

Abstract
The expanding availability of large-scale genomic data and the growing interest in uncovering gene-disease associations call for efficient tools to visualize and evaluate gene expression and genetic variation data. Here, we developed a comprehensive pipeline that was implemented as an interactive Shiny application and a standalone desktop application. NeuroVar is a tool for visualizing genetic variation (single nucleotide polymorphisms and insertions/deletions) and gene expression profiles of biomarkers of neurological diseases. Data collection involved filtering biomarkers related to multiple neurological diseases from the ClinGen database. NeuroVar provides a user-friendly graphical user interface to visualize genomic data and is freely accessible on the project’s GitHub repository (https://github.com/omicscodeathon/neurovar).
Statement of need
Disease biomarkers are genes or molecules that indicate the presence or severity of a disease. Their identification provides important insights into disease etiology and can facilitate the development of new treatments and therapies [1]. Integrating multi-omics data, such as gene expression and genetic variations, has emerged as a powerful approach for biomarker discovery.
Several genomics studies have discovered multiple genetic variations linked to numerous neurological conditions that are complex diseases with a significant level of heterogeneity, such as Alzheimer’s disease [2] and Parkinson’s disease [3]. Some studies have also used genetic variants to detect the presence of human disorders [4].
The discovered biomarkers are extensively documented in various scientific publications and are accessible through databases like the Clinical Genome (ClinGen) database. ClinGen stores a vast amount of genomic data, including a comprehensive dataset of biomarkers associated with multiple diseases, such as various neurological disorders [5].
Multiple computational tools have been developed in recent years to analyze genomic data, including gene expression data analysis [6, 7], identification of potential inhibitors for therapeutic targets [8], and comparative analysis of molecular and genetic evolution [9]. However, there is still a need for a specialized tool that focuses on filtering critical disease biomarkers, as this will help in studies that work on finding genes that are involved in diseases using transcriptomic data generated from sequencing experiments [10–13]. Such a tool would help users identify phenotypic subtypes of diseases in their patients, thereby facilitating more accurate diagnoses and personalized treatment plans.
In this study, we developed a novel tool named “NeuroVar” to analyze biomarker data for neurological diseases specifically, including gene expression profiles and genetic variations such as single nucleotide polymorphisms (SNPs) and nucleotide insertion and/or deletion (Indels).
Implementation
Data collection
The ClinGen database [5] provides a dataset of biomarkers of multiple diseases from which we filtered data of all the available neurological syndromes (eleven) and non-neurological diseases with neurological manifestations (seven).
Software development
Two versions of the tool were developed: an R shiny and a desktop application.
The shiny application was developed using multiple R packages, including Shiny (RRID:SCR_001626) [14] and shinydashboard [15]. Other R packages are used for data manipulation, including dplyr (RRID:SCR_016708) [16], readr [17], tidyverse (RRID:SCR_019186) [18], purrr (RRID:SCR_021267) [19], vcfR (RRID:SCR_023453) [20], bslib [21], stringr (RRID:SCR_022813) [22], data.table [23], fs [24], DT [25], sqldf [26], and ggplot2 (RRID:SCR_014601) [27].
For the stand-alone desktop application, the wxPython framework [28] was used to build a similar GUI. A variety of Python libraries were employed, including Pandas (RRID:SCR_018214) [29], MatPlotLib (RRID:SCR_008624) [30], and NumPy (RRID:SCR_008633) [31]. After testing, the application was packaged as an installer using cx_Freeze [32]. Finally, it was distributed as a zip file to be downloaded.
Pipeline validation and case study
To validate the pipeline, a case study was performed on the public dataset SRP149638 [33] available on the SRA database [34]. The dataset corresponds to RNA sequencing data from the peripheral blood mononuclear cells from healthy donors and Amyotrophic Lateral Sclerosis (ALS) patients. The ALS patients involved in the study have mutations in the FUS, TARDBP, SOD1, and VCP genes.
The file’s preprocessing, genetic expression analysis, and variant calling were performed using the Exvar R package [35]. The Exvar package uses the rfastp package [36] and the gmapR package [37] for preprocessing fastq files, the GenomicAlignments package (RRID:SCR_024236) [38], and the DESeq2 packages (RRID:SCR_015687) [39] for gene expression data analysis, as well as the VariantTools [40] and VariantAnnotation (RRID:SCR_000074) [41] packages for variant calling.
