

Abstract
Aim
Social anxiety disorder (SAD) is a common disorder characterized by excessive fear of scrutiny and embarrassment, leading to severe distress and avoidance behaviors or dysfunctions. SAD and other relevant diseases have been reported to be associated with a higher risk of aging‐related diseases, such as Alzheimer's disease, Parkinson's disease, and diabetes mellitus. Recently, epigenetic clock analysis, which measures biological aging based on comprehensive DNA methylation (DNAm) status, has been widely conducted. We conducted epigenetic clock analyses in patients with SAD and controls, examining various epigenetic age acceleration and DNAm‐based predictive values of aging‐related proteins (GrimAge components and GrimAge2 components), including leptin level.
Methods
We used the publicly available DNAm dataset, GSE164056, which consists of 66 patients with SAD and 77 controls of Caucasian descent aged between 18 and 50 years. We conducted regression analyses investigating the association between SAD and various indices of epigenetic aging, using age and sex as covariates.
Results
None of the epigenetic clocks showed significant differences in age acceleration. Of the DNAm‐based predictive values of aging‐related proteins, leptin level in GrimAge components (q = 0.0123) and GrimAge2 components (q = 0.0123) were significantly lower in patients with SAD than in controls.
Conclusions
The results of this study suggested that leptin may be involved in SAD pathogenesis as an aging‐related protein. Therefore, further studies with different designs are required.
Keywords: aging, anxiety, hypothalamus, leptin, methylation
Short abstract
Social anxiety disorder (SAD) is characterized by excessive fear of scrutiny and embarrassment, leading to severe distress and avoidance behaviors or dysfunctions. We examined various epigenetic age acceleration and predictive values of aging‐related protein levels based on DNA methylation status. Leptin level was significantly lower in patients with SAD than in controls.
1. INTRODUCTION
Social anxiety disorder (SAD) is characterized by excessive fears of scrutiny, embarrassment, and humiliation in social situations, leading to severe distress and avoidance behaviors or dysfunctions. It is a common disorder that is estimated to occur in 3–7% of the adult population in the U.S. each year. SAD usually develops during childhood or adolescence and can subsequently lead to major depression, extensive dysfunction, and poor quality of life, especially if left untreated. 1 Both genetic and environmental factors have been reported to be implicated in the pathogenesis of SAD. 2 Functional neural circuits involving neurohormones and neurotransmitter systems, such as dopamine, glutamate, and oxytocin, as well as the amygdala, insular cortex, and prefrontal cortex, have been reported to be implicated in the pathogenesis of SAD. 3 , 4 , 5 , 6 Hypothalamic–pituitary axis (HPA) reactivity has also been suggested to be involved and is reported to influence avoidance behavior. 5 The average age at onset of SAD is younger, and the morbidity period is often longer.
Recently, aging research and the potential effects of epigenetic changes on aging through DNA methylation (DNAm) at cytosine‐phosphate‐guanine (CpG) sites have gained attention. 7 , 8 Several “epigenetic clocks” have been developed to predict biological aging based on DNAm patterns. 9 , 10 , 11 , 12 , 13 Estimates of epigenetic age acceleration have been associated with psychiatric disorders, 14 , 15 , 16 , 17 , 18 neurodegenerative disorders, 19 , 20 and all‐cause mortality. 10
The first generation of epigenetic clocks, including HorvathAge (based on 353 CpG sites), 21 HannumAge (based on 71 CpG sites), 9 and SkinBloodAge (based on 391 CpG sites), 22 were developed to predict chronological age itself. PhenoAge, developed by Levine et al. 11 based on 513 CpG sites, is associated with various aging outcomes, including all‐cause mortality, cancer, physical functioning, coronary heart disease, and Alzheimer's disease.
GrimAge, developed by Lu et al. 12 based on 1030 CpG sites, incorporates several compositions, based on chronological age, sex, seven DNAm‐based predictive values of aging‐related plasma proteins' levels (adrenomedullin [ADM], beta‐2‐microglobulin [B2M], Cystatin C, growth differentiation factor‐15 [GDF15], leptin, plasminogen activation inhibitor‐1 [PAI1], and tissue inhibitor of metalloproteinases‐1 [TIMP1]), and DNAm‐based smoking pack‐years (DNAmPACKYRS). More recently, Lu et al. 13 developed GrimAge2, an enhanced version of GrimAge, by incorporating hemoglobin A1c and C‐reactive protein levels into its components. These components are called GrimAge components and GrimAge2 components. For instance, leptin, one of the proteins comprising these components, plays a protective role against aging‐related diseases such as metabolic syndrome, diabetes, and cardiovascular disease. 23
DNAm‐based telomere length (DNAmTL) was developed by Lu et al. 24 based on 140 CpG sites and evaluates telomere lengths. DunedinPACE, developed based on 173 CpG sites, is a measure to quantify changes in the pace of biological aging according to the prediction of several physical function biomarkers. 25 Each algorithm differs in the type of epigenetic data on which it is trained. These estimators capture certain biological aspects of aging and have been successfully applied in the study of various psychiatric disorders. 26
Social anxiety disorder is closely associated with highly neurotic personality traits. 27 , 28 Neuroticism has been reported to increase the risk of future occurrence of aging‐related diseases, such as Alzheimer's disease and Parkinson's disease. 29 , 30 Young cancer survivors are more likely to develop SAD. 31 , 32 Generalized anxiety disorder, which is categorized as the same type of neurosis as SAD, has been reported to be associated with a higher risk of aging‐related diseases, such as diabetes mellitus and stroke. 33 Social anxiety is also frequently comorbid with major depressive disorder (MDD), 34 and we have previously found abnormalities in several epigenetic age acceleration and GrimAge components in patients with MDD. 17 Furthermore, social anxiety, especially among adolescents, increases the risk of suicide attempts, suicidal ideation, and suicide deaths. 35 Regarding suicide, we previously found significant shortening of telomere length, an indicator of aging, in suicide decedents. 36 Like these, SAD and various concomitant symptoms are reportedly associated with aging. To our knowledge, no study has performed an epigenetic clock analysis of SAD. We conducted biological age analyses based on the DNAm status of patients with SAD and healthy controls to investigate the association between this disease and aging from an epigenetic perspective. Additionally, we searched for specific findings in SAD by comparing our results of previous epigenetic clock study on MDD, which is often comorbid with SAD.
