

Abstract
Objectives:
Myositis is a heterogeneous family of diseases including dermatomyositis (DM), immune-mediated necrotizing myopathy (IMNM), antisynthetase syndrome (AS), and inclusion body myositis (IBM). Myositis-specific autoantibodies define different subtypes of myositis. For example, patients with anti-Mi2 autoantibodies targeting the CHD4/NuRD complex (a transcriptional repressor) have more severe muscle disease than other DM patients. This study aimed to define the transcriptional profile of muscle biopsies from anti-Mi2-positive DM patients.
Methods:
RNA sequencing was performed on muscle biopsies (n=171) from patients with anti-Mi2-positive DM (n=18), DM without anti-Mi2 autoantibodies (n=32), AS (n=18), IMNM (n=54), and IBM (n=16) as well as 33 normal muscle biopsies. Genes specifically upregulated in anti-Mi2-positive DM were identified. Muscle biopsies were stained for human immunoglobulin and protein products corresponding to genes specifically upregulated in anti-Mi2-positive muscle biopsies.
Results:
A set of 135 genes, including SCRT1 and MADCAM1, was specifically overexpressed in anti-Mi2-positive DM muscle. This set was enriched for CHD4/NuRD-regulated genes and included genes that are not otherwise expressed in skeletal muscle. The expression levels of these genes correlated with anti-Mi2 autoantibody titers, markers of disease activity, and with the other members of the gene set. In anti-Mi2-positive muscle biopsies, immunoglobulin was localized on the myonuclei, MAdCAM-1 protein was present in the cytoplasm of perifascicular fibers, and SCRT1 protein was localized to myofiber nuclei.
Conclusions:
Based on these findings, we hypothesize that anti-Mi2 autoantibodies could exert a pathogenic effect by entering damaged myofibers, inhibiting the CHD4/NuRD complex, and subsequently derepressing the unique set of genes defined in this study.
Keywords: Myositis, RNA-sequencing, NURD complex, anti-Mi2, dermatomyositis
INTRODUCTION
Myositis is a family of autoimmune systemic disorders affecting not only the muscle, but also other organs and systems, such as the skin, the lungs, and/or the joints. Most myositis patients have a unique myositis-specific autoantibody (MSA). The MSAs so clearly define unique subsets of myositis patients that some authors have hypothesized that these autoantibodies may be causally linked to the pathogenesis of the disease.[1–4] However, the pathogenic role of autoantibodies in myositis patients remains unproven.
Among dermatomyositis (DM) patients, the four most prevalent MSAs are anti-Mi2, anti-NPX2, anti-TIF1γ, and anti-MDA5 autoantibodies. Of note, although DM patients with other autoantibodies may also have severe muscle disease, as a group, those with anti-Mi2 autoantibodies have the most severe muscle disease, with weaker muscles, higher serum muscle enzyme levels, and more prominent myofiber necrosis than other DM patients. Furthermore, in anti-Mi2-positive patients, autoantibody levels correlate with DM disease activity.[5–8]
Anti-Mi2 autoantibodies recognize chromodomain helicase DNA-binding (CHD) proteins, a family of ATP-dependent chromatin remodelers that are functionally critical subunits of the nucleosome remodeling and deacetylase (CHD4/NuRD) complex, a well-described transcriptional repressor.[9, 10] Although CHD3 and CHD4 are the most common target antigens tested in anti-Mi2 serologic assays, CHD3, CHD4, and CHD5 share significant sequence homology and are likely recognized by autoantibodies in anti-Mi2-positive DM patients.
In the past, our group identified several specific transcriptional features in muscle biopsies from patients with different types of myositis.[11] For example, we showed that MADCAM1 is uniquely overexpressed in muscle biopsies from anti-Mi2-positive DM patients. In the current study, we have focused our analysis on DM patients with anti-Mi2 autoantibodies, allowing us to show that muscle biopsies from these patients have a unique transcriptional profile, with overexpression of a set of more than one hundred genes related to the CHD4/NuRD complex. We also show that the expression level of each gene in the set is associated with the level of circulating anti-Mi2 autoantibodies and with markers of disease activity. Furthermore, the expression level of the members of this gene set is mutually correlated. Based on these observations, along with our finding that human immunoglobulin is present within muscle fibers of anti-Mi2-positive DM patients, we hypothesize that disruption of CHD4/NuRD complex function by intracellular anti-Mi2 autoantibodies could derepress gene expression and explain the observed transcriptional pattern.
METHODS
Patients
In this study, we included muscle biopsies from myositis patients enrolled in institutional review board-approved (IRB) longitudinal cohorts from the National Institutes of Health in Bethesda, MD; the Johns Hopkins Myositis Center in Baltimore, MD; the Vall d’Hebron Hospital, and the Clinic Hospital in Barcelona if they fulfilled Lloyd’s criteria for inclusion body myositis,[12] or they fulfilled the Casal and Pinal criteria for other types of myositis,[1] and were positive for one of the following MSA: anti-Mi2, anti-Jo1, anti-NXP2, anti-TIF1γ, anti-MDA5, anti-SRP or anti-HMGCR. Autoantibody testing was performed using one or more of the following techniques: ELISA, immunoprecipitation of proteins generated by in vitro transcription and translation (IVTT-IP), line blotting (EUROLINE myositis profile), or immunoprecipitation from 35S-methionine-labeled HeLa cell lysates. Patients were classified as antisynthetase syndrome if they were positive for anti-Jo1 autoantibodies, as DM if they tested positive for anti-Mi2, anti-NXP2, anti-MDA5, or anti-TIF1γ, and as immune-mediated necrotizing myositis if they had autoantibodies against SRP or HMGCR. The histologically normal muscle biopsies were obtained from the Johns Hopkins Neuromuscular Pathology Laboratory (n=12), the Skeletal Muscle Biobank of the University of Kentucky (n=8), and the National Institutes of Health (n=13). The normal muscle biopsies from the Johns Hopkins Neuromuscular Pathology Laboratory were obtained for clinical purposes but did not show any histological abnormality; the rest of the normal biopsies were obtained from healthy volunteers. All biopsies were from subjects enrolled in institutional review board (IRB)-approved longitudinal cohorts in the different hospitals.
Out of the 18 anti-Mi2 patients included in this study, 11 were previously included in our earlier publication,[11] and the remaining 7 patients were recently added to our cohort. This larger number of biopsies provided greater power to study gene expression differences between the different groups and antibody-defined subgroups in the current manuscript.
Standard protocol approvals and patient consent
This study was approved by the Institutional Review Boards of the National Institutes of Health, the Johns Hopkins Myositis Center, the Clinic, and the Vall d’Hebron Hospitals. Written informed consent was obtained from each participant. All methods were performed in accordance with the relevant guidelines and regulations.