Results
Supported disease
NeuroVar integrates biomarkers of multiple neurological diseases, including epilepsy, ALS, intellectual disability, autism spectrum disorder, brain malformation syndrome, syndromic disorders, cerebral palsy, RASopathy, aminoacidopathy, craniofacial malformations, Parkinson’s disease, and PHARC syndrome. It also integrates seven non-neurological diseases with neurological manifestations: peroxisomal disorders, hereditary cancer, mitochondrial disease, retina-related disorders, general gene curation, hearing loss, and fatty acid oxidation disorders. Each disease syndrome includes multiple disease types; for example, sixteen types of ALS disorder are integrated.
Operation and implementation
The desktop and Shiny applications have the same user interface; however, the implementation is different.
The Shiny application is platform-independent, while the desktop application is optimized for the Windows operating system. The necessary library requirements for the tool are automatically installed in both versions. The amount of RAM used depends on the servers or the machine being used, and the only prerequisites for using the tool are having R installed for the shiny application and having Python installed for the desktop application.
The tool is compatible with RNA sequencing data. The input data files should be in CSV format for gene expression data and VCF (Variant Call Format) format for genetic variants data. Guidance of the files’ organization is available in the tool’s Github repository in detail (path: omicscodeathon/neurovar/demonstration_data).
Detailed guidelines for installing and using both versions of the application are provided in the project’s GitHub repository.
The application’s usage
The application dashboard includes three pages. The first page, named “Biomarker”, provides data on the disease’s biomarkers. Initially, the user should select the target disease syndrome and the specific disease subtypes from the provided list (Figure 1).
Figure 1.

The layout of the “Biomarker” page. The user is requested to define the target disease, disease type, and gene of interest.
Next, a list of biomarkers is provided with additional data, including the gene’s mode of inheritance, description, type, and transcripts. Also, a link for the official online report validating the gene’s association with the disease is provided (Figure 2).
Figure 2.

The output of the “Biomarker” page. The output includes two tables detailing key information about the selected gene.
The second page, named “Expression”, is used to visualize the biomarkers expression profile. After importing a CSV file and identifying the key columns, the log2FC value and adjusted p-value are requested to define the differential expression profile. By default, the adjusted p-value is set to less than 0.01, and the logFC value is set to less or more than 2 (Figure 3).
Figure 3.

The layout of the “Expression” page. The user is requested to upload the data file and select the p-value and the log-FC value required to construct the differential expression profile.
As a result, the expression profiles of the target disease biomarkers (previously selected) are summarized in a table and represented in a volcano plot (Figure 4).
Figure 4.

The output of the “Expression” page. As output, a summary of the genes’ expression profiles is displayed in a table and a volcano plot.
The third page, named “Variants”, allows the visualization of SNPs and Indels data. The user is requested to define the path to the directory containing the VCF files. The files are expected to be divided into two folders, named “controls” and “patients”, containing the VCF files of the controls and patients, respectively. The user needs to define the variant type as SNPs or Indels (Figure 5).
Figure 5.

The layout of the “Variant” page. The user is prompted to specify the path to the data-containing folder and the data type.
The VCF files are processed and annotated, and then the variants in the target disease biomarkers are filtered and resumed in a table comparing the reference genome, the control group, and the patients’ group (Figures 6 and 7).
Figure 6.

The output of the “Variant” page. Table summarizing the SNPs in the target disease’s biomarkers.
Figure 7.

The output of the “Variant” page. Table summarizing the INDELs in the target disease’s biomarkers.
Case study results
To validate the pipeline, we conducted a case study using the public dataset that provides RNA sequencing data of ALS patients who were declared to carry mutations in the FUS, TARDBP, SOD1, and VCP genes [33].
Initially, we used NeuroVar to explore the roles of the genes FUS, TARDBP, SOD1, and VCP in ALS. Our findings confirmed that FUS, TARDBP, and SOD1 are recognized ALS biomarkers, while VCP is not. ALS has 26 subtypes, with FUS being a biomarker for type 6, SOD1 for type 1, and TARDBP for type 10, suggesting that the patients in the study may represent a mixture of these ALS subtypes.
Next, we investigated whether mutations in these genes impacted their expression profiles. Using an adjusted p-value threshold of 0.05 and a log fold change (logFC) cutoff of 2, we found that out of 21 known ALS biomarkers, only one gene—TUBA4A—was differentially expressed. Notably, none of the four genes (FUS, TARDBP, SOD1, and VCP) showed differential expression.