2. METHODS
2.1. Publicly available DNAm dataset (GSE164056)
We used publicly available DNAm data from the Gene Expression Omnibus database. The GSE164056 dataset was generated by Wiegand et al. and consists of 66 patients with SAD and 77 controls of Caucasian descent between the ages of 18 and 50 years (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164056, last accessed on December 8, 2023). All participants were assessed with the Structured Clinical Interview for DSM 4th edition. 37 The severity of social anxiety was evaluated using the Liebowitz Social Anxiety Scale (LSAS). 38 All of the participants provided written informed consent, which was approved by the University of Tübingen local ethics committee in accordance with the Declaration of Helsinki. DNA was extracted using the QIAamp Blood Maxi Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Genomic DNA was bisulfite‐converted using the EZ DNA Methylation™ Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer's instructions. DNAm was determined using the Infinium MethylationEPIC Kit (Illumina, San Diego, CA, USA).
2.2. Evaluation of epigenetic age acceleration and DNAm‐based aging‐predictive factors
As epigenetic aging evaluation, six epigenetic clocks (HorvathAge, HannumAge, SkinBloodAge, PhenoAge, GrimAge, and GrimAge2), DNAmTL, GrimAge components, and GrimAge2 components were calculated using an online DNAm age calculator (https://horvath.genetics.ucla.edu/html/dnamage/ last accessed on July 23, 2024). 12 , 13 , 21 Furthermore, we calculated epigenetic age acceleration. AgeAccelHorvath, AgeAccelHannum, AgeAccelSkinBlood, AgeAccelPheno, AgeAccelGrim, and AgeAccelGrim2 were defined as the residual from regressing each DNAmAge on the chronological age. Positive and negative values indicated whether the epigenetic age is higher or lower than the expected age (based on chronological age), respectively. The age‐adjusted estimate of DNAmTL (DNAmTLadjAge) was defined as the residual calculated by regressing DNAmTL on chronological age. Positive and negative values indicated whether DNAmTLadjAge was longer or shorter than the expected DNAmTL (based on chronological age), respectively. We calculated DunedinPACE with R package DunedinPACE. In addition, we could calculate predicted values for multiple cell compositions (CD8+ T cell, CD4+ T cell, Natural killer cell, B cell, Monocyte, Granulocyte, and PlasmaBlast) by using the Houseman method 21 , 39 in this calculator.
2.3. Statistical analysis
Statistical analyses were performed using R version 4.3.1 (R Development Core Team, Vienna, Austria). In the demographic data, continuous variables were compared with Mann–Whitney U test and categorical variables with Fisher's exact test between patients and controls. The correlation between chronological age and DNAmAge was tested using the Spearman correlation analysis. For each epigenetic age acceleration, GrimAge component, and GrimAge2 component, the association with phenotype (SAD or control) was tested using multiple regression analyses with age and sex as confounders. In addition, as additional analyses, since cell compositions 40 and smoking status 41 , 42 could also affect DNA methylation, we also conducted multiple regression analyses, including DNAmPACKYRS, a predictor of cumulative smoking, and multiple cell compositions (CD8+ T cell, CD4+ T cell, Natural killer cell, B cell, Monocyte, Granulocyte, and Plasmablast) as additional confounding factors. Also, we compared the results of this analyses for SAD and the results of our previous epigenetic clock analyses for Caucasian patients with MDD, which has a high comorbidity rate with SAD. 34 We used the cohorts of GSE125105 dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE125105, last accessed on July 23, 2024) 43 , 44 consisting of patients with MDD and controls and having been used in our previous analyses for MDD. 18 Furthermore, among patients with SAD, the association between the LSAS score and each epigenetic age acceleration, GrimAge component, and GrimAge2 component was tested using multiple regression analysis, adjusting for age and sex as confounding factors. For multiple testing, we calculated q values by adjusting the p values using the Benjamini Hochberg method. Statistical significance was defined as q < 0.05. Dummy variables for phenotype and sex were assigned as follows: sex: male = 0, female = 1; phenotype: control = 0, SAD = 1.
3. RESULTS
There were no significant differences in age or sex between the patients with SAD and controls. The LSAS scores were significantly higher in patients with SAD than in controls (p = 1.40 × 10−22) (Table 1). We could obtain the predicted values of ADM, B2M, Cystatin C, GDF15, Leptin, PAI1, and TIMP1 levels, and DNAmPACKYRS as GrimAge components and the predicted values of ADM, Cystatin C, GDF15, Leptin, PAI1, and TIMP1 levels as GrimAge2 components. To be consistent with the notation in the online calculator, the components of GrimAge are denoted as DNAmADM, DNAmB2M, DNAmCystatinC, DNAmGDF15, DNAmLeptin, DNAmPAI1, DNAmTIMP1 and DNAmPACKYRS, and the components of GrimAge2 are denoted as DNAmadm, DNAmCystatin_C, DNAmGDF_15, DNAmleptin, DNAmpai_1, and DNAmTIMP_1. Demographic data, epigenetic clock, DNAmTL, epigenetic age acceleration, GrimAge components, and GrimAge2 components are listed in Table 1.
TABLE 1.