Anti-Mi2 autoantibody titers
Quantitative anti-Mi2 autoantibody ELISA was performed as previously described.[5, 6] In short, 96-well ELISA plates were coated overnight at 4°C with 100ng of Mi2β protein (Abcam, ab124864) diluted in PBS. Replicate wells were incubated with phosphate-buffered saline (PBS) alone. After washing the plates, human serum samples, diluted 1:400 in PBS with 0.05% Tween (PBS-T), were added to the wells (1 hour, 37°C). Then, HRP-labeled goat anti-human antibody (Jackson ImmunoResearch 109–036-088; 1:10,000) was added to each well (30 minutes, 37° C). Color development was performed using SureBlue™ peroxidase reagent (KPL) and absorbance at 450 nm was measured. For each sample, the background absorbance from the PBS-coated wells was subtracted from that of the corresponding Mi2β-coated wells. Test sample absorbances were normalized from an arbitrary positive anti-Mi2 patient, which was used as a reference serum included in every ELISA. The cutoff for a negative anti-Mi2 autoantibody titer was set at 0.17 arbitrary units (mean absorbance plus 3 standard deviations of a healthy control cohort), as previously reported.[5, 6]
RNA sequencing
Bulk RNAseq was performed on frozen muscle biopsy specimens as previously described.[11, 13–16] Briefly, RNA was extracted with TRIzol (Thermo Fisher Scientific). Libraries were either prepared with the NeoPrep system according to the TruSeq Stranded mRNA Library Prep protocol (Illumina, San Diego, CA) or with the NEBNext Poly(A) mRNA Magnetic Isolation Module and Ultra™ II Directional RNA Library Prep Kit for Illumina (New England BioLabs, ref. #E7490, and #E7760).
Pathology and immunofluorescence
Slides with muscle biopsies processed for clinical purposes were stained for hematoxylin and eosin, Gomori trichrome, CD56, membrane attack complex, NADH, and COX and then digitized using a Leica Slide Scanner SCN400F.
For immunofluorescence, 10μm unfixed sections were immunoreacted overnight at 4° in a humidified chamber using a mixture of the following primary antibodies: MAdCAM-1 (Invitrogen, PA5–98417, 1:100), Laminin (Millipore, MAB 1914P, 1:200), and SCRT1 (Sigma, HPA045265, 1:100). The sections were then washed in phosphate-buffered saline and immunoreacted using a mixture of the following fluorochrome-conjugated secondary antibodies: Goat-anti-Rabbit IgG 555 (Invitrogen, A21429, 1:200), Goat-anti-Rat IgG 647 (Invitrogen, A48265, 1:100), and Goat-anti-Human IgG 488 (Invitrogen, A11013, 1:200). The sections were then washed in washing buffer, incubated for 15 min at room temperature in Hoechst 33258 (Abcam, ab228550, 1:4000) to stain the cell nuclei, rinsed in phosphate-buffered saline, and cover-slipped using Prolong Diamond Antifade mountant. Relevant sections were stained exclusively with Goat-anti-Rabbit IgG and Goat-anti-Rat IgG secondary antibodies, without the addition of any primary antibody, resulting in no detectable signal. All sections were imaged using a high-resolution immunofluorescence confocal microscope Leica SP8.
Statistical and bioinformatic analysis
Reads were demultiplexed using bcl2fastq/2.20.0 and preprocessed using fastp/0.21.0. The abundance of each gene was determined using Salmon/1.5.2 and quality control was summarized using multiqc/1.11. Counts were normalized using the Trimmed Means of M values (TMM) from edgeR/3.34.1 for graphical analysis. Differential expression was performed using limma/3.48.3. Enrichment analysis with human pathways was performed using clusterProfiler/4.6.0 and the Reactome dataset.
For visualization of data, we used both the R and Python programming languages. The Benjamini-Hochberg correction was used to adjust for multiple comparisons, and a corrected value of p (q value) 0.05 was considered statistically significant.
To determine whether genes specifically upregulated in muscle biopsies from anti-Mi2-positive DM patients are enriched for genes repressed by the CHD4/NuRD complex, we first identified the homologous mouse genes corresponding to the human anti-Mi2 gene set (https://www.informatics.jax.org/homology.shtml). Next, we utilized five distinct databases derived from CHD4 knockout mouse models to identify genes that are upregulated when CHD4 is depleted. Specifically, we identified CHD4/NuRD-repressed genes by analyzing publicly available transcriptomic data from skeletal muscle samples derived from Chd4mck mice, as well as heart muscle samples from Chd4corin, Chd4myh6, and 9.5, 10, and 10.5E Chd4nkx mice.[17, 18] We then performed a one-sided Fisher’s exact test to compare the proportion of CHD4/NuRD-repressed genes among the upregulated genes from anti-Mi2-positive DM muscle with a mouse homolog to the proportion of CHD4/NuRD-repressed genes among the rest of the mouse genes with a human homolog that were studied in the abovementioned studies.[17, 18]
RESULTS
A set of genes is specifically overexpressed in patients with anti-Mi2-positive DM
To define the transcriptomic profile of anti-Mi2 myositis muscles, we performed bulk RNA sequencing on RNA obtained from 171 muscle biopsies. This included biopsies from 18 anti-Mi2-positive DM patients, 32 DM patients with other MSAs (14 with anti-NXP2, 12 with anti-TIF1, and 6 with anti-MDA5 autoantibodies), 18 AS patients, 54 IMNM patients (44 with anti-HMGCR and 10 with anti-SRP autoantibodies), 16 IBM patients, as well as 33 histologically normal muscle biopsies.
We observed that the most significantly differentially expressed genes in patients with anti-Mi2 dermatomyositis compared to all of the other muscle biopsies included in the study were a) not expressed at detectable levels in non-anti-Mi2 muscle biopsies or normal skeletal muscle, b) were all overexpressed, and c) were unrelated to the interferon pathway (Figure 1). Of note, interferon induced transmembrane protein 5 (IFITM5) was identified as one of the genes specifically overexpressed in anti-Mi2 DM muscle biopsies. While its name suggests otherwise, IFITM5 has not been shown to be interferon inducible.[19] It should also be noted that the same genes were specifically upregulated in quadriceps, deltoid, and biceps muscle biopsies from anti-Mi2-positive DM patients. Interestingly, expression levels of these genes were higher in the biceps and deltoid than in the quadriceps (Supplementary Figure 4).
Figure 1.