Finally, we examined the types of mutations present in these genes. We detected 23 SNPs across seven biomarkers: DAO (all ALS types), FIG4 (ALS type 11), ERBB4 (ALS type 19), TUBA4A (ALS type 22), KIF5A (ALS type 25), C9orf72 (ALS type 1), and TBK1 (ALS type 4). No indels were detected in any of the biomarkers. Interestingly, the biomarkers FUS, TARDBP, and SOD1 exhibited neither SNPs nor Indels, suggesting that the mutations in these genes may be due to other types of genomic changes.
A demonstration video describing how to visualize the demonstration data using neurovar is available on GitHub (Figure 8).
Figure 8.
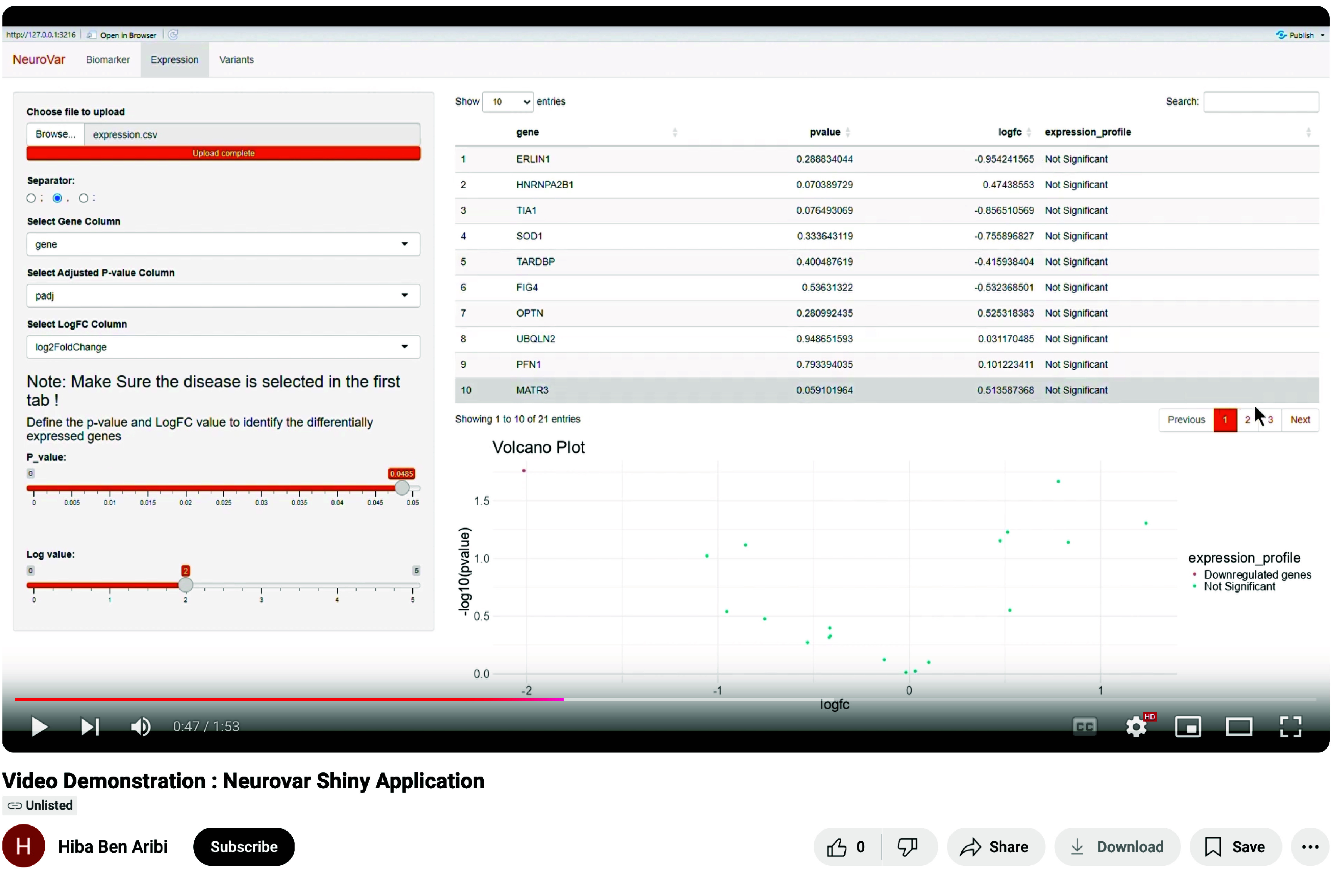
Video demonstration of the NeuroVar Shiny Application [44]. https://youtu.be/cYZ8WOvabJs?si=W7v3AZ_pAsXt7ZsI.
