalis). It could be seen that the CY3 compound has inhibitory effects up to > 50 μg/mL concentration. It was effective as the standard antibiotic, including fluconazole, against all pathogens tested (Candida albicans, Candida parapsilosis, and Candida tropicalis) with up to > 50 μg/mL concentration.
Cytotoxicity
Using an MTT assay, assessed the cytotoxicity of the synthesized CY3 compound on two cell lines including Vero and HepG2. The results were then compared in terms of cytotoxic concentrations of two concentrations (Vero cell line CC50 at 200 µM and HepG2 cell line CC50 at 200 µM) with those of the standard drug, including podophylotoxin, to assess their effectiveness.
Computational screening
According to the available drug-likeness and medicinal chemistry rules, an orally active drug must have less than two violations. The results revealed that the synthesized CY3 compound, has not exhibited any violations, indicating that it complies with the drug-likeness and medicinal chemistry rules, with a Brenk and Leadlikeness alert. The obtained results suggest that this CY3 compound has the potential to be an orally active molecule. Similarly, ADMET and bioactivity score analysis also show that this molecule has a good ADME and bioactivity score with lower toxicity. BAR and BOILED-Egg analysis also favour that this compound easily absorbs human intestinal and cross-blood–brain barriers (Supplementary information).
Molecular docking analysis
Here, molecular docking analysis was performed by using three well-known docking tools: AD4, Vina, and iGEMDOCK. Initially, blind docking analysis was performed to identify the most favourable target protein. Therefore, five well-known bacterial target proteins including DNA gyrase, GlmS, DHFR, DHSS, and UDPPS were selected. The obtained results revealed that the synthesized compound showed potential inhibitory action against two target proteins, DHFR and DHSS, respectively. Then performed MGD to identify the most promising binding pocket. Therefore, multiple grids were used to cover all the amino acids in the pocket, each with variable sizes and centres, to identify the active pocket. Here, the obtained results confirm that grid number IV, is the most suitable for analysing potential binding interactions. Ultimately, conducted a target docking analysis to validate the molecular docking analysis. Thus, carried out multiple times docking analyses to ensure the accuracy of the results. Then proceeded to compare the obtained results with the reference and standard drugs. The CY3 compound had binding energies of − 7.07, − 7.80, and − 124.73 kcal/mol respectively for DHFR and − 7.05, − 8.30, and − 118.79 kcal/mol respectively for DHSS. It also had inhibition constants of 6.60 and 6.78 µM for DHFR and DHSS. On the other hand, the CY3 compound was examined against five types of bacteria (S. aureus, E. coli, K. pneumoniae, P. aeruginosa, and A. baumannii) in vitro to find their antibacterial potential. It could be seen that the CY3 compound shows in vitro inhibitory effects up to > 100 μg/mL concentration. Therefore, both the in vitro experimental results and the docking results obtained exhibit significant variability. The discrepancies between the docking inhibition constants and the in vitro inhibitory effects of the CY3 compound may be due to several common factors, such as binding affinity, protein conformation, solubility, metabolic factors, and assay conditions. As a result, their dual inhibitory potential confirms their antibacterial action. Dihydrofolate reductase (DHFR) inhibitors impede cell growth by inhibiting the synthesis of DNA, RNA, and proteins. Similarly, dehydrosqualene synthase (DHSS) is an enzyme in Staphylococcus aureus that catalyses the conversion of farnesyl diphosphate into dehydrosqualene (MeOH). The involvement of presqualene diphosphate (PSPP), dehydrosqualene (DHS) products, and various inhibitors further complicates the mechanisms of action for both DHSS and DHFR inhibitors27–29.
