

Summary
DNA methylation plays a critical role in establishing and maintaining cellular identity. However, it is frequently dysregulated during tumor development and is closely intertwined with other genetic alterations. Here, we leveraged multi-omic profiling of 687 tumors and matched non-involved adjacent tissues from kidney, brain, pancreas, lung, head and neck, and endometrium to identify aberrant methylation associated with RNA and protein abundance changes and build a Pan-Cancer catalog. We uncovered lineage-specific epigenetic drivers including hypomethylated FGFR2 in endometrial cancer. We showed that hypermethylated STAT5A is associated with pervasive regulon downregulation and immune cell depletion, suggesting that epigenetic regulation of STAT5A expression constitutes a molecular switch for immunosuppression in squamous tumors. We further demonstrated that methylation subtype-enrichment information can explain cell-of-origin, intra-tumor heterogeneity, and tumor phenotypes. Overall, we identified cis-acting DNA methylation events that drive transcriptional and translational changes, shedding light on the tumor’s epigenetic landscape and the role of its cell-of-origin.
eTOC blurb
Liang et al. catalog pan-cancer DNA methylation with concordant transcriptional and translational changes, revealing lineage-specific epigenetic driver FGFR2 hypomethylation in uterine corpus endometrial carcinoma, and STAT5 hypermethylation as an immunosuppression switch in squamous tumors. They also identify methylation-driven subtypes associated with cell-of-origin, tumor heterogeneity, tumor phenotype, and links to therapeutic potential.
Graphical Abstract
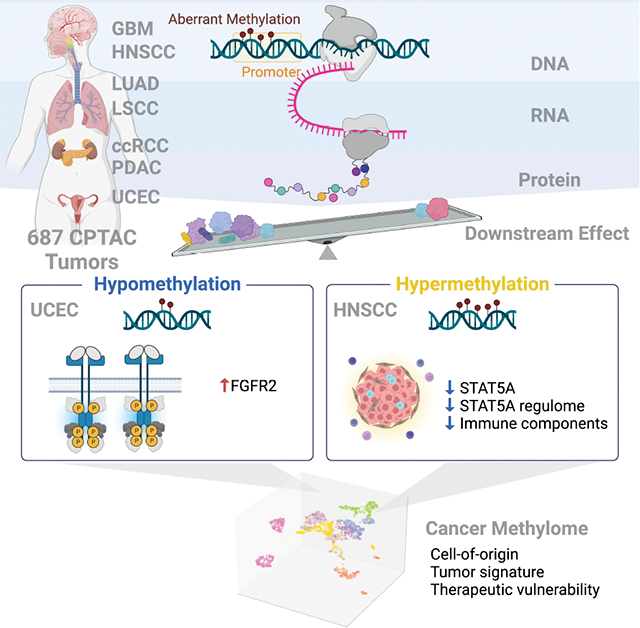
Introduction
Cytosine methylation is an epigenetic modification that confers stability and flexibility in the spatiotemporal gene regulation of many biological processes, including establishment and maintenance of cell identity. Aberrant DNA methylation is a hallmark of human cancer development and progression1–3. Aberrant DNA methylation has been observed in the global hypomethylation of repetitive sequences and the gene-specific hypermethylation of numerous CpG islands (CGI)4,5. Such changes within promoter regions can silence tumor suppressor genes or deregulate oncogenes. Moreover, the widespread changes in DNA methylation patterns usually arise in the early stages of tumorigenesis, suggesting a driving role of aberrant DNA methylation6. Given the reversible and dynamic nature of DNA methylation, treating cells with DNA demethylating agents might reprogram neoplastic cells back toward a normal state7. Delineating the functional consequences of aberrant DNA methylation is critical for improving cancer diagnosis, prognosis, and treatment.
Identifying DNA methylation patterns with functional roles in cancer and distinguishing them from tissue-specific epigenetic footprints remains challenging8. While some analytical approaches facilitate exploring the connection between DNA methylation and gene expression changes9,10, most studies have focused solely on transcriptome expression. We propose aggregating multi-omic data to comprehensively understand how tumor-specific methylation impacts both transcription and translation. By leveraging proteomic data as a direct measure of biological activity, we aim to discover DNA methylation drivers and gain insight of their role underlying tumor development.