The demographic data for patients and controls.
| Controls | Patients | p Value | |
|---|---|---|---|
| n = 77 | n = 66 | ||
| Age, years [IQR] | 24 [22–28] | 24 [20–28.75] | 0.434* |
| Male, n (%) | 29 (37.7) | 20 (30.3) | 0.328** |
| Male | n = 29 | n = 20 | |
| Age, years [IQR] | 26 [24–28] | 27.5 [21.75–31.25] | 0.669* |
| Female | n = 48 | n = 46 | |
| Age, years [IQR] | 24 [21–27] | 23 [20–26.75] | 0.468* |
| LSAS | 10 [4–19] | 72 [47.75–91.75] | 1.40 × 10−22 * |
| Epigenetic Clock | |||
| HorvathAge, median [IQR] | 36.02 [32.23–38.68] | 35.03 [31.40–39.89] | |
| HannumAge, median [IQR] | 19.5 [16.10–22.06] | 18.68 [16.19–21.91] | |
| SkinBloodAge, median [IQR] | 26.04 [21.90–28.87] | 24.35 [20.60–29.18] | |
| PhenoAge, median [IQR] | −51.46 [−54.98 – −47.79] | −51.62 [−55.37 – −47.34] | |
| GrimAge, median [IQR] | 158.3 [155.3–161.6] | 156.7 [155.1–160.9] | |
| GrimAge2, median [IQR] | 123.7 [121.3–126.9] | 122.9 [120.9–126.5] | |
| DNAmTL, median [IQR] | 7.996 [7.896–8.111] | 8.029 [7.910–8.118] | |
| Age acceleration | |||
| AgeAccelHorvath, median [IQR] | 0.1624 [−2.5117–2.5630] | −0.7956 [−2.8512–2.4867] | |
| AgeAccelHannum, median [IQR] | 0.1228 [−1.5849–1.7260] | −0.2395 [−1.6476–1.4322] | |
| AgeAccelSkinBlood, median [IQR] | 0.5905 [−1.8190–1.8554] | −1.0208 [−1.8787–1.7839] | |
| AgeAccelPheno, median [IQR] | −0.2437 [−2.1032–2.3366] | 0.4141 [−2.92393–2.19582] | |
| AgeAccelGrim, median [IQR] | 0.1317 [−1.1595–1.0166] | −0.2829 [−1.2980–1.0362] | |
| AgeAccelGrim2, median [IQR] | −0.3206 [−1.0402–1.0509] | 0.02259 [−1.73342–1.09955] | |
| DNAmTLAdjAge, median [IQR] | −0.008025 [−0.0933831–0.0838027] | 0.0073217 [−0.0733179–0.0666366] | |
| DunedinPACE, median [IQR] | 0.8903 [0.8470–0.9544] | 0.8955 [0.8365–0.9479] | |
| GrimAge component | |||
| DNAmADM, median [IQR] | 894.3 [889.3–900.9] | 895.1 [887.8–900.3] | |
| DNAmB2M, median [IQR] | 4 925 092 [4890497–4 974 689] | 4 923 587 [4867137–4 973 240] | |
| DNAmCystatinC, median [IQR] | 1 120 082 [1110581–1 131 725] | 1 112 580 [1106972–1 130 566] | |
| DNAmGDF15, median [IQR] | 2969 [2919–3007] | 2961 [2921–3027] | |
| DNAmLeptin, median [IQR] | 110 847 [110402–111 235] | 110 483 [110106–110 921] | |
| DNAmPAI1, median [IQR] | 120 673 [119865–121 451] | 120 242 [119417–121 111] | |
| DNAmTIMP1, median [IQR] | 33 270 [32891–33 873] | 33 091 [32677–33 784] | |
| DNAmPACKYRS, median [IQR] | 101.50 [99.26–103.56] | 101.71 [99.47–105.56] | |
| GrimAge2 component | |||
| DNAmadm, median [IQR] | 863.4 [855.2–869.7] | 862.5 [855.3–868.7] | |
| DNAmCystatin_C, median [IQR] | 1 108 161 [1097476–1 124 090] | 1 100 931 [1093208–1 120 628] | |
| DNAmGDF_15, median [IQR] | 3030 [2961–3070] | 3010 [2969–3071] | |
| DNAmleptin, median [IQR] | 104 703 [104232–105 265] | 104 503 [103972–104 944] | |
| DNAmpai_1, median [IQR] | 112 086 [111327–113 062] | 111 837 [110934–112 571] | |
| DNAmTIMP_1, median [IQR] | 32 483 [31832–32 962] | 32 188 [31731–32 920] |
Abbreviations: ADM, adrenomedullin; B2M, beta 2 microglobulin; DNAm, DNA methylation; DNAmPACKYRS, DNA methylation‐based smoking pack‐years; DNAmTLAdjAge, age‐adjusted estimate of DNA methylation‐based telomere length; GDF15, growth differentiation factor‐15; LSAS, the Liebowitz Social Anxiety Scale; PAI1, plasminogen activation inhibitor‐1; TIMP1, tissue inhibitor of metalloproteinases‐1.
The p values were calculated with Mann–Whitney U test.
The p value was calculated with Fisher's exact test.
To confirm the suitability of the epigenetic clocks themselves for the data of this cohort, we found significant correlations between chronological age and each epigenetic clock and DNAmTL in the cohort (HorvathAge: rho = 0.808, q = 3.99 × 10 −34 ; HannumAge: rho = 0.855, q = 7.51 × 10 −42 ; SkinBloodAge: rho = 0.890, q = 1.93 × 10 −49 ; PhenoAge: rho = 0.789, q = 1.66 × 10 −31 ; GrimAge: rho = 0.907, q = 3.16 × 10 −54 ; GrimAge2: rho = 0.863, q = 2.94 × 10 −43 ; DNAmTL: rho = −0.668, q = 7.98 × 10 −20 ) (Figure S1).