Conclusion
In conclusion, a revised and efficient protocol advocating a simple route is demonstrated for the synthesis of CY3 compound in good yield using the Claisen-Schmidt condensation reaction. Here, the CY3 compound was successfully synthesized by using the substituted aldehyde and acetophenone. Subsequently, a thorough analysis of the structure of this compound was performed using FTIR, 1H NMR, 13C NMR, and ES-MS. Also, screened the synthesized compound for their in vitro pharmacological activities. It effectively interacted with microbes such as S. aureus (ATCC29213), E. coli (ATCC25922), K. pneumoniae (ATCC27736), P. aeruginosa (ATCC27853), and A. baumannii (ATCC1605) as references and standard drugs including colistin, levofloxacin, and vancomycin. The CY3 compound was effective at inhibiting the growth of all five pathogens. Furthermore, AD4, Vina, and iGEMDOCK were used to find the more promising binding interaction. The CY3 compound shows the binding energies of − 7.07, − 7.80, and − 124.73 kcal/mol respectively for DHFR and − 7.05, − 8.30, and − 118.79 kcal/mol respectively for DHSS. The inhibition constant 6.60 and 6.78 µM respectively for DHFR and DHSS, which correlate the results for its antibacterial action. Here, also tested the antifungal activity of the synthesised CY3 compound against three fungal strains: Candida albicans (ATCC 90028), Candida parapsilosis (ATCC 22019), and Candida tropicalis (ATCC 750). It could be seen that the CY3 compound has inhibitory effects up to > 50 μg/mL concentration. In this study, also evaluated the cytotoxicity of the synthesized CY3 compound on two cell lines, Vero and HepG2. The results were then compared in terms of cytotoxic concentrations at two concentrations (Vero cell line CC50 at 200 µM and HepG2 cell line CC50 at 200 µM). As bacterial growth inhibitors, the results of α,β-unsaturated carbonyl compound synthesis look promising and open new drug discovery paths.
Supplementary Information
Acknowledgements
The authors acknowledge the Division of Medicinal and Process Chemistry, Microbiology and Laboratory Animal Facility Division CSIR-CDRI, Lucknow, India for providing the facilities to carry out research work and financial support. The manuscript bears communication number IU/R&D/2024-MCN0002805 from Integral University, Lucknow.
Abbreviations
- CY3
3-(2-Bromo-5-fluorophenyl)-1-(thiophen-2-yl) prop-2-en-1-one
- PAINS
Pan Assay Interference Structures
- AD4
AutoDock
- GlmS
Glucose 6-phosphate synthase
- DHFR
Dihydrofolate reductase
- DHSS
Dehydrosqualene synthase
- UDPPS
Undecaprenyl pyrophosphate synthase
- WHO
World health organization
- PDB
Protein data bank
- ppm
Parts per million
- Hz
Hertz
- TSB
Tryptic soy broth
- DMSO
Dimethyl sulfoxide
- MIC
Minimum inhibitory concentration
- CLSI
Clinical and Laboratory Standard Institute
- CC50
50% Cytotoxic concentration
- MW
Molecular weight
- RB
Rotatable bonds
- OHNH
Hydrogen bond donors
- ON
Hydrogen bond acceptor sites
- TPSA
Topological polar surface area
- log P
Partition coefficient
- Ro5
Lipinski’s rule of five
- DDD
Drug design and development
- BAS
Bioactivity score
- ADMET
Absorption, distribution, metabolism, excretion, and toxicity
- BOILED-Egg
Brain or IntestinaL EstimateD
- BAR
Bioavailability radar
- PASS
Prediction of activity spectra for substances
- MGD
Multi-grid docking
Author contributions
ARK, MBL, and IA planned and supervised the experiments. CSY synthesized, characterized compounds and prepare the initial draft of the manuscript, while IA and MN assisted in the manuscript’s writing and review. IA and NA carried out computational studies and molecular docking analyses. RG arrange chemicals, consumables, and edited the manuscript. All authors discussed the results and contributed to the final manuscript.
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Iqbal Azad, Email: iazad@iul.ac.in.
Abdul Rahman Khan, Email: arahman@iul.ac.in.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-79747-8.
