

Abstract
Rare or de novo structural variation, primarily in the form of copy number variants, is detected in 5%–10% of autism spectrum disorder (ASD) families. While complex structural variants involving duplications can generally be detected using microarray or short-read genome sequencing (GS), these methods frequently fail to characterize breakpoints at nucleotide resolution, requiring additional molecular methods for validation and fine-mapping. Here, we use Oxford Nanopore Technologies PromethION long-read GS to characterize complex genomic rearrangements (CGRs) involving large duplications that segregate with ASD in five families. In total, we investigated 13 CGR carriers and were able to resolve all breakpoint junctions at nucleotide resolution. While all breakpoints were identified, the precise genomic architecture of one rearrangement remained unresolved with three different potential structures. The findings in two families include potential fusion genes formed through duplication rearrangements, involving IL1RAPL1–DMD and SUPT16H–CHD8. In two of the families originating from the same geographical region, an identical rearrangement involving ANK2 was identified, which likely represents a founder variant. In addition, we analyze methylation status directly from the long-read data, allowing us to assess the activity of rearranged genes and regulatory regions. Investigation of methylation across the CGRs reveals aberrant methylation status in carriers across a rearrangement affecting the CREBBP locus. In aggregate, our results demonstrate the utility of nanopore sequencing to pinpoint CGRs associated with ASD in five unrelated families, and highlight the importance of a gene-centric description of disease-associated complex chromosomal rearrangements.
Autism spectrum disorder (ASD), which is observed in >1% of individuals worldwide, is a highly variable neurodevelopmental condition characterized by impaired social interaction and communication, and repetitive or restricted behaviors. The heritability of ASD is estimated to be 70%–80% (Sandin et al. 2017). Studies show that the genetic contribution to ASD is very heterogeneous and that the contribution of genetic variation to ASD risk ranges from variants with very small effects (Weiner et al. 2017) to nearly Mendelian forms caused by structural variation (SV) or de novo mutations (Wilfert et al. 2021). Currently, there are more than 100 genes associated with ASD (Rolland et al. 2023). Rare or de novo single nucleotide variants (SNVs) and SVs are found in up to 15% of ASD families, but for the remaining >80% of families, the underlying genetic contributors remain to be resolved.
SVs are genomic alterations that involve changes in the DNA sequence of at least 50 bp, including deletions, duplications, insertions, inversions, and translocations. SVs are common (∼20,000/individual) and constitute the largest proportion of genetic variation between individuals (Feuk et al. 2006). Established risk-conferring SVs are identified in 6%–7% of ASD families, with de novo events identified more frequently in simplex families. Meanwhile, rare inherited SVs are more frequently identified in multiplex families, often inherited from an unaffected parent (Wilfert et al. 2021). While most SVs are <10 kbp in size, large and rare SVs are more frequently found in individuals with ASD than in the general population (Pinto et al. 2010). SVs spanning hundreds of kilobase pairs often impact more than one gene, and it is not uncommon that such events are associated with additional phenotypes.
Complex genomic rearrangements (CGRs) are SVs that involve multiple breakpoints and/or are made up of more than one SV in cis (Schuy et al. 2022). This complexity makes their identification and characterization significantly more challenging (Nazaryan-Petersen et al. 2018). CGRs can be caused by various mechanisms, including chromoanasynthesis, which is characterized by chains of duplications and deletions. Chromoanasynthesis is believed to take place during DNA replication and is commonly caused through a series of fork-stalling and template-switching events (Zepeda-Mendoza and Morton 2019). Previously, CGRs were considered to be infrequent occurrences in germline cells. However, thanks to advancements in genome sequencing (GS), we now understand that CGRs are not only abundant but in fact are present across all genomes (Collins et al. 2020). Recent studies have also shown that CGRs are common in rare diseases (Lindstrand et al. 2019; Plesser Duvdevani et al. 2020), potentially contributing to the phenotypic variability between individuals with similar copy number variants (CNVs) (Marshall et al. 2008). Although CGRs are important contributors to genetic disorders, they are still poorly understood. This lack of understanding is partially due to the low-resolution CNV analyses commonly used in genetic diagnostics, including exome sequencing and microarray, as well as the complexity of resolving CGRs using short-read GS (srGS) (Schuy et al. 2022). CGRs involving large duplicated segments are particularly challenging to resolve using srGS, since paired-end mapping cannot span the duplicated segments and coverage analysis often cannot give precise breakpoint junctions (BPJs). Breakpoints resolved at nucleotide resolution are necessary to fully resolve the structure of CGRs involving duplications. Identification of the breakpoints is also important to infer the potential consequences of the duplication, which can position genes in new regulatory environments or lead to the formation of fusion transcripts. Fine-mapping the breakpoints of complex duplications can additionally facilitate inference of the mutational mechanism that drove their formation (Carvalho and Lupski 2016). While array methods and srGS struggle to resolve CGRs, it is less challenging using long-read GS (lrGS) approaches.
In this study, we employed Oxford Nanopore Technologies (ONT) lrGS to analyze five families where one or more children were diagnosed with ASD. All included families harbor an inherited CGR involving duplications which had been identified by srGS, but where the architecture of the genomic rearrangement and breakpoints could not be fully elucidated from srGS data (Trost et al. 2022). Using ONT lrGS, we resolve the CGRs structure and study the effects on methylation patterns to unravel both disease-causing and CGR-forming mechanisms.
Results
In total, 20 samples were sequenced, including probands and parents, as well as several affected and unaffected siblings. The pedigrees for the families are shown in Figure 1. The lrGS generated an average of 77 Gb data per sample, with an average read-length of 12 kbp; resulting in an average coverage of 25×. Running our SV pipeline, we identified 14,109–21,831 SVs per sample. Focusing on the previously identified CGRs, all breakpoints were spanned by nanopore reads, and rearrangements in four of the five families could be fully resolved (Table 1). In the five families, we detect four unique CGRs, with one rearrangement shared between family 1 and family 2, and the remainder unique to each family. Breakpoint spanning reads displayed in Integrative Genomics Viewer (IGV) format are shown in Supplemental Figure S1 for each unique CGR. The CGRs are all complex duplications, consisting of duplicated fragments that are chained together by two BPJs. None of the CGRs include deletions. The largest CGR is present in families 1 and 2 and spans nearly 5 Mb, while the smallest CGR, found in family 4, consists of <30 kbp of duplicated sequence. Most of the CGRs affect only a few genes, however, all rearrangements affect at least one gene previously associated with ASD (Table 1).
Figure 1.

Families selected for whole genome nanopore sequencing. Pedigrees of the families included in the study. Filled symbols indicate individuals diagnosed with ASD. Individuals selected for sequencing are denoted with an asterisk (*), and individuals previously shown to carry chromosomal rearrangements are marked with a hashtag (#).