Here, we integrated multi-omic data from 687 patients across seven cancer types from the Clinical Proteomic Tumor Analysis Consortium (CPTAC)11, and systematically examined the impact of cis-acting aberrant DNA methylation events on information flow, features, and functional consequences. Our analysis identified common and tissue-specific epigenetic events, including significant alterations in cancer genes affecting hallmark pathways. Distinct cancer subtypes were characterized by unique methylation patterns, validated by RNA and protein signatures reflecting each subtype’s molecular characteristics. Moreover, we identified putative druggable genes tightly regulated by DNA methylation, offering potential targets for tailored therapeutic interventions. This comprehensive catalog advances our understanding of DNA methylation-mediated tumorigenesis and offers insights towards the development of epigenetic therapies.
Results
Pan-Cancer landscape of DNA methylation and associated functional changes
To construct a landscape of cancer methylomes and associated functional changes, we collected 687 human tumors with available DNA methylation profiles (Infinium EPIC array), gene expression (RNA-seq), and protein abundance (mass spectrometry) across seven cancer types from CPTAC. This cohort was comprised of 107 clear cell renal cell carcinomas (ccRCC), 94 glioblastomas (GBM), 104 head and neck squamous cell carcinomas (HNSCC), 107 lung squamous cell carcinomas (LSCC), 102 lung adenocarcinomas (LUAD), 79 pancreatic ductal adenocarcinomas (PDAC), and 94 uterine corpus endometrial carcinomas (UCEC). We also collected DNA methylation data from matched normal adjacent tissues (NAT) including kidney, head and neck, pancreas, and lung from CPTAC. For GBM and UCEC datasets that lacked DNA methylation data from NATs, we acquired the corresponding DNA methylome from The Cancer Genome Atlas (TCGA) and the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) datasets in the form of Infinium HumanMethylation BeadChip (HM450) (Table S1, STAR Methods). All DNA methylation profiles were processed from raw array data, with standardized processing, quality control, and batch correction procedures (STAR methods). This expanded dataset allowed us to estimate methylation among reference tissues and to interrogate aberrant DNA methylation in a tissue-specific context (Figure S1).
First, to estimate how methylome, transcriptome, and proteome are correlated, we quantified the pairwise correlation between data sets for the 12,943 genes where all measurements are available using linear models, taking into account cancer type and tumor purity as covariates (STAR method, Figure 1A and Table S1). Consistent with expectations, RNA expression positively correlated with protein abundance, and promoter DNA methylation negatively correlated with RNA expression or protein abundance, indicating that promoter hypermethylation leads to gene silencing, while hypomethylation enhances gene expression (Figure 1A). Focusing on the 10,844 genes showing significant mRNA-protein correlation, we categorized them into groups based on the correlation between promoter methylation and RNA/protein expression. The correlation coefficients varied in strength and direction (Figure 1B), with 64.2% of promoter methylation showing no correlation, 18.4% correlated with RNA expression, and 3.5% correlated with protein abundance (Figure 1C). Only 13.9% of the promoter methylation exhibited correlation with both RNA expression and protein abundance (Figure 1C). For example, MXRA5 and MNDA were correlated with RNA expression and protein abundance, respectively, while CARD11 and MGMT showed correlations with both RNA expression and protein abundance (Figure 1D). When parallel correlation tests were conducted within single cancer types without considering tumor purity, the fractions of promoters correlated with both RNA and protein abundance were even lower. This suggests that pan-cancer analysis with a larger sample size enhances discovery power (Table S1).
Figure 1. Correlations between promoter DNA methylation, transcriptome, and proteome.

(A) Left: A schematic of tumor types collected for this study. Right: Density plot showing the distribution of adjusted R values between protein abundance and RNA expression (yellow), promoter methylation and RNA expression (orange) or protein abundance (green).
(B) Scatter plot showing adjusted R values distribution for genes with promoter methylation correlated with RNA (left, orange), protein (middle, green), or both RNA and protein (right, blue).
(C) Upsetplot showing the breakdown of genes based on their correlation with promoter methylation and RNA/protein expression.
(D) Examples with distinct correlations between promoter methylation, RNA expression, and protein abundance. Each dot represents one tumor, with the solid line representing the correlation between scaled RNA and promoter methylation, and the dashed line representing the correlation between scaled protein abundance and promoter methylation. Significant correlations are highlighted (orange: RNA only; green: protein only, blue and purple: RNA and protein).
(E) Examples of anti-correlated genes with promoter hypermethylation, exhibiting upregulation at the RNA level and downregulation at the protein level.
(F) Pathway enrichment analysis of the 31 anti-correlated genes with promoter hypermethylation, showing upregulation at the RNA level and downregulation at the protein level. Pathway with FDR P-value <0.05 is highlighted in bold.
See also Figures S1, S2, and Table S1.
