

Abstract
We investigated the efficiency of target-enriched long-read sequencing (TELSeq) for detecting antimicrobial resistance genes (ARGs) and mobile genetic elements (MGEs) within complex matrices. We aimed to overcome limitations associated with traditional antimicrobial resistance (AMR) detection methods, including short-read shotgun metagenomics, which can lack sensitivity, specificity, and the ability to provide detailed genomic context. By combining biotinylated probe-based enrichment with long-read sequencing, we facilitated the amplification and sequencing of ARGs, eliminating the need for bioinformatic reconstruction. Our experimental design included replicates of human fecal microbiota transplant material, bovine feces, pristine prairie soil, and a mock human gut microbial community, allowing us to examine variables including genomic DNA input and probe set composition. Our findings demonstrated that TELSeq markedly improves the detection rates of ARGs and MGEs compared to traditional sequencing methods, underlining its potential for accurate AMR monitoring. A key insight from our research is the importance of incorporating mobilome profiles to better predict the transferability of ARGs within microbial communities, prompting a recommendation for the use of combined ARG–MGE probe sets for future studies. We also reveal limitations for ARG detection from low-input workflows, and describe the next steps for ongoing protocol refinement to minimize technical variability and expand utility in clinical and public health settings. This effort is part of our broader commitment to advancing methodologies that address the global challenge of AMR.
Antimicrobial resistance genes (ARGs) are the genetic foundation of antimicrobial resistance (AMR), a phenomenon that diminishes the efficacy of antibiotics, posing a significant threat to global public health. To detect AMR, the prevailing technique involves culturing specific bacteria, then subjecting these isolates to various antibiotic concentrations to observe their growth or inhibition. Additionally, culture-independent diagnostic testing (CIDT) methods are employed, using primers to identify specific ARGs in samples without the need for culturing. However, both approaches come with notable drawbacks: Culture-based tests are time-consuming and generally limited to a narrow spectrum of bacterial species, whereas CIDT methods often lack sensitivity, are limited in the number and specificity of ARGs they can detect within a single workflow, and do not provide the genomic context for the ARGs identified (Loman et al. 2013; Ko et al. 2022).
Target enrichment assays have recently been described as a new culture-independent method for detecting ARGs. These assays use biotinylated probes that hybridize to ARG targets, allowing for amplification before sequencing (Noyes et al. 2017; Lanza et al. 2018). Target enrichment probes differ from PCR primers in several important ways. First, probe sets can be designed to simultaneously target megabases worth of reference sequences; second, probes are longer than PCR primers, i.e., typically 120 bp; third, probes will hybridize to targets even when the sequences contain many mismatches; and lastly, probes capture not only the hybridized target but also nucleic acids that flank the target.
Target enrichment protocols have been designed to work with short-read sequencing platforms, such as Illumina. However, the use of short-read sequencing hinders precise analysis of the regions flanking ARGs owing to the short length of the reads. Bioinformatic assembly steps can be taken to reconstruct longer contiguous sequences, though this process can be highly error prone. For example, it has been previously described that assembled sequences are fragmented, especially within ARG-containing regions, thus making it difficult or impossible to identify genomic regions that neighbor ARGs (Abramova et al. 2023). Nonetheless, identifying the genomic region that flanks the ARGs is valuable, since some mobile genetic elements (MGEs) can rapidly mobilize ARGs between distantly related bacteria. Understanding the profile of ARG-flanking MGEs is fundamental for estimating the likelihood that an ARG may be acquired by pathogenic bacteria, thus generating clinically relevant AMR information. We recently demonstrated that target enrichment can be combined with long-read sequencing to capture both ARGs and flanking MGEs, without the need for bioinformatic sequence reconstruction, i.e., assembly (Slizovskiy et al. 2022). We call this workflow target-enriched long-read sequencing (TELSeq). This previously published work used a probe set that targeted ∼7000 ARGs, but no MGEs; therefore, the detected flanking MGEs were captured only due to a “bystander” effect. It is unknown whether targeting MGEs and ARGs would increase the sensitivity and utility of TELSeq in profiling MGE-colocalized ARGs, allowing for inference of mobility potential. Furthermore, this previous work used a molecular workflow that required a relatively high amount of input genomic DNA (gDNA), whereas lower-input workflows (i.e., 200–1000 ng) have since been developed, but not yet used in a TELSeq workflow. Finally, our previous proof-of-concept study encompassed a relatively small number of pilot samples, and thus did not support a full statistical analysis of factors that might influence the workflow's performance. These technical considerations are critical for moving TELSeq beyond research applications, and into clinical and public health practice.
The objective of this study was to identify technical factors that impact TELSeq's performance, particularly turnaround time, accuracy, sensitivity, specificity, and reproducibility. To achieve this objective, we designed an experiment in which technical replicates of challenging sample matrices were subjected to two technical variables in a fully crossed design (Fig. 1): (1) enrichment platform (i.e., high vs. low gDNA input); and (2) probe set, i.e., ARGs only, MGEs only, or ARGs and MGEs combined. Results of TELSeq were formally compared to results obtained from a standard, nonenriched workflow. Our results demonstrate that TELSeq continues to outperform nonenriched long-read sequencing by increasing the detection of ARGs and MGEs across multiple, complex sample types. However, the performance of the low-input enrichment protocol was highly variable, thus limiting its utility for sample matrices with very low amounts of gDNA. When used with PacBio sequencing, TELSeq is recommended for generating detailed, contextualized ARG sequence data without the need for bioinformatic reconstruction, particularly for samples with moderate-to-high amounts of gDNA.
Figure 1.
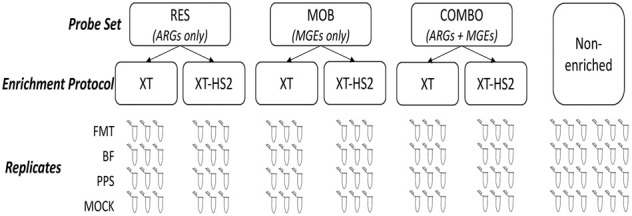
Experimental design overview.
Results
Study overview
The sample types used for this experiment included human fecal microbiota transplant (FMT) material, bovine feces (BF), pristine prairie soil (PPS), and a commercial mock human gut microbial community standard (MOCK). Replicates of each sample were subjected to target enrichment using three different probe sets: resistome enrichment (“RES” probes), mobilome enrichment (“MOB” probes), or resistome–mobilome combined enrichment (“Combo” probes). Each sample was sequenced using two enrichment platforms: TELSeq-XT-HS2 (XT-HS2) intended for samples with low gDNA inputs (<200 ng); and TELSeq-XT (XT) intended for samples with high gDNA inputs (>200 ng). For each sample type, nine technical replicates were used as input for TELSeq sequencing using either XT-HS2 or XT platforms (total nsample = 72). Alongside replicates prepared for sequencing via the XT-HS2 or XT enrichment platforms, triplicates of each sample type were sequenced via nonenriched long-read circular consensus sequencing (CCS, PacBio) (nsample = 24).
TELSeq improves the sequencing yield of resistomes and mobilomes even with low DNA input
As compared to nonenriched libraries, TELSeq libraries contained a higher proportion of reads originating from ARGs and/or MGEs. As expected, XT sequencing in particular led to a consistent increase in the detection of reads containing ARGs in comparison to nonenriched sequencing; these increases were similar to previously reported results for both short- and long-read enriched libraries (Noyes et al. 2017). Specifically, the overall median ARG on-target percentage in FMT went from 0.1% in nonenriched to 2.9% in XT libraries; in BF from 0.2% to 16.6%; in MOCK from 2.4% to 20.7%; and lastly, for PPS from 0% to 0.2% (Fig. 2). The magnitude of the increased resistome on-target rate relative to the nonenriched samples was most pronounced when RES or Combo probes were implemented and the TELSeq-XT protocol was notably more efficient in sequencing ARG-containing reads than traditional nonenriched sequencing methods. In addition, the XT enrichment protocol increased the MGE on-target rate, which increased from 1% in nonenriched BF libraries to 4.0% in BF XT libraries; from 2.2% to 13.2% in FMT libraries; from 4% to 4.9% in MOCK libraries; and from 1.1% to 17.1% in PPS libraries. For the XT enrichment protocol, the gains in MGE sequencing relative to the nonenriched protocol depended on the sample type and probe set used. For instance, in BF and PPS libraries, RES and Combo ARG–MGE probe systems resulted in increased MGE recovery, while in FMT and MOCK libraries, MOB and Combo probes yielded increased MGE recovery (Fig. 2).