Table 1.
Summary of the detected rearrangements
| Families | Copy number profile | HGVS | Duplicated genes |
|---|---|---|---|
| 1,2 | DUP-TRIP-DUP | chr4:g.113954280_qterdelins[ chr4:g.113912749_113915113inv; chr4:g.111100841_qter] |
ELOVL6, ENPEP, PITX2, FAM241A, AP1AR, TIFA, ALPK1, NEUROG2, ZGRF1, LARP7, hsa-miR-297, ANK2 |
| 3a | DUP-NML-DUP | chrX:g.29570408_qterdelins[ chrX:g.33038509_33426916inv; chrX:g.29020059_qter] |
IL1RAPL1, DMD |
| 4 | DUP-NML-DUP | chr14:g.21879481_qterdelins[ chr14:g.21501001_21502976; chr14:g.21849620_qter] |
RNASE13, SUPT16H, CHD8 |
| 5 | TRIP-QUINT-TRIP | chr16:g.3899656_qterdelins[ chr16:g.3689781_3762460inv; chr16:g.3688503_3899656; chr16:g.3689781_3762460inv; chr16:g.3688503_qter] |
DNASE1, TRAP1, CREBBP |
Genes associated with autism in bold. (HGVS) Human Genome Variation Society.
aCGR architecture not conclusively resolved.
Analysis of the BPJs of the CGRs (Table 2) shows that all BPJs except one carry microhomologies. The longest microhomology is observed in the CGR of family 3 and consists of 175 bp. The majority of BPJs carry microhomologies that are 3 nt long. Only one BPJ is lacking microhomologies, this BPJ was found in the CGR of families 1 and 2, and consists of an 8 nt stretch of seemingly random sequence. Two BPJs are located within matching repeats: BPJ 1 in family 3, as well as BPJ 1 in family 5. In both cases, the matched repeats are Alu elements. None of the BPJs carry signatures of small deletions.
Table 2.
Breakpoint junction characteristics of the four CGRs
| Families | BPJ | Chr | PosA | PosB | GeneA | GeneB | INS | Homology | PosA repeat | PosB repeat |
|---|---|---|---|---|---|---|---|---|---|---|
| 1,2 | 1 | 4 | 111100841 | 113912749 | ELOVL6 | ANK2 | – | AA | – | MLT1A |
| 1,2 | 2 | 4 | 113915113 | 113954280 | ANK2 | ANK2 | 8 nt | – | AluSz | – |
| 3 | 1 | X | 29020059 | 33038509 | IL1RAPL1 | DMD | – | 175 nt | AluSx3 | AluSz |
| 3 | 2 | X | 29570408 | 33426916 | IL1RAPL1 | – | – | AAT | LTR48B | – |
| 4 | 1 | 14 | 21501001 | 21879481 | RNASE13 NDRG2 | CHD8 | – | TGAA | L1MD2 | – |
| 4 | 2 | 14 | 21502976 | 21849620 | RNASE13 NDRG2 | SUPT16H | – | GTT | – | – |
| 5 | 1 | 16 | 3688503 | 3689781 | – | – | – | GGC | AluY | AluJo |
| 5 | 2 | 16 | 3762460 | 3899656 | TRAP1 | CREBBP | – | TCA | – | FLAM_C |
(INS) Insertion.
Discovery of a founder CGR in the Newfoundland population
Analysis of the lrGS data of families 1 and 2 shows that the identified ANK2 rearrangements are nearly identical, sharing the exact same breakpoint features (Tables 1, 2), and overall structure (Fig. 2A,B). No blood connection between family 1 and family 2 could be established; however, both originate from Newfoundland, Canada, indicating that the CGR could be a founder variant in that region (Rahman et al. 2003; Kaurah et al. 2007; Chan et al. 2022). A haplotype analysis was conducted using 2701 SNVs located within the duplicated region, as well as upstream and downstream segments. Specifically, SNVs were obtained by computing the intersect of SNVs within the duplicated region (4:111100841–113954280) that were shared by all the carriers in family 1. Subsequently, we evaluated the proportion of those SNVs that were present in the other individuals (Fig. 2C). The ANK2 CGR carriers of family 2 carry 96% of the SNVs identified in family 1, a significant overrepresentation of shared alleles (P = 0.01471, Wilcoxon rank-sum test). In comparison, analysis of SNVs in upstream and downstream segments of the same size show lower levels of shared SNVs, although there is a tendency toward elevated sharing with members of family 2 (Supplemental Fig. S2). Variant sharing genome-wide is not increased between family 1 and family 2, when compared with the remaining families (P = 0.0882, Wilcoxon rank-sum test). The high degree of variant sharing within the duplicated regions suggests that the CGR in both families descend from a common ancestor.
Figure 2.

Rearrangement overlapping ANK2. A CGR partially overlapping ANK2 was identified in two unrelated Newfoundland families. (A) A schematic representation of the rearrangement. (B) IGV screenshots showcase the breakpoint region at the start of segment B (Chr 4: 111,100 kbp) in the mothers of families 1 and 2. (C) Fraction of ANK2 haplotype SNVs present in the CGR genomic region indicates a common ancestor in families 1 and 2.
The CGR of families 1 and 2 includes a triplicated region of 2 kbp (blue fragment C, Fig. 2A) and a duplication of 5 Mb (blue fragments B and D, Fig. 2A). The rearrangement duplicates 12 genes, including the first two exons of ANK2, which has previously been associated with ASD. Notably, the duplication also includes a miRNA (hsa-miR-297). Performing a target prediction analysis in miRDB (https://www.mirdb.org), we find 599 potential targets of this miRNA. Assessment of the function of these targets through analysis in STRINGdb (https://string-db.org/) reveals that these 599 targets are enriched in genes expressed in the central nervous system (n = 237, false discovery rate [FDR] = 8 × 10−7) and brain (n = 233, FDR = 1 × 10−6), as well as genes associated with ASD (monarch, HP:0000717, n = 15, FDR = 0.0077).