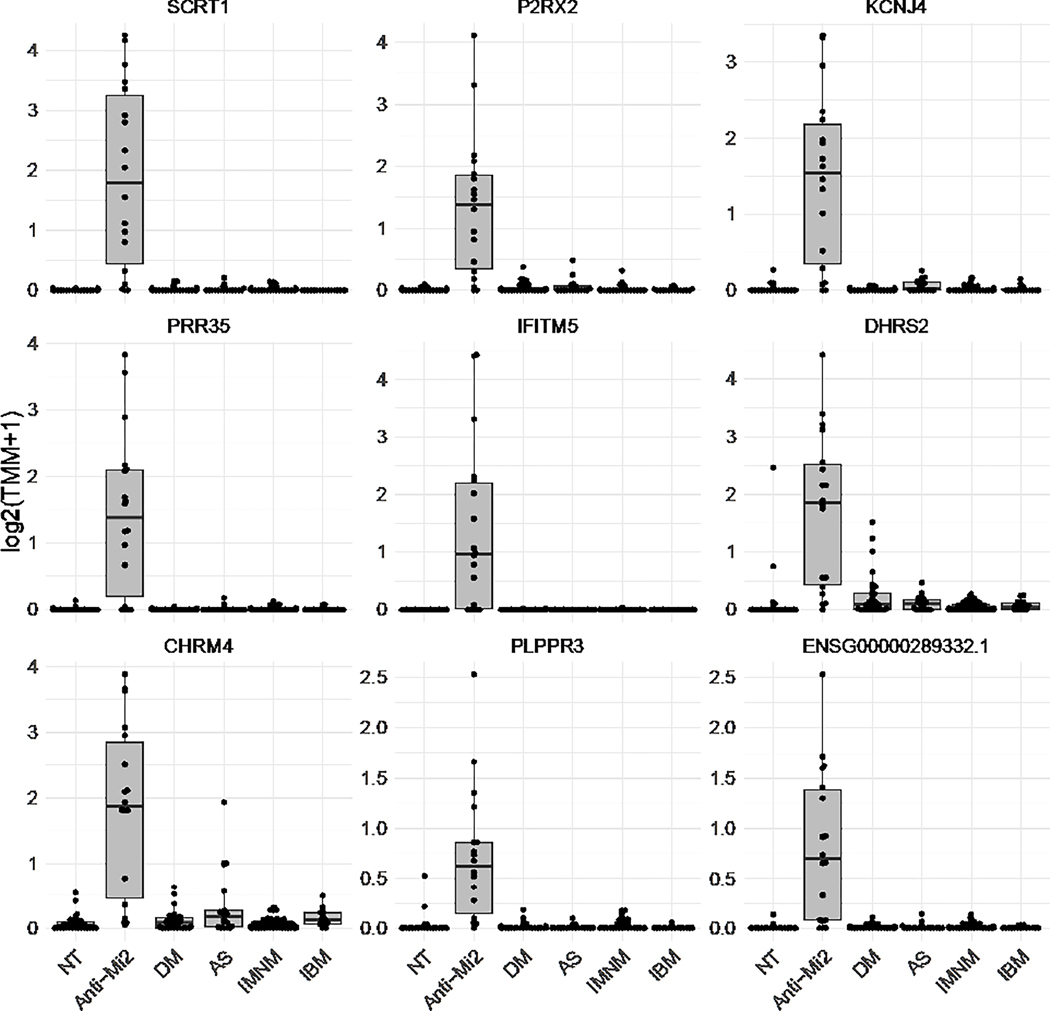
The most differentially overexpressed genes in anti-Mi2 dermatomyositis muscle compared to all the other muscle biopsies included in the study (histologically normal muscle biopsies [NT], non-anti-Mi2 dermatomyositis [DM], antisynthetase syndrome [AS], immune-mediated necrotizing myositis [IMNM], and inclusion body myositis [IBM]).
To formally define this set of genes we calculated the intersection of the differentially overexpressed genes (q-value < 0.05) between the anti-Mi2 DM group and each of the other study groups (Figures 2 and 3, Supplementary Table 1, Supplementary Figure 1). Of note, only one differentially expressed gene was simultaneously under-expressed in anti-Mi2-positive DM muscle compared to each of the other study groups (Supplementary Figure 2). Performing the same analysis in other DM-specific autoantibody groups revealed no genes that were specifically underexpressed or overexpressed (Supplementary Figures 1 and 2).
Figure 2.
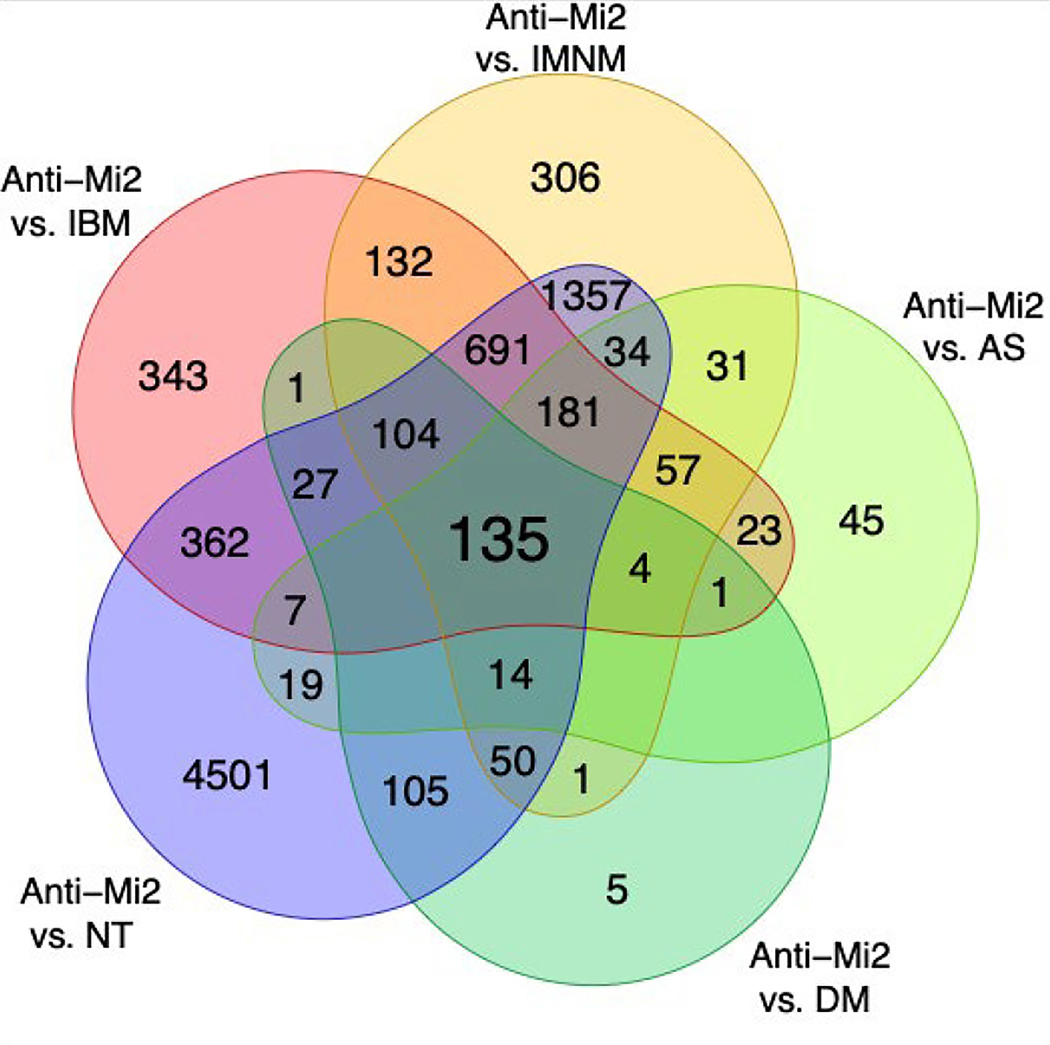
Venn diagram showing the number of genes that were differentially overexpressed (q-value < 0.05) in DM patients with anti-Mi2 autoantibodies compared to other myositis patients and normal muscle biopsies.