Figure 2.

Boxplots summarizing library input and quality parameters (A) and resistome or mobilome on-target statistics (B), reported by sample type, enrichment protocol, and probe set for TELSeq and PacBio (i.e., nonenriched) sequencing methods.
When the low-input XT-HS2 platform was used, both ARG- and MGE-based on-target sequencing rates in BF, FMT, and MOCK libraries were similar or greater than what was achieved via nonenriched sequencing (Fig. 2), again supporting the efficacy of the target enrichment protocol as compared to standard nonenriched sequencing. However, the relative increases in XT-HS2 on-target rates were more moderate as compared to the increases generated by the XT protocol. For example, the median ARG on-target rate for BF was 4.3% for XT-HS2 libraries, compared to 16.6% for XT libraries; 0.9% versus 2.9% for the FMT libraries; 2.5% versus 20.7% for the MOCK libraries; and 0.0% versus 0.2% for the PPS libraries. As compared to XT libraries, probe set design was less influential in the ARG sequencing rate. The variable performance of XT-HS2 can be explained by variability in the concentration of the final libraries. More specifically, all 12 of the libraries generated by XT-HS2 and ARG probes contained <10 ng/μL of prepared DNA, as did all nine of the PPS XT-HS2 libraries. Within the 36 XT libraries, only one had concentration <10 ng/μL, and the mean was 83.5 ng/μL (median 94.4, range 0.36–120.6) (Fig. 2). Additionally, the median DNA quality as determined by the 260 nm:280 nm NanoDrop ratio was notably lower across all samples for the XT-HS2 libraries as compared to the XT libraries, with libraries subjected to the combination ARG–MGE probes attaining proportionally higher quality. We note that although both enrichment protocols led to an increase in the amount of technical duplicates, this effect was more pronounced in XT protocol, with median duplicate levels ranging from 0.9% of all reads in the PPS libraries to 7.0% of all reads in the MOCK libraries (Fig. 2).
Although the on-target sequencing favored the XT protocol over XT-HS2, this may have also been driven by differences in sequencing yield, which was higher for XT libraries (Fig. 2). One way to circumvent the yield problem is to analyze the relative abundance of individual ARGs, rather than total resistome on-target rates. Hence, we considered the variation in the relative abundance of the resistome and mobilome across the various enrichment protocols and probe systems (Fig. 3). To remove potential bias associated with variable gene lengths and variable numbers of reads, the abundance was normalized by both gene length and sequencing yield. Based on analysis using zero-adjusted gamma regression, TELSeq BF, FMT, and PPS libraries had a consistently higher relative ARG group abundance using XT protocols (log10 effect estimate [SE] = 5.40 [0.38], 5.66 [0.28], 2.62 [0.27], respectively; all sample type P < 0.001) and XT-HS2 protocols (2.29 [0.81], P = 0.005; 6.09 [0.33], P < 0.001; 3.49 [0.63], P < 0.001, respectively), across all enriched replicates (Fig. 3). Enrichment in BF, FMT, and PPS libraries also yielded greater MGE accession relative abundance as compared to nonenriched sequencing using XT (0.59 [0.17], 3.29 [0.12], 5.66 [0.07], respectively; all sample type P < 0.01) or XT-HS2 (2.99 [0.21], 4.41 [0.13], 6.88 [0.10], respectively; all sample type P < 0.001). This result was consistent with our analysis of the median on-target rate, again showing that both the XT and XT-HS2 protocols successfully enriched for ARGs and MGEs.
Figure 3.

Violin plots showing the log10 relative abundance of ARG groups (A) and MGE accessions (B) normalized by gene length and sequencing yield, for BF, FMT, and PPS samples, colored by enrichment protocol and probe set. Technical replicates of TELSeq libraries are shown individually, while the 24 nonenriched replicates are summarized into a single violin plot.
While both XT and XT-HS2 resulted in a consistent increase in relative resistome abundance as compared to nonenriched sequencing, there was no uniform pattern across probe sets (Fig. 3A). For PPS samples, we note that the relative abundance of detected ARGs was higher in XT versus XT-HS2 libraries especially when used with Combo probes (P < 0.001) which lead to a >450-fold improvement relative to nonenriched libraries. Moreover, the PPS libraries appeared to be fairly divergent to the FMT and BF libraries in that the variation in the relative abundance of PPS libraries was narrower, exhibiting a more pronounced tendency to cluster around the mean. For BF samples, significant ARG recovery (P < 0.01) was achieved for XT protocol using either the RES or MOB probe sets which yielded approximately threefold and ∼45-fold increased relative abundance over that of nonenriched libraries. However, the XT-HS2 low-input protocol achieved comparable increases in recovery via the Combo and MOB probe sets, yielding an average increase of ∼10-fold and ∼200-fold relative to nonenriched libraries (P < 0.01). For FMT samples, the XT-HS2 RES libraries had a smaller range in the relative resistome abundance than XT but this range was not significantly different from XT when the Combo and MOB probe sets were used. Such results are meaningful since they demonstrate that upon normalizing for sequencing yield, XT-HS2 increased the resistome abundance in comparison to the nonenriched libraries when employed with either the Combo or MOB probe set, but it was unsuccessful in yielding high ARG abundance when employed with the RES probe set, perhaps because of the high number of failed RES replicates.
Similar to the relative resistome abundance, Figure 3B demonstrates that both XT and XT-HS2 yielded a significant increase in the relative mobilome abundance in comparison to the unenriched data. Among BF samples, TELSeq-XT and XT-HS2 libraries subjected to RES probes yielded the highest mobilome abundance increase (>900-fold and 39-fold, respectively, P < 0.001), and this was followed by TELSeq-XT-HS2 libraries subjected to Combo probes (>750-fold, P < 0.001). Probe systems using MOB and Combo, when used with the XT protocol, did not produce significant increases in mobilome abundance, but did produce increases when used as part of the low-input XT-HS2 platform (P < 0.01). In FMT samples, mobilome relative abundance was improved compared to nonenriched libraries, most prominently when the RES probes were deployed in XT libraries (P = 0.02), or when Combo probes are utilized in XT-HS2 libraries (P = 0.006). While MOB probes marginally improved MGE recovery as compared to nonenriched PPS libraries, as with FMT, RES probes when deployed in XT or Combo probes when deployed in XT-HS2 protocols in PPS samples resulted in the most significant increases in sequenced MGE relative abundance. It is possible that the higher relative abundance of MGEs detected with the RES and Combo probes compared to the MOB probes in all samples could be attributed to the bystander effect, i.e., if the MGEs were flanking targeted ARGs, and thus were captured by the RES probes.
Detected resistome and mobilome diversity differs by sample type and enrichment protocol
Principal component analysis indicated that enrichment protocol was associated with significant differences in resistome and mobilome beta-diversity (Fig. 4). These differences were statistically significant for resistome analysis in the FMT (ANOSIM R = 0.34, P = 0.001), BF (ANOSIM R = 0.22, P = 0.006), and PPS (ANOSIM R = 0.35, P = 0.004) samples (Fig. 4A–C). Similar trends were observed for mobilome beta-diversity (Fig. 4D–F), with significant differences by enrichment protocol for FMT (ANOSIM R = 0.22, P = 0.005), BF (ANOSIM R = 0.17, P = 0.007), and PPS (ANOSIM R = 0.27, P = 0.001). Across all three sample types, the XT-HS2 protocol resulted in a relatively large amount of variability in beta-diversity across replicates and probe sets. The variability in the beta-diversity for the XT and nonenriched data sets was significantly influenced by the sample type. Nonenrichment led to very low variability in beta-diversity for the PPS and BF data sets, but higher variability in the FMT data set. Conversely, the XT protocol produced low beta-diversity variability in the BF and FMT data sets, but high variability in the PPS data set. The ARG group tetQ had one of the largest loading vectors across all three samples, suggesting that it played a large role in differentiating the resistome composition generated by the three enrichment protocols. Across all sample types, the tetQ loading variable was associated with the XT and XT-HS2 data sets, suggesting that the use of enrichment may have been crucial for detecting this ARG group.
