

Abstract
Selectivity in radical chain oligomerizations involving [1.1.1]propellane – i.e., to make [n]staffanes – has been notoriously challenging to control when n > 1 is desired. Herein, we report selective syntheses of SF5- and CF3SF4-containing [2]staffanes from SF5Cl and CF3SF4Cl, demonstrating cases whereby oligomerization is preferentially truncated after incorporation of two bicyclopentane (BCP) units. Synthetic and computational studies suggest this phenomenon can be attributed to alternating radical polarity matching. In addition, single-crystal X-ray diffraction (SC-XRD) data reveal structurally interesting features of the CF3SF4-containing [2]staffane in the solid state.
Keywords: pentafluorosulfanylation, [1.1.1]propellane, radical chain oligomerization, staffanes, strain-release
Introduction
In various radical additions of X–Y across [1.1.1]propellane (1), functionalized oligomers known as [n]staffanes – with n > 1, where n denotes the number of individual [1.1.1]bicyclopentane (BCP) linkers – are often observed and swiftly devalorized as side-products [1]. However, targeted synthesis of functionalized [n]staffanes as rigid "molecular spacers" as proposed by Kaszynski and Michl [2–4] could facilitate new developments in nanotechnology [5], liquid crystal design [6–10], and the study of energy-transfer [11–12] or electron-transfer [13–17] processes. We also posit that lower-order [n]staffanes (i.e., n = 2 or 3) are potentially valuable C(sp3)-rich bioisosteres [18–19] that have been seemingly overlooked in the medicinal chemistry arena, in stark contrast to single BCP units over the past 12 years [20–24].
One plausible explanation for the paucity of applications of [n]staffanes in materials or biological settings is a synthetic accessibility issue. For instance, dimerization of substituted BCPs [25–27] or photochemical appendage of 1 onto an extant BCP [28–31] are relatively effective tactics for the selective assembly of certain [n]staffanes; the main caveat is that multiple synthetic steps are required. On the other hand, while a one-pot radical chain oligomerization is conceptually appealing, radical additions of X–Y across 1 in practice can be challenging to control and often lead to complex mixtures of functionalized [n]staffanes, n = 1–5 (Figure 1, top) [2,31–34]. Even though [n]staffanes are often separable by column chromatography, the yields for a single oligomer across a panoply of different transformations typically range from <1% to ≈30% when n > 1 is desired [35]. To the best of our knowledge, the assembly of functionalized [n]staffanes from 1 in high yield/selectivity and in one step via controlled radical oligomerization remains a synthetic challenge.
Figure 1.

(Top) highlighting selectivity challenges in the synthesis of [n]staffanes using excess [1.1.1]propellane (1). (Bottom) selective synthesis of [2]staffanes bearing the SF5 (2) and CF3SF4 (3) groups (this work).
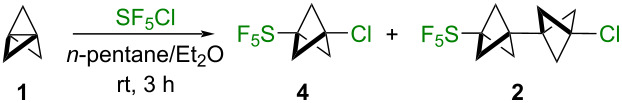
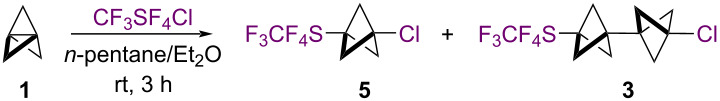
Herein, we report proof-of-concept that our previous work on strain-release pentafluorosulfanylation of 1 [36] using SF5Cl (prepared in house [37] under mild oxidative fluorination conditions [38–44]) can be extended to the selective synthesis of the associated chloropentafluorosulfanylated [2]staffane (SF5-BCP-BCP-Cl, 2), based on alternating radical polarity matching in the chain-propagation steps (Figure 1, bottom) [45–47]. Density functional theory (DFT) calculations provide insight into our observed selectivity, and our hypothesis is bolstered by computation of relative bicyclopentyl radical philicities. In addition, we demonstrate that similar reaction conditions can be applied to the synthesis of the analogous CF3SF4-containing [2]staffane (CF3SF4-BCP-BCP-Cl, 3). Finally, we examined compound 3 by SC-XRD and found that it undergoes a phase transition as a function of rate of cooling; this highlights that the [2]staffanes synthesized during this study are also interesting from a fundamental structural standpoint.
Results and Discussion
Over the past few years, our group has begun to establish strain-release pentafluorosulfanylation as a viable strategy for C(sp3)–SF5 bond formation [35,48]. For instance, in 2022, we reported a method for chloropentafluorosulfanylation of [1.1.1]propellane, i.e., to make SF5-BCP-Cl (4), that ostensibly proceeds through a radical chain propagation mechanism [36]. Under optimized conditions, we obtained product 4 in 86% yield, and the corresponding [2]staffane – SF5-BCP-BCP-Cl (2) – was formed as a minor side-product in 7% yield. While our original goal was to suppress formation of 2, we later pondered whether preferential synthesis of compound 2 would also be possible. Accordingly, we began our screening process by systematically increasing the equivalents of [1.1.1]propellane (1) relative to SF5Cl and evaluating the impact on selectivity (Table 1).
Table 1.