A putative DMD–IL1RAPL1 fusion transcript in an X-linked recessive disorder
The CGR identified in family 3 consists of two duplications, sized 500 and 400 kbp (Table 2). There are three possible solutions to the architecture of this region with the breakpoints identified (Supplemental Fig. S3). While we do have long reads spanning each breakpoint, resolving the order of segments would require reads spanning across the >400 kb duplication, which we unfortunately do not have in the data set. The most parsimonious solution is that the blocks are arranged in a DUP-NML-DUP structure (Fig. 3A), where NML denotes a normal copy number. However, two other possible rearrangements with larger inverted blocks are also possible. The three theoretical structures affect two genes, IL1RAPL1 and DMD. Exon 1 of DMD is located within duplication 2, while IL1RAPL1 spans duplication 1 completely. Analyzing the positioning of these duplications, we note that a potential in-frame DMD–IL1RAPL1 fusion gene is formed (Fig. 3B) for two of the three possible structures. This fusion gene consists of DMD exon 1, and exons 3–11 of IL1RAPL1. The normal DMD copy is unaffected by the CGR, while IL1RAPL1 is truncated. The third potential CGR structure would disrupt the DMD gene. It could potentially create a shorter truncated transcript including exon 1 of DMD and exons 3–6 of IL1RAPL1, but would lack necessary 3′ structures. Primers were designed to target the fusion transcript, but neither the fusion transcript nor DMD is expressed in the lymphoblastoid cell line (LCL).
Figure 3.

Rearrangement overlapping DMD and ILRAPL1. The DMD–IL1RAPL1 fusion gene was detected in family 3. (A) A schematic representation of one of three potential rearrangement architectures, revealing the disruption of IL1RAPL1, and formation of a DMD–IL1RAPL1 fusion gene. (B) The putative DMD–IL1RAPL1 fusion gene, consisting of DMD exon 1, and IL1RAPL1 exons 3–11.
Family 3 consists of seven individuals, with five carrying the CGR including the unaffected mother, two unaffected daughters, and two affected boys, consistent with X-linked recessive inheritance. We next explored differences in methylation of the two alleles in one unaffected daughter. First, we phased the lrGS data and then we analyzed informative methylation sites at the promoter region of DMD. Through this analysis we obtained one informative methylation site having 40× coverage (Supplemental Fig. S4). The paternal allele had a coverage of 12×, with nine reads indicating unmethylated promoter, and three indicating methylated promoter. The maternal allele had a coverage of 28×, with 11 reads indicating unmethylated promoter, and 17 indicating methylated promoter. Applying a χ2 test, we obtain a P-value of 0.038, indicating that the DMD promoter of the maternal allele is preferentially methylated compared to the paternal allele.
Discovery of a novel SUPT16H–CHD8 fusion transcript
In family 4, the CGR involves a duplication of the CHD8 gene. The detailed lrGS analysis reveals a duplicated segment of 30 kbp that spans the 3′ half of CHD8 (exons 10–37) and the first exon of the adjacent SUPT16H gene (Fig. 4A). At the duplication BPJ, there is a 2.3 kbp segment including exon 1 and part of exon 2 of the gene RNASE13 (located 387 kbp proximal) inserted. This rearrangement enables the formation of putative fusion transcripts between SUPT16H–RNASE13 as well as SUPT16H–CHD8. To investigate these putative fusion transcripts further, primer sets were designed corresponding to the possible alternatively spliced transcripts of SUPT16H and CHD8, and between SUPT16H and RNASE13 (Fig. 4B). Polymerase chain reaction (PCR) was run on RNA extracted from LCLs established from the proband. A SUPT16H–RNASE13 transcript is not expressed at detectable levels, but primers in exon 1 of SUPT16H and the CHD8 exons 11–12 junction verify an expressed SUPT16H–CHD8 fusion transcript. Sequencing the product reveals a direct splicing event between exon 1 of SUPT16H and exon 10 of CHD8 (Fig. 4C). Using quantitative reverse transcription polymerase chain reaction (RT-PCR), we observe an ∼1.5-fold increase in expression of the duplicated CHD8 exons in all three duplication carriers and infer that gene expression from the SUPT16H–CHD8 fusion promoter is of similar levels to that of wild-type CHD8 (Fig. 4D). Meanwhile, expression of the CHD8 exons upstream of the duplication breakpoints approximates that of sex-matched and family controls in all three carriers. The predicted protein structure corresponding to the SUPT16H–CHD8 fusion gene encodes a region involved in the interaction of CHD8 with FAM124B, and one partial chromodomain of the two that are present in the wild-type CHD8 protein.
Figure 4.
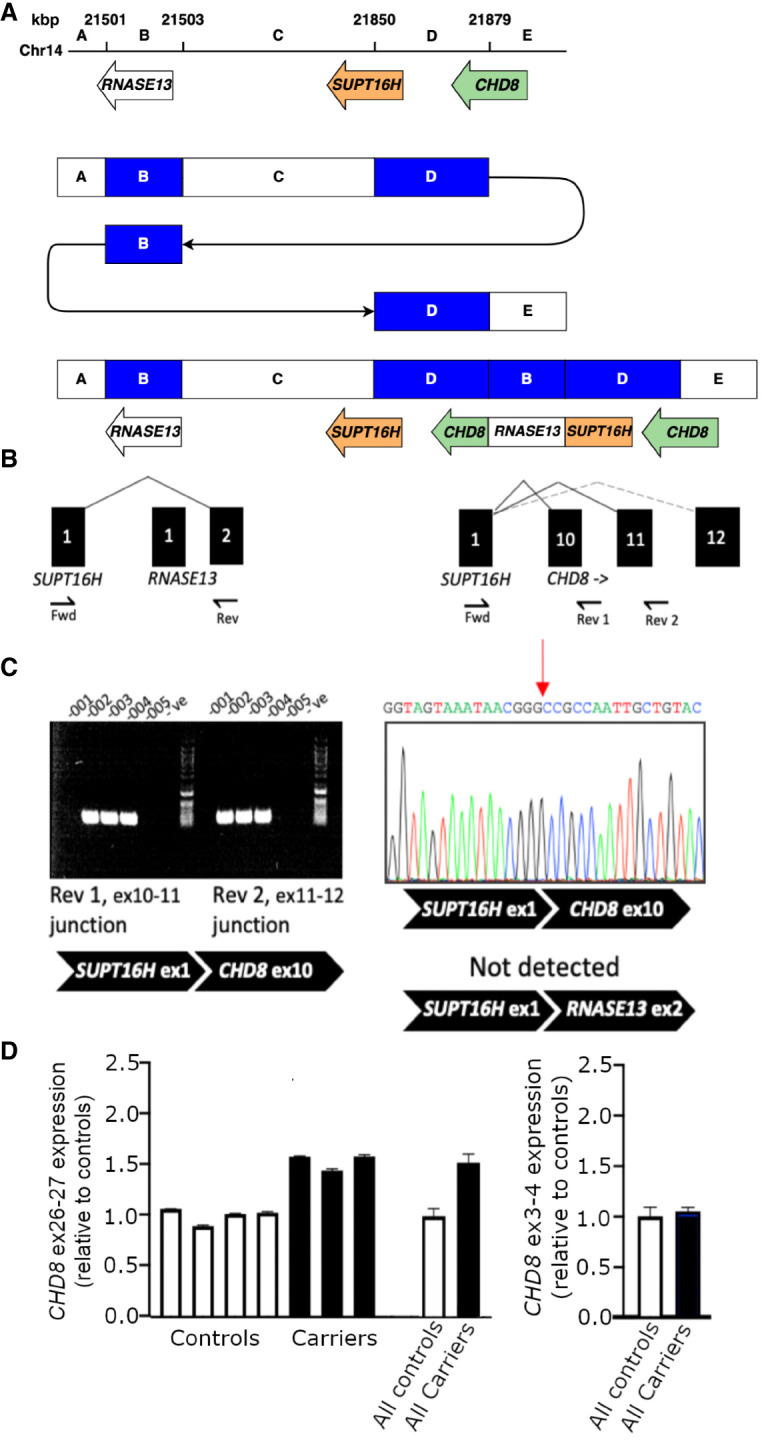
SUTP16H–CHD8 fusion transcript. Functional analysis of the CGR affecting CHD8. (A) Schematic representation of the SUTP16H–CHD8 rearrangement. (B) Alternately spliced products are predicted from the gene fusion. Predicted in-frame (solid line) and frameshift (dotted line) products are indicated. A targeted Fwd–Rev 1 assay was used in addition to a Fwd–Rev 2 assay, the latter being sufficient to detect both potential splicing events. (C) The amplification of PCR products was used to assay SUPT16H–CHD8 exon 10 and exon 11 splicing events. The sequence corresponding to the detected splicing event between SUPT16H exon 1 and CHD8 exon 10 is shown. (D) Average relative fold-change in gene expression of exons within the duplicated 3′ end of CHD8, and upstream exons.