Figure 4.

Resistome and mobilome beta-diversity analysis of Bray–Curtis dissimilarity distances of microbiome samples, including BF, human FMT, and PPS. Group-level ARG and accession-level MGE library compositions are colored by enrichment protocol (XT-HS2 in pink, XT in green, and nonenriched in blue). Arrows in (A–C) indicate the loading vectors for ARG groups. The ARG groups with the longest arrow length are labeled by name.
Pairwise comparison of PERMANOVA results allowed us to identify which enrichment protocols generated significantly different beta-diversity profiles from one another. Based on this analysis, the XT and XT-HS2 protocols did not generate significantly different beta-diversity profiles (all P > 0.05) with the exception of the resistome in PPS libraries and the mobilome in BF (P = 0.045 and 0.006, respectively). In the case of resistome beta-diversity within the PPS samples, although the difference between XT and XT-HS2 was statistically significant, the magnitude of this difference was much smaller than the magnitude observed when comparing XT versus nonenriched and XT-HS2 versus nonenriched (R2 = 13.7% vs. 22% and 32%, respectively). This smaller magnitude of effect indicates that the differences in beta-diversity between XT and XT-HS2 libraries were smaller than the differences between enriched and nonenriched libraries. Indeed, the differences between the enriched and nonenriched libraries were consistently large and statistically significant. In the pairwise comparison of XT versus nonenriched libraries, R2 values for five of the six data sets were >22% (all P < 0.05), indicating that the XT enrichment was associated with a large proportion of the observed variation in beta-diversity. The exception to this pattern was the mobilome beta-diversity in the BF sample, which was not significantly different between the XT and nonenriched libraries (R2 = 10.6%, P = 0.09). For the pairwise comparison of XT-HS2 and nonenriched libraries, R2 values were more variable by sample type, ranging from a low of 11% in the FMT resistome to 32% in the PPS resistome (all P < 0.05). Taken together, these results suggest that the differences in beta-diversity observed by enrichment protocol were associated primarily with differences between TELSeq and nonenriched libraries, with smaller differences between XT and XT-HS2 libraries.
Another way to evaluate composition is through a binary heatmap (Fig. 5). This analysis revealed that the combination of using the XT protocol with the RES probe set led to the most successful ARG enrichment, as we detected nearly all of the resistance mechanisms encountered in the nonenriched libraries, as well as additional mechanisms that were not captured in the nonenriched libraries. Many of the mechanisms detected only within the XT protocol involved resistance to aminoglycosides, beta-lactams, tetracyclines, and trimethoprim (Fig. 5). The Combo probe set was also successful in identifying a high number of resistance mechanisms, with the exception of the PPS sample, which was known to have a very low resistome richness in situ. Additionally, for PPS libraries, the XT-HS2-MOB and XT-MOB protocol and probe combinations were able to reveal many additional resistance mechanisms not detected within the nonenriched libraries, which may be due to the genomic proximity of MGEs with ARGs in situ. We note that the MOB probe sets failed to detect a majority of glycopeptide resistance mechanisms (Fig. 5). Glycopeptide resistance mechanisms are primarily chromosomally located, and therefore would not be as likely to be proximal to MGEs such as plasmid markers and integrative and conjugative elements. Therefore, the lack of glycopeptide ARG detection by the MOB probe sets is consistent with MOB enrichment of MGE-flanked ARGs. The XT-HS2 RES probe set performed unexpectedly poorly across all replicates, including the MOCK replicates. The most immediate cause of this inefficacy was likely the low library concentration generated by the XT-HS2 RES probes (Fig. 2A), and subsequent very low sequencing yield (Fig. 2B). The cause of the failed libraries is unknown, and could be due to systematic failure of the XT-HS2 RES probe set itself, or the inherently stochastic nature of the XT-HS2 enrichment process, which produced high variability in results.
Figure 5.

Binary heatmaps displaying the presence (dark blue) or absence (light blue) of drug-related ARGs at the mechanism level (right-hand axis), sorted by antimicrobial class (left-hand axis), grouped by sample type (BF, FMT, PPS, and MOCK), enrichment platform (TELSeq and nonenriched), and probe set-platform combination (XT-HS2 RES, MOB, and Combo; XT-RES, MOB, and Combo). ARGs associated with biocide and metal resistance are shown in Supplemental Figure S1.
Sample type and sequencing yield outweigh the effect of the probe set on resistome and mobilome richness
To isolate the impacts of the enrichment platform and probe set on ARG and MGE detection using TELSeq, we needed to normalize for sequencing depth due to sequencing throughput disparities across samples, enrichment protocols, and probe sets (Fig. 2). To do this, we rarefied all BF, FMT, and PPS TELSeq libraries that had 5033 or more reads (see Methods). The rarefying procedure, a method used to normalize sequencing data by equalizing the number of reads across samples, did not notably alter the proportion of on-target sequences, affirming the expectation that this process primarily affects the volume of data rather than its qualitative composition (Supplemental Table S1). As anticipated, rarefying led to a reduction in the total amount of sequence data and a lower percentage of duplicate sequences. Nevertheless, despite these data reduction steps, the total base pair throughput from the rarefied XT libraries remained higher than from rarefied XT-HS2 libraries, due to the longer read lengths produced with the XT platform.
Using the rarefied data set, we generated two linear mixed-effect regression (LMER) models, one each for the ARG group and MGE accession richness. Following stepwise backward model selection, the probe set was removed from the final model because its inclusion did not significantly improve model fit. Rather, the two strong explanatory factors in the models included the sample type, with lower resistome richness identified in PPS than in BF and FMT; as well as sequencing yield, which was a positive predictor of both ARG and MGE richness across all samples, as expected (Table 1).
Table 1.
Results from best-fit linear mixed-effect regression models of ARG and MGE richness
| LMER model | Variable(s) | Coefficient | SE | t-Value | 2.50% | 97.50% |
|---|---|---|---|---|---|---|
| ARG richness | (Intercept) | −4.39 | 8.46 | −0.52 | −20.98 | 12.20 |
| REF—BF | — | — | — | — | — | |
| Sample source—FMT | −4.16 | 7.09 | −0.59 | −18.05 | 9.72 | |
| Sample source—PPS | −17.18 | 8.31 | −2.07 | −33.47 | −0.90 | |
| Yield (Mb) | 4.59 | 1.80 | 2.55 | 1.06 | 8.12 | |
| MGE richness | (Intercept) | −29.93 | 10.72 | −2.79 | −50.94 | −8.93 |
| REF—RES | — | — | — | — | — | |
| Probe set—Combo | 16.27 | 8.44 | 1.93 | −0.27 | 32.82 | |
| Probe set—MOB | 2.73 | 8.39 | 0.32 | −13.72 | 19.18 | |
| Yield (Mb) | 12.04 | 1.91 | 6.31 | 8.30 | 15.78 |
TELSeq achieves a detection limit of 0.01% relative abundance within a ground truth mock community sample sequenced to relatively shallow depth
To analyze the MOCK libraries, we identified “ground truth” ARGs (GT-ARGs) contained within reference genomes of the 21 microorganisms comprising the mock community (see Methods). The genomes in this mock community have a known relative abundance (as documented by the manufacturer), which can be used to estimate a limit of detection for ARGs from each genome. Briefly, GT-ARGs consisted of ARGs that were unique to only one organism within the mock community; because of their 1:1 ARG:genome, we were able to use these ARGs as markers of detection for each genome within the community. Of the 21 references, eight did not contain any ARGs that could be considered GT-ARGs (either because they did not contain any ARGs at all or because they did not contain any unique ARG groups). Across all MOCK samples (including nonenriched samples), we identified GT-ARGs from eight of the remaining 13 genomes (Fig. 5). We did not identify any GT-ARGs from Enterococcus faecalis, Clostridium perfringens, Akkermansia muciniphila, and the Escherichia coli strain JM-109. The genomes of E. faecalis and C. perfringens have the lowest relative abundance within the mock community, at 0.001% and 0.0001%, respectively, which could explain their absence within the enriched mock community data. Similarly, A. muciniphila has the next-lowest relative abundance at 1.5% (of all 13 genomes that contained at least one GT-ARG), with the exception of Salmonella enterica.
