

Abstract
Background:
Activated protein C (APC) has anticoagulant and cytoprotective cell signaling activities which often require protease-activated receptor (PAR)1 and PAR3 and PAR cleavages at noncanonical sites (R46-N47 and R41-G42, respectively). Some PAR1-derived peptides(P1) and PAR3-derived peptides(P3), e.g., P1-47-66 and P3-42-65, mimic APC’s cell signaling. In anti-inflammatory assays, these two peptides at low concentrations synergistically attenuate cellular inflammation.
Objective:
To determine whether a P1 peptide covalently linked to a P3 peptide mimics APC’s anti-inflammatory and endothelial barrier stabilization activities.
Methods:
Anti-inflammatory assays employed stimulated THP-1 cells and caspase-1 measurements. Cultured human EAhy926 or murine aortic endothelial cells (EC) exposed to thrombin were monitored for transendothelial electrical resistance (TEER). Bivalent covalently-linked P1:P3 peptides were studied for APC-like activities.
Results:
In anti-inflammatory assays, P1-47-55 was as active as P1-47-66 and some P3 peptides (e.g., P3-44-54 and P3-51-65) were as active as P3-42-65. The bivalent P1:P3 peptide comprising P1-47-55-[Gly(10 residues)]-P3-51-65 (designated “G10 peptide”) was more potently anti-inflammatory than the P1 or P3 peptide alone. In TEER studies of thrombin-challenged EC’s, P1-47-55 and the G10 peptide mimicked APC’s protective actions. In dose-response studies, the G10 peptide was more potent than the P1-47-55 peptide. In murine EC studies, the murine PAR-sequence-derived G10 peptide mimicked murine APC’s activity. Anti-PAR1 and anti-PAR3 antibodies, but not anti-EPCR antibodies, abated G10’s cytoprotection, showing G10’s actions involve PAR1:PAR3. G10 significantly increased survival in murine endotoxemia.
Conclusions:
The PAR-sequence-derived G10 peptide is a bivalent agonist that mimics APC’s cytoprotective anti-inflammatory and endothelial barrier stabilizing actions and APC’s protection against endotoxemic mortality.
Keywords: endothelial cell, inflammasome, protein C, thrombin, proteinase-activated receptor
Visual Abstract
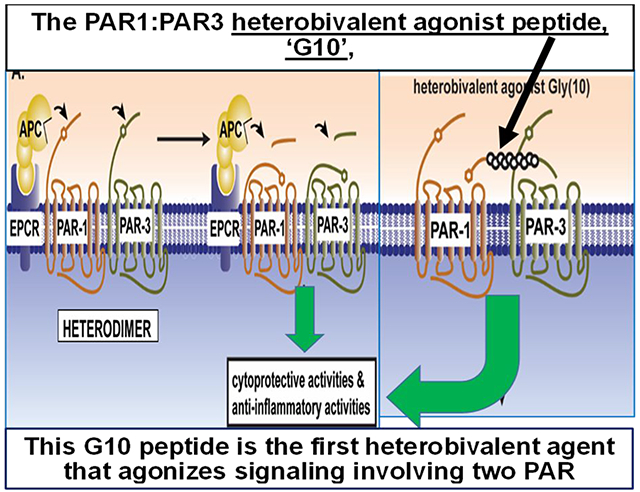
1. INTRODUCTION
The human and murine family of protease activated receptors (PARs) comprise four G protein coupled receptors (GPCRs) that enable proteases to initiate cell signaling and thereby modify cell behavior [1–4]. Proteases activate PARs by cleaving the extracellular N-terminal tail to form a new N-terminus which is a tethered agonist ligand that can bind and selectively activate the parent GPCR via its many typical GPCR transducers, namely G proteins, G protein kinases, and arrestins [1–3]. Synthetic peptides containing the sequence of the N-terminal tethered ligand can also activate their respective PAR, although the synthetic peptide’s signaling effects can sometimes vary from those of the PAR’s N-terminal tethered ligand [1–4].
Activated protein C (APC) is a plasma protease that manifests a broad spectrum of activities that includes its traditional anticoagulant functions as well as multiple cytoprotective effects on a multitude of cell types [5, 6]. The list of cytoprotective actions include anti-apoptotic and anti-inflammatory activities, endothelial barrier stabilization, and favorable alterations of gene expression profiles. The mechanisms for APC’s direct signaling effects in vitro on cells and for APC’s beneficial effects in vivo in preclinical injury models appear to be primarily mediated, at least in part, by PAR1 and PAR3 [5, 6]. Activation by APC of these two GPCRs, PAR1 and PAR3, is based on biased GPCR activation [2–4, 7, 8] due to APC’s cleavages at non-canonical cleavage sites in the PAR1 or PAR3 extracellular N-terminal tail [9–11]. Among the four PARs, PAR1 is the most often studied and exhibits a wide diversity of molecular interactions that underlie its signaling whereas PAR3 is the least studied PAR [2–6, 9–12].
A major insight into mechanisms contributing to APC’s anti-inflammatory actions came from the discovery that APC and the recombinant 3K3A-APC mutant can avert NOD-like receptor protein 3 (NLRP3) inflammasome-induced ischemia-reperfusion that causes cardiac and renal injury in mice [13, 14]. The abilities of the recombinant 3K3A-APC mutant to reduce ocular inflammation in mice [14] and to reduce subarachnoid hemorrhage in rats [15] were related to its ability to suppress NLRP3 inflammasomes. Inflammasomes play a central role in innate immune system-driven inflammation, and the canonical NLRP3 inflammasome is key for innate immune system-driven inflammation [3, 5–7, 16–19]. This inflammasome comprises a large, intracellular cytosomal multiprotein complex that enables the maturation and release of the pro-inflammatory cytokines, interleukin (IL)-1β and IL-18 [7–9, 19–21]. Inflammasome formation involves multiple steps that result in activation of one of its key effectors, the cysteine protease caspase-1, that enables the generation of IL-1β and IL-18 from their precursors [3, 5–7, 10, 13, 16–19, 22]. Bioassays for caspase-1 were useful to monitor the in vitro suppression of NLRP3 inflammasome development by APC in human macrophages [23].
The anti-inflammatory in vivo suppression of the NLRP3 inflammasome in mice by APC requires PAR1 [13], and the in vitro suppression of NLRP3 inflammasome development in human macrophages by APC requires both PAR1 and PAR3 [23]. PAR1-derived and PAR3-derived agonist peptides each mimic this anti-inflammatory action of APC on human macrophages, and, remarkably, the combination of low dose PAR1-derived and PAR3-derived agonist peptides synergistically exerts anti-inflammatory activity [23]. PAR1-derived and PAR3-derived peptides also stabilize endothelial barrier function in vitro and in vivo [5, 6, 9–11]. Many studies showed that PAR1 and PAR3 are required for human or murine APC’s therapeutically protective effects when studied using murine in vivo injury models [5, 6, 9–15, 24–37].
In this report, we extend the studies of PAR1-derived and PAR3-derived peptides that mimic APC’s actions to explore PAR peptide structure-function relationships in anti-inflammatory assays. Here we also show that covalently coupling of a P1 peptide to a P3 peptide yields a novel bivalent PAR1/PAR3-derived peptide that can mimic APC’s in vitro anti-inflammatory and endothelial barrier stabilizing activities and APC’s in vivo ability to increase survival in murine endotoxemic sepsis. We suggest that such orthosteric/allosteric bivalent PAR-derived agonist peptides may be valuable candidates for translation to therapies where APC or APC signaling-selective mutants have been shown to provide therapeutic benefits.
2. METHODS
2.1. Reagents
THP-1 null cells, THP1-defNLRP3 cells, LPS-B5 Ultrapure, PMA, MCC950, Ac-YVAD-CMK, Z-VAD-FMK and Normocin were all purchased from Invivogen (San Diego, CA). The term “THP-1 null” refers to control cells (Invivogen) which manifest very well typical NLRP3 inflammasome activation following appropriate simulation, and hereafter are simply referred to as THP-1 cells [23]. EA.hy926 cells were purchased from ATCC (Atlanta, GA), immortalized mouse primary aortic endothelial cells from Cell Biologics, (Chicago, IL). FAM-FLICA Caspase-1 assay kit from Immunochemistry Technologies (Bloomington, MN), ATP from Biopioneer (San Diego, CA), purified human plasma APC from Enzyme Research Labs (South Bend, IN), and Caspase-1 activity assay from Promega (Madison, WI). Peptides listed in Table 1 were synthesized by Synthetic Biomolecules (San Diego, CA) or by Anaspec (Fremont, CA). Biochemical analyses showed that the G10 peptide was > 95% pure. All other reagents, unless otherwise indicated below, were purchased from Fisher Scientific (Hampton, NH). Recombinant murine APC was purified as described [38]. Murine monoclonal antibodies against endothelial protein C receptor (EPCR) (RCR-252), PAR1 (WEDE-15), and PAR3 (clone 19b) were prepared by the Scripps Research antibody Core facility from hybridoma cell lines (kindly provided by Dr. K. Fukudome (Saga Medical School, Saga, Japan) and Dr. L. Brass (University of Pennsylvania, Philadelphia, PA)).
Table.
Amino acid sequences of peptides derived from protase activated receptor sequences

|
2.2. Cell culture conditions
THP-1 cells were maintained in RPMI 1640 containing 10% heat inactivated fetal bovine serum, Pen-strep (100 U/mL- 100 μg/mL), 2 mM L-glutamate and 100 μg/mL Normocin at 37°C with 5% CO2. Selection pressure was maintained by addition of 200 μg/mL of Hygromycin B Gold to growth medium every other passage. Cell count did not exceed 2 × 106/mL and cells were not passaged beyond 20 times. EA.hy926 and immortalized mouse aortic endothelial cells were grown at 2.5 x 104 cells/500 μL per well to confluence in 8-well E-plate L8 (Agilent Technologies, Santa Clara, CA). For cell impedance measurements, DMEM (Invitrogen) containing 10% fetal calf serum was replaced with serum-free media containing 3% BSA before addition of APC or peptides.