Regarding epigenetic age acceleration, none of the clocks showed a significant association with SAD in multiple regression analysis using age and sex as confounders (AgeAccelHorvath: q = 0.889; AgeAccelHannum: q = 0.649; AgeAccelSkinBlood: q = 0.770; AgeAccelPheno: q = 0.999; AgeAccelGrim: q = 0.770; AgeAccelGrim2: q = 0.585; DNAmTLAdjAge: q = 0.532; DunedinPACE: q = 0.999) (Table 2 and Figure 1). Of the GrimAge components (DNAmADM: q = 0.192; DNAmB2M: q = 0.404; DNAmCystatinC: q = 0.248; DNAmGDF15: q = 0.592; DNAmLeptin: q = 0.0123; DNAmPAI1: q = 0.184; DNAmTIMP1: q = 0.192; DNAmPACKYRS: q = 0.192) and GrimAge2 components (DNAmadm: q = 0.248; DNAmCystatin_C: q = 0.413; DNAmGDF_15: q = 0.889; DNAmleptin: q = 0.0123; DNAmpai_1: q = 0.192; DNAmTIMP_1: q = 0.446), DNAmLeptin, one of the GrimAge components, and DNAmleptin, one of the GrimAge2 components showed significant associations with SAD (Table 2 and Figure 2).
TABLE 2.
The results of multiple regression analyses for the association with phenotype (patients with social anxiety disorder or controls), with age and sex as confounding factors.
| Coefficients | Standard error | t Value | p Value | q Value a | |
|---|---|---|---|---|---|
| Epigenetic age acceleration | |||||
| AgeAccelHorvath | −0.149801 | 0.617335 | −0.24266 | 0.808628 | 0.889491 |
| AgeAccelHannum | −0.313635 | 0.434581 | −0.72170 | 0.471693 | 0.648578 |
| AgeAccelSkinBlood | −0.247913 | 0.513304 | −0.48297 | 0.629874 | 0.769846 |
| AgeAccelPheno | −0.000573 | 0.661913 | −0.00087 | 0.999310 | 0.999310 |
| AgeAccelGrim | −0.129518 | 0.261204 | −0.49585 | 0.620785 | 0.769846 |
| AgeAccelGrim2 | −0.300824 | 0.336144 | −0.89493 | 0.372374 | 0.585159 |
| DNAmTLAdjAge | −0.012544 | 0.012419 | −1.01009 | 0.314210 | 0.531740 |
| DunedinPACE | −0.000073 | 0.019572 | −0.00372 | 0.997036 | 0.999310 |
| GrimAge component | |||||
| DNAmADM | −2.363555 | 1.171159 | −2.01813 | 0.045501 | 0.191822 |
| DNAmB2M | −10442.57 | 7812.820 | −1.33659 | 0.183539 | 0.403785 |
| DNAmCystatinC | −2327.380 | 1398.827 | −1.66381 | 0.098404 | 0.247629 |
| DNAmGDF15 | 4.794037 | 5.722406 | 0.83777 | 0.403600 | 0.591947 |
| DNAmLeptin | −430.9424 | 124.1693 | −3.47060 | 0.000692 | 0.012320 |
| DNAmPAI1 | −534.8867 | 236.1789 | −2.26475 | 0.025075 | 0.183883 |
| DNAmTIMP1 | −88.76374 | 47.00069 | −1.88856 | 0.061034 | 0.191822 |
| DNAmPACKYRS | 1.239601 | 0.633393 | 1.95708 | 0.052342 | 0.191822 |
| GrimAge2 component | |||||
| DNAmadm | −2.265061 | 1.373178 | −1.64950 | 0.101303 | 0.247629 |
| DNAmCystatin_C | −2824.487 | 2224.994 | −1.26944 | 0.206407 | 0.412814 |
| DNAmGDF_15 | 1.987582 | 7.879241 | 0.25226 | 0.801216 | 0.889491 |
| DNAmleptin | −369.8450 | 111.1765 | −3.32665 | 0.001125 | 0.012320 |
| DNAmpai_1 | −448.4068 | 227.1757 | −1.97383 | 0.050383 | 0.191822 |
| DNAmTIMP_1 | −110.2619 | 94.08117 | −1.17199 | 0.243207 | 0.445880 |
Note: These values were calculated by multiple regression analyses examining the association between each item in the first column and the phenotype (patients or controls) using age and sex as confounding factors (Each item in the first column ~ phenotype + age + sex).
Abbreviations: ADM, adrenomedullin; B2M, beta 2 microglobulin; DNAm, DNA methylation; DNAmPACKYRS, DNA methylation‐based smoking pack‐years; DNAmTLAdjAge, age‐adjusted estimate of DNA methylation‐based telomere length; GDF15, growth differentiation factor‐15; PAI1, plasminogen activation inhibitor‐1; TIMP1, tissue inhibitor of metalloproteinases‐1.
The q values were calculated by adjusting the p values using the Benjamini Hochberg method. q < 0.05 is shown in bold.
FIGURE 1.
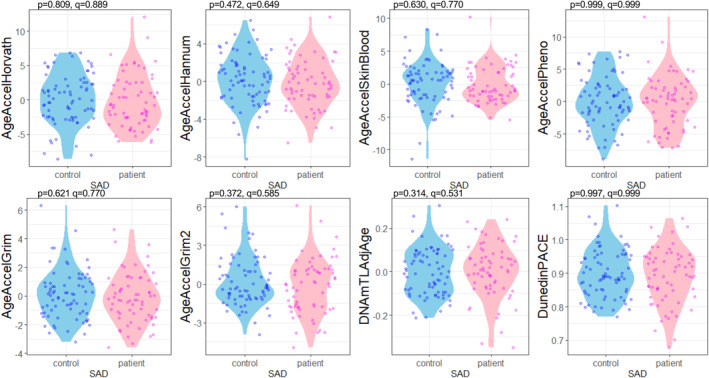
Violin plot showing age acceleration for each group. The p values were calculated by multiple regression analyses to test the association between each epigenetic age acceleration and phenotype (SAD or controls), and the q values were calculated with Benjamini Hochberg adjustment. Blue dots indicate data from the control group, and magenta dots indicate data from the patient group. This diagram was created using the R package ggplot2. DNAmTLAdjAge, age‐adjusted estimate of DNA methylation‐based telomere length; SAD, social anxiety disorder.
FIGURE 2.