A CGR involving a partial triplication of CREBBP results in abnormal methylation patterns
The CGR carried by the proband and his father in family 5 unravel a TRIP-QUINT-TRIP rearrangement with a 70 kbp quintuplication and a 210 kbp triplication overlapping DNASE1, TRAP1 and most of CREBBP (Fig. 5A). Read-depth indicates a total of four copies of a portion of the CREBBP gene, while a small region (labeled C in Fig. 5A) is present in six copies. The CREBBP copy number was also confirmed by digital qPCR. The breakpoint read coverage also supports the proposed structure.
Figure 5.

A CREBBP rearrangement with altered methylation. (A) Schematic of a complex rearrangement affecting CREBBP. (B) CpG methylation across the region in the proband (yellow), the carrier father (orange), and all other samples in this study. Methylation signal expressed as a mean number of methylated calls in rolling windows of 150 CpG sites. Note how hypomethylation regions are associated with gene promoters, and there is an overall decreased methylation level across the rearrangement area in the proband and his father. (C) Analysis of significantly differentially methylated loci between proband and all individuals who do not carry the CREBBP CGR (P < 0.01).
Interestingly, analysis of the CpG methylation across the rearrangement shows decreased methylation across the CGR region in the proband (Fig. 5B), when compared to all individuals from the other families. Specifically, the differential methylation was significant near the proximal BPJ and across the CREBBP gene body (Fig. 5C). The carrier father, who does not show symptoms of ASD, shows an intermediate methylation level between the patient and all other samples across the region. The proband does not show a general deviation across the genome and other probands do not show deviations in methylation across their CGR regions or across the candidate gene promoters (Supplemental Fig. S5).
Discussion
In this study, we use ONT lrGS to resolve neurodevelopmental disorder (NDD)-associated CGRs in five families where the proband has been diagnosed with ASD. The CGRs were all inherited from an unaffected parent (three maternal and two paternal). They were transmitted to six affected children (five males and one female) and six unaffected children (one male and five females). The variants are expected to be unique, or represent very recent founder mutations. None of the variants are present in reference variant databases (gnomAD, 1000 Genomes, or Human Pangenome Reference Consortium data set). All variants overlap genes previously associated with NDD and/or ASD. For four families, we were able to fully resolve the CGR at nucleotide resolution, providing additional insight into the formation and consequences of the rearrangements. For the family with a CGR overlapping IL1RAPL1, all breakpoints could be resolved, but without reads spanning the entire rearrangement (>400 kbp), it is not possible to determine the exact rearrangement structure. Overall, our results show that lrGS is a valid next step to define breakpoints and provide further insight into rearrangements when initial findings by chromosomal microarray (CMA) or srGS identify a CGR that cannot be fully resolved.
Both family 1 and family 2 carry the exact same rearrangement even though we were not able to identify a shared blood relative. SNV analysis indicates that the two families are not closely related, but show increased variant sharing across the duplicated segment. The data point to the CRG as a recent founder mutation, but since there are variants in the 5 Mb duplicated segment of the CGR that differ between the family 1 and family 2, the common ancestor was likely several generations ago. Because de novo mutations have been shown to cluster and are associated with large SVs (Michaelson et al. 2012), it is challenging to estimate its age. In these families, the CGR was inherited from unaffected mothers. Interestingly, we note that ASD or subclinical autistic traits are reported among relatives on the maternal side in both families. Since both carrier mothers showed no developmental or neuropsychiatric symptoms, we conclude that the rearrangement of the ANK2 region is a strong risk factor for ASD. The duplications involved in the CGR of families 1 and 2 affect several genes; however, ANK2 is the only one previously associated with ASD. While the primary association with ASD is with loss of function or deletion of ANK2, there are also reports where individuals with ASD show a gain of ANK2 (Kaminsky et al. 2011; Leppa et al. 2016). Variants in ANK2 have also been associated with cardiac disease, including both arrhythmia and structural heart disease. We note that the proband in family 1 is diagnosed with congenital heart disease, while no heart phenotype is reported in either the carrier mother or family 2. Interestingly, there is also a miRNA (hsa-miR-297) located in the duplicated region (Table 1). Through target enrichment analysis and STRING analysis, we identified a significant enrichment for ASD-related genes and genes expressed in the brain among the targets of this miRNA. This suggests that the gain of an extra hsa-miR-297 copy could be linked to ASD. Emerging research suggests that genetic variation in microRNA plays an important role in ASD (Wong et al. 2022; Garrido-Torres et al. 2024). We, therefore, recommend further studies on microRNA in both ASD and other NDDs.