2.3. Cell treatments with APC or peptides and caspase-1 assays
THP-1 cells were plated at a final concentration of 1 x 106/mL in 96 well plates (200 μL cell solution per well) and incubated with PMA (0.5 μM) for 3 hours 37°C in supplemented RPMI as previously described [23]. Media was subsequently changed every 24 hours for 3 days. Various subsets of wells were selected and cells were treated for 60 min at 37°C in serum-free RPMI with various concentrations of APC or peptides [17,18]. Following DPBS wash, cells were incubated with LPS-B5 Ultrapure (lipopolysaccharide) (1 μg/mL) for 3 hours at 37°C in serum-free RPMI to prime the inflammasome. After a DPBS wash, in order to fully activate caspase-1 activity, cells were incubated with ATP (5 mM), in the presence or absence of 10 μM YVAD (caspase-1 inhibitor), or in the presence or absence of 2.5 μM ZVAD (pan-caspase inhibitor) for 45 minutes at 37°C, prior to using the caspase-1 activity assay following the manufacturer’s (Promega) directions. Caspase activity was monitored as luminescence changes due to substrate hydrolysis and reported as relative luminescent units (RLU) [23]. For some data presentations, RLU values were obtained by subtraction of the caspase activity value (RLU) observed for addition of YVAD (caspase-1 inhibitor) and the result is presented as adjusted net caspase-1 activity. Luminescence was measured using the Tecan Genios Pro plate reader. Active caspase-1 was measured by incubating cells with the fluorescent inhibitor probe FAM-YVAD-FMK to label active caspase-1 fluorescently. Fluorescence at excitation 488 nm and emission at 430 nm and reported as relative fluorescent units (RFU) using the Tecan Genios Pro plate reader.
2.4. Transendothelial electrical resistance (TEER) assay
Integrity of EA.hy926 human endothelial cells monolayers was measured using an electric cell-substrate impedance sensing system (iCelligence, Acea Biosciences, San Diego, CA). An increase or decline in transendothelial electrical resistance (TEER) across the cell monolayers indicated, accordingly, a decrease or increase of endothelial paracellular permeability. EA.hy926 cells were plated at 2.5 x 104 cells/500 μL per well of a E-plate L8 (Agilent Technologies, Santa Clara, CA) using complete DMEM media in presence of 10% FBS. The sensor plates were placed on the iCelligence machine inside a CO2 incubator at 37 °C and allowed to reach confluence to form an endothelial monolayer which produced a plateau on the TEER measurements. Then, the EA.hy926 cells were washed with DPBS buffer and the media was exchanged to DMEM, 3% BSA without FBS. Human or murine APC’s or peptides (Table 1) at varying concentrations or control buffer were added to each well and then incubated for 30 min in the CO2 incubator followed by thrombin addition (0.25 nM final) which produced a sharp decrease of TEER in control wells. Alternatively, mouse primary aortic endothelial cells in place of EAhy926 cells were used and subjected to treatments of thrombin, APC or G10 peptide.
2.5. Murine Endotoxin-induced Sepsis
All animal protocols were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute, La Jolla, CA. C57BL/6J male mice were obtained from the Scripps Research Institute’s rodent breeding colony. 8-11 weeks old mice received an intraperitoneal injection of lipopolysaccharides (LPS) (30 mg/kg) from Escherichia Coli O55:B5 (Sigma-Aldrich). Four hours after LPS injection, mice were anesthetized via isofluorane inhalation and injected intravenously in the retroorbital plexus with 2 mg/kg human G10 peptide dissolved in sterile saline. Mice were followed for survival over 10 days. The husbandry and handling of mice conformed to guidelines set by the Institutional Animal Care Committees, following the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.6. Statistical analysis
Data were analyzed using GraphPad Prism 6. For caspase-1 assays a two-way analysis of variance with Tukey’s post hoc correction or one-way analysis of variance with Welch’s post hoc correction was used to assess statistical significance among parameter across multiple normally distributed cell parameters. Survival curves for the murine sepsis study were analyzed by Kaplan-Meyer Mantel-Cox test. P values of 0.05 or less were considered statistically significant. Each treatment condition was tested in duplicate and caspase measurements were performed at minimum in triplicate. All values are reported as mean ± S.E. of the mean unless otherwise stated. For TEER assays, significant values were calculated using unpaired T-test with non-parametric distributions. ** p< 0.001, ***p<0.0001.
3. RESULTS
3.1. PAR1-derived and PAR3-derived peptides reduce caspase-1 activity
The PAR1-derived peptide comprising residues 47-66, designated P1-47-66 (see Table 1), and the PAR3-derived peptide comprising residues 42-65, designated P3-42-65 (Table 1), each mimics the anti-inflammatory action of APC on human macrophages and on stimulated THP-1 cells to reduce NLRP3-dependent elaboration of caspase-1 [23]. To gain further insights into structural requirements for the anti-inflammatory actions of these peptides, several analogs of each peptide (Table 1) were studied using previously described methods [23] for their ability to reduce caspase-1 activity in stimulated THP-1 cells (Figure 1) similarly to the ability of APC to reduce caspase-1 activity. When the PAR1 peptide sequence was shortened from 47-66 to 47-55, the anti-inflammatory activity was retained (Figure 1A), leading us to further studies using this shortened PAR1 sequence (see below). However, when the PAR1 sequences were shortened in various analogs. i.e., to 47-54, 47-52, 48-66, 49-66, 52-66, or 48-54, there was a significant reduction of the peptide‘s ability to reduce caspase-1 activity (Figure 1A). Replacement of the N-terminal Asn47 residue with Gln in the P1-47-66 peptide had no significant effect but the Ala47 replacement reduced but did not eliminate the peptide’s inhibitory activity under the assay conditions. Replacement of Asn47 with Asp ablated the observed anti-inflammatory activity of P1-47-66. Acetylation of the N-terminal alpha amino group on Asn47 in P1-47-66, designated Ac-P1-47-66, eliminated P1-47-66’s observed activity (Figure 1A). These data show that maintaining the intact N-terminal sequence of PAR1-derived peptides is important for good activity while the C-terminus of the peptide can be shortened until residue 55 but not further. Thus, for further studies, we focused on P1-47-55 or P1-47-66.
Figure 1. PAR1-derived and PAR3-derived peptides of differing sequences vary in their ability to reduce caspase-1 activity in activated THP-1 cells.
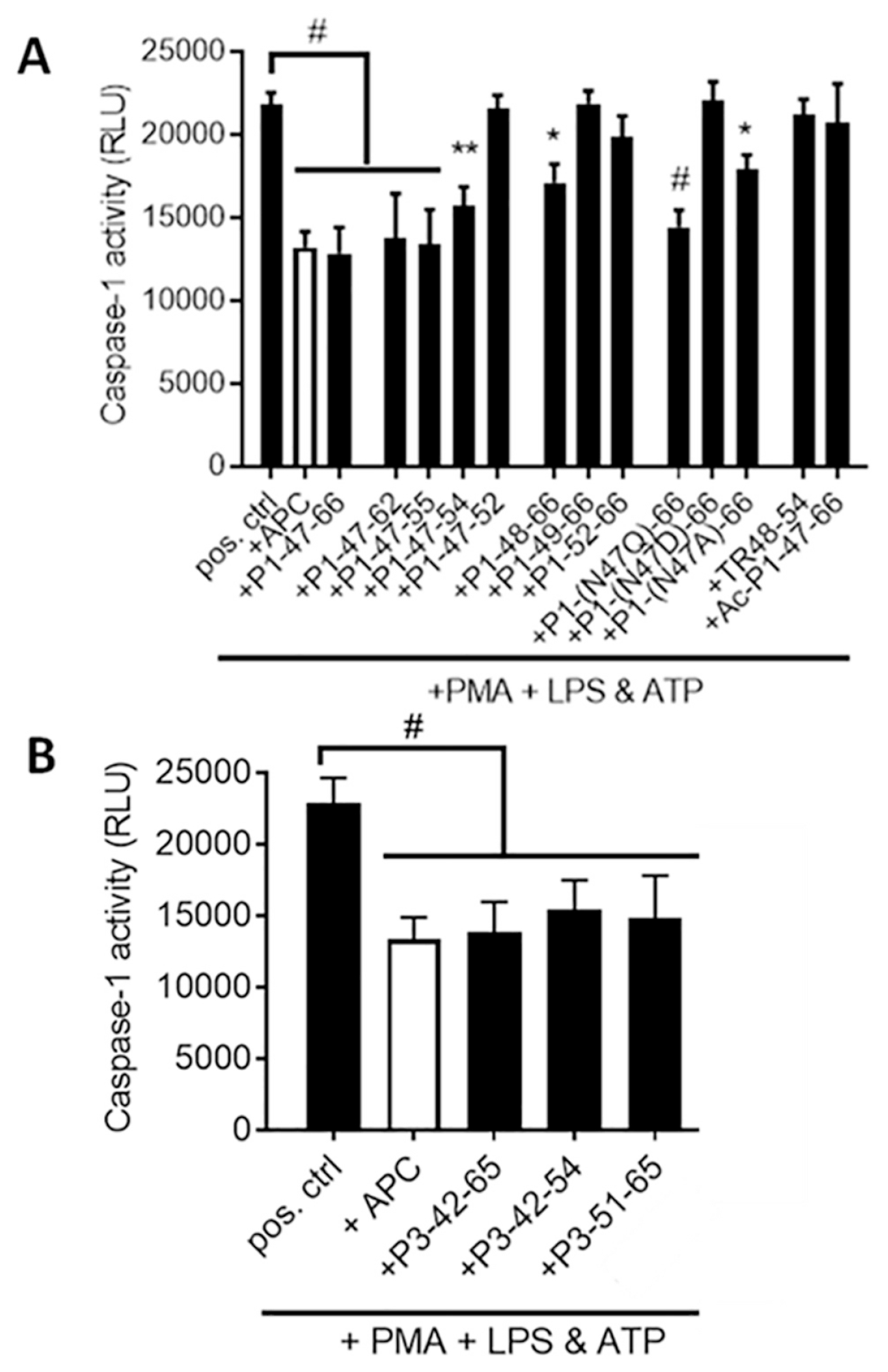
(A) The ability of PAR1-derived peptides (50 μM) and (B) PAR3-derived peptides (50 μM) to inhibit elaboration of caspase-1 activity in activated THP-1 cells was measured using previously described methods [23]. APC (4 μg/ml) is shown for comparison. Data points represent mean ± SEM of at least 3 independent experiments. #P < 0.005, *P < 0.05 (vs. LPS + ATP).