Effect of [1.1.1]propellane (1) equivalents relative to SF5Cl on selectivitya.
| ||||
|
| ||||
| entry | 1 (equiv) | 4 (% yield)b | 2 (% yield)b | 4:2 |
|
| ||||
| 1 | 1.0 | 49% | 4% | 12:1.0 |
| 2 | 2.0 | 74% | 22% | 3.4:1.0 |
| 3 | 3.0 | 44% | 29% | 1.5:1.0 |
| 4 | 4.0 | 54% | 40% | 1.3:1.0 |
| 5 | 6.0 | 48% | 45% | 1.1:1.0 |
| 6 | 8.0 | 43% | 53% | 1.0:1.3 |
| 7 | 10 | 32% | 51% | 1.0:1.6 |
| 8 | 20 | 24% | 72% | 1.0:3.0 |
| 9 c | 6.0 | 30% | 63% (51%) d | 1.0:2.1 |
aA 0.1 M solution of SF5Cl in n-pentane (0.1 mmol) was added to a 0.8 M solution of [1.1.1]propellane in Et2O under Ar atmosphere and stirred at rt for 3 h. bYield determined by 19F NMR; cSF5Cl was added portion-wise. dIsolated yield.
A 0.1 M solution of SF5Cl in n-pentane was added to a 0.8 M solution of 1 in Et2O at room temperature, and the mixture was stirred for 3 hours prior to 19F NMR analysis. Upon increasing from 1.0 to 6.0 equiv of 1, we observed the 4:2 product ratio decreased dramatically from 12:1 to 1.1:1. Using 8.0 equiv of 1, the ratio flipped such that 2 became the major product (i.e., 4:2 = 1:1.3) and was formed in 53% yield. Interestingly, even with 8.0 equiv of 1, 96% of the material balance could be accounted for in the formation of these two products alone by 19F NMR. To the best of our knowledge, this is an exceptionally rare instance whereby oligomerization of 1 appears to be stunted beyond formation of the [2]staffane; only trace yields of putative higher-order staffanes (n > 2) were detected in the crude reaction mixture. Remarkably, using 20 equiv of 1, the observed 4:2 product ratio improved to 1:3, with 2 formed in 72% yield. In an even more extreme case, the [2]staffane still remained the major product formed in 66% yield when using 50 equiv of 1 (19F NMR signals of presumed higher-order staffanes became more apparent, though their combined yield still remained ≈18%).
Ultimately, we found adding SF5Cl portion-wise – to keep the "effective" equiv of 1 higher at any given moment – proved to be a suitable compromise to access 2 in 63% yield by 19F NMR (51% isolated) using 6.0 equiv of 1 (Table 1). We also demonstrated that this procedure can be performed on a 4.0 mmol scale (with respect to SF5Cl) to provide access to ≈0.5 g of 2 in 43% isolated yield. Additional details on reaction optimization can be found in Supporting Information File 1.
Upon increasing the equivalents of 1 during the screening process, we also found that irradiation with white LEDs was not necessary to boost product yields, as it was in our previously reported synthesis of 4 [36]. Both our laboratory [36] and the Qing laboratory [49] have previously observed that SF5Cl additions to 1 can proceed in the absence of light. Note that recent work from the laboratories of Cahard and Bizet [50] suggests that autoxidation of the ethereal solvent could serve as one possible explanation for initial formation of SF5 radicals in the absence of light to initiate a radical chain reaction. It is also well established that [1.1.1]propellane participates in radical-chain reactions (i.e., oligomerization) at room temperature in solution to form unsubstituted [n]staffanes.
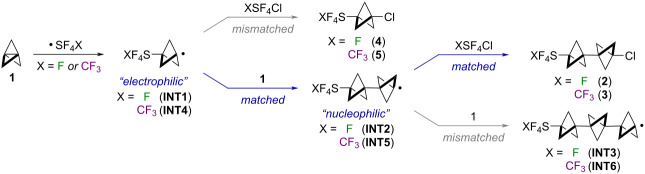
The origin of this innately controlled oligomerization was then investigated through density functional theory (DFT) calculations. The free energy profile of the radical chain propagation sequence was computed at the PWPB95-D4/def2-QZVPP//PCM(Et2O)-ωB97X-D/def2-TZVP level of theory [51–58] (Figure 2). Following addition of an SF5 radical to 1 to form INT1, a Cl atom could be abstracted from SF5Cl via TS1 to form 4 or, alternatively, INT1 could be added to another equiv of 1 via TS2 to form INT2. Although formation of 4 is notably more thermodynamically favorable than INT2 (ΔΔG = −9.2 kcal/mol), a small difference in activation free energy is predicted (ΔΔG‡ = −1.4 kcal/mol). This, at least in part, provides an explanation as to how favoring the path to INT2 may be achieved in practice through increasing concentration of 1.
Figure 2.

Computed free energy profile for the oligomerization of [1.1.1]propellane (1) following SF5 radical addition at PWPB95-D4/def2-QZVPP//PCM(Et2O)-ωB97X-D/def2-TZVP level of theory.