The limited impact of methylation on the proteome may be attributable to biological factors (e.g., translational regulation, tissue-specific expression, protein degradation, and post-translational modifications) or to technical factors resulting from the low detection sensitivity of low-abundance transcripts or proteins12,13. Indeed, we observed a significant decrease in RNA expression and protein abundance in genes where promoter methylation correlated only with RNA expression, compared to those where methylation correlated with both RNA expression and protein abundance (Figure S2A, Wilcoxon P < 2.2e-16). Furthermore, among the 353 genes with promoter methylation correlated solely with protein abundance, we identified 31 genes, including the previously reported MNDA14, with promoter hypermethylation and gene upregulation, indicative of a biological effect rather than a technical artifact (Figure 1D and 1E, Table S1). These genes exhibited enrichment in neutrophil degranulation and glycogen metabolism pathways (Figure 1F and Table S1). The significant correlation between promoter methylation and protein abundance suggests the presence of an additional regulatory layer likely detectable through proteomic data, emphasizing the intricate and context-specific nature of DNA methylation’s impact on gene regulation.
To characterize the prevalence of aberrant DNA methylation across seven cancer types, we next detected recurrent and deregulated DNA methylation in tumors compared to NATs. We then determined the association of DNA methylation with changes in both mRNA expression and protein abundance using the published pipeline RESET9. To minimize batch effects or confounding bias across cancer types, each cohort was analyzed individually. Our analysis aimed to profile aberrant DNA methylation for regions actively contributing to gene regulation15. Therefore, probe sets were limited to those within promoter regions, or 300 bp upstream and downstream of the transcription start sites for the RESET pipeline (Figure S2B and Table S2). Overall, we detected 5570 hypermethylated CpG sites associated with mRNA downregulation of 2549 genes, 889 hypermethylated CpG sites associated with protein downregulation of 425 genes, 537 hypomethylated CpG sites associated with mRNA upregulation of 442 genes, and 166 hypomethylated CpG sites with protein upregulation of 124 genes (FDR < 10%, Figure 2A). No aberrant DNA methylation has been identified in the PDAC cohort, which could be explained by low tumor purity16. Consistent with previous studies, promoter hypermethylation was observed more frequently than promoter hypomethylation in all cancer types9,17. Our results confirmed that several well-known tumor-associated genes are regulated epigenetically, including MLH1 in UCEC18, MGMT in GBM and HNSCC19. However, most identified genes have not been previously implicated in DNA methylation-mediated regulation (Table S2).
Figure 2. The cancer methylome landscape associated with transcriptomic and proteomic change.

(A) RNA expression (upper) and protein abundance (lower) changes in genes between aberrant and normal samples. Y axis corresponds to the statistical significance of aberrant DNA methylation with changed expression and x axis to the median difference of gene expression of samples with or without aberrant DNA methylation. Representative genes are colored based on methylation status: yellow, hypermethylation; blue, hypomethylation. Dot size indicates the number of CGIs associated with expression changes.
(B) Venn diagrams showing the number of hypermethylated (upper) and hypomethylated (lower) genes having significant RNA expression and/or protein abundance changes.
(C) Common and cancer type-specific aberrant methylations of cancer-associated genes. Shading of the filled circle indicates the median methylation difference (upper), RNA expression difference (middle), and protein abundance difference (lower) between aberrant and normal samples at significant CpG sites. Dot size is proportional to the number of samples harboring indicated aberrant DNA methylation events in the cancer cohort.
Next, for individual aberrant DNA methylation events, we examined the concordance between mRNA and protein changes. Out of 964 total hypermethylation and 217 total hypomethylation events, we identified 365 hypermethylation and 74 hypomethylation events associated with both RNA expression and protein abundance changes (Figure 2B and Figure S2C), with a 70.5% validation rate for the genes having available DNA methylation (HM450) and RNA-seq data in TCGA cohorts (FDR < 10%, Table S2). Among them, we observed 98.9% of the aberrant methylation had the same direction of effect across the seven cancer types. About 78.8% of the aberrant methylation have been recovered from the same analysis with tumor purity-adjusted methylation values (FDR < 10%, Table S2), suggesting that our reported events are not being driven by underlying differences in tumor purity. Having constructed a comprehensive map of the cis-acting cancer methylome, we were able to delineate functional impacts of deregulated DNA methylation for a set of genes directly related to tumorigenesis (Table S2). Figure 2C illustrates the median methylation, RNA, and protein differences for cancer-associated genes, presented as the difference between NAT and tumor samples. In line with previous studies, we observed 41 of the aberrant methylation events are cancer type-specific17,20,21. Tumor necrosis receptor FAS in GBM22 and homeobox gene MEIS1 in UCEC23 are two such examples. Overall, these results highlight the context-dependent regulation of DNA methylation and tissue-specific carcinogenesis17. Only 5 of the aberrant methylation events, namely hypomethylated EGFR, and hypermethylated STAT5A, MGMT, CARD11, and TRIM22 are common across cancer types. The generalizability of these aberrant DNA methylation patterns suggests their importance in tumor development.