When the PAR3 peptide sequence was shortened from residues 42-65 to 42-54 or 51-65 and screened for ability to inhibit caspase-1 elaboration, there was no significant decrease in the peptide’s ability to reduce caspase-1 activity relative to the inhibitory activity of APC under the conditions employed (Figure 1B). Thus, it was possible to delete the N-terminal residues 42-50 and maintain activity. Remarkably, this is a striking example for a PAR-agonist peptide that does not require a native-like sequence equivalent to or nearly equivalent to the PAR N-terminus that is created by PAR proteolysis to agonize the PAR response.
Previous studies showed that the 24-mer P3-42-65 peptide reduces caspase-1 activity synergistically when in combination with the 20-mer P1-47-66 peptide [23], as seen in Figure 2A. New studies show that the shortened P3-51-65 peptide that lacks 9 residues of the canonical N-terminal sequence which includes residue-42 similarly showed synergism when tested with P1-47-66 for reducing caspase-1 activity (Figure 2A). Thus, the greatly shortened P3-51-65 peptide, like the canonical P3-42-65, mimics APC anti-inflammatory activity and it also shows greatly enhanced activity when combined with P1-47-66.
Figure 2. The PAR3-derived peptide, P3-51-65, reduces caspase-1 activity synergistically in the presence of P1-47-66 or when it is covalently linked to P1-47-55 in the bivalent G10 peptide.
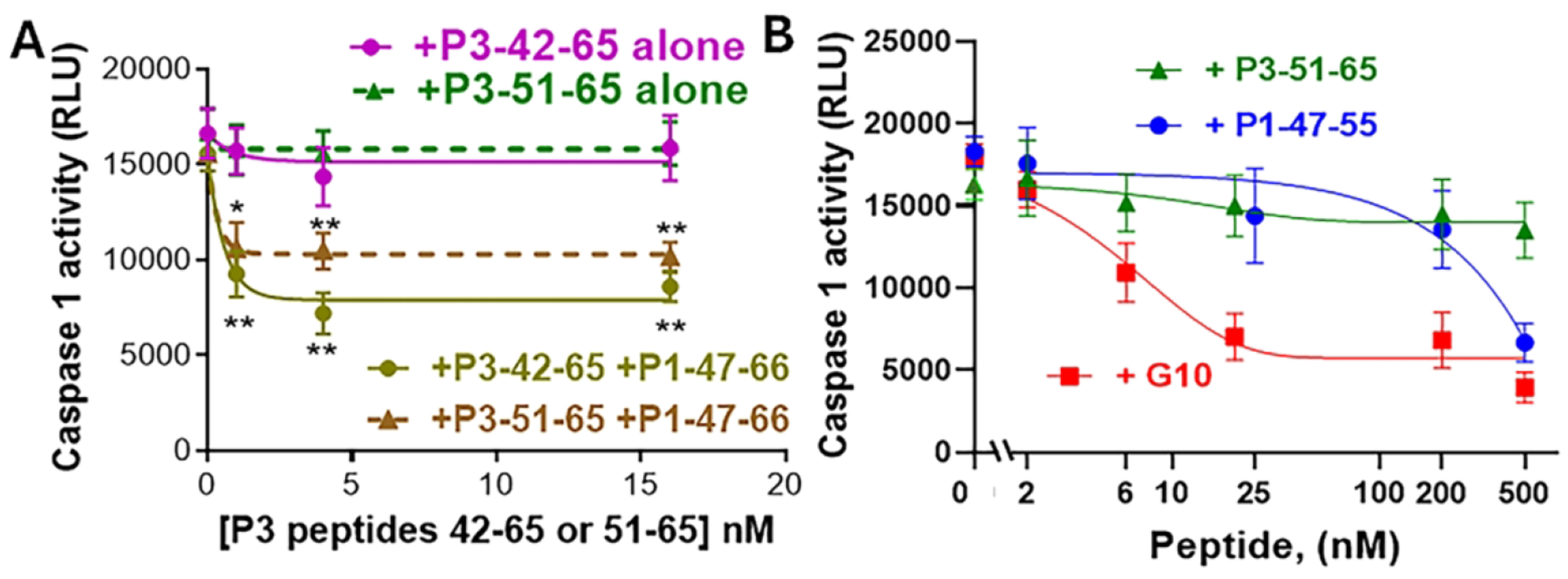
THP-1 cells were plated at a final concentration of 1 x 106/mL in 96 well plates and incubated with PMA (0.5 μM) for 3 hours 37°C in supplemented RPMI. Media was subsequently changed every 24 hours for 3 days. (A) Selected wells were treated with P3-42-65 alone or P3-51-65 alone or with each P3 peptide in the presence of P1-47-66 (1.0 nM). (B) Alternatively, selected wells were treated with P1-47-55 alone or P3-51-65 alone or G10 peptide (0 – 500 nM) (□), as indicated, Then for (A) and (B), cells were washed with DPBS and then incubated with LPS (1 μg/mL) for 3 hours at 37°C in serum-free RPMI. Cells were washed with DPBS and incubated with ATP (5 mM) in the presence or absence of 10 μM YVAD (caspase-1 inhibitor) for 45 minutes at 37°C, prior to performing the caspase-1 activity assay following manufacturer’s directions measuring luminescence (relative units and expressed as relative Caspase-1 activity (RLU). Data points represent mean ± S.D. of at least 3 independent experiments. *P < 0.05, **P<0.005 (vs. LPS + ATP).
3.2. Covalent linkage of the P1-47-55 peptide to the P3-51-65 peptide yields a PAR-derived heterobivalent peptide, G10, that potently reduces caspase-1 activity.
When the shortened P1-47-55 was covalently linked to the shortened P3-51-65 peptide using a 10-mer oligo-Gly peptide to yield the heterobivalent PAR1:PAR3-derived peptide that is labeled the “G10” peptide (Table 1), the bivalent G10 peptide potently reduced caspase-1 activity when compared to either the P1-47-55 or P3-51-65 peptide components alone (Figure 2B). Dose-response data showed that the G10 peptide was at least 10-fold more potent than the P1-47-55 peptide alone and at least 50-fold more potent than the P3-51-65 alone (Figure 2B).
3.3. A PAR1-derived peptide, a PAR3-derived peptide, and a PAR-derived bivalent peptide, G10, inhibit thrombin-induced decrease of endothelial resistance in TEER assays.
An important cytoprotective activity of APC involves its ability to blunt the ability of thrombin (IIa) to disrupt endothelial barrier stability [5, 6]. Studies of various P1 and P3 peptides and of the G10 peptide to blunt thrombin’s endothelial barrier disruption using human EAhy926 cells were made. Peptide dose response studies of peptides ranging from 0 to 50 nM (Figure 3) showed that P1-47-66 at ≤ 50 nM moderately protected endothelial cells (Figure 3A) while P3-42-65 at ≤ 50 nM had almost no effect (Figure 3B). However, G10 at 5 to 50 nM potently blunted thrombin’s barrier disruption effect on endothelial cells (Figure 3C). Additional dose-response studies for the peptide components of G10 ranging from 30 to 300 nM were made (Figure 4). The G10 peptide was significantly more effective than the P1-47-55 alone (Figure 4A) or the oligo-Gly-P1-47-55 peptide alone (Figure 4B). The P3-51-65 alone and the P3-51-65-oligo-Gly peptide alone were much less effective than G10 (compare Figures 4A and 4C). Data from multiple repetitions were used to calculate these peptides’ endothelial protective effects as decrease of area under the curves (AUC) for normalized cell index (NCI) relative to the large effect of thrombin alone. AUC was calculated for NCI values for 0 to 60 min and expressed as relative units of AUC. Dose response data for G10 showed 50% AUC decrease at 30 nM whereas ≥ 300 nM of the P1 or P3 peptides, with or without the 10-mer-oligo-Gly peptide attachment, was needed for 50 % reduction of AUC (Figure 4D). Thus, the attachment of the 10-mer oligo-Gly peptide to P1-47-55 alone or to P3-51-65 alone caused no significant alterations of activity of the P1-47-55 or P3-51-65 peptides alone (Figure 4). Importantly, the bivalent G10 was significantly more potent for endothelial stabilization than G10’s component P1 or P3 peptides.
Figure 3. The PAR1-derived peptide, P1-47-66, the PAR3-derived peptide, P3-42-65, and the PAR1:PAR3-derived bivalent peptide, G10 inhibit thrombin-induced disruption of endothelial barrier integrity.