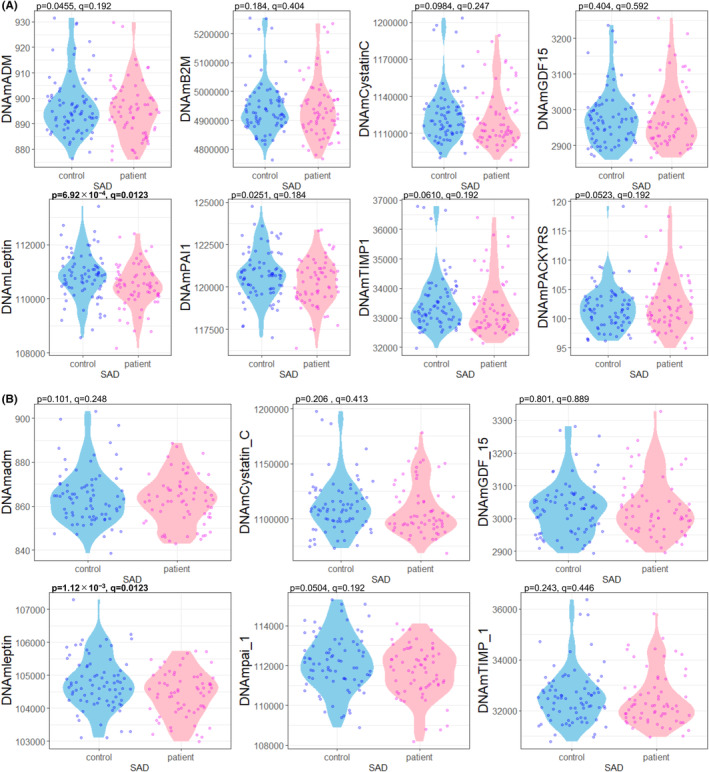
Violin plot showing (A) each GrimAge component and (B) each GrimAge2 component for each group. The p values were calculated by multiple regression analyses to test the association between each component and phenotype (SAD or controls), and the q values were calculated with Benjamini Hochberg adjustment. Blue dots indicate data from the control group, and magenta dots indicate data from the patient group. This diagram was created using the R package ggplot2. ADM, adrenomedullin; B2M, beta 2 microglobulin; DNAm, DNA methylation; DNAmPACKYRS, DNA methylation‐based smoking pack‐years; DNAmTLAdjAge, age‐adjusted estimate of DNA methylation‐based telomere length; GDF15, growth differentiation factor‐15; PAI1, plasminogen activation inhibitor‐1; SAD, social anxiety disorder; TIMP1, tissue inhibitor of metalloproteinases‐1.
In our previous study, which conducted epigenetic clock analyses for MDD, prediction values of Cystatin C levels in GrimAge components and GrimAge2 components showed significant association with MDD in GSE125105 cohort. However, DNAmLeptin in GrimAge or DNAmleptin in GrimAge2 did not significantly associate with MDD in this cohort (Table S1). 18
As additional regression analyses, we included DNAmPACKYRS and multiple cell compositions (CD8+ T cell, CD4+ T cell, Natural killer cell, B cell, Monocyte, Granulocyte, and Plasmablast) as additional confounding factors other than age and sex. As results, no epigenetic age acceleration showed significant association, but DNAmLeptin ( q = 0.0169) and DNAmPAI ( q = 0.048) in GrimAge components, and DNAmleptin ( q = 0.0281) in GrimAge2 components significantly associated with SAD (Table S2). In addition, we investigated correlations between DNAmLeptin or DNAmPAI1 in GrimAge components, and other confounding factors. Only sex was significantly correlated with DNAmLeptin ( q = 1.17 × 10 −5 ), and only DNAmPACKYRS ( q = 0.0407) were significantly correlated with DNAmPAI1 (Table S3).
Furthermore, we investigated the association between LSAS score and each epigenetic age acceleration, GrimAge component, and GrimAge2 component. No age acceleration or components was significantly associated with LSAS score (Table S4 and Figures S2 and S3).
4. DISCUSSION
To our knowledge, this is the first study to conduct epigenetic age analyses of SAD including GrimAge and GrimAge2 components. Epigenetic age acceleration was not significantly different between patients with SAD and controls. Among the GrimAge components and GrimAge2 components, the DNAm‐based prediction values of leptin levels were significantly lower in patients with SAD than in controls, even after adjusting with the Benjamini Hochberg method. Although the DNAm‐based prediction values of leptin level are less correlated with age than the other components, leptin is believed to suppress hunger and is negatively correlated with disease or mortality risk. 12 , 13 Although SAD is often associated with MDD, 34 the results of this study were different from those of our previous epigenetic clock analyses for MDD. 18 Our findings on leptin may be specific to SAD. The fact that SAD has a younger onset, and a longer duration of disease may be partial factors in the specify. 1 Furthermore, the significance of association between predicted values of leptin levels and SAD remained even after including confounding factors such as smoking status and cell composition, and we found PAI1 also had significant association with SAD in additional regression analyses.
Leptin regulates food intake and energy expenditure, 45 mediated by its binding to leptin receptors expressed throughout the central and peripheral nervous systems. In the central nervous system, the leptin receptor gene is highly expressed in the hypothalamic nuclei, including the paraventricular nucleus (PVN), a region that plays important roles in the maintenance of homeostasis, such as driver of the HPA response. 46 , 47 Hyperactivity of the HPA axis results in excessive secretion of adrenocorticotropic hormone (ACTH) and cortisol. Acute elevations in cortisol levels have great importance as a protective response to stress, but long‐term exposure to cortisol can lead to not only physical problems such as obesity, cancer, and cardiovascular disease, but also anxiety and depression through hippocampal and other neurological injuries. 48 , 49 , 50 , 51 Cortisol and ACTH levels were inversely correlated with leptin level. 52 Given these findings, we would expect that while the HPA system in patients with SAD is hyperactive due to various anxiety or stress conditions, leptin is inversely related; however, the results on leptin in anxiety are inconsistent among studies. 53 , 54 , 55 , 56 , 57 , 58 , 59 Due to complex feedback mechanisms, low leptin levels may be the secondary result of abnormal activation of the HPA system, and/or high leptin levels may be the result of increased leptin resistance. This study showed a significant negative association between SAD and leptin based on DNAm status; however, the severity of social anxiety was not significantly associated. The results from the various study designs should be confirmed in future studies.