References
- 1.Kumar, H., Bansal, K. K. & Goyal, A. Synthetic methods and antimicrobial perspective of pyrazole derivatives: An insight. Anti-Infective Agents18, 207–223 (2019). [Google Scholar]
- 2.Martin, H., Kavanagh, K. & Velasco-Torrijos, T. Targeting adhesion in fungal pathogen Candida albicans. Future Med. Chem.13, 313–334 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Ahmad, N., Azad, M. I., Khan, A. R. & Azad, I. Benzimidazole as a promising antiviral heterocyclic Scaffold: A review. J. Sci. Arts21, 273–284 (2021). [Google Scholar]
- 4.Rudrapal, M. et al. Chalcone scaffolds, bioprecursors of flavonoids: Chemistry, bioactivities, and pharmacokinetics. Molecules26, (2021). [DOI] [PMC free article] [PubMed]
- 5.Bandgar, B. P. et al. Synthesis of new olefin chalcone derivatives as antitumor, antioxidant and antimicrobial agents. Med. Chem. Res.21, 4512–4522 (2012). [Google Scholar]
- 6.Schultz, T. W. & Yarbrough, J. W. Trends in structure–toxicity relationships for carbonyl-containing α, β-unsaturated compounds. SAR QSAR Environ. Res.15, 139–146 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Sunasee, R. & Narain, R. Covalent and noncovalent bioconjugation strategies. Wiley Blackwell6(9781118359), 1–75 (2014). [Google Scholar]
- 8.Ahangarpour, M., Kavianinia, I. & Brimble, M. A. Thia-Michael addition: The route to promising opportunities for fast and cysteine-specific modification. Org. Biomol. Chem.21, 3057–3072 (2023). [DOI] [PubMed] [Google Scholar]
- 9.Susilawati, S., Anwar, C., Saleh, I. & Salni, S. (2023) Flavonoid as anti-Candida agents. IJFAC (Indonesian J. Fundam. Appl. Chem.)8, 88–97.
- 10.Pournajaf, A. et al. PCR-based identification of methicillin-resistant Staphylococcus aureus strains and their antibiotic resistance profiles. Asian Pac. J. Trop. Biomed.4, S293–S297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helen, K. & Ashlesha, K. Vancomycin-resistant Staphylococcus aureus: Formidable threat or silence before the storm?. J. Infect. Dis. Epidemiol.5, 1–9 (2019). [Google Scholar]
- 12.Mannion, B. A., Weiss, J. & Elsbach, P. Separation of sublethal and lethal effects of the bactericidal/permeability increasing protein on Escherichia coli. J. Clin. Invest.85, 853–860 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomar, G. et al. Two-phase electrohydrodynamic simulations using a volume-of-fluid approach. J. Comput. Phys.227, 1267–1285 (2007). [Google Scholar]
- 14.Blurock, E. S. Detailed mechanism generation. 1. Generalized reactive properties as reaction class substructures. J. Chem. Inf. Comput. Sci.44, 1336–1347 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Yadav, C. S. et al. Exploring the therapeutic potential of chalcones in oncology: A comprehensive review. Curr. Bioact. Compd.20, (2023).