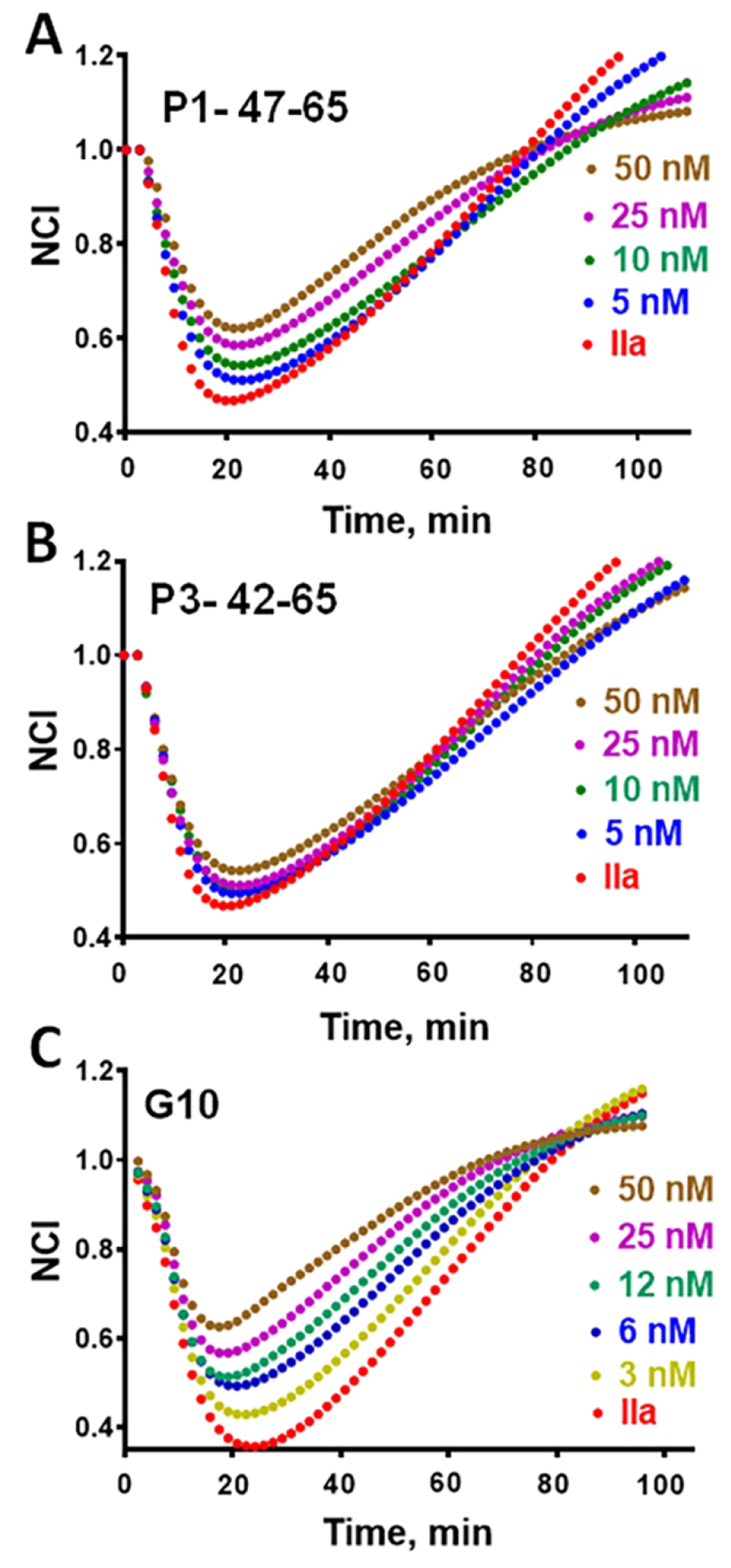
EA.hy926 human endothelial cells were used to collect TEER data as described in Methods and in [30]. Barrier disruption was promoted by addition of thrombin (IIa) (0.25 nM final) in each well except the baseline control. Each peptide (final concentration indicated next to each curve) was added 30 min prior the addition of thrombin. Cell index values were normalized to the value observed at the time of the thrombin addition that was defined as 1.0 on the x-axis and are shown beginning for the time at which thrombin was added and for treatments using (A) P1-47-66 peptide, (B) P3-42-65 peptide, or C) the G10 peptide.
Figure 4. G10 inhibits thrombin-induced disruption of endothelial barrier integrity much better than its individual component P1-47-55 or P3-51-65 peptides.
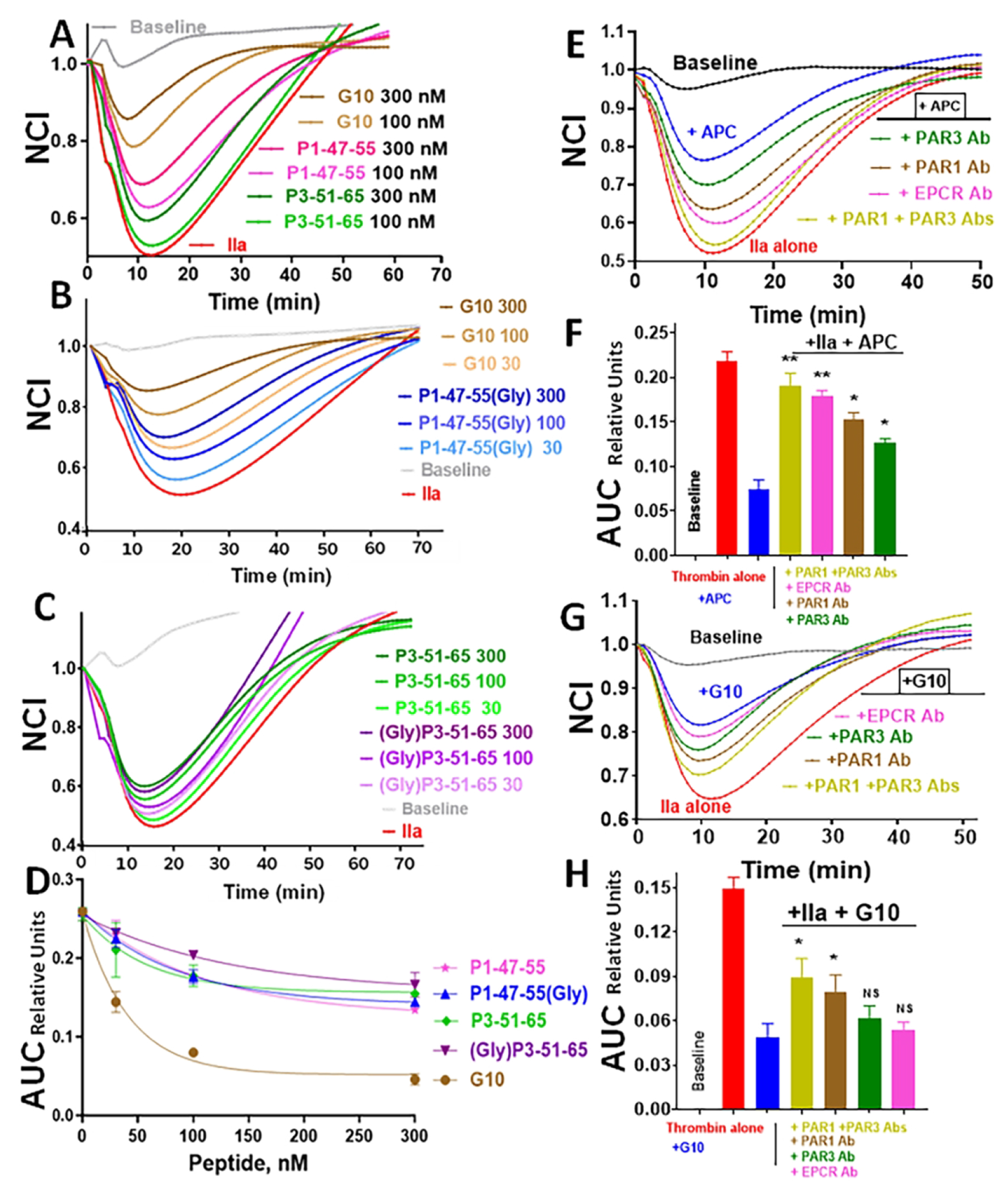
Endothelial barrier integrity assays used EA.hy926 endothelial cells as described above. Cell index values were normalized (NCI on y-axis) to the value observed at the time of the thrombin addition that was defined as 1.0 on the x-axis and are shown beginning for the time at which thrombin was added. Panels A, B and C show data for the individual G10-component peptides, P1-47-55 and P3-51-65 with and without a 10-mer-oligoGly attached, and for G10 at 30, 100, and 300 nM (as indicated) for their ability to inhibit thrombin’s disruption of endothelial barrier integrity. Data are for (A) G10 peptide and P1-47-55, (B) G10 and P1-47-55(Gly) (P1 peptide with C-terminal 10-mer oligo-Gly sequence), and (C) P3-51-65 and (Gly)P3-51-65 (P3 peptide with N-terminal 10-meroligo-Gly sequence). (D) The calculated area between the baseline curve and each peptide’s curve was calculated and defined as Area under the Curve (AUC) in arbitrary relative units for each peptide and plotted to show the relative potency for endothelial barrier stabilization. Data in (D) show average values for duplicate experiments. Statistical values were obtained by using unpaired T-test with non-parametric distributions, the P value for G10 differing from each of the other 4 peptides at each concentration was P< 0.001. For (A), (B), and (C), the “Baseline” control curves are for only buffer additions and the red curves are for thrombin (IIa) addition. (E,F,G,H) For studies to determine whether antibodies against PAR1, PAR3, and EPCR suppress the endothelial barrier protection by APC or G10 peptide, EA.hy926 cells were processed as above, and cells were incubated with antibodies (20 μg/mL final) that were added 30 min prior to treatment with APC (4 μg/mL) or G10 (300 nM). AUC data were generated as described above. **p<0.01; *p<0.05. ns indicates not significant.
3.4. PAR1 and PAR3 are required for the endothelial barrier protective effects of the G10 peptide.
To evaluate the receptor requirements for the G10 peptide’s protective effects on thrombin-challenged endothelial cells, monoclonal antibodies against EPCR, PAR1, and PAR3 were tested for their ability to ablate G10’s and APC’s protective effects. As expected for APC’s actions, each antibody significantly reduced APC’s endothelial barrier stabilization (Figures 4E, 4F). For G10’s protective actions, anti-PAR1 or its combination with anti-PAR3 significantly ablated G10’a protection while other antibodies had no significant effect (Figures 4G, 4H). This indicates that PAR1 is required for G10’s normal level of protective effects on endothelial cells.
3.5. The length of the oligo-Gly linker in bivalent G10 peptide analogs influences G10 bioactivities.
To assess the influence of the length of the oligo-Gly linker component of G10 analogs on their activities, G10 analogs were made that contained 4, 8 or 12 Gly residues (Table 1). Studies of the peptides’ ability to initiate reduction of capase-1 in stimulated THP-1 cells showed that G10 analogs with 8 or 12 Gly residues were similar to G10 (Figure 5A) while the analog with 4 Gly residues for the linker showed essentially no substantial activity at up to 200 nM, a concentration where neither P1-47-55 nor P3-51-65 peptides showed substantial activity (Figure 2B). G10 peptides containing oligo-Gly linker sequences of varying lengths were tested at 300 nM for ability to reduce thrombin-induced disruption of endothelial barriers using EAhy926 cells. As described above. time course plots for the protective effects of each peptide were used to calculate AUC values (Figure 5C). G10 analogs with 8, 10 or 12 Gly residues were more active than the P1-47-55-(10-mer-oligo-Gly) peptide, and G10 containing the 10-mer oligo-Gly linker was the most active. As seen above for suppression of inflammasome generation, the oligo-Gly linker with 4 Gly residues totally failed to achieve the activity gain observed for covalently coupling the P1 and P3 peptides with the 10-mer oligo-Gly linker. Consequently, for a bivalent PAR-derived peptide agonists that mimic APC’s anti-inflammatory and endothelial barrier protective activities, the 10-mer oligo-Gly linker is the optimal linker length.