Discussion and conclusion
NeuroVar is a novel tool for visualizing genetic variation and gene expression data related to neurological diseases. The tool is designed to visualize genetic variation and gene expression data, with a particular emphasis on neurological disorders. This specialization makes it an invaluable resource for researchers and clinicians focused on these conditions. It offers features to filter biomarkers by specific diseases, which aids in confirming gene-disease associations and prioritizing genes for further investigation.
The tool supports eleven neurological syndromes and seven non-neurological diseases with neurological manifestations. While the supported diseases list is currently limited to data from the ClinGen database, it will be frequently updated, and data sources will be expanded to include other databases in the future.
NeuroVar is available as a desktop application and as a Shiny application. Both versions are user-friendly and do not require computational skills to operate them. Additionally, all necessary dependencies are automatically installed with the tools. This dual accessibility of NeuroVar caters to users with varying preferences and technical backgrounds, which makes it more accessible and easier to use than other visualization tools of genetic variant data, such as the command line tool VIVA [42] to analyze VCF files and the “Transcriptomics oSPARC” web tool for gene expression data visualization hosted on the o2S2PARC platform (RRID:SCR_018997) [6].
In addition to its user-friendly design, NeuroVar streamlines the research workflow by eliminating the need for multiple filtering steps across different platforms. By integrating essential functions within a single interface, it allows users to conduct comprehensive analyses without leaving the application, thereby enhancing efficiency and reducing errors. The inclusion of a quick-access library on the first page further aids in referencing important data, making it easier to revisit and validate findings. This centralization of tasks, coupled with a focus on neurological diseases and extensive biomarker information, makes NeuroVar a highly useful tool for advancing research in the field.
Availability of source code and requirements
Project name: NeuroVar
Project home page: https://github.com/omicscodeathon/neurovar
Operating system: Platform independent
Programming language: Python and R
Other requirements: None
License: Artistic license 2.0
RRID: SCR_025640
DOI for the Project’s GitHub Repository: https://doi.org/10.5281/zenodo.13375646
DOI for the Shiny application: https://doi.org/10.5281/zenodo.13375493
DOI for the desktop application: https://doi.org/10.5281/zenodo.13375579
DOI for the data: https://doi.org/10.5281/zenodo.13375591.
Acknowledgements
The authors thank the National Institutes of Health (NIH) Office of Data Science Strategy (ODSS), and the National Center for Biotechnology Information (NCBI) for their immense support before and during the April 2023 Omics codeathon organized by the African Society for Bioinformatics and Computational Biology (ASBCB).
Data availability
The following resources can be accessed in the project’s GitHub repository, https://github.com/omicscodeathon/neurovar:
The open-source code for both the Shiny application and the desktop application.
An installation guide.
A video demonstration.
The processed case study data is available as demonstration data in Zenodo [43].
Data came from ClinVar, and the presented case study was performed on the public dataset SRP149638 from the SRA database.
The open source code of the Shiny application and the desktop application are available in the project’s GitHub Repository: https://github.com/omicscodeathon/neurovar.
Installation Guide, demonstration data, and video demonstration (Figure 8) are also available in the project’s GitHub Repository: https://github.com/omicscodeathon/neurovar.
Snapshots of the project code [45], shiny application code [46], and desktop application code [47] are all in Zenodo.
Abbreviations
ALS: Amyotrophic Lateral Sclerosis; Indel: insertion and/or deletion; logFC: log fold change; SNP: single nucleotide polymorphism; VCF: Variant Call Format.
Declarations
Ethics approval and consent to participate
The authors declare that ethical approval was not required for this type of research.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HBA: Conceptualization, Methodology, Validation, Writing; NA: Data Analysis, Methodology, Validation, Writing; OIA: Resources, Manuscript review, and Project Supervision.