- 16.Zhao, S., Pi, C., Ye, Y., Zhao, L. & Wei, Y. Recent advances of analogues of curcumin for treatment of cancer. Eur. J. Med. Chem.180, 524–535 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Goodarzi, M., Saeys, W., De Araujo, M. C. U., Galvão, R. K. H. & Vander Heyden, Y. Binary classification of chalcone derivatives with LDA or KNN based on their antileishmanial activity and molecular descriptors selected using the Successive Projections Algorithm feature-selection technique. Eur. J. Pharm. Sci.51, 189–195 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Chen, B., Zhu, Z., Chen, M., Dong, W. & Li, Z. Three-dimensional quantitative structure–activity relationship study on antioxidant capacity of curcumin analogues. J. Mol. Struct.1061, 134–139 (2014). [Google Scholar]
- 19.Du, Z. Y. et al. Anti-proliferative, anti-inflammatory and antioxidant effects of curcumin analogue A2. Arch. Pharm. Res.36, 1204–1210 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Bukhari, S., Franzblau, S., Jantan, I. & Jasamai, M. Current prospects of synthetic curcumin Analogs and Chalcone derivatives against mycobacterium tuberculosis. Med. Chem. (Los. Angeles).9, 897–903 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Qin, H. L. et al. Synthesis of α, β-unsaturated carbonyl-based compounds, oxime and oxime ether analogs as potential anticancer agents for overcoming cancer multidrug resistance by modulation of efflux pumps in tumor cells. J. Med. Chem.59, 3549–3561 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Iraji, A. et al. Ugi adducts: Design and synthesis of natural-based & #945;-glucosidase Inhibitors. Lett. Org. Chem.19, 1084–1093 (2022). [Google Scholar]
- 23.Ou, J. L. et al. Structure–activity relationship analysis of curcumin analogues on anti-influenza virus activity. FEBS J.280, 5829–5840 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Correia-Da-Silva, M. et al. Synthesis and characterization of curcumin-chitosan loaded gold nanoparticles by Oryctes rhinoceros’ chitin for cosmeceutical application. Molecules.28, 1799 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salomone, S. et al. Immunosuppressive effects of natural α, β-unsaturated carbonyl-based compounds, and their analogs and derivatives, on immune cells: A review. Front. Pharmacol.1, 22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huo, Z. P., Feng, X. C., Wang, Y., Tian, Y. T. & Qiu, F. Sulfite as the substrate of C-sulfonate metabolism of α, β-unsaturated carbonyl containing andrographolide: analysis of sulfite in rats’ intestinal tract and the reaction kinetics of andrographolide with sulfite. Chin. J. Nat. Med.19, 706–712 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Dhaliwal, J. S. et al. Pharmacotherapeutics applications and chemistry of chalcone derivatives. Molecules.27, 7062 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okolo, E. N. et al. New chalcone derivatives as potential antimicrobial and antioxidant agent. Sci. Rep.11, 1–13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dammene Debbih, O. et al. Hydrazone analogs as DNA gyrase inhibitors and antioxidant agents: Structure-activity relationship and pharmacophore modeling. J. Chem. Sci.136, 1–19 (2024). [Google Scholar]
- 30.Venil, C. K., Velmurugan, P., Dufossé, L., Devi, P. R. & Ravi, A. V. Fungal pigments: Potential coloring compounds for wide ranging applications in textile dyeing. J. Fungi.6, 68 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amslinger, S. The tunable functionality of α, β-unsaturated carbonyl compounds enables their differential application in biological systems. ChemMedChem5, 351–356 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Bara-Estaún, A. et al. Paramagnetic relaxation agents for enhancing temporal resolution and sensitivity in multinuclear FlowNMR spectroscopy. Chem. Eur. J.29, e202300215 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee, J. S., Kim, D. H., Liu, K. H., Oh, T. K. & Lee, C. H. Identification of flavonoids using liquid chromatography with electrospray ionization and ion trap tandem mass spectrometry with an MS/MS library. Rapid Commun. Mass Spectrom.19, 3539–3548 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Holopainen, J. M., Rantamäki, A. H. & Wiedmer, S. K. Melting points-the key to the anti-evaporative effect of the tear film wax esters. Investig. Ophthalmol. Vis. Sci.54, 5211–5217 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Bekiaris, G. et al. Three different Fourier-transform mid-infrared sampling techniques to characterize bio-organic samples. J. Environ. Qual.49, 1310–1321 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Davitt, K. et al. 290 and 340 nm UV LED arrays for fluorescence detection from single airborne particles. Opt. Express13, 9548 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Azad, I. et al. Phenanthridine derivatives as promising new anticancer agents: synthesis, biological evaluation and binding studies. Future Med. Chem.12, 709–739 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Kalusalingam, A., Kalirajan, R., Jubie, S., Gowramma, B. & Suresh, B. Synthesis and biological evaluation of some Chalcone derivatives Synthesis and Biological evaluation of some heterocyclic derivatives of Chalcones. Int. J. ChemTech Res.1, 27–34 (2023). [Google Scholar]
- 39.Lozano, A. I. et al. The role of electron transfer in the fragmentation of phenyl and cyclohexyl boronic acids. Int. J. Mol. Sci.20, 5578 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomberg, A. Gaussian 09W tutorial an introduction to computational chemistry using g09w and Avogadro software.