Noting that our integrative analysis can also reasonably capture the impact of non-cancerous cells in the tumor microenvironment on bulk multi-omics profiles, we also included published single-cell RNA-seq datasets to annotate whether the reported event is likely to be identified in tumor, immune, or stromal cells24–28 (STAR methods). For example, in ccRCC, EGFR was expressed in cancer cells and TRIM22 was expressed in immune cells, indicating the possibility of aberrant DNA methylation affecting different populations of cells in the tumor microenvironment. In total, 190 out of 436 events (43.6%) were annotated for their specific expression (Table S2). While our integrative multi-omic analysis based on bulk tissue provides a reliable estimate across a large number of tumors, the complementary scRNA-seq annotations deepen these analyses by evaluating gene expression patterns of the reported events at the cellular level for future study.
cis-acting aberrant DNA methylation as a possible driver event
DNA methylation-mediated modulation might be an important mechanism affecting the regulation of oncogenes and tumor suppressor genes. To test this hypothesis, we characterized cis-acting aberrant DNA methylation on 299 driver genes29. Since DNA methylation can affect the binding of transcription factors (TFs)30, we examined the number and enrichment of TF binding sites (TFBS) in loci associated with expression changes. All loci were associated with at least one TFBS. Both hypomethylated and hypermethylated CGI sites were characterized by a similar number of TFBS (mean 8.14 v.s. 7.49 TFBS) (Figure 3A). PREP1, a master regulator that functions as a tumor suppressor in maintaining genome stability31, was the most enriched TFBS in hypermethylated loci. This result supports the notion that TFs might serve as both the readers and effectors of aberrant DNA methylation in tumors30, leading to altered expression as revealed by transcriptomic and proteomic data.
Figure 3. Characterization of aberrant methylation in driver genes.

(A) Distribution of the number of transcription factor binding sites for functional hypermethylation (yellow) and hypomethylation (blue). Enriched motif was highlighted in the inset.
(B) Mutual exclusivity and co-occurrence of genomic and epigenomic alterations in driver genes in LSCC.
(C) Violin plot comparing histone H3 acetylation levels between highly hypermethylated tumors and lowly hypermethylated tumors. Boxes represent the interquartile range (IQR), with the median frequency indicated by the horizontal line. Whiskers extend from the boxes to indicate the data range. Statistically significant differences between groups were determined using Wilcoxon rank sum test.
(D) Heatmap of histone sites exhibiting significant differential acetylation (FDR < 0.1) among immune subtypes. The grayscale color scale denotes the hypermethylation frequency in each tumor.
(E) RNA expression (upper) and protein abundance (lower) levels stratified by IDH2 genomic alterations and IDH2 hypomethylation (blue) versus IDH2 normal methylation (gray), with ** denoting Wilcoxon P<0.005. Median values are shown as solid black lines, and first and third quartiles are represented by dashed lines.
(F) Pathway diagram illustrating the average difference in RNA expression (left square) and protein abundance (right square) between IDH2 hypomethylated samples and normal methylated samples in LSCC. The shading of the filled squares indicates the extent of the differences.
(G) Positive (upper) and negative (lower) correlation coefficients of histone acetylation levels and methylation levels at α-KG target genes among IDH1/IDH2 wild-type, IDH1 mutant, IDH2 hypomethylated samples, and IDH2 mutant. The breakdown of each group was shown in the pie chart below. Boxes represent the IQR, with the median correlation value indicated by the horizontal line. Whiskers extend from the boxes to show the data range. Statistically significant differences between groups were determined using FDR-corrected P-values, with **** indicating P < 2.2e-16.
Next, we explored the relationship among genetic alterations, DNA methylation, or histone acetylation. For the driver genes with cis-acting DNA methylations, most methylation events were mutually exclusive with genomic alterations, as exemplified in LSCC (Figure 3B and Figure S3A). For example, the correlation between KLF5 expression and promoter DNA methylation suggests that methylation is the main factor regulating its expression (Figure S3B). Investigating the interplay between DNA methylation and histone acetylation revealed that tumors exhibiting a high frequency of hypermethylation are significantly linked to decreased levels of H3 acetylation (Figure 3C, Wilcoxon P<0.05), indicating an overall repression of genes. Furthermore, a distinct relationship was observed among immune subtypes, hypermethylation frequency, and histone acetylation profiles, where the immune cool group showed an association with tumors displaying high hypermethylation frequency (Figure 3D). The pattern of mutual exclusivity and the interaction between DNA methylation and histone acetylation suggests that DNA methylation contributes to positive selection and histone changes, respectively, potentially playing a driving role in tumorigenesis.