Figure 5. The length of the oligo-Gly linker in G10 peptide analogs can alter the anti-inflammatory activity and the endothelial protective activity of the bivalent PAR1:PAR3-derived bivalent peptides.
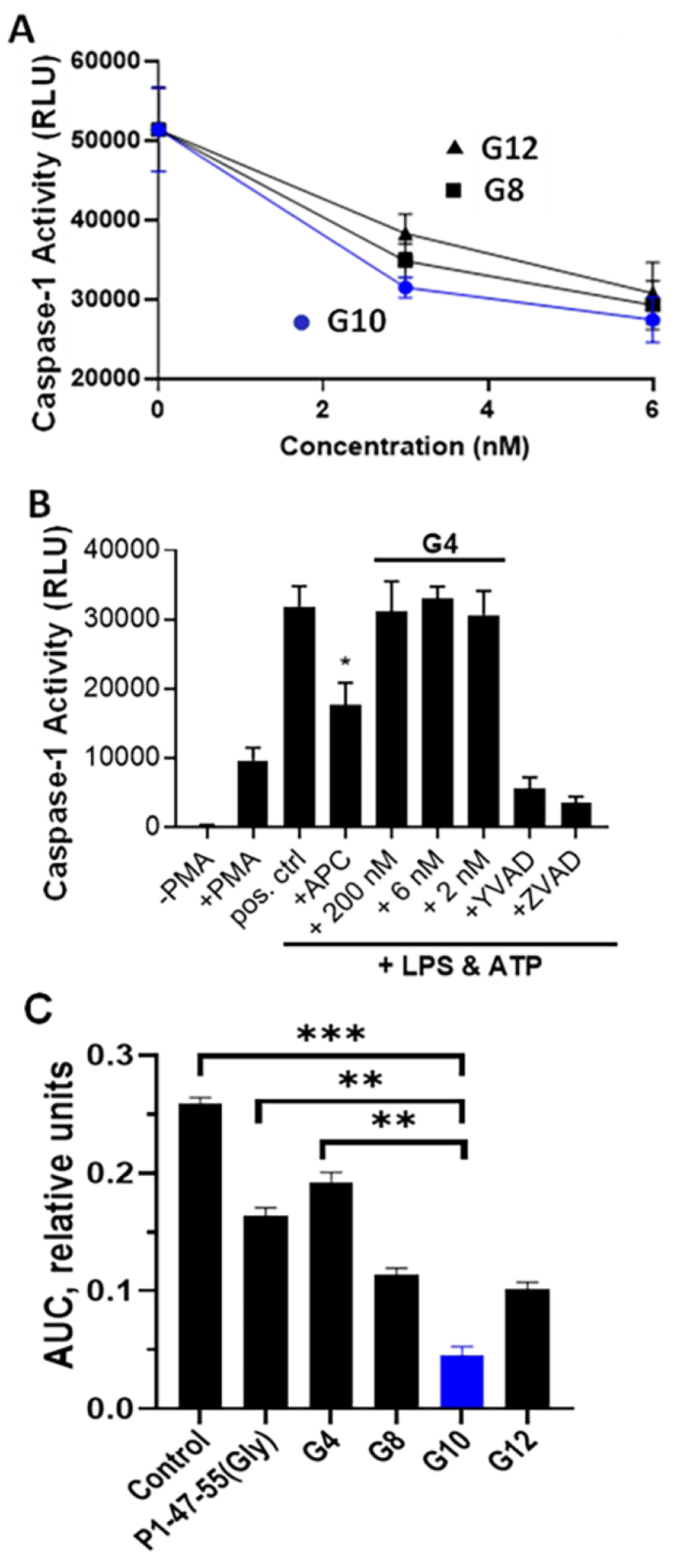
G10 analogs that contained 4, 8 or 12 Gly residues (Table 1) were tested (A, B) for ability to suppress caspase-1 generation in stimulated THP-1 cells, as previously done [23] and as above (Figures 1, 2), and tested (C) for endothelial barrier stabilization using EAhy926 human cells, as above (Figure 4). (A) Anti-inflammatory dose-response data for G10 analogs are seen for those containing 8, 10, or 12 oligo-Gly linkers, designated G8, G10, or G12, in the low dose concentration range where each showed a similar potency. (B) Data for the G10 analog containing only 4 Gly residues, designated G4, showed no detectable reduction of caspase-1 activity at up to 200 nM compared to the positive control of APC (4 ug/ml). Data represent mean ± S.D. of at least 3 independent experiments and * indicates P value < 0.05 vs. positive (pos.) control. (C) G10 analogs were tested at 300 nM for ability to reduce thrombin-induced disruption of endothelial integrity as above (Figure 4). From time course plots for this activity of each peptide at 300 nM, the diminished area under the curve (AUC) was calculated and is shown. The AUC value designated “Control” was for no peptide addition. The P1-47-57-oligo-10-Gly peptide (designated P1-47-55(10Gly)) (300 nM final) was a positive reference control. Statistical values were obtained using unpaired T-test with non-parametric distributions. ** p< 0.01, ***p<0.001.
3.6. A murine G10 peptide, like the human G10 peptide, mimics APC endothelial cell barrier protection.
Murine recombinant APC’s mimicked multiple cytoprotective activities of human APC both in vitro and in vivo, and many studies showed that PAR1 and PAR3 enabled murine APC’s protective effects [5, 6, 9–15, 24–37]. Since it’s desirable to be aware of potential species-based differences in a compound’s bioactivity, studies of mouse (m)G10, an analog of human (h)G10 (Table 1), were made using murine aortic endothelial cells (Figure 6). Both mAPC and hAPC dose-dependently reduced thrombin-induced disruption of murine endothelial barrier function, and mAPC appeared 3-fold to 5-fold more potent than hAPC on these murine cells based on semi-quantitative estimates, including AUC data comparisons (Figure 6B) for time course data (Figure 6A). Similar to murine and human APC’s, both mG10 and hG10 peptides blunted thrombin-induced endothelial barrier disruption in murine endothelial cells (Figure 6C, 6D). Similar to mAPC versus hAPC, mG10 appeared approximately 3-fold to 5-fold more potent than hG10 on these murine cells.
Figure 6. Using mouse sequence PAR1-derived peptide, a mouse PAR3-derived peptide, and a mouse PAR-derived bivalent peptide, G10, inhibits thrombin-induced decrease of endothelial resistance in TEER assays.
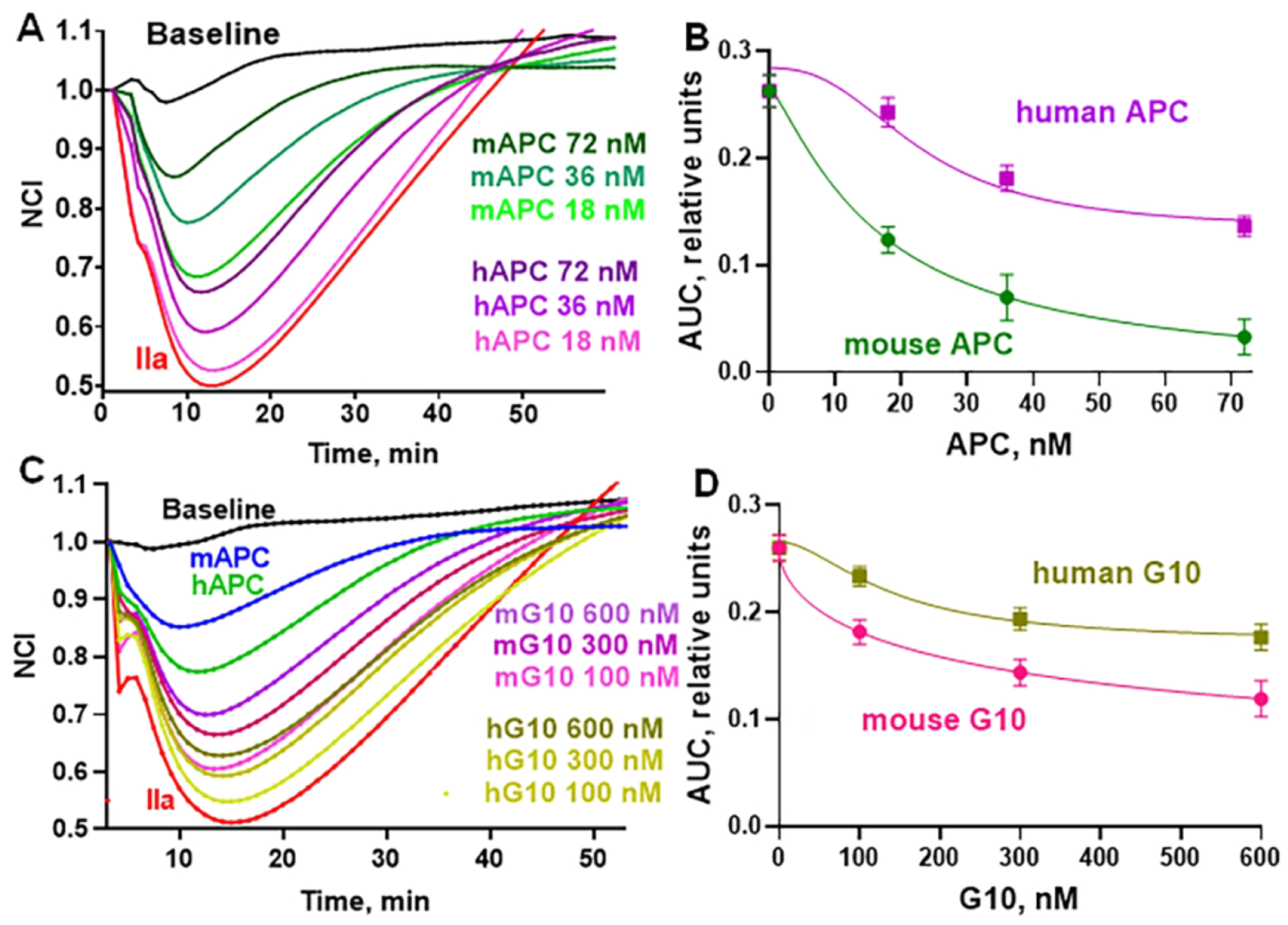
Endothelial cell barrier integrity was monitored following disruption by addition of thrombin (0.25 nM final) to cultured murine aortic endothelial cells as described above for human EA.hy926 cells and in Methods. The ability of mouse (m) APC, human (h) APC, mG10, and hG10 to counteract thrombin-induced endothelial barrier disruption was determined. Cell index values were normalized (NCI) to a value defined as 1.0 for the time of the thrombin addition, as above. (A) mAPC and hAPC were evaluated at 72, 36, and 18 nM (values indicated in figure) for reduction of thrombin-induced barrier disruption. (B) AUC data for mAPC and hAPC were calculated from data in panel A. (C) Peptides of mG10 sequence and hG10 sequence (Table 1) were tested at 600, 300, and 100 nM for reduction of thrombin-induced barrier disruption. Curves are shown for mAPC and hAPC (72 nM). (D) AUC data for G10’s were calculated from data in panel C. For (A) and (C), the “Baseline” control curves are for only buffer additions and the red curves are for thrombin (IIa) addition.