Funding
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
References
- 1.Hirschhorn JN, Lohmueller KE, Byrne E et al. A comprehensive review of genetic association studies. Genet. Med., 2002; 4(2): 45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Kunkle BW, Grenier-Boley B, Sims R et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet., 2019; 51: 414–430. doi: 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li W, Fu Y, Halliday GM et al. PARK genes link mitochondrial dysfunction and Alpha-Synuclein pathology in sporadic Parkinson’s disease. Front. Cell Dev. Biol., 2021; 9: 612476. doi: 10.3389/fcell.2021.612476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wesonga RM, Awe OI. . An assessment of traditional and genomic screening in newborns and their applicability for Africa. Inform. Med. Unlocked, 2022; 32: 101050. doi: 10.1016/j.imu.2022.101050. [DOI] [Google Scholar]
- 5.Rehm HL, Berg JS, Brooks LD et al. ClinGen—the clinical genome resource. N. Engl. J. Med., 2015; 372(23): 2235–2242. doi: 10.1056/NEJMsr1406261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aribi HB, Ding M, Kiran A. . Gene expression data visualization tool on the o2S2PARC platform. F1000Research, 2022; 11: 1267. doi: 10.12688/f1000research.126840.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Die JV, Elmassry MM, LeBlanc KH et al. geneHummus: an R package to define gene families and their expression in legumes and beyond. BMC Genom., 2019; 20: 591. doi: 10.1186/s12864-019-5952-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogbodo UC, Enejoh OA, Okonkwo CH et al. Computational identification of potential inhibitors targeting cdk1 in colorectal cancer. Front. Chem., 2023; 11: 1264808. doi: 10.3389/fchem.2023.1264808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Awe OI, En najih N, Nyamari MN et al. Comparative study between molecular and genetic evolutionary analysis tools using African SARS-CoV2 variants. Inform. Med. Unlocked, 2023; 36: 101143. doi: 10.1016/j.imu.2022.101143. [DOI] [Google Scholar]
- 10.El Abed F, Baraket G, Nyamari MN et al. Differential expression analysis of miRNAs and mRNAs in epilepsy uncovers potential biomarkers. bioRxiv. 2023; 10.1101/2023.09.11.557132. [DOI]
- 11.Chikwambi Z, Hidjo M, Chikondowa P et al. Multi-omics data integration approach identifies potential biomarkers for Prostate cancer. bioRxiv. 2023; 10.1101/2023.09.11.557129. [DOI]
- 12.Nyamari MN, Omar KM, Fayehun AF et al. Expression level analysis of ACE2 receptor gene in African-American and non-African-American COVID-19 patients. bioRxiv. 2023; 10.1101/2023.09.11.557129. [DOI]
- 13.Nzungize L, Kengne-Ouafo JA, Wesonga MR et al. Transcriptional profiles analysis of COVID-19 and malaria patients reveals potential biomarkers in children. bioRxiv. 2022; 10.1101/2022.06.30.498338. [DOI]
- 14.Chang W, Cheng J, Allaire J et al. Shiny: web application framework for R. R package version 1.7.4.9002. 2023; https://shiny.rstudio.com/.
- 15.Chang W, Borges Ribeiro B. . Create dashboards with ‘Shiny’. 2023; http://rstudio.github.io/shinydashboard/.
- 16.Wickham H, François R, Henry L et al. dplyr: a grammar of data manipulation. 2022; https://github.com/tidyverse/dplyr.
- 17.Wickham H, Hester J, Bryan J. . readr: read rectangular text data. 2022; https://readr.tidyverse.org, https://github.com/tidyverse/readr.
- 18.Wickham H, Averick M, Bryan J et al. Welcome to the tidyverse. J. Open Source Softw., 2019; 4(43): 1686. doi: 10.21105/joss.01686. [DOI] [Google Scholar]
- 19.Wickham H, Henry L. . purrr: functional programming tools. 2023; https://purrr.tidyverse.org.
- 20.Knaus BJ, Grünwald NJ. . VCFR: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour., 2017; 17(1): 44–53. ISSN 757. doi: 10.1111/1755-0998.12549. [DOI] [PubMed] [Google Scholar]
- 21.Sievert C, Cheng J. . bslib: custom ‘bootstrap’ ‘sass’ themes for ‘shiny’ and ‘rmarkdown’. 2023; https://github.com/rstudio/bslib.
- 22.Wickham H. . stringr: simple, consistent wrappers for common string operations. 2022; https://stringr.tidyverse.org, https://github.com/tidyverse/stringr.
- 23.Dowle M, Srinivasan A, Gorecki J et al. Data.table: extension of ‘data.frame’. 2023; https://github.com/Rdatatable/data.table.
- 24.Hester J, Wickham H, Csárdi G. . Fs: cross-platform file system operations based on ‘libuv’. 2023; https://fs.r-lib.org.