Comparison to NATs showed DNA hypomethylation is the main perturbation occurring in the IDH2 gene, found in 6 of 107 LSCC tumors (Figure 3E and Figure S3C). Since overexpression of IDH2 contributes to altered energy metabolism32,33, we examined the metabolic activity of those cancers in samples with or without IDH2 hypomethylation. The results showed preferential upregulation of genes involved in cancer metabolism, including KDMs, ALKBHs, TETs, and MTOR, with the specific genes and extent of their expression changes varying slightly across different pathways (Figure 3F). To detect the downstream metabolic remodeling effect of IDH2 hypomethylation, we investigated the relationship between IDHs alterations and histone acetylation profiles. A total of 393 patients with available acetylation and methylation data were categorized into four groups: IDH1/IDH2 wild-type (n=374), IDH1 mutants (n=11), IDH2 hypomethylated samples (n=5), and IDH2 mutants (n=3). We found that IDH2 hypomethylation, similar to IDH1 and IDH2 mutants, can significantly impact the correlations between histone acetylation and DNA methylation levels at 2-hydroxyglutarate (α-KG) target genes (Figure 3G and Table S2) and IDH2 target genes (Figure S3D and Table S2). For example, significantly lower levels of H1 K168K, H1 K75K, and H2 K86K were observed in IDH1/2 mutant and IDH2 hypomethylated samples compared to IDH1/2 wild types and IDH2 normal methylated samples, respectively (Figure S3E). Despite the distinct nature of IDH2 hypomethylation compared to IDH1 and IDH2 mutations, the similarity in correlation patterns suggests a potential metabolic convergence of these alterations across cancer types. The combined methylation and acetylation profiling revealed driver mutation-independent IDH2 activation.
Hypomethylated RTKs are newly identified driver events
We found that several receptor tyrosine kinases (RTKs), including FGFR2 and EGFR, are frequently hypomethylated in UCEC and across cancer types, respectively (Figure 2C). To dissect the contribution of hypomethylated RTKs to oncogene activation, we examined the relationship between promoter methylation and genetic alteration. Specifically, we identified mutations, fusions, and copy number variations (CNVs), and their effects on RTK RNA and protein levels.
About 63.2% (12 of 19) of FGFR2 missense and indel mutations were activating mutations that enabled high-grade inflammation and cell proliferation without hypomethylation34–39 (Figure 4A). However, we identified 9 UCEC tumors carrying cis-acting hypomethylated FGFR2, 8 of which had co-occurring genomic alterations. Furthermore, unsupervised clustering of DNA methylation data across 94 CPTAC UCEC tumors and 43 TCGA normal samples from the same organ type revealed that FGFR2 hypomethylated cases formed a distinct cluster with lower DNA methylation than normal tissues (Figure 4B). Specifically, one CGI (cg10314760) within the FGFR2 promoter displayed a strong correlation between promoter hypomethylation and active gene expression both at the RNA and protein levels (Figure 4C). Our results suggest that promoter methylation is a major factor modulating expression of FGFR2, and that FGFR2 hypomethylation represents another mechanism of RTK activation potentially commensurate with activating mutations.
Figure 4. Collaborative effects of FGFR2 mutations and hypomethylation on FGFR2 upregulation.

(A) Lolliplot showing missense mutations of FGFR2 in UCEC samples. The amino acids and types of mutations are labeled. Positions that are recurrently mutated are highlighted with the number of occurrences. The FGFR2 functional domains are colored.
(B) Unsupervised clustering of UCEC tumors (upper) and normal adjacent tissues (lower) based on DNA methylation of the FGFR2 promoter.
(C) Correlation of methylation with gene expression (upper) and protein abundance (lower). Samples are colored based on genetic and/or epigenetic alterations of FGFR2. Tumors harboring FGFR2 hypomethylation are highlighted by large dot size.
(D) RNA expression (upper) and protein abundance (lower) levels stratified by FGFR2 genomic alterations and FGFR2 hypomethylation (blue) versus FGFR2 normal methylation (gray), with ** denoting Wilcoxon P<0.005. Median values are shown as solid black lines, and first and third quartiles are represented by dashed lines.
See also Figure S4.