DM: dermatomyositis with autoantibodies other than anti-Mi2, AS: antisynthetase syndrome, IBM: inclusion body myositis, IMNM: immune-mediated necrotizing myositis, NT: histologically normal biopsies.
Figure 3.
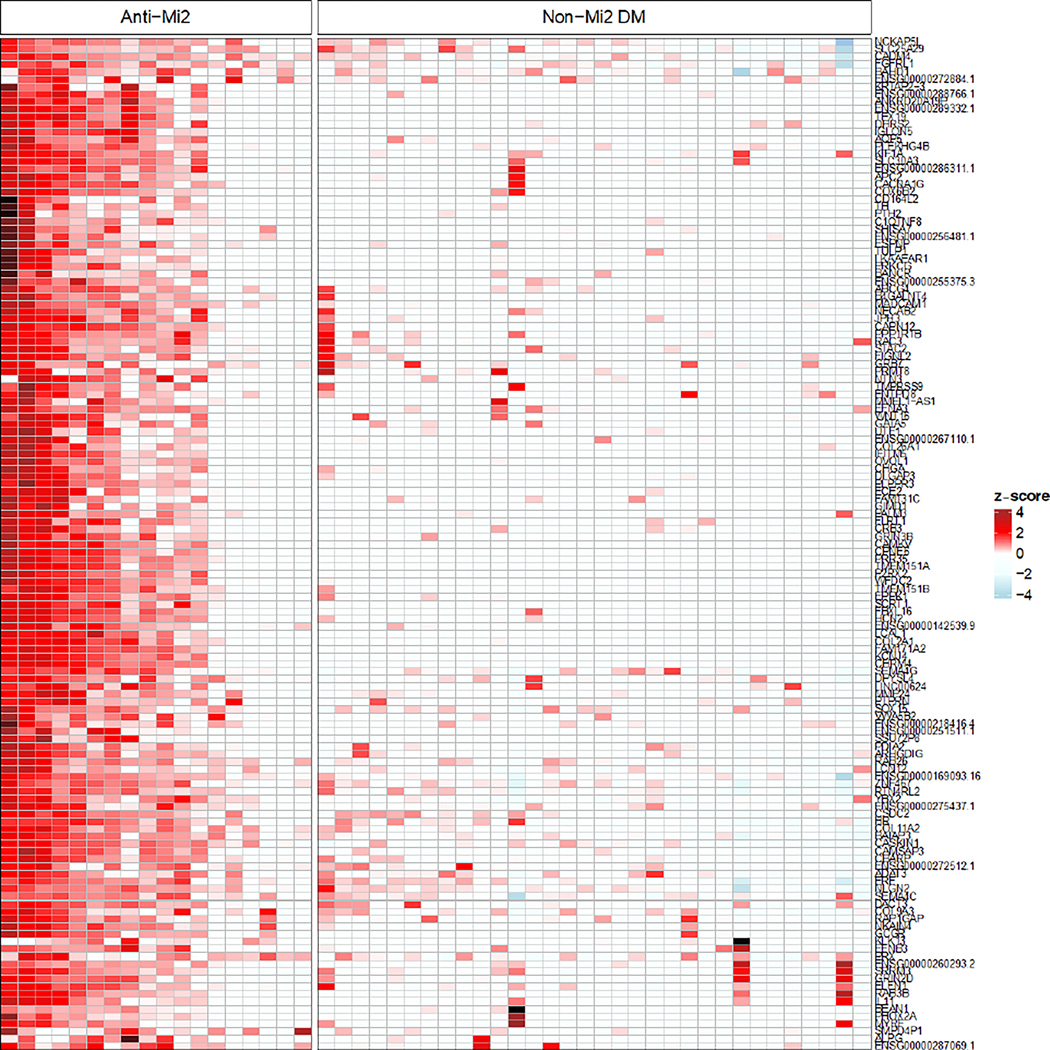
Normalized expression (z-score) of the specifically overexpressed genes in patients with anti-Mi2-positive dermatomyositis (DM) compared with non-Mi2-positive DM.
The upregulated genes in anti-Mi2-positive DM muscle biopsies were involved in a heterogeneous set of cellular pathways. Accordingly, enrichment analysis using this set of genes showed only a very limited number of pathways, with borderline levels of signification (Supplementary Figure 3).
To explore whether this set of genes was overexpressed in a coordinated manner, we performed a correlation analysis. Indeed, there was a marked positive correlation between the expression level of nearly all the genes in the set. This implies that the transcriptional derepression of these genes is tightly coordinated and raises the possibility of a single causal mechanism (Figure 4).
Figure 4.
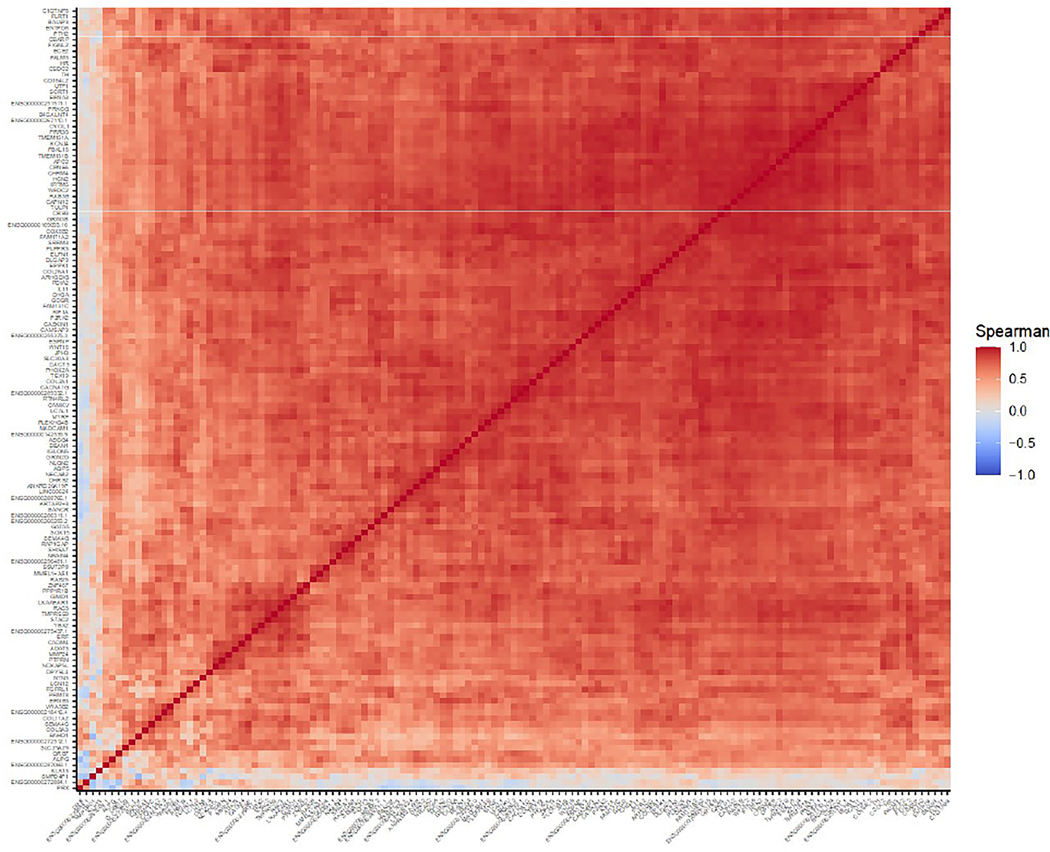
Correlation of expression levels of the specifically overexpressed genes in patients with anti-Mi2-positive dermatomyositis.