Subsequently, INT2 could abstract a Cl atom from SF5Cl via TS3 to form 2 or add to a third equiv of 1 through TS4, leading to INT3. Once again, Cl atom abstraction is thermodynamically (ΔΔG = −11.7 kcal/mol) and kinetically (ΔΔG‡ = −2.8 kcal/mol) favored. However, the ΔΔG‡ is notably greater in the second product-determining step than the first product-determining step, which is consistent with our experimental observation that 2 forms preferentially over further oligomerization.
For another point of comparison, we examined the reactivity of 1 with CF3SF4Cl. This reagent is known to behave comparably to SF5Cl in radical chain reactions [17,59–61] and can also be prepared conveniently in house [62]. In an analogous equivalents screen, we found that the 5:3 product ratio shifts from 7.7:1 using 1.0 equiv of 1 to 1:2.1 using 20 equiv of 1 (Table 2). In the latter scenario, 96% of the material balance could be accounted for in the formation of 5 and 3, indicating oligomerization is likewise stunted beyond incorporation of two BCP units. Also similar to pentafluorosulfanylation conditions, we found that adding CF3SF4Cl portion-wise to 6.0 equiv of 1 enables access to 3 in 60% yield by 19F NMR (53% isolated). Interestingly, we observed that aryl-SF4Cl compounds do not follow the same selectivity trend as SF5Cl and CF3SF4Cl additions, suggesting that the controlled oligomerization phenomenon is quite sensitive to changes in the fluorinated sulfur reagent scaffold (see Supporting Information File 1 for more details).
Table 2.
Effect of [1.1.1]propellane (1) equivalents relative to CF3SF4Cl on selectivity.a
| ||||
|
| ||||
| entry | 1 (equiv) | 5 (% yield)b | 3 (% yield)b | 5:3 |
|
| ||||
| 1 | 1.0 | 80% | 10% | 7.7:1.0 |
| 2 | 2.0 | 71% | 21% | 3.4:1.0 |
| 3 | 3.0 | 65% | 31% | 2.1:1.0 |
| 4 | 4.0 | 55% | 40% | 1.4:1.0 |
| 5 | 6.0 | 49% | 40% | 1.2:1.0 |
| 6 | 8.0 | 39% | 46% | 1.0:1.2 |
| 7 | 10 | 33% | 62% | 1.0:1.9 |
| 8 | 20 | 31% | 65% | 1.0:2.1 |
| 9 c | 6.0 | 35% | 60% (53%) d | 1.0:1.7 |
aA 0.1 M solution of CF3SF4Cl in n-pentane (0.03 mmol) was added to a 0.8 M solution of [1.1.1]propellane in Et2O under Ar atmosphere and stirred at rt for 3 h. bYield determined by 19F NMR. cCF3SF4Cl was added portion-wise. dIsolated yield.
This second instance of controlled oligomerization of 1 using CF3SF4Cl was also studied at the PWPB95-D4/def2-QZVPP//PCM(Et2O)-ωB97X-D/def2-TZVP level of theory (Figure 3). Addition of a CF3SF4 radical to 1 affords INT4, which can either abstract a Cl atom from CF3SF4Cl via TS5 to make 5 or add to another equiv of 1 via TS6 to access INT5. As anticipated, chlorination of the radical is thermodynamically favored over addition to 1 (ΔΔG = −10.7 kcal/mol). It is also predicted here that the free energy of activation is lower for chlorination, albeit only by 0.4 kcal/mol. This is consistent with the notion that the kinetic preference can be overcome by increasing the concentration of 1 relative to CF3SF4Cl. In the second product-determining step, Cl atom abstraction by INT5 via TS7 to make 3 is kinetically favorable over addition of a third equiv of 1 via TS8 to access INT6, although the preference is not as large as for formation of 2 (ΔΔG‡ = −1.3 kcal/mol).
Figure 3.

Computed free energy profile for the oligomerization of [1.1.1]propellane (1) following CF3SF4 radical addition at the PWPB95-D4/def2-QZVPP//PCM(Et2O)-ωB97X-D/def2-TZVP level of theory.
Thus, the predicted trend for both SF5Cl and CF3SF4Cl additions across 1 indicates a stronger preference for Cl atom abstraction over continued oligomerization in the second product-determining step than in the first – in line with our experimental observations. One possible explanation for this phenomenon is rooted in better radical polarity matching after incorporation of the second BCP unit [37–38]. That is, the carbon-centered radicals in both INT1 and INT4 are closer to strong electron-withdrawing groups than are the radical centers in INT2 and INT5, rendering INT1 and INT4 relatively more electrophilic. Inductive effects drop off steeply with distance, and it is also established that a substituent (or, e.g., a radical or cation) on the transannular carbon atom of a bicyclopentyl moiety can interact through space [35,63–64]. The consequence is ostensibly that more "nucleophilic" INT2 and INT5 are better matched for Cl atom abstraction from the "electrophilic" reagent (SF5Cl or CF3SF4Cl).
To test this hypothesis, we examined computed trends in various electronic parameters for the INT1–INT3 and the INT4–INT6 series (Table 3). For instance, across several charge models (i.e., Hirshfeld [65], NPA [66–68], and CHELPG [69]), the charge (q) on the carbon atom on which the radical is centered becomes more negative (or less positive) the farther it is from either the SF5 or CF3SF4 substituent, consistent with the notion that it becomes more nucleophilic. Moreover, the Δq is largest between the first two intermediates in both series – INT1 vs INT2 and INT3 vs INT4 – indicating that the most dramatic change in bicyclopentyl radical philicity would arise after incorporation of the second BCP unit.