To distinguish the oncogenic effects of FGFR2 hypomethylation from co-occurring aberrations, we stratified UCEC tumors by the type of FGFR2 genomic alteration and examined FGFR2 expression accordingly. We found that the median of FGFR2 RNA expression or protein abundance are consistently higher in tumors with hypomethylated FGFR2 than that in tumors with normal methylated FGFR2. Higher expression of FGFR2 is significant in the group with CNVs at the cognate locus (Wilcoxon P < 0.005) (Figure 4D). Validation in the TCGA UCEC cohort confirmed our findings. FGFR2 exhibited hypomethylation at CGI site (cg10314760) in 21 of 174 tumor samples, strongly correlating with active gene expression (Figure S4A). Consistent correlation between FGFR2 hypomethylation and upregulation was observed in samples with wild type FGFR2, shallow deletion of FGFR2, and shallow amplification of FGFR2 (Figure S4B). This result suggests that, whereas activating mutations, amplifications, and promoter hypomethylation enable FGFR2 upregulation to differing extents, co-occurring FGFR2 hypomethylated sites result in even more profound expression changes than amplification alone. These results emphasize the important role of promoter hypomethylation in contributing to oncogenic gain-of-function. Similarly, hypomethylated EGFR was associated with EGFR upregulation in HNSCC, LSCC, and ccRCC (Figure S4C-D). Our results are consistent with the pan-cancer analysis suggesting that tumors harbor multiple aberrations within individual oncogenes such as FGFR2 or EGFR, likely conferring enhanced oncogenicity in combination40. Overall, although recurrent gain-of-function genomic alterations in RTKs have long been known to promote a variety of cancers41, our results reveal that RTK hypomethylation is also a bona fide epigenetic driver across several cancer types.
Hypermethylation of STAT5A is associated with pervasive changes in STAT5A regulon activity
Altered expression of TF can disrupt the activity of its regulon, a group of genes that are regulated by a common regulatory element. To estimate the impact of aberrant DNA methylation on regulon activity, we first identified 14 cis-acting hypermethylation events at TFs, and then tested their association with corresponding regulons, such as receptors, activator, repressor, and target genes involved in the same pathway (Fisher’s exact test, FDR P<0.1, Table S3). We identified significant associations between hypermethylated TFs and low regulon activity at both RNA expression and protein abundance levels. Since STAT5A regulon comprises the largest number of interacting genes, we therefore focus on STAT5A for the downstream analysis.
STAT5A controls cell identity, cytotoxicity, and cell survival; dysregulation of those processes can contribute to tumorigenesis42. Unsupervised clustering of STAT5A-interacting proteins, using expression data from HNSCC tumors divided samples into two groups: those with high regulon activity and those with low regulon activity (Figure 5A). Notably, samples with hypermethylated STAT5A were significantly enriched in the regulon-low group (Fisher’s exact test, P=7.8E-05). The same pattern was observed in protein abundance (Figure 5B, Fisher’s exact test, P=0.025). Furthermore, exome sequencing of these tumors did not identify any distinct, recurrent coding sequence mutations in STAT5A-interacting genes (Table S3), suggesting that other genetic drivers were not involved. Additionally, STAT5A phosphorylation was not significantly associated with either STAT5A methylation status or STAT5A regulon activity (Figure S5A). Altogether, the enrichments we observed indicate that aberrant methylation of STAT5A leads to pervasive regulon changes in HNSCC. Similar to HNSCC, hypermethylated STAT5A was associated with low regulon activity in LSCC (Figure S5B).
Figure 5. STAT5A hypermethylation associated with pervasive STAT5A regulon changes.

(A) Unsupervised clustering of STAT5A regulon genes using Pearson correlation of scaled RNA sequencing data. Annotations denote STAT5A expression and methylation levels. Mean activity indicates the overall sum of regulon activity. The color scale is proportional to expression (red: upregulation; blue: downregulation).
(B) Unsupervised clustering of STAT5A regulon genes using Pearson correlation of scaled global proteome data.
(C) Violin plot comparing regulon activity in hypermethylated STAT5A (yellow) and normally methylated STAT5A (gray) samples. Median values are shown as solid black lines, and first and third quartiles are represented by dashed lines. Statistical significance was determined using a Wilcoxon rank-sum test.
(D) Pathway members and interactions in the STAT5A regulon. The mean expression differences between STAT5A hypermethylated samples and normally methylated samples are indicated by shading of the filled squares.