PAI1 is an important inhibitor of tissue plasminogen activator and is increased in a variety of clinical situations related to aging. 60 , 61 PAI1 has also been reported to effect on brain neurons adversely. 62 In our previous study, DNAm‐based predictive value of PAI1 level was significantly elevated in patients with autism spectrum disorders than controls. 63
This study had several limitations. The sample in this study was obtained only from single cohort of Caucasians, and it is unknown whether the results can be adapted to other cohorts or racial groups. Among epigenetic clocks, there are several negative values that are often far from the actual age. However, in relative terms, all clocks showed a significant correlation with the actual age (Figure S1). The GrimAge and GrimAge2 components are protein predictors based on DNAm status and require actual test values. GrimAge components and GrimAge2 components reflect levels of proteins that were selected as reliable aging predictors by machine learning when GrimAge and GrimAge2 were developed, respectively. 12 , 13 However, in this study, the prediction values of leptin and PAI1 levels were not significantly correlated with actual age in the cohort. We thought the reason is that GrimAge indicates susceptibility to mortality diseases such as cardiovascular disease and life expectancy rather than actual chronological age. We were unable to obtain background information that might have affected the DNAm status, such as actual smoking history. 64 For smoking, we substituted DNAmPACKYRS. as a confounding factor for additional regression analyses.
5. CONCLUSION
Despite these limitations, we believe this study provides findings on SAD from a biological perspective based on DNAm status. Although we did not observe a significant acceleration of DNA methylation age in patients with SAD, we observed significant differences in leptin levels. We hypothesize that leptin is centrally involved in the pathogenesis of SAD as one of the aging‐related proteins. We believe that this study provides a basis for future studies on the pathogenesis of SAD. Further studies are required from various perspectives, including biological aging.
AUTHOR CONTRIBUTIONS
Nobuhiko Noguchi: Data curation, Formal analysis, Investigation, Visualization, Writing – original draft. Toshiyuki Shirai: Data curation, Formal analysis, Investigation, Visualization, Writing – original draft. Akira Suda: Data curation, Formal analysis. Saki Hattori: Data curation, Formal analysis. Masatoshi Miyauchi: Data curation, Formal analysis. Satoshi Okazaki: Data curation, Formal analysis. Junichi Fujita: Data curation, Formal analysis. Takeshi Asami: Data curation, Formal analysis. Ikuo Otsuka: Conceptualization, Methodology, Data curation, Formal analysis, Writing – review & editing. Akitoyo Hishimoto: Conceptualization, Supervision, Writing – review & editing.
FUNDING INFORMATION
This study was supported by a grant from SENSHIN Medical Research Foundation (Ikuo Otsuka) and by JSPS KAKENHI [Grant Numbers JP21K07545 (Ikuo Otsuka) and JP20KK0194 (Akitoyo Hishimoto)].
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
All the participants whose data were recorded in GSE164056 provided written informed consent which was approved by the University of Tübingen local ethics committee in accordance with the Declaration of Helsinki.
Approval of the Research Protocol by an Institutional Reviewer Board: All the participants whose data were recorded in GSE164056 participated in the research approved by the University of Tübingen local ethics committee.
Informed Consent: All the participants provided written informed consent.
Registry and the Registration No. of the Study/Trial: N/A.
Animal Studies: N/A.
Supporting information
Data S1:
Noguchi N, Shirai T, Suda A, Hattori S, Miyauchi M, Okazaki S, et al. Biological aging analysis based on DNA methylation status for social anxiety disorder. Neuropsychopharmacol Rep. 2024;44:774–783. 10.1002/npr2.12487
Nobuhiko Noguchi and Toshiyuki Shirai have equally contributed to this work.
DATA AVAILABILITY STATEMENT
We used the publicly available GSE164056 dataset, which was generated in a previous study 37 and can be found in the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164056).
REFERENCES
- 1. Ruscio AM, Brown TA, Chiu WT, Sareen J, Stein MB, Kessler RC. Social fears and social phobia in the USA: results from the National Comorbidity Survey Replication. Psychol Med. 2008;38(1):15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ. The genetic epidemiology of phobias in women. The interrelationship of agoraphobia, social phobia, situational phobia, and simple phobia. Arch Gen Psychiatry. 1992;49(4):273–281. [DOI] [PubMed] [Google Scholar]
- 3. Schneier FR, Abi‐Dargham A, Martinez D, Slifstein M, Hwang DR, Liebowitz MR, et al. Dopamine transporters, D2 receptors, and dopamine release in generalized social anxiety disorder. Depress Anxiety. 2009;26(5):411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sareen J, Campbell DW, Leslie WD, Malisza KL, Stein MB, Paulus MP, et al. Striatal function in generalized social phobia: a functional magnetic resonance imaging study. Biol Psychiatry. 2007;61(3):396–404. [DOI] [PubMed] [Google Scholar]
- 5. Ziegler C, Dannlowski U, Bräuer D, Stevens S, Laeger I, Wittmann H, et al. Oxytocin receptor gene methylation: converging multilevel evidence for a role in social anxiety. Neuropsychopharmacology. 2015;40(6):1528–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoge EA, Pollack MH, Kaufman RE, Zak PJ, Simon NM. Oxytocin levels in social anxiety disorder. CNS Neurosci Ther. 2008;14(3):165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. López‐Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186(2):243–278. [DOI] [PubMed] [Google Scholar]