3.7. The G10 peptide improves survival in murine endotoxemic sepsis.
To evaluate the ability of peptide G10 to mimic APC’s ability to improve survival in endotoxin-treated mice, animals that had been treated with E.coli-derived LPS received an intravenous injection of 2 mg/kg of human G10 peptide at 4 hours after LPS. Animals were then followed for survival over ten days (Figure 7). The G10 peptide significantly increased the survival rate from 31% seen for untreated mice to 53% seen for the G10 peptide-treated group (p=0.018). Thus, peptide G10’s in vivo treatment mimics sigmaling-selective APC’s in vivo survival effects in murine endotoxin-treated mice.[39, 40].
Figure 7. Human G10 peptide treatment improves survival in a mouse model of bacterial endotoxemia.
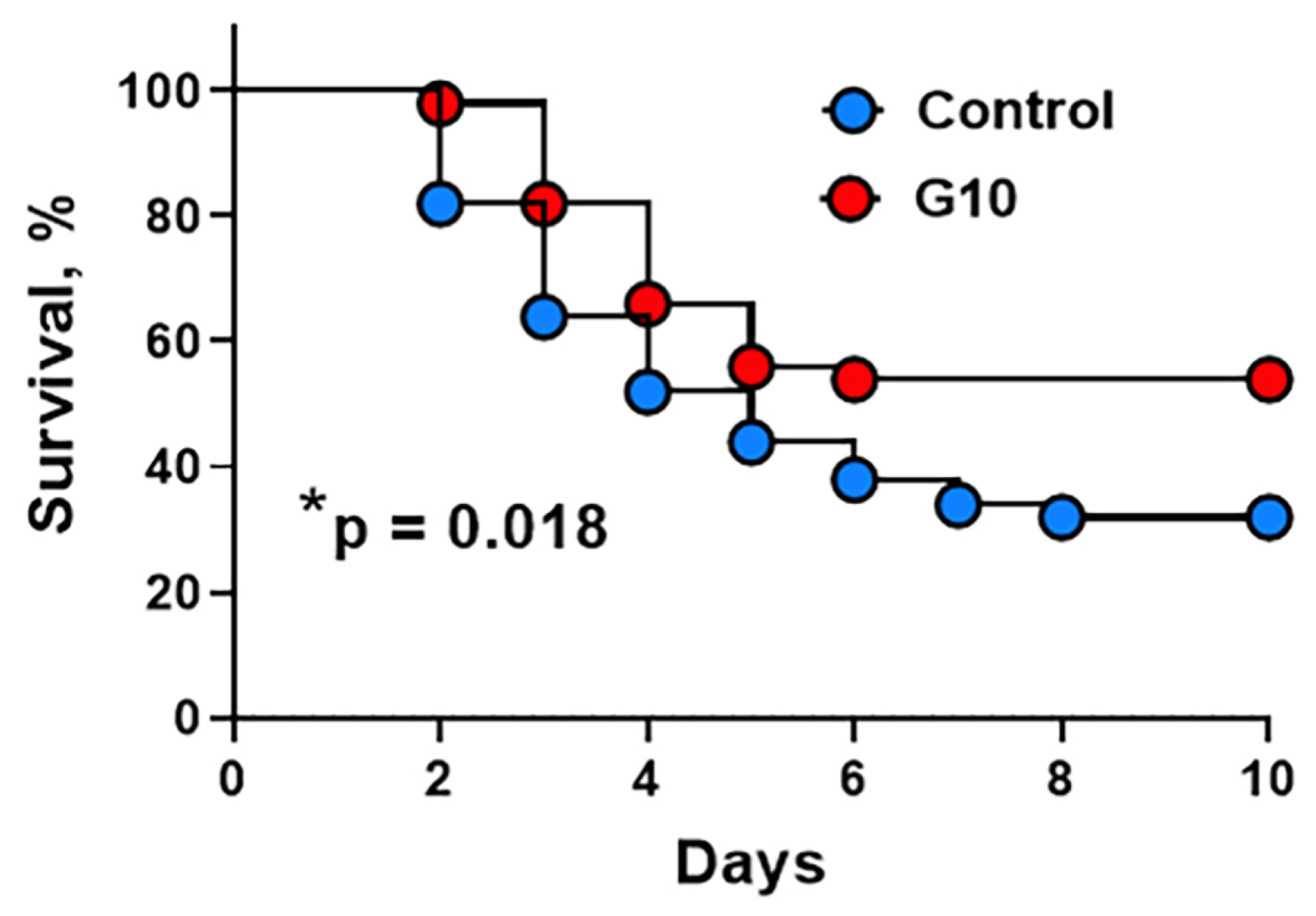
C57BL/6J mice were injected intraperitoneally with 30 mg/kg of LPS. Four hours after the LPS injection, mice were left untreated or injected intravenously with human G10 peptide (2 mg/kg) and followed for survival. The G10 peptide treatment increased the survival rate from 31% of untreated mice to 53% of the G10-treated group. Cumulative results were derived from three independent experiments. (Total n = 50 mice for each group). p=0.018.
4. DISCUSSION
Agonism of PARs following proteolysis of the extracellular N-terminal tail is driven by the newly revealed N-terminal or related synthetic peptides, e.g., for APC, PAR1 peptides (P1) beginning with N47 or PAR3 peptides (P3) beginning with G42. This report extends previous studies that showed that the P1-47-66 and P3-42-65 peptides each can mimic APC in that each is anti-inflammatory by reducing NLRP3-driven caspase-1 activity in stimulated human THP-1 cells [23]. Structure-activity relationship studies here show that shorter P1 and P3 peptides, namely P1-47-55 and P3-51-65, each mimics APC’s anti-inflammatory activity on THP-1 cells. For anti-inflammatory activity, the P1 peptide required a native Asn47 or the very similar Gln47 residue, consistent with this P1-47-65 being an orthosteric agonist for biased PAR1 signaling, resembling the effect caused by APC’s cleavage of PAR1 at Arg46. Remarkably, the P3-51-65 lacks the 10 N-terminal G42-E50 sequence, indicating it cannot function as a typical PAR orthosteric agonist. This suggests that the ability of P3-51-65 to agonize cell signaling is based on its ability to function as an allosteric factor that influences PAR signaling. The endothelial cell study that discovered the existence of PAR1:PAR3 heterodimers concluded that PAR3 functions as an allosteric modulator of PAR1 signaling [41]. Since the anti-inflammatory activity of P3-42-65 required both PAR1 and PAR3, the P3 N-terminal peptides, in particular P3-51-65 which here exerts anti-inflammatory activity like P3-42-65 but which lacks the canonical N-terminal orthostatic agonist sequence beginning with G42, are presumed to function as allosteric activators of the PAR1:PAR3 heterodimer. The potent G10 peptide is the first example of covalent linkage of two PAR peptide agonists that target two PARs; notably, in this case, one agonist targets an orthosteric site on PAR1 for peptides that begin with N47 while the other is an allosteric PAR3-related agonist that targets sites on one or more receptors that remain unidentified. In contrast to beneficial agonists, antagonists may be useful, and a PAR-targeting bivalent antagonizing compound was described which involved linking two non-peptide antagonists that target PAR1 and PAR2 [42]. G10 is an orthosteric/allosteric bivalent agonist, a type of GPCR bivalent ligand that was identified and highlighted for other GPCRs more than a decade ago but that has not previously been identified for PARs until now [43, 44].
The bivalent PAR1:PAR3-derived G10 peptide formed by covalent linkage of P1-47-55 to P3-51-65 via a 10-mer oligo-Gly peptide mimics APC’s anti-inflammatory activity with a potency that is an order of magnitude more potent than either of the constituent P1 or P3 peptides alone. Optimization of the length of the oligo-Gly linker for G10 analogs showed that an optimal linkage length involved 10 Gly residues that is approximately 35 Å long and that when the linker was only 4 Gly residues that is approximately 12 Å long, the benefits of bivalency for G10 were ablated. In endothelial barrier stabilization assays, attachment of a 10-mer oligo-Gly peptide to either P1-47-55 alone or P3-51-65 alone had no effect. Thus, it’s the linkage per se, not the Gly residues, that improves activity of G10.
To gain insights into whether the G10 peptide requires PAR1, PAR3, or EPCR for its beneficial actions on endothelial cells, anti-receptor antibodies were tested for their ability to reduce G10’s activity. Anti-PAR1 antibodies and the combination of anti-PAR1 and anti-PAR3 antibodies; however, neither anti-EPCR nor anti-PAR3 antibodies alone significantly ablated G10’s actions. Previous studies of individual P1-47-66 and P3-42-65 peptides using the same anti-receptor antibodies previously showed that anti-PAR1 antibodies reduced the anti-inflammatory activity of both of those isolated P1 and the P3 peptides while anti-EPCR showed no inhibition [23]. Thus, like the isolated P1 peptide, G10 requires PAR1 for induction of its normal level of cell signaling. Nonetheless, one cannot exclude the possibility that G10 might also act on other receptors that are not PAR’s to contribute to some of its actions, and such effects might involve either the P1 or the P3 components of G10. In summary, PAR1 appears to be central for G10’s mechanism of action.