Fusion genes, commonly found in cancers, have a less understood role in ASD, and only a few cases have been reported in the literature (Holt et al. 2012). Here, we report two potential fusion genes: DMD–IL1RAPL1 in family 3 and CHD8–SUPT16H in family 4. A fusion of DMD–IL1RAPL1 has been reported previously in an individual with ASD (Jin et al. 2000); however, its contribution to the participant's phenotype remains uncertain as it could potentially be explained by the IL1RAPL1 truncation alone. IL1RAPL1 is involved in synaptogenesis and both nonsynonymous and loss-of-function variants have been associated with both ASD and intellectual disability (Piton et al. 2008; Ramos-Brossier et al. 2015). While there were three possible solutions to the family 3 rearrangement, we argue that either of the two that do create an in-frame DMD–IL1RAPL1 fusion is more likely. The proband phenotype would be expected from an IL1RAPL1 disruption, while loss of function of the DMD gene (created by the third possible solution), does not match the phenotype of the patient. CHD8–SUPT16H fusions have not been previously reported in ASD, but there are numerous reports of microduplications overlapping the two genes based on CMA analysis (Smol et al. 2020). However, based on CMA data alone it is not possible to characterize fusion genes. Given that both CHD8 (Dingemans et al. 2022) and SUPT16H (Bina et al. 2020) are previously reported to be associated with ASD, with numerous reports of truncating sequence variants, we propose that this fusion gene could play an important role in the dysregulation of these genes. Overall, our findings suggest that gene fusions from CGRs could be important contributors to ASD risk, and underscores the necessity of lrGS to fully resolve such events.
The occurrence of CGRs can be explained by three major mechanisms: chromothripsis, chromoplexy, and chromoanasynthesis collectively called chromoanagenesis. The characteristics of the CGR BPJs may give clues as to which specific mechanism was involved (Zepeda-Mendoza and Morton 2019). The four unique CGRs all share a distinct pattern with two clustered copy number gains and two BPJs (Figs. 2–5). All cases show mutational signatures suggestive of a replication-based mechanism; microhomology was observed in seven BPJs (range 2–175 nt, median 3 nt, Table 2), and in the only BPJ lacking microhomology an 8 nt poly(A) stretch was present (families 1, 2 CGR BPJ 2, Table 2). Such findings suggest that the rearrangement formed through fork stalling and template switching (FoSTes) (Lee et al. 2007), which commonly underlie chromoanasynthesis. Short microhomologies can also be consistent with nonhomologous end-joining (NHEJ) that is involved in chromothripsis and chromoplexy. And although NHEJ by itself would not generate large duplications, previous work has shown that FoSTes and NHEJ may operate simultaneously and independently (Nazaryan-Petersen et al. 2018). Of note, the majority (81%, 13/16) of the breakpoints are located within protein-coding genes (Table 2). This may be due to our selection criteria, but could also be a result of the formation process if the CGRs formed during the active transcription of these genes. We have previously reported that multiple types of chromoanagenesis, such as chromoplexy and chromoanasynthesis, may happen simultaneously (Eisfeldt et al. 2021).
Each of the five families studied here carried a CGR initially detected with CMA or srGS and characterized here in detail with lrGS. Our results show that using lrGS alone, without any additional data, the breakpoints could be fully resolved at nucleotide resolution, showing the power of lrGS for resolving CGRs. We performed a comprehensive genomic analysis including structural variant calling, methylation analysis, and phasing. The significance of resolving these CGRs varied among the families. For families 3 and 4, the discovery of potential fusion genes, made possible by lrGS, provided further insight into a possible disease mechanism. For families 3 and 5, the additional layer of epigenetic information provided by nanopore sequencing gave further information about the consequences of the rearrangements. In family 3, the DMD–IL1RAPL1 rearrangement on Chr X seems to lead to a skewed methylation pattern in the female carriers. In family 5, we see a significant difference in methylation across the CREBBP region in the rearrangement carriers. While CREBBP duplication has previously been associated with ASD (Demeer et al. 2013), it is possible that the level of methylation functions as a modifier of risk, potentially contributing to the difference in phenotype between the proband and the CGR carrier father. While the methylation signals identified in families 3 and 5 are interesting to observe, we acknowledge that we cannot conclude from our data whether the differential methylation has any consequences for gene function or for the phenotype of the affected individuals. However, lrGS methylation signals represent a relatively new type of data that will become increasingly generated in rare disease patients, and hence it is important to report these observations to help build a knowledge base for future interpretation.
For ONT lrGS analysis, our workflow was based on a combination of previously published tools and required only a single SV caller. In contrast, the phasing and methylation analyses appear less mature. We utilized Whatshap (Garg et al. 2016) for phasing, and despite its effective performance across regions of normal copy number, it was unable to phase the CGRs in a reliable manner, likely because it assumes diploid and even copy numbers across the genome. Consequently, we applied a custom haplotype analysis in families 1 and 2 (Fig. 2C). We also attempted de novo assembly using Shasta, but only part of the genomes could be successfully assembled and phased. We believe the limited success with assembly is due to the relatively high error rate of the ONT data. To uncover methylation patterns in the ONT lrGS data we utilized Nanopolish, with Integrative Genomics Viewer (IGV) used for visualization. While this workflow was adequate for our targeted analyses, we acknowledge that there are newer and more comprehensive tools now available. Even so, we do see a need for a more comprehensive lrGS differential methylation tool for rare disease analysis, as well as large reference data sets for calibration and filtering. Looking at the methylation across the genome, there are regions showing large variation in the population and others where all samples are similar. A high-quality population reference methylation map should ideally contain thousands of samples, which would facilitate the identification of true outliers and could be used for de novo methylation differentiation discovery. Differentially methylated regions may in turn highlight loci where regulatory mutations are located. Overall, we note that the development of lrGS analysis tools is currently a rapidly changing and maturing field, but further development of standardized and benchmarked pipelines for clinical use is still warranted. Haplotype-resolved de novo assembly could potentially address some remaining challenges, as it facilitates phased variant discovery of all variant types and allows for graph genome analyses (Cheng et al. 2021).
There is a general agreement in the field that lrGS has matured to a point where it has the potential to replace srGS for whole genome sequencing (WGS) analysis in rare diseases. However, several challenges remain for the widespread adoption of lrGS in clinical diagnostics. It is still prohibitively costly for routine analysis for most applications. While new pipelines are now emerging, the field is still immature when it comes to using the full information content in lrGS for clinical interpretation. The amount and quality of DNA may also be an obstacle for some applications, although our experience is that standard extractions work well enough for lrGS. Currently, for most healthcare systems, the potential increase in diagnostic yield does not outweigh the difference in price in terms of implementing lrGS as a first-tier genomic test. However, based on data quality, there is no doubt that future diagnostics will be performed using some form of lrGS. We argue though, based on this study as well as our experience from sequencing larger rare disease cohorts, that there is a role for lrGS in the clinic already today, and that is to sequence probands (or trios) with an NDD diagnosis where CGRs have been identified in prior analyses (srGS or CMA). As shown in this study, lrGS is excellent for resolving CGRs, identifying breakpoints, and giving the additional layer of methylation data. Full resolution of CGRs is important for clinical interpretation and carries value in providing a conclusive answer for the family. This would be the first step toward clinical use of lrGS, with the next step being a second-tier test for all probands where srGS is negative.