However, we did identify GT-ARGs from S. enterica and Clostridium difficile, which have relative abundances of 0.01% and 1.5%, respectively. The GT-ARGs within S. enterica were primarily detected only in enriched samples (and not within nonenriched samples), with the exception of corB and corC, which were detected across nonenriched and enriched replicates. This suggests that TELSeq's limit of detection for the sequencing depth used in this study was ∼0.01% relative abundance, although performance at this level was highly variable across TELSeq replicates (Fig. 6). We identified GT-ARGs from three of the four E. coli strains with GT-ARGs, despite the fact that all four strains are present within the mock community at the same relative abundance of 2.8%. Therefore, the limit of detection due to in situ relative abundance is not the only factor determining false-negative findings within the mock community. Other factors may include variability in the true composition of the mock community, as the manufacturer's documentation states that each organism in the community can contain up to 15% deviation from the targeted average relative abundance. Other factors that impact detection include the extraction efficiency of each organism; the efficiency of the baits in binding to each GT-ARG (which can be impacted by GC content); and the inherent stochasticity of the molecular workflow, particularly the numerous steps in which the mock community cells, DNA and sequencing libraries are subsampled, any one of which can lead to loss of GT-ARGs and thus absence within the final sequencing pool. A notable result was the lack of any GT-ARGs within the XT-HS2 RES replicates. This was likely due to the very low sequencing yield of these samples, as shown in Figure 2A, and discussed above in relation to Figure 5.
Figure 6.

Heatmap of alignments to GT-ARGs (y-axis), by replicate (x-axis). Replicates are grouped by enrichment protocol and probe set (x-axis), and each GT-ARG is listed along with the bacterial species from which it originated (y-axis). Gray cells indicate GT-ARGs with no alignments in the relevant replicate.
Discussion
Our results confirm previous work demonstrating the high sensitivity of molecular target enrichment for ARG and MGE detection within metagenomic samples (Noyes et al. 2017; Lanza et al. 2018; Macedo et al. 2021). We further confirm our previous finding that target enrichment continues to perform well when combined with long-read sequencing (Slizovskiy et al. 2022). A major advancement of the work presented here is the use of a low-input, rapid target enrichment workflow (XT-HS2). Compared to the previous workflow (XT), XT-HS2 accommodates a wider range of sample types (including samples with very low microbial biomass), and reduces bench time from >24 h to <8 h. We demonstrate that XT-HS2 can provide comparable results to XT, particularly in terms of the resistome and mobilome composition of generated data (Fig. 4). However, we also found that the XT-HS2 protocol was more likely to yield libraries with very low DNA concentration, particularly for the ARG probe set (across all replicates of all four samples) and for the PPS sample (Figs. 2, 3). In these cases, the use of XT-HS2 led to low sequence output, low on-target efficiency (Fig. 2), and lower sensitivity to detect ARGs and MGEs (Fig. 5). In total, 21 of the 36 XT-HS2 libraries had a final library concentration of <10 ng/μL, whereas only one of the 36 XT libraries met this threshold. Therefore, the decision to use XT-HS2 should be balanced between constraints to DNA input and turnaround time, and the likely variability in library quality. Future studies should address potential optimizations to the XT-HS2 protocol, which could minimize low-yield issues and improve replicability. For example, the XT-HS2 protocol may perform more robustly when used with sequencing platforms that accommodate low-input library preparation, such as Illumina and Oxford Nanopore Technologies.
A major open question for resistome studies is how to best integrate mobilome profiles (Oh et al. 2018; Slizovskiy et al. 2020; Brown et al. 2022). This is an important question, because ARG mobility is a critical component of predicting whether specific ARGs may transfer from commensal bacteria to pathogens within a microbial community. Our previous work demonstrated that the long reads generated by TELSeq can be used to colocalize ARGs with MGEs, without the need for bioinformatic reconstruction, i.e., without de novo assembly or metagenome-assembled genome methods (Slizovskiy et al. 2022). In this previous research, a probe set tailored exclusively for ARGs was used, prompting an inquiry into whether a specialized probe set for MGEs might better facilitate the identification of MGEs, including those that may harbor ARGs. Our results presented here demonstrate that a combined ARG–MGE probe set performed comparably to the ARG-only and MGE-only probe sets, both in terms of on-target efficiency (Fig. 2) and recovered diversity (Figs. 3, 5). The use of a combined probe set streamlines the laboratory workflow and reduces costs because each sample is subjected to only one enrichment, instead of two in parallel. Therefore, we recommend the use of a combined probe set in future studies that aim to analyze the resistome and mobilome in tandem. We note that future studies should carefully consider the design of combined probe sets, and in particular, the amount of sequence homology between accessions within and across the ARG and MGE reference databases used for probe design. In the current study, we utilized ARG and MGE reference databases without modification, which meant that some ARG accessions were contained within the full-length MGE accessions, likely resulting in some overlap between the RES and MOB probe sets. A combined probe set designed using efficient bait design algorithms would circumvent this problem because such algorithms remove redundancy due to homologous sequences across accessions (Alanko et al. 2022).
TELSeq offers significant advantages, including enhanced detection sensitivity for ARGs and MGEs across diverse samples. This method eliminates the need for complex bioinformatic sequence reconstruction, providing detailed genomic context by capturing both ARGs and flanking MGEs. TELSeq demonstrates high sensitivity and versatility across various sample types, such as human FMT material, BF, PPS, and a mock human gut microbial community. The use of combined ARG–MGE probes streamlines laboratory workflows and reduces costs by avoiding parallel enrichments. However, the method also has disadvantages, such as generating highly uneven coverage across targeted genomes, requiring high DNA input for the XT protocol, and exhibiting technical variability, particularly with the XT-HS2 protocol. This variability necessitates careful optimization and extensive replication, which can be resource-intensive. Additionally, the method is not suitable for robust variant calling due to the shallow, fragmented nature of the data, and its success heavily depends on the design of the biotinylated probes. Despite these challenges, TELSeq remains a powerful tool for resistome and mobilome profiling.
To date, the TELSeq protocol has been used solely for research purposes. When designing a protocol for nonresearch purposes such as human medicine or surveillance, it is critical to understand and minimize technical sources of variability. Our experimental design included triplicates, which is an expensive study design decision; however, the replicates allowed us to look at technical variability in the workflow (Fig. 1). We found that triplicates did not always yield the same presence/absence profiles for targeted ARGs, including in nonenriched data sets (Fig. 5). This variability could be due to differences in sequencing yield between replicates, which particularly impacts richness estimates. However, on-target rates and relative abundance distributions for detected targets should be less impacted by yield because these metrics are normalized to sequencing depth. Indeed, in some cases, ARG and MGE profiles were very consistent across replicates, particularly for the XT enrichment protocol within the FMT and PPS samples (Fig. 3). However, in other cases, interreplicate variability was much higher, and this seemed to be associated with low-yield XT-HS2 libraries (Figs. 2, 3). Another important factor for nonresearch applications is the availability of controls that can be used for quality control. In this study, we used a commercially available mock community of intact cells as a positive control, which yielded several important findings. First, we found that the limit of detection of TELSeq is in the range of 0.01%, although this value would likely change based on sequencing depth and probe binding efficiency. Second, we discovered important limitations in using a mock community as a positive control in the context of resistome analysis. In particular, we found that multiple genomes in the mock community contained the same ARGs. Due to the fragmented state of both enriched and nonenriched metagenomic data sets, it is challenging to conclusively trace ARGs back to their original bacterial host genomes. As a result, the majority of ARGs within the mock community could not serve as a reliable “ground truth” due to the inability to determine their relative abundance distributions at the genomic level. Consequently, our analysis was restricted to employing GT-ARGs (ground truth ARGs), which are ARGs found in a single genome. Nevertheless, calculating the expected relative abundances of these GT-ARGs proved difficult, as the denominator representing the total community's abundance was unclear. The manufacturer's stated 15% deviation in relative abundance compounds this problem. In summary, our results from the mock community analysis confirm previous discussions about the necessity and challenge of developing controls for metagenomic workflows (Sczyrba et al. 2017; Meyer et al. 2022). Future research should concentrate on addressing this complex, technical problem.