Overexpression of anti-Mi2-specific genes in muscle correlates with autoantibody titers and with markers of disease activity
We next investigated the relationship between anti-Mi2 autoantibody titers and the expression of the set of genes specifically upregulated in muscle biopsies from patients with these autoantibodies. Among the 18 anti-Mi2-positive DM patients whose muscle biopsies were included in this study, 15 had serum samples available for this study and were tested using a previously established quantitative anti-Mi2 autoantibody ELISA [5, 6]. Remarkably, anti-Mi2 autoantibody titers were robustly correlated with the expression levels of the set of genes specifically upregulated in muscle biopsies from patients with anti-Mi2 autoantibodies (Figure 5).
Figure 5.
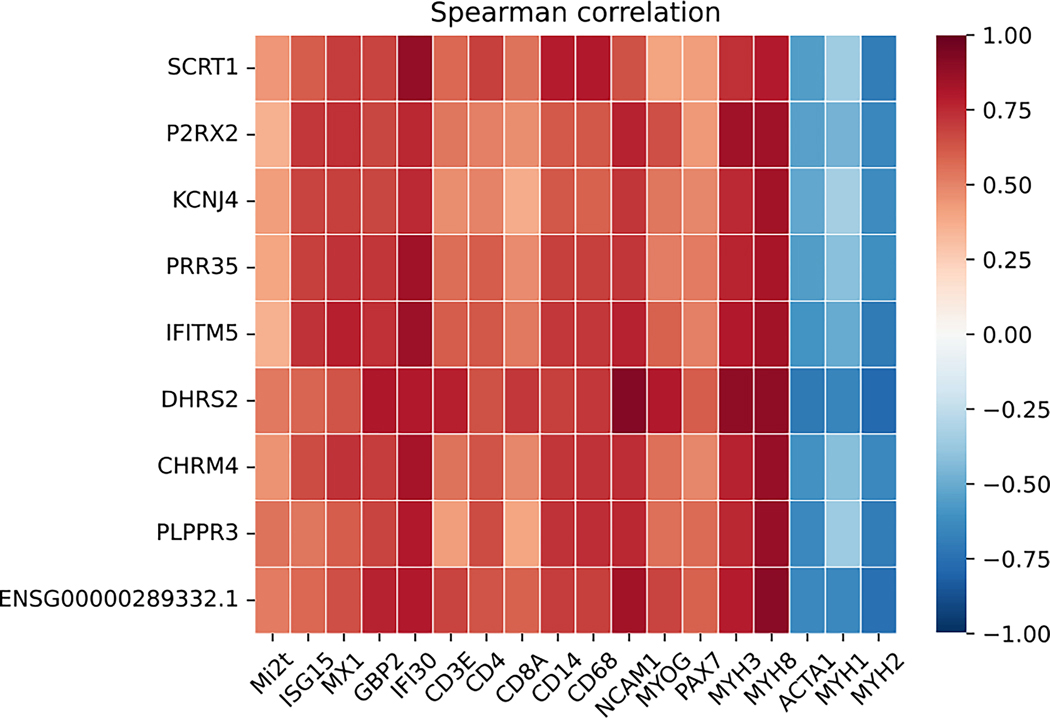
Correlation of the most differentially specifically overexpressed genes in patients with anti-Mi2-positive dermatomyositis and: titer of anti-Mi2 autoantibodies by ELISA (Mi2t), type 1 interferon-inducible genes (ISG15, MX1), type 2 interferon-inducible genes (GBP2, IFI30), T-cell markers (CD3E, CD4, CD8), macrophages (CD14, CD68), markers of muscle differentiation (NCAM1, MYOG, PAX7, MYH3, MYH8), and structural mature muscle proteins (ACTA1, MYH1, MYH2).
We next analyzed the relationship between the expression of the anti-Mi2-specific genes and previously established transcriptomic markers of disease activity. Indeed, there was a strong positive correlation between the expression of the set anti-Mi2-specific genes with type 1 interferon-inducible genes (ISG15, MX1), type 2 interferon-inducible genes (GBP2, IFI30), T-cell markers (CD3E, CD4, CD8), macrophage markers (CD14, CD68), and markers of muscle differentiation (NCAM1, MYOG, PAX7, MYH3, MYH8). Conversely, there was a negative correlation between the expression of the set of anti-Mi2-specific genes with the expression of genes encoding structural proteins found in mature muscle (ACTA1, MYH1, MYH2).
Next, we investigated the relationship between muscle biopsy features and the expression levels of genes specifically upregulated in anti-Mi2-positive DM patients. Using SCRT1 as a representative gene based on its especially high levels of expression only in anti-Mi2-positive DM patients, we found a significant association between its expression level and the extent of necrosis and inflammation observed in the biopsy samples (Figure 6). Taken together with the transcriptomic analysis (Figure 5), these data suggest that the derepression of anti-Mi2-specific genes is intimately linked with the intensity of muscle disease in patients with anti-Mi2-positive DM.
Figure 6.