Table 3.
Key indices computed to compare reactivity. Partial charges (q) and condensed Fukui functions are evaluated at the reacting carbon or chlorine atom and are in units of elementary charge (e).a
| ||||||
|
| ||||||
| compound | q(Hirshfled) (e) |
q(NPA) (e) |
q(CHELPG) (e) |
f 0 (e)b | ω (eV)c | N (eV)d |
|
| ||||||
| 1 | −0.090 | −0.069 | −0.255 | 0.235 | 0.82 | 1.70 |
| SF5Cl | −0.055 | −0.160 | 0.030 | 0.534 | 2.48 | −0.74 |
| CF3SF4Cl | −0.061 | −0.150 | 0.056 | 0.527 | 2.52 | −0.66 |
| INT1 | −0.019 | 0.092 | −0.125 | 0.307 | 2.70 | 2.95 |
| INT2 | −0.053 | 0.073 | −0.196 | 0.347 | 1.78 | 3.58 |
| INT3 | −0.060 | 0.065 | −0.214 | 0.345 | 1.66 | 3.77 |
| INT4 | −0.019 | 0.092 | −0.144 | 0.301 | 2.74 | 2.97 |
| INT5 | −0.053 | 0.073 | −0.193 | 0.346 | 1.78 | 3.59 |
| INT6 | −0.060 | 0.064 | −0.214 | 0.345 | 1.65 | 3.77 |
aCalculations performed at the PCM(Et2O)-ωB97X-D/def2-TZVP level of theory. bRadical Fukui function. cElectrophilicity index. dNucleophilicity index.
In addition to charge models, we evaluated global reactivity indices (ω: electrophilicity index [70] and N: nucleophilicity index [71]) within the conceptual density functional theory (CDFT) framework [72–74]. The data show that BCP has a higher N value – thus stronger nucleophilic tendency – compared to both SF5Cl and CF3SF4Cl. Conversely, comparison of ω values shows significantly higher electrophilicity of SF5Cl and CF3SF4Cl compared to BCP. These results, coupled with decreasing ω and increasing N when more BCP units are incorporated, lend qualitative support to our radical polarity matching hypothesis. Moreover, assessment of radical Fukui functions (f0) [75] indicates that both SF5Cl and CF3SF4Cl are intrinsically more susceptible to radical attack than 1, which is consistent with the lower computed barriers for Cl atom abstraction in each case.
These computed trends also potentially account for the fact that selectivity for the [2]staffane (i.e., truncated oligomerization) using aryl-SF4Cl reagents was not observed. On the basis of CDFT results, a model aryl-SF4Cl compound (i.e., 5-chloropyrimidyl-SF4Cl) was determined to be significantly less "electrophilic" than SF5Cl or CF3SF4Cl, consistent with a reduction in the radical polarity matching effect (see Supporting Information File 1 for details). This was difficult to predict or rationalize based on calculation of free energies of activation alone. Overall, our results suggest that this alternating polarity matching effect is subtle and subject to mitigation yet can lead to desirable products if employed thoughtfully.
Lastly, following our synthetic and computational studies, accessing a CF3SF4-containing [2]staffane in good yield and for the first time created an opportunity for structural analysis. We previously reported and contextualized single-crystal X-ray diffraction (SC-XRD) data on 2 [36]; thus, we proceeded to grow crystals of 3 suitable for X-ray analysis through slow evaporation of ethyl acetate.
To our surprise, an initial measurement of 3 at 90 K revealed an unusually large unit cell (a = 7.14 Å, b = 21.38 Å, and c = 44.04 Å). Following structure solution and refinement [76], we found that 3 crystallizes in an orthorhombic space group P212121 with five symmetry independent moieties (Z' = 5) and with no solvent present in the unit cell as an inversion twin (Figure 4). After close examination of the model, we noticed that the c-axis was roughly divisible by five with a substructure of Z' = 1. This suggested that the Z' = 5 unit cell may be due to a phase transition caused by anisotropic contraction [77].
Figure 4.

(A) The molecular structure of 3 at 90 K with 5 independent moieties in the asymmetric axis viewed along the b-axis. (B) The asymmetric unit of 3 at 90 K viewed along the c-axis. Thermal displacement ellipsoids depicted at 50% probability.
In fact, we confirmed that a phase transition had occurred following structure determination at 240 K [78–79]. The X-ray data revealed that 3 crystallizes in the centrosymmetric orthorhombic space group Pnma in the high temperature phase (HTP) with cell axes a = 21.56 Å, b = 8.95 Å, and c = 7.28 Å, in contrast to P212121 in the low temperature phase (LTP). Note that the b-axis is roughly 1/5 of the c-axis observed at 90 K (the axial rearrangement is due to the change in space group). To discern the approximate temperature of the phase transition, the unit cell was measured in 20 K increments upon cooling from 260 K down to 100 K; additional details are reported in Supporting Information File 1. Interestingly, the original LTP unit cell was not detected; instead, only the reduced cell observed in the HTP was found at all temperatures. However, after warming the same crystal of 3 back to room temperature, it was rapidly cooled to 100 K under a stream of N2 and the larger, disordered cell was observed once more [80]. (These observations also prompted us to measure a structure of 2 at 240 K; in this case, the unit cell is virtually identical at both high and low temperatures, indicating no phase transition had occurred – see Supporting Information File 1 for details.)