Methods
Sample collection and storage
Fresh feces were collected from a healthy human donor participating in the University of Minnesota School of Medicine Fecal Microbiota Transplant program (“FMT” sample). Donor enrollment criteria were followed as part of the Investigational New Drug Application 15071. Strict donor exclusion criteria based on biometric and clinical variables were followed as previously described (Hamilton et al. 2012). Previous stool samples collected from this donor had tested negative for viral, parasitic (including Giardia and Cryptosporidium), and specific culturable vancomycin-resistant Enterococci, methicillin-resistant Staphylococcus aureus (MRSA), carbapenem-resistant Enterobacteriaceae, E. coli O157:H7, Salmonella spp., Shigella spp., and Yersinia spp. pathogens. The fecal sample used in the current study was collected in a single-use toilet hat and was allotted into multiple 50 mL conical polypropylene tubes and immediately transferred to a −80°C freezer for storage after 30 min of initial collection. All donor-specific activities were approved by the University of Minnesota Institutional Review Board.
Fresh feces were collected from a periparturient Holstein Friesian dairy cow (“BF” sample) during a routine field medical examination for herd health (e.g., metritis, metabolic disorders, infectious diseases, etc.) on a commercial Midwestern U.S. farm. This cow was deemed systemically healthy and was kept in a freestall alongside other cows that were not exposed to oral or systemic antimicrobial drugs. Approximately 250 g of fresh feces were collected per rectum and placed into 50 mL conical vials and immediately placed on ice. Samples were transferred to a −80°C freezer for storage within 2 h of collection. All cattle handling procedures and sample collection were performed by veterinarians in accordance with the University of Minnesota Institutional Animal Care and Use Committee (IACUC).
Historically PPS sample was collected in 2019 from a strip of perennial grasses and forbs in Mower County, Minnesota, USA. The Tripoli clay loam (Fine-loamy, mixed, superactive, mesic, Typic Endoaquolls; pH = 6.2; OM = 9.6%; moisture = 58.0%; P:K = 0.052), was composited from three points within 10 m of each other following collection at a depth of 0−20 in on a 0%−2% gradient footslope. The homogenized sample was sealed in an air-tight container and stored at 4°C during chemical and physical characterization. A subsample of this soil was transferred to a −80°C freezer for storage before gDNA extraction.
gDNA extraction
Before extraction, 50 g portions of the BF and FMT, and 250 g portions of PPS were thawed at room temperature. Following a 5 min manual homogenization, the samples were disbursed in 0.25 g aliquots into PowerBead Pro tubes containing zirconium beads and 800 μL lysis buffer for extraction using the Qiagen DNEasy PowerSoil Pro kit (Qiagen 163044275). Molecular-grade sterile water was placed into four randomly selected tubes to serve as negative controls (extraction blanks). After 6 sec of vortexer-mediated homogenization, samples were placed on a 115 V Mini-Beadbeater-96 (BioSpec Products) for mechanical lysis. Sample bead beating proceeded at 2400 rpm for 30 sec for a total of three rounds with a 2 min pause on ice between each round, to prevent overheating. The remainder of the extraction procedure followed the PowerSoil Pro recommendations with inhibitor removal steps, and the final 50 μL of eluted gDNA was stored at −20°C.
For the ZymoBIOMICS mock community, all extraction procedures, including additional preextraction steps to ensure that the final gDNA maintained optimal fragment length and species representation after isolation from the native DNA/RNA shield storage solution, were followed according to the methods outlined by Nicholls et al. (2019). The standard was divided into ten 75 μL aliquots, each centrifuged at 8000×g for 3.5 min before removing and retaining the supernatant, which contained lysed Gram-negative species in the DNA/RNA shield storage solution. The pellets were resuspended in 700 μL lysis buffer and were transferred to zirconium-containing PowerBead Pro tubes of the Qiagen DNEasy PowerSoil Pro kit (Qiagen 163044275). Molecular-grade sterile water was added to four randomly selected tubes to serve as negative controls (extraction blanks). After 6 sec of vortexer-mediated homogenization, samples were placed on a 115 V Mini-Beadbeater-96 (BioSpec Products) for mechanical lysis. Sample bead beating proceeded at 2400 rpm for 5 min for a total of four rounds with a 5 min pause on ice between each round. The resulting ∼500 μL of supernatant was retained and recombined with the supernatant retained earlier, and the samples were subjected to the remaining recommended procedures of the PowerSoil Pro kit, including inhibitor removal steps. The final 50 μL of eluted gDNA was stored at −20°C.
Quantitation of all isolated gDNA was performed using the Qubit 4 Fluorometer (Invitrogen) using the dsDNA high-sensitivity assay kit. Electrophoretic assessment of DNA quality was performed using a genomic screen tape and reagents on a 4200 TapeStation (Agilent). All extraction blanks contained no quantifiable gDNA and therefore were not carried forward into library preparation, targeted enrichment, and sequencing. All sample processing occurred in a Class II Biological Safety Cabinet and was performed by a single laboratory technician following standard decontamination practices using 70% ethanol and irradiation.
Probe design
Targeted enrichment was performed using a custom-designed biotinylated cRNA probe panel for selective hybridization and capture. For probe design, a comprehensive list of predefined publicly available unique nucleotide sequences was compiled for 7868 ARGs from MEGARes v2.0 (Doster et al. 2020), including accessions for: drug resistance, metal resistance, multicompound resistance, and biocide resistance; and for 738 MGE accessions, including full-length sequences for: (1) integrative and conjugative elements (ICE) from ICEberg v2.1 (Liu et al. 2019) and (2) plasmid replicons of Enterobacteriaceae and Gram-positive bacteria from PlasmidFinder v2.1 (Carattoli et al. 2014). These ARG and MGE sequences comprised 8.55 Mb of total sequence. Full-length MGE accessions from ICEberg and PlasmidFinder were utilized, and some of these accessions contained ARGs. Posthoc analysis using alignment with minimap2 showed that 980 of the 7868 ARGs contained in MEGARes v2.0 were also contained within the 738 MGE accessions (at a coverage of 100%), meaning that there was some overlap between the three resulting probe panels, described below. The CATCH pipeline (Metsky et al. 2019) was used to generate custom probe panels using the following parameters: probe stride: 120; probe length: 120; mismatches: 5; extension coverage: 100. Probes were manufactured by Agilent with additional “bait-boosting” to amplify GC-rich regions (defined as GC >65%) for fast hybridization reactions to produce a final panel of probes that were then manufactured. Probes were stored at −80°C before use. Three probe panels were designed to test the efficiency of either resistome and mobilome recovery by TELSeq:
MOB panel consisted of the probe set generated using only all mobilome reference databases (i.e., ICEberg and PlasmidFinder) as sequence input. The MOB panel contained 55,710 unique 120-mer oligos providing 100% horizontal coverage across 15,343,808 MGE bases, with a median 1.75 depth of probe coverage for every nucleotide.
RES panel consisted of the probe set generated using only the MEGARes resistome reference database for sequence input to formulate a final 16,007 120-mer oligos providing 100% horizontal coverage across 8,106,325 ARG bases, with a median 1.83 depth of probe coverage for every nucleotide.
Combo panel consisted of the probe set generated using all resistome and mobilome accessions concatenated into a singular FASTA file (i.e., 8606 accessions) and used as input to formulate a final 71,302 unique 120-mer oligos providing 100% horizontal coverage across 23,450,133 bases, with a median 1.81 depth of probe coverage for every nucleotide.