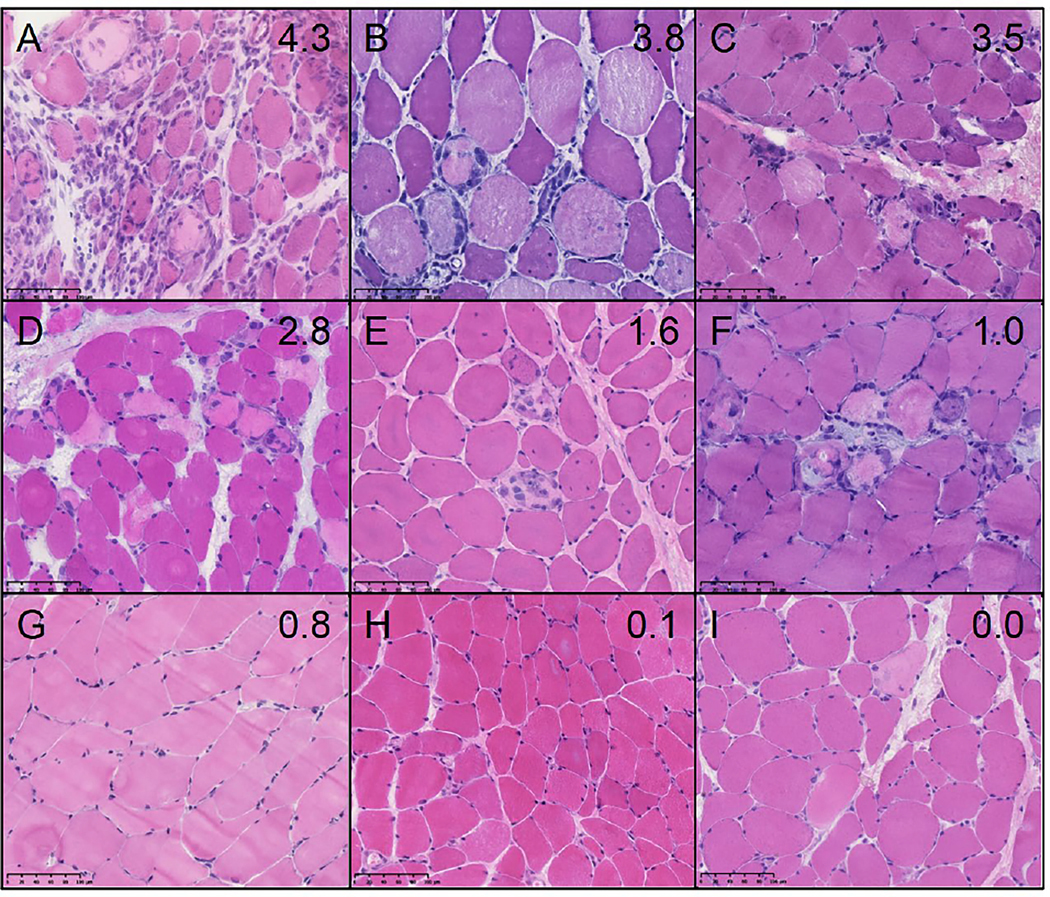
The expression levels of SCRT1 correlate with the intensity of myofiber necrosis and inflammation. Representative hematoxylin and eosin staining of anti-Mi2 dermatomyositis muscle biopsies sorted according to the RNA expression levels of SCRT1. Biopsies A-C have a high expression of SCRT1 (over 3 log2[TMM+1]), D-F have an intermediate expression (between 1 and 3 log2[TMM+1]), and G-I have a low expression (<1 log2[TMM+1]). The individual expression levels of SCRT1 (log2[TMM+1]) are indicated in the top right corner of each image.
The set of anti-Mi2-specific genes is enriched in genes regulated by the CHD4/NuRD complex
To explore whether the gene set specifically upregulated in anti-Mi2-positive DM muscle is enriched in CHD4/NuRD-regulated genes, we pooled five different publicly available datasets from mouse models in which CHD4 had been conditionally knocked-out in cardiac or skeletal muscle and used these to identify genes regulated by the CHD4/NuRD complex. A one-sided Fisher’s exact test revealed that 54% (61/114) of the anti-Mi2-specific genes with a corresponding mouse homolog were regulated by the CHD4/NuRD complex, compared to only 26% (4804/18354) of non-anti-Mi2-specific genes (p=6e-10, Supplementary Figure 2). These results were the same even when comparing the anti-Mi2-specific gene set to each mouse dataset separately (all p<0.05). Signification increased by restricting the analysis to those genes upregulated by CHD4/NuRD (p=5e-17) and disappeared by limiting the analysis to genes downregulated by CHD4/NuRD (all p>0.05). Taken together, this analysis suggests that overexpression of the anti-Mi2-specific gene set may be related to impaired function of the CHD4/NuRD complex.
Immunoglobulin deposition and expression of anti-Mi2-specific gene expression in skeletal muscle cells of anti-Mi2 patients
To further characterize the expression of anti-Mi2-specific genes, we used antibodies against MAdCAM-1 and SCRT1 to localize these proteins in muscle biopsies from anti-Mi2-positive DM patients (Figure 7, Supplementary Figures 5 and 6, corresponding to panel B in Figure 6). Our results showed that MAdCAM-1 protein was predominantly expressed in the cytosol of perifascicular myofibers (Figures 7A and B). In contrast, SCRT1 protein was primarily expressed in the myonuclei (Figures 7C and D). SCRT1 protein was also present in the cytoplasm surrounding the nuclei of severely damaged fibers (Figure 7E).
Figure 7.
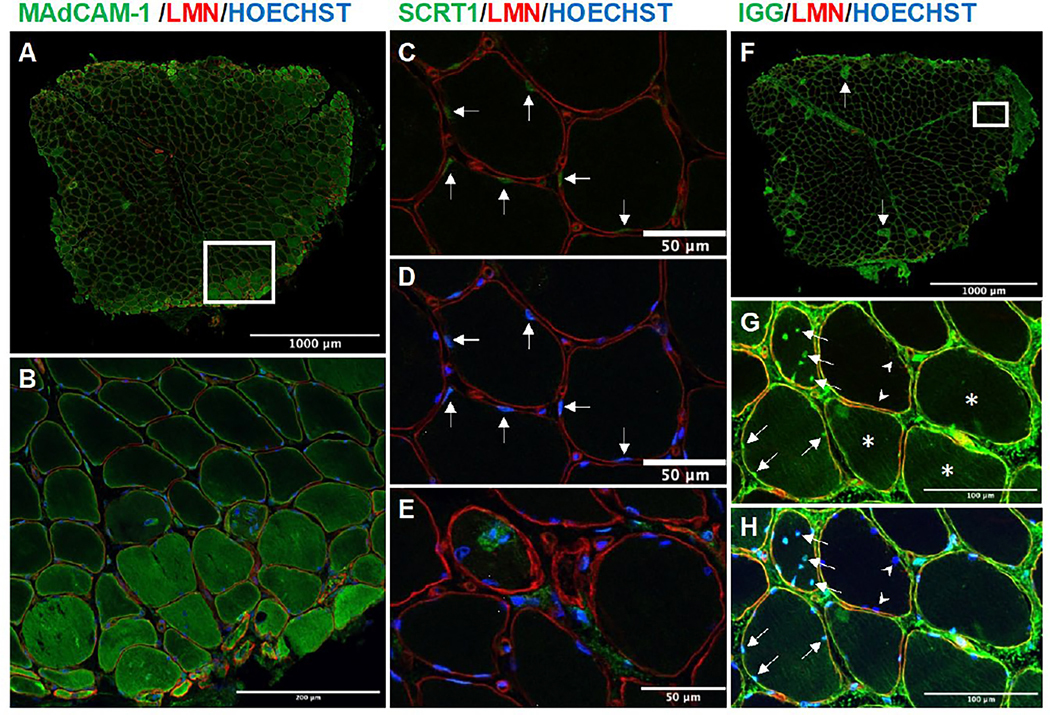
Confocal microscopy images of the muscle biopsy corresponding to panel 6B showing immunofluorescence staining of MAdCAM-1 (A-B), SCRT1 (C-E), and human immunoglobulin (F-H) co-stained with laminin and Hoechst. MAdCAM-1 was predominantly expressed in the perifascicular region (A and B), with varying levels in different muscle fibers (B). SCRT1 was primarily expressed in the nuclei of muscle fibers (arrows C [substracted Hoechst] and D), in the cytoplasm surrounding the nuclei of severely damaged fibers (E). Human immunoglobulin deposition was most prominent in muscle fibers that were MAC-positive (arrow in F, corresponding MAC staining in Supplementary Figure 5). However, healthy-looking muscle fibers showed lower levels of immunoglobulin deposition (asterisks in G). Notably, a significant number of cell nuclei displayed evidence of immunoglobulin deposition, even in cases where the cytoplasmic deposition was absent (indicated by the arrow in G [subtracted Hoechst] and H). Of note, adjacent fibers did not exhibit any staining for nuclear immunoglobulin, as indicated by the arrowheads in G and H. Supplementary stainings for this biopsy are included in Supplementary Figure 5 and the individual channels of each region of interest are included in Supplementary Figure 6. B was obtained from the region marked with a square in A, and G-H was obtained from the region marked with a square in F.