- 41.Damasceno, M. V. A. et al. Modulation of the NLO properties of p-coumaric acid by the solvent effects and proton dissociation. J. Mol. Liq.394, 123587 (2024). [Google Scholar]
- 42.Seenithurai, S. & Chai, J. D. TAO-DFT with the polarizable continuum model. Nanomaterials13, 1593 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hadigheh Rezvan, V. Molecular structure, HOMO–LUMO, and NLO studies of some quinoxaline 1,4-dioxide derivatives: Computational (HF and DFT) analysis. Results Chem.7, 101437 (2024). [Google Scholar]
- 44.Yu, J., Su, N. Q. & Yang, W. Describing chemical reactivity with frontier molecular orbitalets. JACS Au2, 1383–1394 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lata, M. et al. Evolutionary and in silico guided development of novel peptide analogues for antibacterial activity against ESKAPE pathogens. Curr. Res. Microb. Sci.4, 100183 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li, Z. et al. Insights into the antifungal properties of myxobacteria outer membrane β-1,6-glucanase. J. Agric. Food Chem.71, 9656–9666 (2023). [DOI] [PubMed] [Google Scholar]
- 47.Clinical and Laboratory Standards Institute (CLSI). Reference method for broth dilution. Ref. method broth dilution Antifung. susceptibility Test. yeasts. Approv. Stand. 3th ed.28, 0–13 (2008).
- 48.Klepser, M. E., Ernst, E. J., Lewis, R. E., Ernst, M. E. & Pfaller, M. A. Influence of test conditions on antifungal time-kill curve results: Proposal for standardized methods. Antimicrob. Agents Chemother.42, 1207–1212 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pandey, A. R. et al. Antileishmanial evaluation of triazole-butenolide conjugates: Design, synthesis, in vitro screening, SAR and in silico ADME predictions. RSC Med. Chem.14, 1131–1142 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Azad, I., Nasibullah, M., Khan, T., Hassan, F. & Akhter, Y. Exploring the novel heterocyclic derivatives as lead molecules for design and development of potent anticancer agents. J. Mol. Graph. Model.81, 211–228 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.7, 1–13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang, H. et al. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics35, 1067–1069 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Daina, A. & Zoete, V. A BOILED-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem11, 1117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choudhury, S. S. et al. Synthesis of α, β-unsaturated ketones in water: The Claisen-Schmidt condensation revisited. ACS Sustain. Chem. Eng.10, 14271–14279 (2022). [Google Scholar]
- 55.Slassi, S., Aarjane, M. & Amine, A. Synthesis, spectroscopic characterization (FT-IR, NMR, UV-Vis), DFT study, antibacterial and antioxidant in vitro investigations of 4,6-bis((E)-1-((3-(1H-imidazol-1-yl)propyl)imino)ethyl)benzene-1,3-diol. J. Mol. Struct.1255, 132457 (2022). [Google Scholar]
- 56.Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.46, 3–26 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Ghose, A. K. & Crippen, G. M. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci.27, 21–35 (1987). [DOI] [PubMed] [Google Scholar]
- 58.Ghose, A. K., Viswanadhan, V. N. & Wendoloski, J. J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem.1, 55–68 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.