- 25.Xie Y, Cheng J, Tan X et al. DT: a wrapper of the JavaScript library ‘DataTables’. 2023; https://github.com/rstudio/DT.
- 26.Grothendieck G. . sqldf: manipulate R data frames using SQL. 2017; Retrieved from https://cran.r-project.org/web/packages/sqldf/index.html.
- 27.Wickham H. . ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag, 2016. ISBN 978-3-319-24277-4, https://ggplot2.tidyverse.org/. [Google Scholar]
- 28.Talbot H. . WxPython, a GUI toolkit. Linux J., 2000; 2000(74): 5. https://dl.acm.org/citation.cfm?id=349312. [Google Scholar]
- 29.McKinney W. . Data structures for statistical computing in python. In: Proceedings of the 9th Python in Science Conference. vol. 445, 2010; pp. 51–56, doi: 10.25080/Majora-92bf1922-00a. [DOI] [Google Scholar]
- 30.Hunter JD. . Matplotlib: a 2D graphics environment. Comput. Sci. Eng., 2007; 9(3): 90–95. doi: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 31.Harris CR, Millman KJ, Van Der Walt SJ et al. Array programming with NumPy. Nature, 2020; 585(7825): 357–362. doi: 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duarte M. . GitHub - marcelotduarte/cx_Freeze: create standalone executables from Python scripts, with the same performance and is cross-platform. 2023; https://github.com/marcelotduarte/cx_Freeze.
- 33.Zucca S, Gagliardi S, Pandini C et al. RNA-Seq profiling in peripheral blood mononuclear cells of amyotrophic lateral sclerosis patients and controls. Sci. Data, 2019; 6(1): 1–8. doi: 10.1038/sdata.2019.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leinonen R, Sugawara H, Shumway M. . International nucleotide sequence database collaboration. The sequence read archive. Nucleic Acids Res., 2011; 39(Database issue): D19–D21. doi: 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ben Aribi H, Dixon I, Abassi N et al. Exvar: an R package for gene expression and genetic variation data analysis and visualization. 2023; https://github.com/omicscodeathon/Exvar.
- 36.Wang W, Carroll T. . Rfastp: an ultra-fast and all-in-one fastq preprocessor (quality control, adapter, low quality and polyX trimming) and UMI sequence parsing). R package (Version 1.6.0). Bioconductor. 2022; 10.18129/B9.bioc.Rfastp. [DOI]
- 37.Barr C, Wu T, Lawrence M. . gmapR: an R interface to the GMAP/GSNAP/GSTRUCT suite (Version 1.40.0). Bioconductor. 2022; 10.18129/B9.bioc.gmapR. [DOI]
- 38.Lawrence M, Huber W, Pagès H et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol., 2013; 9(8): e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Love MI, Huber W, Anders S. . Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 2014; 15: 550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawrence M, Degenhardt J, Gentleman R. . VariantTools: tools for exploratory analysis of variant calls (Version 1.40.0). Bioconductor. 2022; 10.18129/B9.bioc.VariantTools. [DOI]
- 41.Obenchain V, Lawrence M, Carey V et al. VariantAnnotation: a Bioconductor package for exploration and annotation of genetic variants. Bioinformatics, 2014; 30(14): 2076–2078. doi: 10.1093/bioinformatics/btu168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tollefson GA, Schuster J, Gelin F et al. VIVA (visualization of variants): a VCF file visualization tool. Sci. Rep., 2019; 9(1): 12648. doi: 10.1038/s41598-019-49114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ben Aribi H, Abassi N, Awe OI. . NeuroVar demonstration data [data set]. Zenodo. 2024; 10.5281/zenodo.13375591. [DOI]
- 44.Ben Aribi H. . Video demonstration: neurovar shiny application. Youtube. 2024; https://www.youtube.com/watch?v=cYZ8WOvabJs.
- 45.Ben Aribi H, Abassi N, Awe OI. . NeuroVar: a genetic expression and variation data visualization tool for neurological diseases’ biomarkers. Zenodo. 2024; 10.5281/zenodo.13375646. [DOI]
- 46.Ben Aribi H, Abassi N, Awe OI. . NeuroVar shiny application. Zenodo. 2024; 10.5281/zenodo.13375493. [DOI]
- 47.Ben Aribi H, Abassi N, Awe OI. . NeuroVar desktop application. Zenodo. 2024; 10.5281/zenodo.13375579. [DOI]