In conclusion, our study emphasizes the critical role of lrGS technologies, such as ONT lrGS, in elucidating the complexities of CNVs associated with ASD. We show that lrGS is a powerful strategy to fully resolve complex rearrangements using standardized pipelines, and that the results provide both BPJ information and methylation data that is valuable for interpretation of the variants. However, our work also underscores the need for more developed and accessible bioinformatic tools and workflows to fully leverage these technologies, particularly for tasks like phasing, methylation analysis, and de novo assembly. The ongoing development of such resources, is crucial for enhancing our understanding of SV and its role in ASD, along with other neurodevelopmental disorders. As we further refine these techniques and expand their application, we can anticipate significant strides in ASD research and, ultimately, patient care.
Methods
Participant material
The study focused on five families enrolled in the Autism Speaks MSSNG project, where DNA samples were previously sequenced using Illumina or Complete Genomics srGS (Yuen et al. 2017). Individuals were noted as having a diagnosis of ASD in two ways; (i) met the diagnostic criteria of the autism diagnostic interview-revised (ADI-R) or autism diagnostic observation schedule (ADOS), or (ii) received a clinical diagnosis from an expert physician following the diagnostic and statistical manual of mental disorders (DSM) 4 or 5 criteria (Yuen et al. 2017). Clinical information about each proband and their family members is summarized in Supplemental Table S1. Samples selected for sequencing are listed in Supplemental Table S2, along with their MSSNG SampleID and EGA file name information. Informed consent was obtained for all individuals, as approved by the research ethics boards at The Hospital for Sick Children in Toronto, McMaster University in Hamilton, Memorial University in St. John's, and McGill University in Montreal, where the families were recruited. Families were selected from the MSSNG samples based on the following criteria: originating from either of the four cohorts listed above, carried a complex genome rearrangement that included large duplicated segments, was inherited from an unaffected parent, and carried a rearrangement that had not been fully resolved with srGS using standard SV pipelines. A limited number of families fulfilled all these criteria. The pedigrees of the families are shown in Figure 1.
Epstein–Barr virus–transformed LCLs were previously established by The Centre for Applied Genomics (TCAG) biobanking facility for all samples included in the study. Cells were grown using standard protocols and pelleted for extraction of DNA and RNA. DNA was extracted with the Nanobind CBB Big DNA Kit (Part Number NB-900-001-01) according to the high molecular weight (HMW) protocol for Cultured Mammalian Cells in the Nanobind CBB Big DNA Kit Handbook v1.8. RNA was isolated from cell pellets using the Qiagen RNeasy micro kit and was kept at −80°C until use. First-strand cDNA was synthesized using the Quantabio qScript XLT cDNA SuperMix. The reverse transcription reaction was performed using 2000 ng of RNA per sample, following the manufacturer's protocol. cDNA samples were diluted 1:10 to a concentration of 10 ng/μL and stored at −20°C until use. Transcript abundance was quantitated from cDNA using TaqMan gene expression assays with droplet digital PCR (ddPCR). To limit the detection of genomic DNA, TaqMan assays with a probe spanning an exon junction of the target transcript were selected, and specificity for cDNA was confirmed using noRT controls. Reactions were multiplexed with a TBP endogenous control assay which was chosen for its stable expression levels in LCLs (Lossos et al. 2003). All samples were analyzed with triplicate ddPCR reactions.
For validation of the SUPT16H–CHD8 fusion transcript, primers were designed to amplify the exon junction of the predicted fusion (forward primer sequence: 5′ CTC TGG ACA AAG ACG CTT ATT AT 3′; reverse primer sequence: 5′ GAT GCA GTT CTG CCT GTT ATA C 3′). Forward and reverse primers anneal to exon 1 of SUPT16H and the exons 11–12 junction of CHD8, respectively. PCR reactions were performed using 20 ng of cDNA per sample and following standard protocols. The PCR product was visualized on a 2% agarose gel and was validated by Sanger sequencing.
Long-read sequencing and analysis
Nanopore sequencing was performed at National Genomics Infrastructure (NGI) Uppsala, using the PromethION platform (ONT, Oxford, UK). The sequencing library was prepared using the LSK-109 ligation kit and sequencing was performed using the R9.4 flow cell, with one flow cell per sample. Bases were called using the Guppy base caller (https://nanoporetech.com). Methylated bases were detected using Nanopolish (Loman et al. 2015), and visualized using Integrative Genomic Viewer (IGV) (Thorvaldsdóttir et al. 2013).
The resulting lrGS data were aligned to hg19 using minimap2 (Li 2018), and SVs were called using Sniffles (Sedlazeck et al. 2018), setting the minimum read support parameter to three reads. For downstream filtering and annotation purposes, we used hg19, as some annotations used by our clinical collaborators have not yet been updated to work with later assemblies. To ensure that the use of an older assembly did not influence the results, we performed liftover (https://genome.ucsc.edu/cgi-bin/hgLiftOver) and converted all breakpoint regions ±5 kbp between hg19 and GRCh38, requiring >99% identity and no rearrangements. All regions could be converted properly, indicating that for the regions analyzed in our study, the choice of assembly would not affect the conclusions of the study. SV calls shorter than 2 kbp were considered indels (insertions/deletions) and were, therefore, removed. Lastly, the SVDB toolkit (Eisfeldt et al. 2017) was used to construct a frequency database from noncarriers of the known duplications in each family. This database was utilized to remove common variation and sequencing artifacts from the variant lists of the affected individuals.
SNVs were called using BCFtools (Li 2011), which was run according to the recommendations for ONT lrGS data (-X ont). Phasing was performed using Whatshap (Garg et al. 2016), which was run using high-quality heterozygote SNVs (quality >30, read support >10) found in gnomAD (allele frequency >0.05).
Strand-aggregated methylation frequencies were created from Nanopolish data using modkit 0.3.0 (https://github.com/nanoporetech/modkit), with a GENCODE Release 19 (GRCh37.p13) GFF3 file. The resulting modkit pileups, DDS files, together with the gene annotations were input to a custom R script, where differentially methylated loci (DML) were calculated using DSS_2.48.0 (https://www.bioconductor.org/packages/devel/bioc/vignettes/DSS/inst/doc/DSS.html#12_Citation). Smoothing was done over 500 bp windows (per default) and DML was called using a P-value threshold of 0.01. The R script used to calculate and plot methylation (Fig. 5B,C) is available at GitHub (https://github.com/fellen31/nanopore_paper_figure/blob/master/nanopore_paper_figure.Rmd) and as Supplemental Code.