Accordingly, we gather that the rate of cooling plays an important role whereby rapid cooling effectively "shocks" the crystal of 3, compressing the unit cell isotropically, and ultimately leads to more disorder in the asymmetric unit [81]. This unexpected observation suggests that CF3SF4-containing [2]staffanes, in particular, warrant additional studies and may be of interest, e.g., in liquid crystal design.
Conclusion
Suppressing [n]staffane formation beyond n = 1 in radical chain reactions involving [1.1.1]propellane (1) tends to be more manageable than controlling oligomerization. However, under the right circumstances, alternating radical polarity matching throughout the chain propagation steps could be one way to theoretically "switch off" oligomerization beyond formation of a [2]staffane. Using this logic, our synthetic and computational study demonstrates that selective one-pot syntheses of [2]staffanes can be achieved when employing reagents that serve as radical sources of "extreme" electron-withdrawing groups (e.g., SF5 or CF3SF4), which impact relative philicities of the bicyclopentyl radical intermediates. Over the course of this study, we also found that the SF5- and CF3SF4-containing [2]staffanes reported herein are structurally interesting in their own right. Future work will examine potential applications of 2 and 3 and explore tactics for C–Cl bond functionalization.
Supporting Information
Experimental procedures, characterization data, NMR spectra, computational details, and X-ray crystallographic experimental details.
LTP X-ray crystal structure of compound 3 (2357079.cif), HTP X-ray crystal structure of compound 3 (2357080.cif) and X-ray crystal structure of compound 2 at 240 K (238115.cif).
Acknowledgments
C.R.P. and Y.K. thank Dr. Nils Trapp (ETH Zürich) for helpful discussions. The authors also thank the UC Davis core NMR facility.
This article is part of the thematic issue "Organofluorine chemistry VI" and is dedicated to Professor Josef Michl who recently passed away.
Funding Statement
C.R.P. thanks the NIH/NIGMS (R35GM150861) and the University of California, Davis for financial support. W.Y.K thanks the Croucher Foundation for financial support through a doctoral scholarship. W.Y.K. and D.J.T. thank the NSF ACCESS program (CHE030089) for computational support. The authors also thank the NSF (CHE1531193) for the Dual Source X-ray Diffractometer.
Contributor Information
Dean J Tantillo, Email: djtantillo@ucdavis.edu.
Cody Ross Pitts, Email: crpitts@ucdavis.edu.
Data Availability
All experimental data that supports the findings of this study are available in the published article and/or the supporting information to this article; coordinates for computed structures are openly available in ioChem-BD at https://doi.org/10.19061/iochem-bd-6-384.
References
- 1.Shire B R, Anderson E A. JACS Au. 2023;3:1539–1553. doi: 10.1021/jacsau.3c00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaszynski P, Friedli A C, Michl J. J Am Chem Soc. 1992;114:601–620. doi: 10.1021/ja00028a029. [DOI] [Google Scholar]
- 3.Levin M D, Kaszynski P, Michl J. Chem Rev. 2000;100:169–234. doi: 10.1021/cr990094z. [DOI] [PubMed] [Google Scholar]
- 4.Dilmaç A M, Spuling E, de Meijere A, Bräse S. Angew Chem, Int Ed. 2017;56(21):5684–5718. doi: 10.1002/anie.201603951. [DOI] [PubMed] [Google Scholar]
- 5.Kaszynski P, Michl J. J Am Chem Soc. 1988;110:5225–5226. doi: 10.1021/ja00223a070. [DOI] [Google Scholar]
- 6.Friedli A C, Lynch V M, Kaszynski P, Michl J. Acta Crystallogr, Sect A: Found Crystallogr. 1990;46:377–389. doi: 10.1107/s0108768189014096. [DOI] [Google Scholar]
- 7.Kaszynski P, Friedli A C, McMurdie N D, Michl J. Mol Cryst Liq Cryst (1969-1991) 1990;191:193–197. doi: 10.1080/00268949008038593. [DOI] [Google Scholar]