In a wide variety of preclinical injury model studies, wild type APC and signaling-selective APC mutants have very beneficial effects [5, 6]. For an initial test of G10’s ability to mimic APC’s in vivo beneficial actions, endotoxin-treated mice were treated with G10 and monitored for survival. The G10 peptide significantly increased the survival rate from 31% to 53%. Thus, G10 mimicked APC’s enhanced survival in this murine sepsis model [39, 40]. Several murine endotoxemia issues merit further studies including exploration of different G10 doses and modes of its administration (e.g., multiple doses, continuous infusion, intraperitoneal administration, etc.) as more information is needed to paint a more complete picture for G10’s efficacy in this preclinical murine injury model.
What are the mechanisms by which G10, an orthosteric/allosteric bivalent PAR ligand, mimics APC’s activities? First, PAR1 and PAR3 are broadly found in cells and tissues where APC exerts cytoprotective activities, and, second, PAR1 and PAR3 form heterodimers [41]. Influenced by the law of parsimony (Occam’s razor), we speculate that G10 binds to the PAR1:PAR3 heterodimer and stabilizes and/or engenders one or more active PAR1 conformations and one or more active PAR3 conformations that exert the cytoprotective biased activities which follow APC’s cleavages of PAR1 and PAR3. Although murine PAR3 lacks the Arg41 residue where APC cleaves human PAR3 [11], we know that for many murine in vitro and in vivo studies, murine PAR1 and PAR3 are essential for normal APC cytoprotective actions [5, 6, 9–15, 24–37]. Therefore, for murine endothelial cells studied here, we speculate that the allosteric P3 peptide component of G10, like human P3-42-65 alone which mimicked APC’s in vivo endothelial barrier stabilization in mice [10, 11], employs both the orthosteric P1-derived and the allosteric P3-derived components to achieve effective cell signaling.
Understanding G10’s mechanisms from a broad perspective requires a deconvolution of the signaling mechanisms initiated by PAR-derived N-terminal peptides, e.g., for biased signaling by P1-47-66 and P1-47-55, and of how P3-42-65 and P3-51-65 alter signaling. It’s noteworthy that P3-42-65 is an allosteric activator, not simply an allosteric modulator, when it is given in vivo to mice to reduce vascular leakage in the absence of APC or any P1 peptide [10, 11]. The discovery of biased signaling for GPCR’s [3, 8], including for PAR1 and PAR3 [9, 10], emphasizes the roles for multiple conformational states for PAR1 and PAR3. Each GPCR’s interactome may include multiple cellular proteins, including other GPCRs in heterodimer or homodimer complexes, different G proteins, arrestins, G protein-coupled kinases, or other membrane components (e.g., EPCR, apoER2, Tie 2, integrins, etc [2–6, 12, 24, 45–49]. Allosteric interactions involving one or more of these types of proteins may stabilize a given GPCR, e.g., PAR1 or PAR3, in different conformations [2–8, 11,24, 50–53]. Notably, there are multiple heterodimer and homodimer combinations for each of the 4 PARs in addition to the PAR1:PAR3 [2–4, 24, 45, 46, 54]. Thus, the potential sites for modulations of cell signaling by the PAR1-derived and/or PAR3-derived components of G10 may very well extend beyond the PAR1:PAR3 heterodimer to include intermolecular partners of either PAR1 or PAR3. One remarkable example for how reactions centered around certain receptors, e.g., PAR1 and EPCR, can have different signaling outcomes is the discovery that when a recombinant enzymatically inactive protein C is present, the signaling initiated by thrombin resembles that of APC, not of thrombin [55, 56]. Given the complexities of GPCR allostery, at this point, there are no data and no reasonable speculations to support hypothesizing that G10’s mechanisms extend beyond the PAR1:PAR3 heterodimer, although it is most likely that they do.
The fact that the orthosteric/allosteric bivalent G10 functions as a PAR agonist that mimics APC’s in vitro and in vivo beneficial activities implies a potentially strong translational value for G10 or similar PAR1:PAR3-derived bitopic peptides. Peptides and peptidomimetics that agonize or antagonize GPCRs currently include many approved drugs as well as drugs currently in clinical trials [52, 57–60]. Thus, G10 or similar orthosteric/allosteric bitopic peptides that mimic APC’s cytoprotective cell signaling activities could fit into current major trends for drug development.
In summary, covalent linkage of a PAR1 agonist peptide to a PAR3 allosteric peptide yielded the first orthosteric/allosteric bivalent PAR-derived compound that mimics APC’s beneficial cell signaling activities and APC’s in vivo improvement of survival in endotoxemic sepsis. This G10 peptide and similar PAR-derived bitopic agonist peptides point towards a path to future peptide drugs that mimic APC’s multiple beneficial therapeutic actions.
ACKNOWLEDGEMENTS
We are grateful to Dr. L. Brass (University of Pennsylvania, Philadelphia, PA) for the gift of the PAR1 and PAR3 antibodies and Dr. K Fukudome (Saga Medical School, Saga, Japan) for the EPCR antibody.
FUNDING
This research was supported by grants from the National Institutes of Health (R01HL142975 (J.H.G. and L.O.M.), R01NS117827 (J.H.G.), R01HL172048 (R.A.) and R01HL104165 (L.O.M.)) and the American Heart Association 18POST34020053 (L.D.H.).
Footnotes
DECLARATION OF COMPETING INTERESTS
L.D.H., L.O.M., and J.H.G. are co-inventors for Scripps-owned patents related to some studies in this report. L.O.M. and J.H.G. are on the Scientific Advisory Board of Novapep Lty. R.A. and J.A.F. have no competing interest.
REFERENCES
- 1.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005; 3: 1800–14. 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 2.Han X, Nieman MT, Kerlin BA. Protease-activated receptors: An illustrated review. Res Pract Thromb Haemost. 2021; 5: 17–26. 10.1002/rth2.12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peach CJ, Edgington-Mitchell LE, Bunnett NW, Schmidt BL. Protease-activated receptors in health and disease. Physiol Rev. 2023; 103: 717–85. 10.1152/physrev.00044.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Donnell JS, Fleming H, Noone D, Preston RJS. Unraveling coagulation factor-mediated cellular signaling. J Thromb Haemost. 2023. 10.1016/j.jtha.2023.06.019. [DOI] [PubMed] [Google Scholar]
- 5.Griffin JH, Zlokovic BV, Mosnier LO. Activated protein C: biased for translation. Blood. 2015; 125: 2898–907. 10.1182/blood-2015-02-355974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffin JH, Zlokovic BV, Mosnier LO. Activated protein C, protease activated receptor 1, and neuroprotection. Blood. 2018; 132: 159–69. 10.1182/blood-2018-02-769026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010; 9: 373–86. 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov. 2018; 17: 243–60. 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood. 2012; 120: 5237–46. 10.1182/blood-2012-08-452169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burnier L, Mosnier LO. Novel mechanisms for activated protein C cytoprotective activities involving noncanonical activation of protease-activated receptor 3. Blood. 2013; 122: 807–16. 10.1182/blood-2013-03-488957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stavenuiter F, Mosnier LO. Noncanonical PAR3 activation by factor Xa identifies a novel pathway for Tie2 activation and stabilization of vascular integrity. Blood. 2014; 124: 3480–9. 10.1182/blood-2014-06-582775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willis Fox O, Preston RJS. Molecular basis of protease-activated receptor 1 signaling diversity. J Thromb Haemost. 2020; 18: 6–16. 10.1111/jth.14643. [DOI] [PubMed] [Google Scholar]
- 13.Nazir S, Gadi I, Al-Dabet MM, Elwakiel A, Kohli S, Ghosh S, Manoharan J, Ranjan S, Bock F, Braun-Dullaeus RC, Esmon CT, Huber TB, Camerer E, Dockendorff C, Griffin JH, Isermann B, Shahzad K. Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood. 2017; 130: 2664–77. 10.1182/blood-2017-05-782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palevski D, Ben-David G, Weinberger Y, Haj Daood R, Fernandez JA, Budnik I, Levy-Mendelovich S, Kenet G, Nisgav Y, Weinberger D, Griffin JH, Livnat T. 3K3A-Activated Protein C Prevents Microglia Activation, Inhibits NLRP3 Inflammasome and Limits Ocular Inflammation. Int J Mol Sci. 2022; 23. 10.3390/ijms232214196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan A, Pan X, Wen X, Nie X, Li Y. Activated protein C overexpression suppresses the pyroptosis of subarachnoid hemorrhage model cells by regulating the NLRP3 inflammasome pathway. Exp Ther Med. 2021; 22: 1391. 10.3892/etm.2021.10827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015; 21: 677–87. 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018; 17: 588–606. 10.1038/nrd.2018.97. [DOI] [PubMed] [Google Scholar]
- 18.Palazon-Riquelme P, Lopez-Castejon G. The inflammasomes, immune guardians at defence barriers. Immunology. 2018; 155: 320–30. 10.1111/imm.12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E. The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol. 2015; 10: 395–424. 10.1146/annurev-pathol-012414-040431. [DOI] [PubMed] [Google Scholar]