- 8. Mkrtchyan GV, Abdelmohsen K, Andreux P, Bagdonaite I, Barzilai N, Brunak S, et al. ARDD 2020: from aging mechanisms to interventions. Aging. 2020;12(24):24484–24503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda SV, et al. Genome‐wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, et al. DNA methylation age of blood predicts all‐cause mortality in later life. Genome Biol. 2015;16(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging. 2018;10(4):573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging. 2019;11(2):303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu AT, Binder AM, Zhang J, Yan Q, Reiner AP, Cox SR, et al. DNA methylation GrimAge version 2. Aging. 2022;14(23):9484–9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fries GR, Bauer IE, Scaini G, Wu MJ, Kazimi IF, Valvassori SS, et al. Accelerated epigenetic aging and mitochondrial DNA copy number in bipolar disorder. Transl Psychiatry. 2017;7(12):1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Han LKM, Aghajani M, Clark SL, Chan RF, Hattab MW, Shabalin AA, et al. Epigenetic aging in major depressive disorder. Am J Psychiatry. 2018;175(8):774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Okazaki S, Otsuka I, Horai T, Hirata T, Takahashi M, Ueno Y, et al. Accelerated extrinsic epigenetic aging and increased natural killer cells in blood of suicide completers. Prog Neuropsychopharmacol Biol Psychiatry. 2020;98:109805. [DOI] [PubMed] [Google Scholar]
- 17. Shindo R, Tanifuji T, Okazaki S, Otsuka I, Shirai T, Mouri K, et al. Accelerated epigenetic aging and decreased natural killer cells based on DNA methylation in patients with untreated major depressive disorder. NPJ Aging. 2023;9(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tanifuji T, Okazaki S, Otsuka I, Mouri K, Horai T, Shindo R, et al. Epigenetic clock analysis reveals increased plasma cystatin C levels based on DNA methylation in major depressive disorder. Psychiatry Res. 2023;322:115103. [DOI] [PubMed] [Google Scholar]
- 19. Horvath S, Ritz BR. Increased epigenetic age and granulocyte counts in the blood of Parkinson's disease patients. Aging. 2015;7(12):1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Levine ME, Lu AT, Bennett DA, Horvath S. Epigenetic age of the pre‐frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer's disease related cognitive functioning. Aging. 2015;7(12):1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horvath S, Oshima J, Martin GM, Lu AT, Quach A, Cohen H, et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford progeria syndrome and ex vivo studies. Aging. 2018;10(7):1758–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dong M, Ren J. What fans the fire: insights into mechanisms of leptin in metabolic syndrome‐associated heart diseases. Curr Pharm Des. 2014;20(4):652–658. [DOI] [PubMed] [Google Scholar]
- 24. Lu AT, Seeboth A, Tsai PC, Sun D, Quach A, Reiner AP, et al. DNA methylation‐based estimator of telomere length. Aging. 2019;11(16):5895–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Belsky DW, Caspi A, Corcoran DL, Sugden K, Poulton R, Arseneault L, et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. Elife. 2022;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ryan CP. “epigenetic clocks”: theory and applications in human biology. Am J Hum Biol. 2021;33(3):e23488. [DOI] [PubMed] [Google Scholar]
- 27. Stein MB, Stein DJ. Social anxiety disorder. Lancet. 2008;371(9618):1115–1125. [DOI] [PubMed] [Google Scholar]
- 28. Cremers HR, Roelofs K. Social anxiety disorder: a critical overview of neurocognitive research. Wiley Interdiscip Rev Cogn Sci. 2016;7(4):218–232. [DOI] [PubMed] [Google Scholar]
- 29. Terracciano A, Aschwanden D, Stephan Y, Cerasa A, Passamonti L, Toschi N, et al. Neuroticism and risk of Parkinson's disease: a meta‐analysis. Mov Disord. 2021;36(8):1863–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Terracciano A, Aschwanden D, Passamonti L, Toschi N, Stephan Y, Luchetti M, et al. Is neuroticism differentially associated with risk of Alzheimer's disease, vascular dementia, and frontotemporal dementia? J Psychiatr Res. 2021;138:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schilstra CE, Fardell JE, Ellis SJ, Jones KM, Anazodo AC, Trahair TN, et al. Social anxiety symptoms in survivors of childhood and adolescent cancer. J Adolesc Young Adult Oncol. 2022;11(2):129–137. [DOI] [PubMed] [Google Scholar]
- 32. Deschênes SS, Burns RJ, Schmitz N. Associations between diabetes, major depressive disorder and generalized anxiety disorder comorbidity, and disability: findings from the 2012 Canadian community health survey – mental health (CCHS‐MH). J Psychosom Res. 2015;78(2):137–142. [DOI] [PubMed] [Google Scholar]
- 33. Li X, Wang X. Relationships between stroke, depression, generalized anxiety disorder and physical disability: some evidence from the Canadian community health survey‐mental health. Psychiatry Res. 2020;290:113074. [DOI] [PubMed] [Google Scholar]
- 34. Shalom JG, Shaul‐Tsoran I, Strauss AY, Huppert JD, Andersson G, Aderka IM. Mediation of social anxiety and depression during internet‐delivered treatment for social anxiety disorder. Cogn Behav Ther. 2024;53(4):436–453. [DOI] [PubMed] [Google Scholar]
- 35. Leigh E, Chiu K, Ballard ED. Social anxiety and suicidality in youth: a systematic review and meta‐analysis. Res Child Adolesc Psychopathol. 2023;51(4):441–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Otsuka I, Izumi T, Boku S, Kimura A, Zhang Y, Mouri K, et al. Aberrant telomere length and mitochondrial DNA copy number in suicide completers. Sci Rep. 2017;7(1):3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wiegand A, Kreifelts B, Munk MHJ, Geiselhart N, Ramadori KE, MacIsaac JL, et al. DNA methylation differences associated with social anxiety disorder and early life adversity. Transl Psychiatry. 2021;11(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rytwinski NK, Fresco DM, Heimberg RG, Coles ME, Liebowitz MR, Cissell S, et al. Screening for social anxiety disorder with the self‐report version of the Liebowitz social anxiety scale. Depress Anxiety. 2009;26(1):34–38. [DOI] [PubMed] [Google Scholar]