Resolving the complex genomic rearrangements
The structure of the CGRs was solved manually according to a previously published procedure (Eisfeldt et al. 2021). The structure of a CGR is obtained by finding the DNA fragments (segments) involved in the CGR, as well as their order and orientation. The search for the CGR structure is initiated by finding a set of BPJs defining the segments of the CGR; these BPJs are the calls found in the filtered Sniffles VCF, as described previously. The calls are inspected in IGV (Supplemental Fig. S1).
Next, the segments are defined; each BPJ consists of two breakpoints (called position A and position B, Table 2), the segments are the genomic regions located between these breakpoints; in CGRs involving only one chromosome, the first segment, A is located between the p-terminus and the first breakpoint detected, fragment A is arbitrarily decided to have normal (+) orientation, fragments inverted relatively to A is decided to have inverted (−) orientation. Each segment is associated with a copy number, which is obtained by dividing the read-depth of the segment to the median genomic read-depth and multiplying by 2 (i.e., the ploidy of the human genome), the obtained copy number is verified based on the allele frequencies of SNV within the segments, as well as the number of split reads at the BPJ.
Lastly, the structure of the rearrangement is determined; this is done by forming a chain of segments. The segments are linked by the BPJs, and the number of copies of each segment is decided according to their copy numbers. The chain starts at segment A, and is followed by the segment sharing BPJ with segment A; this walk continues until all fragments are spent according to their copy number. Notably, not all walks result in unique chains: resulting in multiple potential CGR solutions. A CGR may need to be revisited multiple times to account for junctions missed by the callers.
Data access
All raw sequencing data generated in this study have been submitted to the European Genome-phenome Archive (EGA; https://ega-archive.org/) under accession number EGAS50000000390. Sample IDs and file names are summarized in Supplemental Table S2.
Supplemental Material
Acknowledgments
We thank the families for their participation in this long-term study which involved multiple sample collections to be tested on different technologies. We acknowledge Mayada Elsabbagh (McGill University) and Peter Szatmari (McMaster University) for clinical work-up and referral of families. Library preparation and sequencing were performed at the SciLifeLab National Genomics Infrastructure in Uppsala (Uppsala Genome Center). Computational analyses were performed on resources provided by SNIC through the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under the SNIC projects sens2019551. The project was partially funded by a SciLifeLab Technology Development Project Grant (L.F.), the Swedish Research Council Dnr 2017-01861 (L.F.) and Dnr 2019-02078 (A.L.), and Hjärnfonden FO2021-0302 (L.F.). A special thank you to Stiftelsen Bergmangårdarna for providing accommodation and an environment for writing and analysis.
Author contributions: A.L., S.W.S., and L.F. conceptualized the study. J.E., A.L., S.W.S., and L.F. were major contributors to writing the manuscript. E.J.H., J.H., B.A.F., and S.W.S. provided patient samples and clinical information of patients. E.J.H. and J.H. performed functional wet laboratory analyses. J.E. and F.L. performed bioinformatics analysis. All authors have read, edited, and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.279263.124.
Freely available online through the Genome Research Open Access option.
Competing interest statement
The authors declare no competing interests.
References
- Bina R, Matalon D, Fregeau B, Tarsitano JJ, Aukrust I, Houge G, Bend R, Warren H, Stevenson RE, Stuurman KE, et al. 2020. De novo variants in SUPT16H cause neurodevelopmental disorders associated with corpus callosum abnormalities. J Med Genet 57: 461–465. 10.1136/jmedgenet-2019-106193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CMB, Lupski JR. 2016. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17: 224–238. 10.1038/nrg.2015.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AJS, Engchuan W, Reuter MS, Wang Z, Thiruvahindrapuram B, Trost B, Nalpathamkalam T, Negrijn C, Lamoureux S, Pellecchia G, et al. 2022. Genome-wide rare variant score associates with morphological subtypes of autism spectrum disorder. Nat Commun 13: 6463. 10.1038/s41467-022-34112-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Concepcion GT, Feng X, Zhang H, Li H. 2021. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18: 170–175. 10.1038/s41592-020-01056-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, Khera AV, Lowther C, Gauthier LD, Wang H, et al. 2020. A structural variation reference for medical and population genetics. Nature 581: 444–451. 10.1038/s41586-020-2287-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeer B, Andrieux J, Receveur A, Morin G, Petit F, Julia S, Plessis G, Martin-Coignard D, Delobel B, Firth HV, et al. 2013. Duplication 16p13.3 and the CREBBP gene: confirmation of the phenotype. Eur J Med Genet 56: 26–31. 10.1016/j.ejmg.2012.09.005 [DOI] [PubMed] [Google Scholar]
- Dingemans AJM, Truijen KMG, van de Ven S, Bernier R, Bongers EMHF, Bouman A, de Graaff-Herder L, Eichler EE, Gerkes EH, De Geus CM, et al. 2022. The phenotypic spectrum and genotype-phenotype correlations in 106 patients with variants in major autism gene CHD8. Transl Psychiatry 12: 421. 10.1038/s41398-022-02189-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisfeldt J, Vezzi F, Olason P, Nilsson D, Lindstrand A. 2017. TIDDIT, an efficient and comprehensive structural variant caller for massive parallel sequencing data. F1000Res 6: 664. 10.12688/f1000research.11168.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisfeldt J, Pettersson M, Petri A, Nilsson D, Feuk L, Lindstrand A. 2021. Hybrid sequencing resolves two germline ultra-complex chromosomal rearrangements consisting of 137 breakpoint junctions in a single carrier. Hum Genet 140: 775–790. 10.1007/s00439-020-02242-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuk L, Carson AR, Scherer SW. 2006. Structural variation in the human genome. Nat Rev Genet 7: 85–97. 10.1038/nrg1767 [DOI] [PubMed] [Google Scholar]
- Garg S, Martin M, Marschall T. 2016. Read-based phasing of related individuals. Bioinformatics 32: i234–i242. 10.1093/bioinformatics/btw276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido-Torres N, Guzmán-Torres K, García-Cerro S, Pinilla Bermúdez G, Cruz-Baquero C, Ochoa H, García-González D, Canal-Rivero M, Crespo-Facorro B, Ruiz-Veguilla M. 2024. miRNAs as biomarkers of autism spectrum disorder: a systematic review and meta-analysis. Eur Child Adolesc Psychiatry 33: 2957–2990. 10.1007/s00787-023-02138-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt R, Sykes NH, Conceição IC, Cazier JB, Anney RJL, Oliveira G, Gallagher L, Vicente A, Monaco AP, Pagnamenta AT. 2012. CNVs leading to fusion transcripts in individuals with autism spectrum disorder. Eur J Hum Genet 20: 1141–1147. 10.1038/ejhg.2012.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Gardner RJ, Viswesvaraiah R, Muntoni F, Roberts RG. 2000. Two novel members of the interleukin-1 receptor gene family, one deleted in Xp22.1-Xp21.3 mental retardation. Eur J Hum Genet 8: 87–94. 10.1038/sj.ejhg.5200415 [DOI] [PubMed] [Google Scholar]
- Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, et al. 2011. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med 13: 777–784. 10.1097/GIM.0b013e31822c79f9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaurah P, Macmillan A, Boyd N, Senz J, Luca D, Chun A, Suriano N, Zaor G, Lori S, Manen V, et al. 2007. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 297: 2360. 10.1001/jama.297.21.2360 [DOI] [PubMed] [Google Scholar]
- Lee JA, Carvalho CMB, Lupski JR. 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247. 10.1016/j.cell.2007.11.037 [DOI] [PubMed] [Google Scholar]
- Leppa VMM, Kravitz SNN, Martin CLL, Andrieux J, Le Caignec C, Martin-Coignard D, DyBuncio C, Sanders SJJ, Lowe JKK, Cantor RMM, et al. 2016. Rare inherited and de novo CNVs reveal complex contributions to ASD risk in multiplex families. Am J Hum Genet 99: 540–554. 10.1016/j.ajhg.2016.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27: 2987–2993. 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrand A, Eisfeldt J, Pettersson M, Carvalho CMB, Kvarnung M, Grigelioniene G, Anderlid BM, Bjerin O, Gustavsson P, Hammarsjö A, et al. 2019. From cytogenetics to cytogenomics: whole-genome sequencing as a first-line test comprehensively captures the diverse spectrum of disease-causing genetic variation underlying intellectual disability. Genome Med 11: 68. 10.1186/s13073-019-0675-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Quick J, Simpson JT. 2015. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 12: 733–735. 10.1038/nmeth.3444 [DOI] [PubMed] [Google Scholar]
- Lossos IS, Czerwinski DK, Wechser MA, Levy R. 2003. Optimization of quantitative real-time RT-PCR parameters for the study of lymphoid malignancies. Leukemia 17: 789–795. 10.1038/sj.leu.2402880 [DOI] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, et al. 2008. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 82: 477–488. 10.1016/j.ajhg.2007.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, et al. 2012. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151: 1431–1442. 10.1016/j.cell.2012.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazaryan-Petersen L, Eisfeldt J, Pettersson M, Lundin J, Nilsson D, Wincent J, Lieden A, Lovmar L, Ottosson J, Gacic J, et al. 2018. Replicative and non-replicative mechanisms in the formation of clustered CNVs are indicated by whole genome characterization. PLoS Genet 14: e1007780. 10.1371/journal.pgen.1007780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al. 2010. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466: 368–372. 10.1038/nature09146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piton A, Michaud JL, Peng H, Aradhya S, Gauthier J, Mottron L, Champagne N, Lafrenière RG, Hamdan FF, Joober R, et al. 2008. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum Mol Genet 17: 3965–3974. 10.1093/hmg/ddn300 [DOI] [PubMed] [Google Scholar]
- Plesser Duvdevani M, Pettersson M, Eisfeldt J, Avraham O, Dagan J, Frumkin A, Lupski JR, Lindstrand A, Harel T. 2020. Whole-genome sequencing reveals complex chromosome rearrangement disrupting NIPBL in infant with Cornelia de Lange syndrome. Am J Med Genet A 182: 1143–1151. 10.1002/ajmg.a.61539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman P, Jones A, Curtis J, Bartlett S, Peddle L, Fernandez BA, Freimer NB. 2003. The Newfoundland population: a unique resource for genetic investigation of complex diseases. Hum Mol Genet 12: R167–R172. 10.1093/hmg/ddg257 [DOI] [PubMed] [Google Scholar]
- Ramos-Brossier M, Montani C, Lebrun N, Gritti L, Martin C, Seminatore-nole C, Toussaint A, Moreno S, Poirier K, Dorseuil O, et al. 2015. Novel IL1RAPL1 mutations associated with intellectual disability impair synaptogenesis. Hum Mol Genet 24: 1106–1118. 10.1093/hmg/ddu523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland T, Cliquet F, Anney RJL, Moreau C, Traut N, Mathieu A, Huguet G, Duan J, Warrier V, Portalier S, et al. 2023. Phenotypic effects of genetic variants associated with autism. Nat Med 29: 1671–1680. 10.1038/s41591-023-02408-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, Reichenberg A. 2017. The heritability of autism spectrum disorder. JAMA 318: 1182–1184. 10.1001/jama.2017.12141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuy J, Grochowski CM, Carvalho CMB, Lindstrand A. 2022. Complex genomic rearrangements: an underestimated cause of rare diseases. Trends Genet 38: 1134–1146. 10.1016/j.tig.2022.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, Von Haeseler A, Schatz MC. 2018. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 15: 461–468. 10.1038/s41592-018-0001-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smol T, Thuillier C, Boudry-Labis E, Dieux-Coeslier A, Duban-Bedu B, Caumes R, Bouquillon S, Manouvrier-Hanu S, Roche-Lestienne C, Ghoumid J. 2020. Neurodevelopmental phenotype associated with CHD8-SUPT16H duplication. Neurogenetics 21: 67–72. 10.1007/s10048-019-00599-w [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B, Thiruvahindrapuram B, Chan AJS, Engchuan W, Higginbotham EJ, Howe JL, Loureiro LO, Reuter MS, Roshandel D, Whitney J, et al. 2022. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell 185: 4409–4427.e18. 10.1016/j.cell.2022.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner DJ, Wigdor EM, Ripke S, Walters RK, Kosmicki JA, Grove J, Samocha KE, Goldstein JI, Okbay A, Bybjerg-Grauholm J, et al. 2017. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet 49: 978–985. 10.1038/ng.3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfert AB, Turner TN, Murali SC, Hsieh PH, Sulovari A, Wang T, Coe BP, Guo H, Hoekzema K, Bakken TE, et al. 2021. Recent ultra-rare inherited variants implicate new autism candidate risk genes. Nat Genet 53: 1125–1134. 10.1038/s41588-021-00899-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A, Zhou A, Cao X, Mahaganapathy V, Azaro M, Gwin C, Wilson S, Buyske S, Bartlett CW, Flax JF, et al. 2022. MicroRNA and MicroRNA-target variants associated with autism spectrum disorder and related disorders. Genes (Basel) 13: 1329. 10.3390/genes13081329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen RKC, Merico D, Bookman M, Howe JL, Thiruvahindrapuram B, Patel RV, Whitney J, Deflaux N, Bingham J, Wang Z, et al. 2017. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 20: 602–611. 10.1038/nn.4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepeda-Mendoza CJ, Morton CC. 2019. The iceberg under water: unexplored complexity of chromoanagenesis in congenital disorders. Am J Hum Genet 104: 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.