Since samples with hypermethylated STAT5A were significantly associated with lower regulon activity at both RNA and protein levels (Figure 5C), we hypothesized that STAT5A-interacting components (i.e., receptors, kinases, repressors, co-activators, and target genes) would be downregulated in samples with hypermethylated STAT5A. Among the target genes, we observed significant downregulation of IRF1, PRF1, IFNG, IL2RA, and IL6ST (Wilcoxon P<0.05) at either/both RNA and protein levels (Figure 5D). These observations link hypermethylated STAT5A to the regulation of cytokine production, cytotoxicity, cell proliferation, and interferon signaling.
Hypermethylated STAT5A is associated with immune cell depletion in squamous tumors
Recent study has shown that STAT5A-mediated interferon signaling regulates the expression of CD274 (encodes PD-L1) and PDCD1LG2 (encodes PD-L2), reflecting the clinical significance of STAT5A signaling in immunotherapy43. STAT5A target genes are directly implicated in immune response (e.g., IL2RA, IRF1, and IFNG) and their low expression implies alteration of normal immune function and homeostasis44. We therefore focused on characterizing the immune component of HNSCC and LSCC tumors to understand how hypermethylated STAT5A affects the tumor microenvironment.
To explore the microenvironment of HNSCC and LSCC tumors, we stratified transcriptome data from cell mixtures into multiple immune cell types using xCell45. Consensus clustering of 64 different immune-related cell types identified four major immune clusters, including immune-cold, immune-cool, immune-warm, and immune hot subtypes, which harbored general characteristics of immunosuppressive to inflammatory microenvironment28,46,47 (Table S3). STAT5A hypermethylated samples were significantly enriched in the immune-cool group both in HNSCC and LSCC (Figure 6A and Figure S6A, Fisher’s exact test, P<0.00001 and P=0.0002, respectively). STAT5A hypermethylated samples displayed significantly decreased expression of genes associated with immune effectors and dendritic cells (Figure 6B and Figure S6B, Wilcoxon P<0.05). Interestingly, for squamous tumors with available histone acetylation data, we found samples with hypermethylated STAT5A have significantly lower acetylation of histone H3 lysine 14 (H3K14)(FDR=0.05, Figure S6C), representing an overall gene repression48. Our findings suggested that samples with hypermethylated STAT5A are significantly associated with immune cell depletion in squamous tumors.
Figure 6. Functional impact of STAT5A hypermethylation on immune cell depletion in HNSCC.
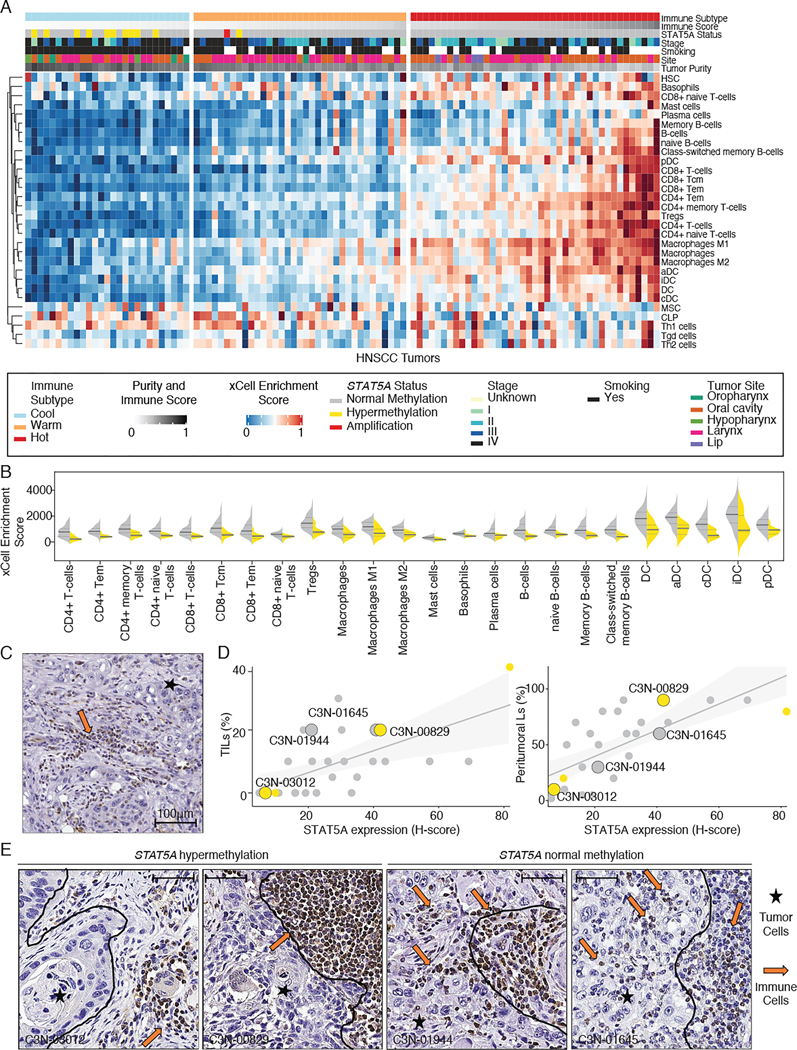
(A) Heatmaps showing distinct immune subtypes of HNSCC tumors derived from xCell enrichment scores. The top panel shows the immune score, DNA methylation status of STAT5A, immune subtype, and tumor stage.