- 39. Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Loyfer N, Magenheim J, Peretz A, Cann G, Bredno J, Klochendler A, et al. A DNA methylation atlas of normal human cell types. Nature. 2023;613(7943):355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li JL, Jain N, Tamayo LI, Tong L, Jasmine F, Kibriya MG, et al. The association of cigarette smoking with DNA methylation and gene expression in human tissue samples. Am J Hum Genet. 2024;111(4):636–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fragou D, Pakkidi E, Aschner M, Samanidou V, Kovatsi L. Smoking and DNA methylation: correlation of methylation with smoking behavior and association with diseases and fetus development following prenatal exposure. Food Chem Toxicol. 2019;129:312–327. [DOI] [PubMed] [Google Scholar]
- 43. Arloth J, Eraslan G, Andlauer TFM, Martins J, Iurato S, Kühnel B, et al. DeepWAS: multivariate genotype‐phenotype associations by directly integrating regulatory information using deep learning. PLoS Comput Biol. 2020;16(2):e1007616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moore SR, Halldorsdottir T, Martins J, Lucae S, Müller‐Myhsok B, Müller NS, et al. Sex differences in the genetic regulation of the blood transcriptome response to glucocorticoid receptor activation. Transl Psychiatry. 2021;11(1):632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Friedman J. The long road to leptin. J Clin Invest. 2016;126(12):4727–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Elmquist JK, Bjørbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395(4):535–547. [PubMed] [Google Scholar]
- 47. Herman JP, Tasker JG. Paraventricular hypothalamic mechanisms of chronic stress adaptation. Front Endocrinol. 2016;7:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature. 2011;476(7361):458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Russell G, Lightman S. The human stress response. Nat Rev Endocrinol. 2019;15(9):525–534. [DOI] [PubMed] [Google Scholar]
- 50. Buitelaar JK. The role of the HPA‐axis in understanding psychopathology: cause, consequence, mediator, or moderator? Eur Child Adolesc Psychiatry. 2013;22(7):387–389. [DOI] [PubMed] [Google Scholar]
- 51. Faravelli C, Lo Sauro C, Lelli L, Pietrini F, Lazzeretti L, Godini L, et al. The role of life events and HPA axis in anxiety disorders: a review. Curr Pharm Des. 2012;18(35):5663–5674. [DOI] [PubMed] [Google Scholar]
- 52. Licinio J, Mantzoros C, Negrão AB, Cizza G, Wong ML, Bongiorno PB, et al. Human leptin levels are pulsatile and inversely related to pituitary‐adrenal function. Nat Med. 1997;3(5):575–579. [DOI] [PubMed] [Google Scholar]
- 53. Ozmen S, Şeker A, Demirci E. Ghrelin and leptin levels in children with anxiety disorders. J Pediatr Endocrinol Metab. 2019;32(10):1043–1047. [DOI] [PubMed] [Google Scholar]
- 54. Naufel MF, Boldarine VT, Oyama LM, do Nascimento CMO, Silva dos Santos GM, Hachul H, et al. Age and leptinemia association with anxiety and depression symptoms in overweight middle‐aged women. Menopause. 2019;26(3):317–324. [DOI] [PubMed] [Google Scholar]
- 55. Cernea S, Both E, Huţanu A, Şular FL, Roiban AL. Correlations of serum leptin and leptin resistance with depression and anxiety in patients with type 2 diabetes. Psychiatry Clin Neurosci. 2019;73(12):745–753. [DOI] [PubMed] [Google Scholar]
- 56. Bellisario V, Panetta P, Balsevich G, Baumann V, Noble J, Raggi C, et al. Maternal high‐fat diet acts as a stressor increasing maternal glucocorticoids' signaling to the fetus and disrupting maternal behavior and brain activation in C57BL/6J mice. Psychoneuroendocrinology. 2015;60:138–150. [DOI] [PubMed] [Google Scholar]
- 57. Ambrus L, Westling S. Leptin, anxiety symptoms, and hypothalamic‐pituitary‐adrenal Axis activity among drug‐free, female suicide attempters. Neuropsychobiology. 2019;78(3):145–152. [DOI] [PubMed] [Google Scholar]
- 58. Yamauchi T, Inoue K, Iwasaki S, Muramatsu T, Hayashi T, Kiriike N. Intracerebroventricular administration of leptin increases anxiety‐like behavior in female rats after semi‐starvation – implications for anxiety in eating disorders. Osaka City Med J. 2009;55(1):9–18. [PubMed] [Google Scholar]
- 59. Stroe‐Kunold E, Buckert M, Friederich HC, Wesche D, Kopf S, Herzog W, et al. Time course of leptin in patients with anorexia nervosa during inpatient treatment: longitudinal relationships to BMI and psychological factors. PLoS One. 2016;11(12):e0166843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vaughan DE. PAI‐1 and atherothrombosis. J Thromb Haemost. 2005;3(8):1879–1883. [DOI] [PubMed] [Google Scholar]
- 61. Vaughan DE, Rai R, Khan SS, Eren M, Ghosh AK. Plasminogen activator inhibitor‐1 is a marker and a mediator of senescence. Arterioscler Thromb Vasc Biol. 2017;37(8):1446–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau‐dependent pathology and cognitive decline. Nature. 2018;562(7728):578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Okazaki S, Kimura R, Otsuka I, Funabiki Y, Murai T, Hishimoto A. Epigenetic clock analysis and increased plasminogen activator inhibitor‐1 in high‐functioning autism spectrum disorder. PLoS One. 2022;17(2):e0263478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jamieson E, Korologou‐Linden R, Wootton RE, Guyatt AL, Battram T, Burrows K, et al. Smoking, DNA methylation, and lung function: a Mendelian randomization analysis to investigate causal pathways. Am J Hum Genet. 2020;106(3):315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1:
Data Availability Statement
We used the publicly available GSE164056 dataset, which was generated in a previous study 37 and can be found in the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164056).