- 20.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001; 29: 301–5. 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Libby P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J Am Coll Cardiol. 2017; 70: 2278–89. 10.1016/j.jacc.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Brien M, Moehring D, Munoz-Planillo R, Nunez G, Callaway J, Ting J, Scurria M, Ugo T, Bernad L, Cali J, Lazar D. A bioluminescent caspase-1 activity assay rapidly monitors inflammasome activation in cells. J Immunol Methods. 2017; 447: 1–13. 10.1016/j.jim.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Healy LD, Fernandez JA, Mosnier LO, Griffin JH. Activated protein C and PAR1-derived and PAR3-derived peptides are anti-inflammatory by suppressing macrophage NLRP3 inflammasomes. J Thromb Haemost. 2021; 19: 269–80. 10.1111/jth.15133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee-Rivera I, Lopez E, Lopez-Colome AM. Diversification of PAR signaling through receptor crosstalk. Cell Mol Biol Lett. 2022; 27: 77. 10.1186/s11658-022-00382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo H, Liu D, Gelbard H, Cheng T, Insalaco R, Fernandez JA, Griffin JH, Zlokovic BV. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron. 2004; 41: 563–72. 10.1016/s0896-6273(04)00019-4. [DOI] [PubMed] [Google Scholar]
- 26.Guo H, Singh I, Wang Y, Deane R, Barrett T, Fernandez JA, Chow N, Griffin JH, Zlokovic BV. Neuroprotective activities of activated protein C mutant with reduced anticoagulant activity. Eur J Neurosci. 2009; 29: 1119–30. 10.1111/j.1460-9568.2009.06664.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Guo H, Wang Y, Singh I, Liu D, Fernandez JA, Griffin JH, Chow N, Zlokovic BV. Species-dependent neuroprotection by activated protein C mutants with reduced anticoagulant activity. J Neurochem. 2009; 109: 116–24. 10.1111/j.1471-4159.2009.05921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo H, Zhao Z, Yang Q, Wang M, Bell RD, Wang S, Chow N, Davis TP, Griffin JH, Goldman SA, Zlokovic BV. An activated protein C analog stimulates neuronal production by human neural progenitor cells via a PAR1-PAR3-S1PR1-Akt pathway. J Neurosci. 2013; 33: 6181–90. 10.1523/JNEUROSCI.4491-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madhusudhan T, Wang H, Straub BK, Grone E, Zhou Q, Shahzad K, Muller-Krebs S, Schwenger V, Gerlitz B, Grinnell BW, Griffin JH, Reiser J, Grone HJ, Esmon CT, Nawroth PP, Isermann B. Cytoprotective signaling by activated protein C requires protease-activated receptor-3 in podocytes. Blood. 2012; 119: 874–83. 10.1182/blood-2011-07-365973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinha RK, Wang Y, Zhao Z, Xu X, Burnier L, Gupta N, Fernandez JA, Martin G, Kupriyanov S, Mosnier LO, Zlokovic BV, Griffin JH. PAR1 biased signaling is required for activated protein C in vivo benefits in sepsis and stroke. Blood. 2018; 131: 1163–71. 10.1182/blood-2017-10-810895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Zhao Z, Rege SV, Wang M, Si G, Zhou Y, Wang S, Griffin JH, Goldman SA, Zlokovic BV. 3K3A-activated protein C stimulates postischemic neuronal repair by human neural stem cells in mice. Nat Med. 2016; 22: 1050–5. 10.1038/nm.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazic D, Sagare AP, Nikolakopoulou AM, Griffin JH, Vassar R, Zlokovic BV. 3K3A-activated protein C blocks amyloidogenic BACE1 pathway and improves functional outcome in mice. J Exp Med. 2019; 216: 279–93. 10.1084/jem.20181035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Kisler K, Nikolakopoulou AM, Fernandez JA, Griffin JH, Zlokovic BV. 3K3A-Activated Protein C Protects the Blood-Brain Barrier and Neurons From Accelerated Ischemic Injury Caused by Pericyte Deficiency in Mice. Front Neurosci. 2022; 16: 841916. 10.3389/fnins.2022.841916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren D, Fedorova J, Davitt K, Van Le TN, Griffin JH, Liaw PC, Esmon CT, Rezaie AR, Li J. Activated Protein C Strengthens Cardiac Tolerance to Ischemic Insults in Aging. Circ Res. 2022; 130: 252–72. 10.1161/CIRCRESAHA.121.319044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Livnat T, Weinberger Y, Fernandez JA, Bashir A, Ben-David G, Palevski D, Levy-Mendelovich S, Kenet G, Budnik I, Nisgav Y, Griffin JH, Weinberger D. Activated Protein C (APC) and 3K3A-APC-Induced Regression of Choroidal Neovascularization (CNV) Is Accompanied by Vascular Endothelial Growth Factor (VEGF) Reduction. Biomolecules. 2021; 11. 10.3390/biom11030358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huuskonen MT, Wang Y, Nikolakopoulou AM, Montagne A, Dai Z, Lazic D, Sagare AP, Zhao Z, Fernandez JA, Griffin JH, Zlokovic BV. Protection of ischemic white matter and oligodendrocytes in mice by 3K3A-activated protein C. J Exp Med. 2022; 219. 10.1084/jem.20211372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinberger Y, Budnik I, Nisgav Y, Palevski D, Ben-David G, Fernandez JA, Margalit SN, Levy-Mendelovich S, Kenet G, Weinberger D, Griffin JH, Livnat T. 3K3A-Activated Protein C Inhibits Choroidal Neovascularization Growth and Leakage and Reduces NLRP3 Inflammasome, IL-1beta, and Inflammatory Cell Accumulation in the Retina. Int J Mol Sci. 2023; 24. 10.3390/ijms241310642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandez JA, Xu X, Liu D, Zlokovic BV, Griffin JH. Recombinant murine-activated protein C is neuroprotective in a murine ischemic stroke model. Blood Cells Mol Dis. 2003; 30: 271–6. 10.1016/s1079-9796(03)00034-2. [DOI] [PubMed] [Google Scholar]
- 39.Kerschen E, Hernandez I, Zogg M, Jia S, Hessner MJ, Fernandez JA, Griffin JH, Huettner CS, Castellino FJ, Weiler H. Activated protein C targets CD8+ dendritic cells to reduce the mortality of endotoxemia in mice. J Clin Invest. 2010; 120: 3167–78. 10.1172/JCI42629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kerschen EJ, Fernandez JA, Cooley BC, Yang XV, Sood R, Mosnier LO, Castellino FJ, Mackman N, Griffin JH, Weiler H. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007; 204: 2439–48. 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci U S A. 2007; 104: 5662–7. 10.1073/pnas.0700763104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Majewski MW, Gandhi DM, Rosas R Jr., Kodali R, Arnold LA, Dockendorff C. Design and Evaluation of Heterobivalent PAR1-PAR2 Ligands as Antagonists of Calcium Mobilization. ACS Med Chem Lett. 2019; 10: 121–6. 10.1021/acsmedchemlett.8b00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valant C, Robert Lane J, Sexton PM, Christopoulos A. The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2012; 52: 153–78. 10.1146/annurev-pharmtox-010611-134514. [DOI] [PubMed] [Google Scholar]
- 44.Valant C, Sexton PM, Christopoulos A. Orthosteric/allosteric bitopic ligands: going hybrid at GPCRs. Mol Interv. 2009; 9: 125–35. 10.1124/mi.9.3.6. [DOI] [PubMed] [Google Scholar]
- 45.Han X, Nieman MT. The domino effect triggered by the tethered ligand of the protease activated receptors. Thromb Res. 2020; 196: 87–98. 10.1016/j.thromres.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin H, Liu AP, Smith TH, Trejo J. Cofactoring and dimerization of proteinase-activated receptors. Pharmacol Rev. 2013; 65: 1198–213. 10.1124/pr.111.004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang XV, Banerjee Y, Fernandez JA, Deguchi H, Xu X, Mosnier LO, Urbanus RT, de Groot PG, White-Adams TC, McCarty OJ, Griffin JH. Activated protein C ligation of ApoER2 (LRP8) causes Dab1-dependent signaling in U937 cells. Proc Natl Acad Sci U S A. 2009; 106: 274–9. 10.1073/pnas.0807594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Minhas N, Xue M, Fukudome K, Jackson CJ. Activated protein C utilizes the angiopoietin/Tie2 axis to promote endothelial barrier function. FASEB J. 2010; 24: 873–81. 10.1096/fj.09-134445. [DOI] [PubMed] [Google Scholar]
- 49.Cao C, Gao Y, Li Y, Antalis TM, Castellino FJ, Zhang L. The efficacy of activated protein C in murine endotoxemia is dependent on integrin CD11b. J Clin Invest. 2010; 120: 1971–80. 10.1172/JCI40380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Changeux JP, Christopoulos A. Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell. 2016; 166: 1084–102. 10.1016/j.cell.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 51.Wootten D, Christopoulos A, Marti-Solano M, Babu MM, Sexton PM. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat Rev Mol Cell Biol. 2018; 19: 638–53. 10.1038/s41580-018-0049-3. [DOI] [PubMed] [Google Scholar]
- 52.Kunze G, Isermann B. Targeting biased signaling by PAR1: function and molecular mechanism of parmodulins. Blood. 2023; 141: 2675–84. 10.1182/blood.2023019775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shpakov AO. Allosteric Regulation of G-Protein-Coupled Receptors: From Diversity of Molecular Mechanisms to Multiple Allosteric Sites and Their Ligands. Int J Mol Sci. 2023; 24. 10.3390/ijms24076187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nieman MT. Protease-activated receptors in hemostasis. Blood. 2016; 128: 169–77. 10.1182/blood-2015-11-636472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bae JS, Yang L, Manithody C, Rezaie AR. The ligand occupancy of endothelial protein C receptor switches the protease-activated receptor 1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood. 2007; 110: 3909–16. 10.1182/blood-2007-06-096651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roy RV, Ardeshirylajimi A, Dinarvand P, Yang L, Rezaie AR. Occupancy of human EPCR by protein C induces beta-arrestin-2 biased PAR1 signaling by both APC and thrombin. Blood. 2016; 128: 1884–93. 10.1182/blood-2016-06-720581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Michael E, Covic L, Kuliopulos A. Lipopeptide Pepducins as Therapeutic Agents. Methods Mol Biol. 2022; 2383: 307–33. 10.1007/978-1-0716-1752-6_21. [DOI] [PubMed] [Google Scholar]
- 58.Flaumenhaft R, De Ceunynck K. Targeting PAR1: Now What? Trends Pharmacol Sci. 2017; 38: 701–16. 10.1016/j.tips.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davenport AP, Scully CCG, de Graaf C, Brown AJH, Maguire JJ. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat Rev Drug Discov. 2020; 19: 389–413. 10.1038/s41573-020-0062-z. [DOI] [PubMed] [Google Scholar]
- 60.Di Pizio A, Bermudez M, De Graaf C, Jockers R. Editorial: Peptide-binding GPCRs coming of age. Front Endocrinol (Lausanne). 2023; 14: 1189508. 10.3389/fendo.2023.1189508. [DOI] [PMC free article] [PubMed] [Google Scholar]