- 8.Friberg S E, Kayali I, Kaszynski P, Michl J. Langmuir. 1992;8:996–998. doi: 10.1021/la00039a041. [DOI] [Google Scholar]
- 9.Janecki T, Shi S, Kaszynski P, Michl J. Collect Czech Chem Commun. 1993;58:89–104. doi: 10.1135/cccc19930089. [DOI] [Google Scholar]
- 10.Messner M, Kozhushkov S I, de Meijere A. Eur J Org Chem. 2000;(7):1137–1155. doi: 10.1002/1099-0690(200004)2000:7<1137::aid-ejoc1137>3.0.co;2-2. [DOI] [Google Scholar]
- 11.Zimmerman H E, Goldman T D, Hirzel T K, Schmidt S P. J Org Chem. 1980;45:3933–3951. doi: 10.1021/jo01308a001. [DOI] [Google Scholar]
- 12.Obeng Y S, Laing M E, Friedli A C, Yang H C, Wang D, Thulstrup E W, Bard A J, Michl J. J Am Chem Soc. 1992;114:9943–9952. doi: 10.1021/ja00051a029. [DOI] [Google Scholar]
- 13.Joran A D, Leland B A, Geller G G, Hopfield J J, Dervan P B. J Am Chem Soc. 1984;106:6090–6092. doi: 10.1021/ja00332a062. [DOI] [Google Scholar]
- 14.Leland B A, Joran A D, Felker P M, Hopfield J J, Zewail A H, Dervan P B. J Phys Chem. 1985;89:5571–5573. doi: 10.1021/j100272a002. [DOI] [Google Scholar]
- 15.Murthy G S, Hassenrück K, Lynch V M, Michl J. J Am Chem Soc. 1989;111:7262–7264. doi: 10.1021/ja00200a057. [DOI] [Google Scholar]
- 16.Yang H C, Magnera T F, Lee C, Bard A J, Michl J. Langmuir. 1992;8(11):2740–2746. doi: 10.1021/la00047a026. [DOI] [Google Scholar]
- 17.Mazières S, Raymond M K, Raabe G, Prodi A, Michl J. J Am Chem Soc. 1997;119:6682–6683. doi: 10.1021/ja971059h. [DOI] [Google Scholar]
- 18.Lovering F, Bikker J, Humblet C. J Med Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 19.Tsien J, Hu C, Merchant R R, Qin T. Nat Rev Chem. 2024;8:605–627. doi: 10.1038/s41570-024-00623-0. [DOI] [PubMed] [Google Scholar]
- 20.Stepan A F, Subramanyam C, Efremov I V, Dutra J K, O’Sullivan T J, DiRico K J, McDonald W S, Won A, Dorff P H, Nolan C E, et al. J Med Chem. 2012;55(7):3414–3424. doi: 10.1021/jm300094u. [DOI] [PubMed] [Google Scholar]
- 21.Locke G M, Bernhard S S R, Senge M O. Chem – Eur J. 2019;25:4590–4647. doi: 10.1002/chem.201804225. [DOI] [PubMed] [Google Scholar]
- 22.Mykhailiuk P K. Org Biomol Chem. 2019;17:2839–2849. doi: 10.1039/c8ob02812e. [DOI] [PubMed] [Google Scholar]
- 23.Tse E G, Houston S D, Williams C M, Savage G P, Rendina L M, Hallyburton I, Anderson M, Sharma R, Walker G S, Obach R S, et al. J Med Chem. 2020;63:11585–11601. doi: 10.1021/acs.jmedchem.0c00746. [DOI] [PubMed] [Google Scholar]
- 24.Cuadros S, Goti G, Barison G, Raulli A, Bortolato T, Pelosi G, Costa P, Dell'Amico L. Angew Chem, Int Ed. 2023;62:e202303585. doi: 10.1002/anie.202303585. [DOI] [PubMed] [Google Scholar]
- 25.Rehm J D D, Ziemer B, Szeimies G. Eur J Org Chem. 2001:1049–1052. doi: 10.1002/1099-0690(200103)2001:6<1049::aid-ejoc1049>3.0.co;2-v. [DOI] [Google Scholar]
- 26.Mazal C, Paraskos A J, Michl J. J Org Chem. 1998;63:2116–2119. doi: 10.1021/jo971419j. [DOI] [Google Scholar]
- 27.Bunz U, Szeimies G. Tetrahedron Lett. 1990;31:651–652. doi: 10.1016/s0040-4039(00)94592-1. [DOI] [Google Scholar]
- 28.Blokhin A V, Tyurekhodzhaeva M A, Sadovaya N K, Zefirov N S. Russ Chem Bull. 1989;38:1779. doi: 10.1007/bf00956980. [DOI] [Google Scholar]
- 29.Sadovaya N K, Blokhin A V, Tyurekhodzhaeva M A, Grishin Y K, Surmina L S, Koz'min A S, Zefirov N S. Russ Chem Bull. 1990;39(3):637–638. doi: 10.1007/bf00959608. [DOI] [Google Scholar]
- 30.Cheng X-Y, Du F-S, Li Z-C. J Polym Sci (Hoboken, NJ, U S) 2023;61:472–481. doi: 10.1002/pol.20220635. [DOI] [Google Scholar]
- 31.Bunz U, Szeimies G. Tetrahedron Lett. 1989;30(16):2087–2088. doi: 10.1016/s0040-4039(01)93718-9. [DOI] [Google Scholar]
- 32.Bunz U, Polborn K, Wagner H-U, Szeimies G. Chem Ber. 1988;121:1785–1790. doi: 10.1002/cber.19881211014. [DOI] [Google Scholar]
- 33.Sadovaya N K, Blokhin A V, Surmina L S, Tyurekhodzhaeva M A, Koz'min A S, Zefirov N S. Russ Chem Bull. 1990;39(10):2224. doi: 10.1007/bf01557749. [DOI] [Google Scholar]