Finally, a prerequisite for potential interaction between anti-Mi2 autoantibodies and their intracellular cognate antigen is the penetration of the autoantibodies through the cell membrane. To explore this, we stained muscle biopsies from patients with anti-Mi2-positive DM with anti-human IgG. We found that cytoplasmic staining for human immunoglobulin was highest in muscle fibers that were diffusely positive for MAC (Figure 7F and Supplementary Figure 6). However, even healthy-looking muscle fibers without significant MAC staining showed lower levels of immunoglobulin deposition (Figure 7G). Most significantly, many nuclei from cells that did not show cytoplasmic MAC or immunoglobulin deposition, stained positive for human immunoglobulin (Figure 7G and H).
DISCUSSION
In this study, we describe a highly coordinated program of transcriptional derepression that occurs specifically in the muscles of DM patients with anti-Mi2 autoantibodies. We identified a set of 135 genes whose expression levels were linked to markers of disease severity as well as to the titer of anti-Mi2 autoantibodies. Furthermore, we found that: a) the anti-Mi2-specific gene set is enriched for genes regulated by the CHD4/NuRD complex, b) immunoglobulin is present within the nuclei and cytoplasm of muscle fibers from anti-Mi2-positive DM patients, and c) proteins encoded by the Mi2-specific genes are expressed within muscle fibers from anti-Mi2-positive patients.
Given that the autoantigens of anti-Mi2 autoantibodies (i.e., CHD3/4) are components of the transcriptional repressor CHD4/NuRD, we hypothesized that anti-Mi2 autoantibodies may penetrate the sarcolemma of previously damaged muscle fibers, bind to CHD proteins, and interfere with the CHD4/NURD complex. As the CHD4/NuRD complex serves to inhibit gene expression, we propose that disrupting its function might derepress the set of genes defined in this study, which could have a toxic effect on muscle fibers.
Of note, Preusse et al. (2021) recently conducted a study comparing muscle biopsies from anti-Mi2-positive and anti-TIF1γ-positive DM patients using the nCounter PanCancer Immune Profiling Panel, which included 770 genes.[20] Interestingly, IL11, the only gene in their panel that was present in our set of anti-Mi2-specific genes, was overexpressed with one of the highest fold changes in their dataset.
This study has several limitations, first, we noted that a number of the anti-Mi2-positive DM patients included in this study did not display the specific transcriptional phenotype observed in the majority of these patients (Figure 3). However, these patients were also those who had the lowest anti-Mi2 autoantibody titers and the least inflammatory muscle biopsies (Figure 5). We propose that the lack of expression of the transcriptional program associated with anti-Mi2 autoantibodies in these patients simply reflects a very mild phenotype. Alternatively, these patients could constitute a distinct pathogenic subtype within the larger group of anti-Mi2-positive DM patients. Second, different methods were used to screen sera for myositis-specific autoantibodies at the different study sites. That being said, out of the 18 anti-Mi2 patients included in this study, serum was available from 15 and we verified the presence of anti-Mi2 autoantibodies in all of these using our in-house ELISA.[5, 6] Third, we utilized the Casal and Pinal autoantibody-based classification criteria as inclusion criteria for the non-IBM myositis patients. These classification criteria are sensitive, specific, and have the advantage of recognizing the different autoantibody-positive subgroups as distinct disease entities. However, besides the external validation included in the original study, they have only been externally validated for patients with anti-MDA5-positive dermatomyositis.[21]
These limitations notwithstanding, this study demonstrates that anti-Mi2 autoantibodies are not just a useful biomarker for DM but define a distinct subtype of DM with a unique set of upregulated genes. Furthermore, our data raise the possibility that anti-Mi2 autoantibodies could play a direct pathogenic role in the development of anti-Mi2 dermatomyositis by disrupting the CHD4/NuRD complex and activating a set of genes that could be toxic to the muscle fibers. However, further investigations will be necessary to determine whether anti-Mi2 autoantibodies upregulate these genes by directly binding to CHD4/NuRD and whether overexpression of these genes contributes to myofiber damage in anti-Mi2 DM patients. Such experiments could include performing viability studies and transcriptomic analyses on muscle fibers that have been exposed to anti-Mi2 autoantibodies from DM patients.
Supplementary Material
KEY MESSAGES.
What is already known about this subject?
Anti-Mi2 dermatomyositis is characterized by more severe muscle involvement and more prominent myofiber necrosis than other types of dermatomyositis.
Anti-Mi2 autoantibodies target functional subunits of the CHD4/NuRD complex, a transcriptional repressor.
What does this study add?
Muscle biopsies from patients with anti-Mi2-positive dermatomyositis are characterized by the overexpression of a set of more than 100 genes, some of them not usually expressed in skeletal muscle.
The expression levels of these genes are mutually correlated, associated with the titers of anti-Mi2 autoantibodies and with markers of disease activity.
This set of genes is enriched in genes that are known to be regulated by the CHD4/NuRD complex.
Members of this gene set are expressed at the protein level in the muscle fibers of anti-Mi2-positive dermatomyositis patients.
Immunoglobulin is localized to the cytoplasm and nuclei of the myofibers in anti-Mi2-positive dermatomyositis patients.
How might this impact on clinical practice?
This study confirms that anti-Mi2 autoantibodies define a distinct subtype of dermatomyositis.
Our data suggest the possibility that anti-Mi2 autoantibodies could exert a pathogenic effect by entering damaged myofibers, inhibiting the CHD4/NuRD complex, and subsequently derepressing the genes defined in this study.
Funding:
This study was funded, in part, by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health. This work was also supported by the Peter Buck and the Huayi and Siuling Zhang Discovery Fund.
Footnotes
Competing interests: None.