TELSeq library preparation and enrichment
gDNA purification and preparation
Aliquots of the 50 µL gDNA eluates were subjected to 0.6 (vol/vol) AMPure XP bead-based purification (Agencourt Biosciences Corp.) and an elution time of 10 min in room temperature. Transposable element (TE) buffer (1×, pH 8.0) was used to reconstitute all gDNA into the solution. Purified aliquots were subjected to further DNA fragmentation to achieve an ∼5 kb insert range. For all samples (i.e., BF, FMT, PPS, and MOCK), technical triplicate libraries were prepared using two equimolar gDNA aliquot pools of each sample type to achieve ∼4 μg gDNA input for an ∼5 kb insert range. Fragmentation proceeded for all triplicates using COVARIS miniTUBEs (COVARIS Inc.) and mechanical fragmentation using an M220 COVARIS focused ultrasonicator (COVARIS Inc.) with the following settings: peak power 6 W, duty factor 20%, cycles/burst 900, 500 sec, 4°C. All libraries were then subjected to 0.8 (vol/vol) AMPure XP bead purification (Agencourt Biosciences Corp.) and electrophoretic verification using an Agilent TapeStation 4200 (Agilent).
Sheared and purified aliquots were subjected to further DNA size selection by removing fragments <1 kb using a 0.75% agarose gel DF cassette (cat no. BLF7510) with S1 marker (Sage Science Inc.) on a BluePippin pulse-field electrophoretic size selector (Sage Science Inc.). All aliquots were eluted using 10 µL of supplied elution buffer to obtain a final volume of 40 µL.
TELSeq library construction
Samples were subjected to two targeted DNA enrichment protocols based on modifications to the Agilent high gDNA input SureSelectXT and low gDNA input SureSelectXT-HS2 systems (Agilent). For libraries subjected to SureSelectXT system (2–4 μg gDNA), procedures for DNA end-repair, adapter ligation, precapture amplification, bead-based purification, and quality control were performed as previously described (Slizovskiy et al. 2022). Following lyophilization and redilution to the appropriate volume, triplicate libraries across all sample sources were then hybridized with the addition of equimolar quantities of custom-designed biotinylated probes from panels of either RES, MOB, or Combo, along with 10% RNase block solution. Incubation proceeded for 16 h at 65°C with the heated lid set to 75°C to minimize evaporation and maintain the integrity of longer fragments. Subsequent capture steps were performed using per-protocol conditioned MyONE streptavidin T1 beads (Invitrogen Co). Hybridization was facilitated by placing samples on a plate mixer (1200 rpm) for 5 min at room temperature. The mixer was paused and incubation proceeded for an additional 55 min at room temperature, while manually mixing (i.e., pipetting up and down 10 times) every 7 min in order to maintain capture and amplification of larger fragments for long-read library preparation. Following hybridization steps, we performed capture steps using per-protocol reagents and sample-to-reagent ratios, as well as temperatures and durations. We performed three additional bead-washing cycles to optimize the retention of probe-mediated captured DNA fragments. All subsequent steps, including postcapture indexing and amplification, bead-based purification, and quality control were performed as previously described (Slizovskiy et al. 2022).
For libraries subjected to SureSelectXT-HS2 system (600–1000 ng gDNA), libraries were subjected to DNA end-repair and dA-tailing using modified thermocycling parameters: (1) 20°C for 15 min; (2) 68°C for 15 min; and unique molecular oligos (UMIs) were ligated per manufacturer's protocol. AMPure XP bead purification using a 0.8 (vol/vol) bead:sample mixture was performed and eluted libraries were immediately subjected to prehybridization PCR amplification using Herculase II DNA polymerase with dNTPs in buffer. Each library was amplified according to the following thermocycling program: (1) 2 min at 96°C; (2) 10 cycles of the following: 20 sec at 96°C, 30 sec at 65°C, and 6 min at 72°C; (3) 10 min at 72°C, and ramped down to 4°C. Precapture amplified libraries were subjected to 0.8 (vol/vol) AMPure XP bead purification (Agencourt Biosciences Corp.) and electrophoretic verification using an Agilent TapeStation 4200 (Agilent).
The amplified gDNA in each library was subjected to hybridization to equimolar amounts of one of the three custom-designed biotinylated probe sets which were suspended in combination with RNase, 25% (vol/vol) index blockers, and fast hybridization buffer. Following initial thermocycling for 5 min at 95°C and 10 min at 65°C, libraries began hybridizing using 60 cycles of: (1) 1 min at 62°C; (2) 3 sec at 37°C. Following this high-temperature cycling, the hybridization reaction was allowed to continue for another 1 h held constantly at 21°C. Subsequent capture steps were performed using per-protocol conditioned MyONE streptavidin T1 beads (Invitrogen Co). Hybridization was facilitated by placing samples on a plate mixer (1200 rpm), and incubation proceeded for 55 min at room temperature. After the first 25 min of vortexing, the incubation was paused in order to manually mix the hybridized library with the capture beads (i.e., slowly pipetting up and down 10 times) to maintain capture and amplification of larger fragments for long-read library preparation. Resuspension and bead washing in a series of wash buffers was conducted per protocol for five total washes using Wash Buffer 2.
Postcapture amplification steps were performed after combining captured libraries with the appropriate volume of amplification reagent, and the following modified thermocycler program was used: 2 min hold at 96°C followed by 18 cycles of ramping between 96°C (20 sec), 65°C (30 sec), and 72°C (6 min). Libraries were held for an additional 5 min at 72°C. Indexed and amplified captured libraries were eluted from the streptavidin-coated beads following resuspension and incubation at room temperature in a magnetic rack for 10 min. The amplified captured libraries were subjected to 0.8 (vol/vol) AMPure XP bead purification (Agencourt Biosciences Corp.), and bead elution in 1× TE was performed following 10 min of incubation at room temperature. Final electrophoretic verification using an Agilent TapeStation 4200 (Agilent) was performed for all libraries.
Circular consensus sequencing
All prepared SMRTbell templates were prepared according to the manufacturer's protocol for TELSeq and nontargeted long-read libraries, and these were pooled and sequenced using two serial runs of equal sequencing depth on a Pacific Biosciences Sequel 6.0 system (Pacific Biosciences) using two Sequel SMRT cells (1 M with 3.0 chemistry). A 20 h movie runtime was used for each cell. PacBio CCS reads (minPassess = 3; MinAccuracy = 90%) were generated using SMRT Link v 7.0.
Bioinformatic analysis
Rarefaction, quality filtering, and deduplication
Since the target enrichment protocol uses PCR which introduces the possibility of duplicated reads at higher levels than what is normally expected in nonenriched metagenomic data (Noyes et al. 2017), we used the deduplication procedure of Slizovskiy et al. (2022) to remove duplicate CCS reads produced by both the TELSeq and nonenriched protocols. We briefly describe the procedure and discuss the minor modifications that were made. First, if the sample had <200 reads, then we implemented an unclustered deduplication option that adds all of the reads into one cluster file and keeps the remaining 199 cluster files empty; for samples with >200 reads, the cluster-based deduplication method is the same as described in Slizovskiy et al. (2022). We note that the clustering was based on read length and was achieved through the sklearn.cluster.KMeans class using 200 clusters. This step required parsing the FASTQ files through the use of the SeqIO module from Biopython. All CCS reads in one nonempty cluster file were then pairwise aligned using the BLAST-like alignment tool (BLAT) (Kent 2002). The PSL output files from BLAT were parsed through the SearchIO module from Biopython as follows: for each high-scoring segment pair, if the span of the hit and query was at least 90% of the hit and query read length, respectively, then the reads were considered duplicates. Such reads were aggregated into sets, and when generating the deduplicated FASTQ files, only one random read per set was retained.