- 34.Wiberg K B, Waddell S T. J Am Chem Soc. 1990;112:2194–2216. doi: 10.1021/ja00162a022. [DOI] [Google Scholar]
- 35.Friedli A C, Kaszynski P, Michl J. Tetrahedron Lett. 1989;30:455–458. doi: 10.1016/s0040-4039(00)95226-2. [DOI] [Google Scholar]
- 36.Kraemer Y, Ghiazza C, Ragan A N, Ni S, Lutz S, Neumann E K, Fettinger J C, Nöthling N, Goddard R, Cornella J, et al. Angew Chem, Int Ed. 2022;61:e202211892. doi: 10.1002/anie.202211892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shou J-Y, Xu X-H, Qing F-L. Angew Chem, Int Ed. 2021;60(28):15271–15275. doi: 10.1002/anie.202103606. [DOI] [PubMed] [Google Scholar]
- 38.Pitts C R, Santschi N, Togni A, inventors. Method For Preparing a Polyfluorinated Compound. WO/2019/229103. WO Patent. 2019 Dec 5;
- 39.Pitts C R, Bornemann D, Liebing P, Santschi N, Togni A. Angew Chem, Int Ed. 2019;58:1950–1954. doi: 10.1002/anie.201812356. [DOI] [PubMed] [Google Scholar]
- 40.Häfliger J, Pitts C R, Bornemann D, Käser R, Santschi N, Charpentier J, Otth E, Trapp N, Verel R, Lüthi H P, et al. Chem Sci. 2019;10:7251–7259. doi: 10.1039/c9sc02162k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bornemann D, Pitts C R, Ziegler C J, Pietrasiak E, Trapp N, Kueng S, Santschi N, Togni A. Angew Chem, Int Ed. 2019;58(36):12604–12608. doi: 10.1002/anie.201907359. [DOI] [PubMed] [Google Scholar]
- 42.Brüning F, Pitts C R, Kalim J, Bornemann D, Ghiazza C, de Montmollin J, Trapp N, Billard T, Togni A. Angew Chem, Int Ed. 2019;58(52):18937–18941. doi: 10.1002/anie.201910594. [DOI] [PubMed] [Google Scholar]
- 43.Ragan A N, Kraemer Y, Kong W-Y, Prasad S, Tantillo D J, Pitts C R. Angew Chem, Int Ed. 2022;61:e202208046. doi: 10.1002/anie.202208046. [DOI] [PubMed] [Google Scholar]
- 44.Kraemer Y, Bergman E N, Togni A, Pitts C R. Angew Chem, Int Ed. 2022;61:e202205088. doi: 10.1002/anie.202205088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tyurekhodzhaeva M A, Bratkova A A, Blokhin A V, Brel V K, Koz'min A S, Zefirov N S. J Fluorine Chem. 1991;55(3):237–240. doi: 10.1016/s0022-1139(00)82351-9. [DOI] [Google Scholar]
- 46.De Vleeschouwer F, Van Speybroeck V, Waroquier M, Geerlings P, De Proft F. Org Lett. 2007;9(14):2721–2724. doi: 10.1021/ol071038k. [DOI] [PubMed] [Google Scholar]
- 47.Domingo L R, Pérez P. Org Biomol Chem. 2013;11:4350–4358. doi: 10.1039/c3ob40337h. [DOI] [PubMed] [Google Scholar]
- 48.Kraemer Y, Buldt J A, Kong W-Y, Stephens A M, Ragan A N, Park S, Haidar Z C, Patel A H, Shey R, Dagan R, et al. Angew Chem, Int Ed. 2024;63:e202319930. doi: 10.1002/anie.202319930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao X, Shou J-Y, Qing F-L. Sci China: Chem. 2023;66:2871–2877. doi: 10.1007/s11426-023-1715-2. [DOI] [Google Scholar]
- 50.Nguyen T M, Popek L, Matchavariani D, Blanchard N, Bizet V, Cahard D. Org Lett. 2024;26:365–369. doi: 10.1021/acs.orglett.3c04043. [DOI] [PubMed] [Google Scholar]
- 51.Goerigk L, Grimme S. J Chem Theory Comput. 2011;7:291–309. doi: 10.1021/ct100466k. [DOI] [PubMed] [Google Scholar]
- 52.Caldeweyher E, Bannwarth C, Grimme S. J Chem Phys. 2017;147:034112. doi: 10.1063/1.4993215. [DOI] [PubMed] [Google Scholar]
- 53.Caldeweyher E, Ehlert S, Hansen A, Neugebauer H, Spicher S, Bannwarth C, Grimme S. J Chem Phys. 2019;150:154122. doi: 10.1063/1.5090222. [DOI] [PubMed] [Google Scholar]
- 54.Weigend F, Ahlrichs R. Phys Chem Chem Phys. 2005;7:3297–3305. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 55.Cancès E, Mennucci B, Tomasi J. J Chem Phys. 1997;107:3032–3041. doi: 10.1063/1.474659. [DOI] [Google Scholar]
- 56.Chai J-D, Head-Gordon M. Phys Chem Chem Phys. 2008;10:6615–6620. doi: 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- 57.Neese F. Wiley Interdiscip Rev: Comput Mol Sci. 2022;12:e1606. doi: 10.1002/wcms.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gaussian 16. Wallingford, CT: Gaussian, Inc.; 2016. [Google Scholar]