Contributorship: All authors contributed to the development of the manuscript, including interpretation of results, substantive review of drafts, and approval of the final draft for submission.
Ethical approval information: All biopsies were from subjects enrolled in institutional review board (IRB)-approved longitudinal cohorts in the National Institutes of Health, the Johns Hopkins, the Clinic Hospital, or the Vall d’Hebron Hospital.
Data sharing statement: Any anonymized data not published within the article will be shared by request from any qualified investigator.
Patient and public involvement: Patients and/or the public were not involved in the design, conduct, reporting, or dissemination plans of this research.
REFERENCES
- 1.Casal-Dominguez M, Pinal-Fernandez I, Pak K, Huang W, Selva-O’Callaghan A, Albayda J, et al. Performance of the 2017 European Alliance of Associations for Rheumatology/American College of Rheumatology Classification Criteria for Idiopathic Inflammatory Myopathies in Patients With Myositis-Specific Autoantibodies. Arthritis Rheumatol. 2022 Mar; 74(3):508–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allenbach Y, Arouche-Delaperche L, Preusse C, Radbruch H, Butler-Browne G, Champtiaux N, et al. Necrosis in anti-SRP(+) and anti-HMGCR(+)myopathies: Role of autoantibodies and complement. Neurology. 2018. Jan 12. [DOI] [PubMed] [Google Scholar]
- 3.Arouche-Delaperche L, Allenbach Y, Amelin D, Preusse C, Mouly V, Mauhin W, et al. Pathogenic role of anti-signal recognition protein and anti-3-Hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: Myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann Neurol. 2017. Apr; 81(4):538–548. [DOI] [PubMed] [Google Scholar]
- 4.Bergua C, Chiavelli H, Allenbach Y, Arouche-Delaperche L, Arnoult C, Bourdenet G, et al. In vivo pathogenicity of IgG from patients with anti-SRP or anti-HMGCR autoantibodies in immune-mediated necrotising myopathy. Ann Rheum Dis. 2019. Jan; 78(1):131–139. [DOI] [PubMed] [Google Scholar]
- 5.Pinal-Fernandez I, Mecoli CA, Casal-Dominguez M, Pak K, Hosono Y, Huapaya J, et al. More prominent muscle involvement in patients with dermatomyositis with anti-Mi2 autoantibodies. Neurology. 2019. Nov 5; 93(19):e1768–e1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinal-Fernandez I, Pak K, Casal-Dominguez M, Hosono Y, Mecoli C, Christopher-Stine L, et al. Validation of anti-Mi2 autoantibody testing by line blot. Autoimmun Rev. 2020. Jan; 19(1):102425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanboon J, Inoue M, Hirakawa S, Tachimori H, Hayashi S, Noguchi S, et al. Pathologic Features of Anti-Mi-2 Dermatomyositis. Neurology. 2021. Jan 19; 96(3):e448–e459. [DOI] [PubMed] [Google Scholar]
- 8.Fornaro M, Girolamo F, Cavagna L, Franceschini F, Giannini M, Amati A, et al. Severe muscle damage with myofiber necrosis and macrophage infiltrates characterize anti-Mi2 positive dermatomyositis. Rheumatology (Oxford). 2021. Jun 18; 60(6):2916–2926. [DOI] [PubMed] [Google Scholar]
- 9.Seelig HP, Moosbrugger I, Ehrfeld H, Fink T, Renz M, Genth E. The major dermatomyositis-specific Mi-2 autoantigen is a presumed helicase involved in transcriptional activation. Arthritis Rheum. 1995. Oct; 38(10):1389–1399. [DOI] [PubMed] [Google Scholar]
- 10.El Abdellaoui-Soussi F, Yunes-Leites PS, Lopez-Maderuelo D, Garcia-Marques F, Vazquez J, Redondo JM, et al. Interplay between the Chd4/NuRD Complex and the Transcription Factor Znf219 Controls Cardiac Cell Identity. Int J Mol Sci. 2022. Aug 24; 23(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinal-Fernandez I, Casal-Dominguez M, Derfoul A, Pak K, Miller FW, Milisenda JC, et al. Machine learning algorithms reveal unique gene expression profiles in muscle biopsies from patients with different types of myositis. Ann Rheum Dis. 2020. Sep; 79(9):1234–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lloyd TE, Mammen AL, Amato AA, Weiss MD, Needham M, Greenberg SA. Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology. 2014. Jul 29; 83(5):426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinal-Fernandez I, Casal-Dominguez M, Derfoul A, Pak K, Plotz P, Miller FW, et al. Identification of distinctive interferon gene signatures in different types of myositis. Neurology. 2019. Sep 17; 93(12):e1193–e1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinal-Fernandez I, Amici DR, Parks CA, Derfoul A, Casal-Dominguez M, Pak K, et al. Myositis Autoantigen Expression Correlates With Muscle Regeneration but Not Autoantibody Specificity. Arthritis Rheumatol. 2019. Aug; 71(8):1371–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amici DR, Pinal-Fernandez I, Mazala DA, Lloyd TE, Corse AM, Christopher-Stine L, et al. Calcium dysregulation, functional calpainopathy, and endoplasmic reticulum stress in sporadic inclusion body myositis. Acta Neuropathol Commun. 2017. Mar 22; 5(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amici DR, Pinal-Fernandez I, Christopher-Stine L, Mammen AL, Mendillo ML. A network of core and subtype-specific gene expression programs in myositis. Acta Neuropathol. 2021. Nov; 142(5):887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomez-Del Arco P, Perdiguero E, Yunes-Leites PS, Acin-Perez R, Zeini M, Garcia-Gomez A, et al. The Chromatin Remodeling Complex Chd4/NuRD Controls Striated Muscle Identity and Metabolic Homeostasis. Cell Metab. 2016. May 10; 23(5):881–892. [DOI] [PubMed] [Google Scholar]
- 18.Wilczewski CM, Hepperla AJ, Shimbo T, Wasson L, Robbe ZL, Davis IJ, et al. CHD4 and the NuRD complex directly control cardiac sarcomere formation. Proc Natl Acad Sci U S A. 2018. Jun 26; 115(26):6727–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho TJ, Lee KE, Lee SK, Song SJ, Kim KJ, Jeon D, et al. A single recurrent mutation in the 5’-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet. 2012. Aug 10; 91(2):343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Preusse C, Eede P, Heinzeling L, Freitag K, Koll R, Froehlich W, et al. NanoString technology distinguishes anti-TIF-1gamma(+) from anti-Mi-2(+) dermatomyositis patients. Brain Pathol. 2021. May; 31(3):e12957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.So H, So J, Lam TT, Wong VT, Ho R, Li WL, et al. Performance of the 2017 European Alliance of Associations for Rheumatology/American College of Rheumatology Classification Criteria in Patients With Idiopathic Inflammatory Myopathy and Anti-Melanoma Differentiation-Associated Protein 5 Positivity. Arthritis Rheumatol. 2022. Sep; 74(9):1588–1592. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.