Resistome and mobilome analysis
For all original libraries and subsamples (which will be collectively referred to as samples from hereon), alignment of the deduplicated CSS reads was done with minimap2 with the -ax flag set to map-pb (Li 2018). The reads were aligned to MEGARes v2.1 (Doster et al. 2020) with all the genes requiring SNP confirmation removed for resistome analysis; and to the combination of ICEberg v2.0 (Liu et al. 2019), PlasmidFinder v2.1 (Carattoli et al. 2014), and ACLAME v0.4 (Leplae et al. 2010) for mobilome analysis. This resulted in two Sequence Alignment Map (SAM) files per sample. To avoid misidentifying ARG sequences as MGE sequences, a new sample-by-sample approach was implemented in a custom Python script whereby the SAM files yielded from a sample were parsed with Pysam (2023) in order to identify MGE alignments that overlapped with ARG alignments. For any pair of ARG and MGE sequences in the SAM file, the MGE sequence was subsequently removed from the downstream mobilome analyses for the sample if there was an overlap in the alignments that covered at least 50% of the smallest sequence.
Next, for each sample, the two SAM files from the alignment step were passed to additional custom Python scripts for mobilome and resistome characterization. This script used a parameter referred to as gene fraction cutoff, defined as the proportion of nucleotides in a reference gene accession that was aligned to by one or more reads in a sample. The first step of this script removed any significantly overlapped MGE reference sequences and small ARG or MGE alignments contained within an MGE or ARG alignment. The next step identified all alignments where the gene fraction cutoff was at least 80% and 50% for resistome and mobilome accessions, respectively. Additionally, mobilome characterizations used ISfinder (Siguier et al. 2006) and ISbrowser (Kichenaradja et al. 2010) to parse PlasmidFinder and ACLAME sequences to identify insertional sequence (IS) and TE families through BLASTN. Sequences were considered to be in an IS or a TE family if and only if the E-value was no >1 × 10−10, and the minimal identity reached 80% over at least 80% of the query length. Any reference sequences that were no longer listed in NCBI were given the label “Unclassified.” The counts of unique resistome and mobilome accessions identified within each sample were used to describe richness and diversity.
Statistical analysis
The resistome relative abundance was calculated for each ARG group within each library i as follows:
where xj is the jth gene accession in a group, n is the number of accessions in a group, and yi is the sequencing yield for each library. Quantifying the relative abundance of the mobilome, for each MGE gene accession x sequenced by the probe-enrichment combination i was calculated by using the formula
where yi is the sequencing yield for each library. Statistical assessment of the differences in the relative abundance of ARG groups and MGE accessions achieved by TELSeq sequencing relative to nonenriched PacBio sequencing (set as reference) was performed using a zero-adjusted gamma distribution parameterized within a generalized additive model using the gamlss (version 4.3-3) package in R (Stasinopoulos and Rigby 2008). This approach was chosen to optimize handling of the response variable with distributions between 0 and 1 especially for highly skewed and kurtotic (near-zero) events as observed in ecological data. Individual models were used for either the resistome or mobilome of BF, FMT, and PPS samples. Explanatory variables included individually the enrichment protocol (XT, XT-HS2, nonenriched), probe type (RES, MOB, or Combo), as well as the interaction of the two variables. Significance across all statistical comparisons was determined with a predefined alpha set at 0.05. The distribution of relative abundances by sample type, enrichment protocol, and probe system for resistome and mobilome genes were summarized in violin plots that are log10-scaled to optimize visualization.
To assess differences in resistome and mobilome richness between the enrichment platform, probe set, and sample type, we used linear mixed-effects regression (LMER) models built using the lmer function of the lme4 library in R (Bates et al. 2015). For these models, only TELSeq libraries were used, as nonenriched libraries were not subjected to the different probe sets and therefore could not be compared by probe set. Additionally, the MOCK sample was not included in the analysis due to its very low richness. Due to large differences in library yield between XT and XT-HS2, we first rarefied the XT and XT-HS2 data sets, as follows: First, four libraries with very low sequencing yield were removed from regression analysis, leaving 50 libraries for rarefying; among these 50 libraries, the lowest library size was 5033 reads, and this library was used to set the rarefying level; next, reads from the remaining 49 libraries were randomly subsampled down to 5033 reads using the seqtk toolkit (Li 2013), to achieve the same read count across all 50 libraries. Rarefied data sets were then subjected to the same deduplication, alignment, resistome, and mobilome analyses as described above for the nonrarefied data sets. All initial, full models contained the same four fixed-effect predictors, namely, sequenced base pairs (i.e., yield), enrichment protocol, probe set, and sample type; in all models, sample ID was specified as a random effect to account for repeated measures by triplicate testing. Model testing was conducted via backward stepwise model reduction (i.e., systematically removing fixed effects) until the lowest possible Akaike information criterion (AIC) was observed. The REML criterion value for each model was retrieved directly from the merMod object given by the lmer function, whereas the AIC value and the R2 values required the merMod's class extractAIC function and MuMIn's (Bartoń 2023) r.squaredGLMM function, respectively. Finally, 95% confidence intervals were calculated for fixed-effect parameters using the Wald method.
Analysis of beta-diversity of ARG and MGEs across all samples was performed following the Hellinger transformation of resistome and mobilome counts stored in the Phyloseq object to reduce the influence of rare accessions. Bray–Curtis distances were calculated and directly used in ordination analysis to the resulting principal components and loadings as implemented in the microViz package (version 0.12.1). Beta-diversity was assessed at the “group” level for ARGs; and at the accession level for MGEs; with a predefined alpha of 0.05 for all statistical tests. Differences in resistome and mobilome composition were assessed independently for each sample type, and statistical differences in the composition according to independent variables including enrichment platform and probe set were assessed via the omnibus analysis of similarities test (ANOSIM). Pairwise permutational multivariate analysis of variance (PERMANOVA) was further performed using adonis2 as implemented in the pairwiseAdonis package (version 0.4) based on 1000 permutations.
Figure generation
All the heatmaps and violin plots were generated with the Python seaborn module (Waskom 2021).
Analysis of mock community ARGs
Reference genomes for the mock community bacteria were downloaded from https://files.zymoresearch.com/protocols/_d6331_zymobiomics_gut_microbiome_standard.pdf and BLAST v2.13.0 was used to identify ARGs within each individual genome, using MEGARes v3.0.0 database as a reference (Bonin et al. 2023). The gene fraction coverage for each ARG was calculated as the proportion of nucleotides within each ARG that matched to the reference ARG. ARGs with ≥95% gene fraction coverage were considered as potential ground truth ARGs (GT-ARGs), with the exception of a single ARG (MEG_3084) in the genome of Veillonella rogosae, which contained a GT-ARG with 93% gene fraction coverage; this GT-ARG was retained. Accessions in MEGARes that contained the flag “RequiresSNPConfirmation” were removed from consideration. The MEGARes ontology was then used to group potential GT-ARGs by their “group”-level classification. The group-level list was then used to generate a dictionary that mapped each of the mock community genomes to its corresponding groups. Groups that were found in more than one genome were then filtered out, leaving only groups that were unique to a single mock community genome. All MEGARes accessions within these remaining “unique” groups were then extracted from the MEGARes v3.0.0 database in order to construct the GT-ARG database. Raw reads from each sample were aligned to the GT-ARG database with minimap2 v2.26 using the “-ax” flag for PacBio HiFi sequence reads as input. The ResistomeAnalyzer from AMR++ was used to calculate the gene fraction for each identified GT-ARG, and GT-ARGs that achieved >95% gene fraction were considered present.
Data access
All raw sequence data and sample metadata generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA1081843. Sample metadata was recorded using the MIMARKS host-associated metagenomic sample guidelines (Bowers et al. 2017). All source code that was used in this study is publicly available at GitHub (https://github.com/jonathan-bravo/TELCoMB) and as Supplemental Code. Custom Python scripts are also available as Supplemental Code.
Supplemental Material
Acknowledgments
Financial support for this work was provided by the National Institute of Health (NIH) National Institute of Allergy and Infectious Disease (NIAID) Grant 1R01AI141810-01 and R01AI173928-01A1. We thank the University of Minnesota Microbiota Therapeutics Program and Drs. Alexander Khoruts and Michael Sadowsky for allowing us to access their FMT donor samples. We thank the University of Minnesota Genomics Center (UMGC) for their assistance with the sequencing phase of this study. We thank Agilent Technologies for their technical guidance in the early planning stages of this research. We are grateful to Hava Blair (University of Minnesota) for obtaining the pristine prairie soil used in this study.