- 59.Ikeda A, Zhong L, Savoie P R, von Hahmann C N, Zheng W, Welch J T. Eur J Org Chem. 2018:772–780. doi: 10.1002/ejoc.201701568. [DOI] [Google Scholar]
- 60.Ikeda A, Capellan A, Welch J T. Org Biomol Chem. 2019;17:8079–8082. doi: 10.1039/c9ob01797f. [DOI] [PubMed] [Google Scholar]
- 61.Deng M, Wilde M, Welch J T. J Org Chem. 2023;88:11363–11366. doi: 10.1021/acs.joc.3c01177. [DOI] [PubMed] [Google Scholar]
- 62.Zhao X, Shou J-Y, Newton J J, Qing F-L. Org Lett. 2022;24:8412–8416. doi: 10.1021/acs.orglett.2c03540. [DOI] [PubMed] [Google Scholar]
- 63.Wu J I-C, Schleyer P v R. Pure Appl Chem. 2013;85(5):921–940. doi: 10.1351/pac-con-13-01-03. [DOI] [Google Scholar]
- 64.Sterling A J, Smith R C, Anderson E A, Duarte F. J Org Chem. 2024;89:9979–9989. doi: 10.1021/acs.joc.4c00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hirshfeld F L. Theor Chim Acta. 1977;44:129–138. doi: 10.1007/bf00549096. [DOI] [Google Scholar]
- 66.Reed A E, Weinstock R B, Weinhold F. J Chem Phys. 1985;83:735–746. doi: 10.1063/1.449486. [DOI] [Google Scholar]
- 67.NBO 7.0. Madison, WI, USA: Theoretical Chemistry Institute, University of Wisconsin; 2018. [Google Scholar]
- 68.Glendening E D, Landis C R, Weinhold F. J Comput Chem. 2019;40:2234–2241. doi: 10.1002/jcc.25873. [DOI] [PubMed] [Google Scholar]
- 69.Breneman C M, Wiberg K B. J Comput Chem. 1990;11:361–373. doi: 10.1002/jcc.540110311. [DOI] [Google Scholar]
- 70.Parr R G, Szentpály L v, Liu S. J Am Chem Soc. 1999;121(9):1922–1924. doi: 10.1021/ja983494x. [DOI] [Google Scholar]
- 71.Domingo L R, Chamorro E, Pérez P. J Org Chem. 2008;73:4615–4624. doi: 10.1021/jo800572a. [DOI] [PubMed] [Google Scholar]
- 72.Lu T, Chen Q. Realization of Conceptual Density Functional Theory and Information‐Theoretic Approach in Multiwfn Program. In: Liu S, editor. Conceptual Density Functional Theory. Vol. 2. Weinheim, Germany: Wiley-VCH; 2022. pp. 631–647. [DOI] [Google Scholar]
- 73.Geerlings P, De Proft F, Langenaeker W. Chem Rev. 2003;103:1793–1874. doi: 10.1021/cr990029p. [DOI] [PubMed] [Google Scholar]
- 74.Lu T, Chen F. J Comput Chem. 2012;33:580–592. doi: 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- 75.Parr R G, Yang W. J Am Chem Soc. 1984;106:4049–4050. doi: 10.1021/ja00326a036. [DOI] [Google Scholar]
- 76.Sheldrick G M. Acta Crystallogr, Sect C: Struct Chem. 2015;71(1):3–8. doi: 10.1107/s2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stephenson G A. J Pharm Sci. 2006;95:821–827. doi: 10.1002/jps.20442. [DOI] [PubMed] [Google Scholar]
- 78.Chen L-Z, Huang D-D, Pan Q-J, Ge J-Z. RSC Adv. 2015;5:13488–13494. doi: 10.1039/c4ra12690d. [DOI] [Google Scholar]
- 79.Tello M J, Lopez-Echarri A, Zubillaga J, Ruiz-Larrea I, Zuniga F J, Madariaga G, Gomez-Cuevas A. J Phys: Condens Matter. 1994;6(34):6751–6760. doi: 10.1088/0953-8984/6/34/007. [DOI] [Google Scholar]
- 80.Izyumov Y A, Syromyatnikov V N. Phase Transitions and Crystal Symmetry. Dordrecht, Netherlands: Kluwer Academic Publishers; 1990. [DOI] [Google Scholar]
- 81.Chattoraj S, Sun C C. Crystals. 2023;13:270. doi: 10.3390/cryst13020270. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, characterization data, NMR spectra, computational details, and X-ray crystallographic experimental details.
LTP X-ray crystal structure of compound 3 (2357079.cif), HTP X-ray crystal structure of compound 3 (2357080.cif) and X-ray crystal structure of compound 2 at 240 K (238115.cif).
Data Availability Statement
All experimental data that supports the findings of this study are available in the published article and/or the supporting information to this article; coordinates for computed structures are openly available in ioChem-BD at https://doi.org/10.19061/iochem-bd-6-384.