Author contributions: Conceptualization was completed by I.B.S., C.B., and N.R.N.; methodology was completed by I.B.S., C.B., and N.R.N.; investigation was by I.B.S. and N.R.N.; formal analysis was completed by all authors; visualization was completed by I.B.S., N.B., J.E.B., N.R.N., and C.B.; supervision was by C.B. and N.R.N.; writing and editing were done by I.B.S., N.B., J.E.B., C.B., and N.R.N.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279226.124.
Freely available online through the Genome Research Open Access option.
Competing interest statement
The authors declare no competing interests.
References
- Abramova A, Karkman A, Bengtsson-Palme J. 2023. Metagenomic assemblies tend to break around antibiotic resistance genes. bioRxiv 10.1101/2023.12.13.571436v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanko JN, Slizovskiy IB, Lokshtanov D, Gagie T, Noyes NR, Boucher C. 2022. Syotti: scalable bait design for DNA enrichment. Bioinformatics 38(Supplement_1): i177–i184. 10.1093/bioinformatics/btac226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoń K. 2023. MuMIn: multi-model inference. https://cran.r-project.org/web/packages/MuMIn/index.html [accessed February 28, 2024].
- Bates D, Mächler M, Bolker B, Walker S. 2015. Fitting linear mixed-effects models using lme4. J Stat Softw 67: 1–48. 10.18637/jss.v067.i01 [DOI] [Google Scholar]
- Bonin N, Doster E, Worley H, Pinnell LJ, Bravo JE, Ferm P, Marini S, Prosperi M, Noyes N, Morley PS, et al. 2023. MEGARes and AMR++, v3.0: an updated comprehensive database of antimicrobial resistance determinants and an improved software pipeline for classification using high-throughput sequencing. Nucleic Acids Res 51: D744–D752. 10.1093/nar/gkac1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, et al. 2017. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol 35: 725–731. 10.1038/nbt.3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CL, Mullet J, Hindi F, Stoll JE, Gupta S, Choi M, Keenum I, Vikesland P, Pruden A, Zhang L. 2022. mobileOG-db: a manually curated database of protein families mediating the life cycle of bacterial mobile genetic elements. Appl Environ Microbiol 88: e0099122. 10.1128/aem.00991-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carattoli A, Zankari E, García-Fernández A, Voldby Larsen M, Lund O, Villa L, Møller Aarestrup F, Hasman H. 2014. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother 58: 3895–3903. 10.1128/AAC.02412-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doster E, Lakin SM, Dean CJ, Wolfe C, Young JG, Boucher C, Belk KE, Noyes NR, Morley PS. 2020. MEGARes 2.0: a database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data. Nucleic Acids Res 48: D561–D569. 10.1093/nar/gkz1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton MJ, Weingarden AR, Sadowsky MJ, Khoruts A. 2012. Standardized frozen preparation for transplantation of fecal microbiota for recurrent Clostridium difficile infection. Am J Gastroenterol 107: 761–767. 10.1038/ajg.2011.482 [DOI] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664. 10.1101/gr.229202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichenaradja P, Siguier P, Pérochon J, Chandler M. 2010. ISbrowser: an extension of ISfinder for visualizing insertion sequences in prokaryotic genomes. Nucleic Acids Res 38(suppl_1): D62–D68. 10.1093/nar/gkp947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko KKK, Chng KR, Nagarajan N. 2022. Metagenomics-enabled microbial surveillance. Nat Microbiol 7: 486–496. 10.1038/s41564-022-01089-w [DOI] [PubMed] [Google Scholar]
- Lanza VF, Baquero F, Martínez JL, Ramos-Ruíz R, González-Zorn B, Andremont A, Sánchez-Valenzuela A, Ehrlich SD, Kennedy S, Ruppé E, et al. 2018. In-depth resistome analysis by targeted metagenomics. Microbiome 6: 11. 10.1186/s40168-017-0387-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leplae R, Lima-Mendez G, Toussaint A. 2010. ACLAME: a classification of mobile genetic elements, update 2010. Nucleic Acids Res 38(suppl_1): D57–D61. 10.1093/nar/gkp938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Seqtk: a fast and lightweight tool for processing FASTA or FASTQ sequences. https://github.com/lh3/seqtk.
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Li X, Xie Y, Bi D, Sun J, Li J, Tai C, Deng Z, Ou H-Y. 2019. ICEberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucleic Acids Res 47: D660–D665. 10.1093/nar/gky1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Constantinidou C, Christner M, Rohde H, Chan JZ-M, Quick J, Weir JC, Quince C, Smith GP, Betley JR, et al. 2013. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA 309: 1502–1510. 10.1001/jama.2013.3231 [DOI] [PubMed] [Google Scholar]
- Macedo G, van Veelen HPJ, Hernandez-Leal L, van der Maas P, Heederik D, Mevius D, Bossers A, Schmitt H. 2021. Targeted metagenomics reveals inferior resilience of farm soil resistome compared to soil microbiome after manure application. Sci Total Environ 770: 145399. 10.1016/j.scitotenv.2021.145399 [DOI] [PubMed] [Google Scholar]
- Metsky HC, Siddle KJ, Gladden-Young A, Qu J, Yang DK, Brehio P, Goldfarb A, Piantadosi A, Wohl S, Carter A, et al. 2019. Capturing sequence diversity in metagenomes with comprehensive and scalable probe design. Nat Biotechnol 37: 160–168. 10.1038/s41587-018-0006-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer F, Fritz A, Deng Z-L, Koslicki D, Lesker TR, Gurevich A, Robertson G, Alser M, Antipov D, Beghini F, et al. 2022. Critical assessment of metagenome interpretation: the second round of challenges. Nat Methods 19: 429–440. 10.1038/s41592-022-01431-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls SM, Quick JC, Tang S, Loman NJ. 2019. Ultra-deep, long-read nanopore sequencing of mock microbial community standards. Gigascience 8: giz043. 10.1093/gigascience/giz043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes NR, Weinroth ME, Parker JK, Dean CJ, Lakin SM, Raymond RA, Rovira P, Doster E, Abdo Z, Martin JN, et al. 2017. Enrichment allows identification of diverse, rare elements in metagenomic resistome-virulome sequencing. Microbiome 5: 142. 10.1186/s40168-017-0361-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh M, Pruden A, Chen C, Heath LS, Xia K, Zhang L. 2018. Metacompare: a computational pipeline for prioritizing environmental resistome risk. FEMS Microbiol Ecol 94: fiy079. 10.1093/femsec/fiy079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pysam 2023. Pysam. https://github.com/pysam-developers/pysam [accessed July 13, 2023].
- Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S, Dröge J, Gregor I, Majda S, Fiedler J, Dahms E, et al. 2017. Critical assessment of metagenome interpretation—a benchmark of metagenomics software. Nat Methods 14: 1063–1071. 10.1038/nmeth.4458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. 2006. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34: D32–D36. 10.1093/nar/gkj014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slizovskiy IB, Mukherjee K, Dean CJ, Boucher C, Noyes NR. 2020. Mobilization of antibiotic resistance: are current approaches for colocalizing resistomes and mobilomes useful? Front Microbiol 11: 1376. 10.3389/fmicb.2020.01376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slizovskiy IB, Oliva M, Settle JK, Zyskina LV, Prosperi M, Boucher C, Noyes NR. 2022. Target-enriched long-read sequencing (TELSeq) contextualizes antimicrobial resistance genes in metagenomes. Microbiome 10: 185. 10.1186/s40168-022-01368-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasinopoulos DM, Rigby RA. 2008. Generalized additive models for location scale and shape (GAMLSS) in R. J Stat Softw 23: 1–46. 10.32614/CRAN.package.gamlss [DOI] [Google Scholar]
- Waskom ML. 2021. seaborn: statistical data visualization. J Open Source Softw 6: 3021. 10.21105/joss.03021 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.