(B) Violin plots comparing xCell enrichment scores of immune effectors and dendritic cells in hypermethylated STAT5A (yellow) and normally methylated STAT5A (gray) samples in HNSCC tumors. Median values are shown as solid black lines, and first and third quartiles are represented by dashed lines. Statistical significance was determined using a Wilcoxon rank-sum test.
(C) Representative image of IHC (immunohistochemistry) staining of STAT5A protein (brown) in HNSCC tumor sample. Scale bar = 100 μm.
(D) Correlation between the quantified STAT5A protein abundance versus the level of tumor-infiltrating lymphocytes (TILs, left panel) or peritumoral lymphocytes (right panel). Samples were colored by the DNA methylation status of STAT5A (yellow: hypermethylation, gray: normal methylation). Samples with representative IHC images are highlighted by large dot size.
(E) IHC staining of STAT5A in HNSCC tumors. Representative tumor cells (stars) and lymphocytes (arrows) are shown. Tumor boundary is indicated by a black line. Scale bar = 100 μm.
Next, to further identify the expression specificity of STAT5A hypermethylation in HNSCC, we evaluated 29 representative HNSCC cases by immunohistochemistry (IHC) markers STAT5A (Table S3). We found prominent STAT5A protein expression in the tumor-infiltrating lymphocytes (TILs) and peritumoral lymphocytes, while STAT5A was minimally expressed or absent in tumor cells (Figure 6C). The overall level of STAT5A abundance in the stained slide was correlated with the level of TILs or peritumoral lymphocytes (Figure 6D). Interestingly, samples with hypermethylated STAT5A showed a distinct boundary between tumor cells and STAT5A-expressing immune cells (Figure 6E, left panels), while tumor samples with normal methylated STAT5A showed a mixture of tumor cells and STAT5A-expressing immune cells (Figure 6E, right panels). We speculated that immune cells with hypermethylated STAT5A might limit the migration of lymphocytes in the tumor microenvironment, however the working model requires further investigation in the future.
Our finding of lower tumor microenvironment factors in STAT5A-hypermethylated samples is in agreement with previous studies showing that hematopoietic stem cell proliferation was severely impaired in Stat5A-deficient mice49–51. It is also consistent with STAT5A being identified as a key tumor suppressor in lymphoma cell lines52. In addition, studies have indicated that the development of HNSCC is closely related to immunosuppression and immune escape53. These findings suggest that STAT5A hypermethylation may mediate the disease-dependent expression of STAT5A-targeted genes54 and contributes to altered tumor immunogenicity.
Aberrant methylation associated with therapeutic vulnerabilities
We next explored if epigenetic features can classify tumors into transcriptionally and translationally distinct subtypes. To identify subtypes based on methylation patterns, we generated methylation profiles from 687 tumors and NATs, and used uniform manifold approximation and projection (UMAP) to reduce methylation signals from 340,000 CGIs into two dimensions55 (Figure 7 and Figure S7A). We found that tumors clustered by organ system, including brain (GBM), kidney (ccRCC), lung (LUAD and LSCC), head and neck (HNSCC), pancreas (PDAC), and uterus (UCEC) (Figure 7A, first column). Additionally, squamous cell cancers, LSCC and HNSCC, formed a distinct cluster adjacent to LUAD. The distinct pattern between chromophobe renal cell carcinomas (C3N-00492 and C3N-01175) and ccRCC samples (Figure S7A) were consistent with their distinct origins56,57. Direct comparisons between tumors and corresponding normal adjacent tissues revealed pronounced methylation differences (Figure 7A, second column). To investigate intrinsic lineage differences in DNA methylation between tumors possessing different cells-of-origin, we examined differentially methylated CGIs in tumor samples compared to NAT samples and found 99 cancer-specific aberrant methylation promoters (Figure 7B and Table S4). Together, the results suggest that the DNA methylome of tumors faithfully reflects cell-of-origin and malignant transformation.
Figure 7. Summary of the cancer methylome for cell-of-origin, tumor signatures, and therapy.
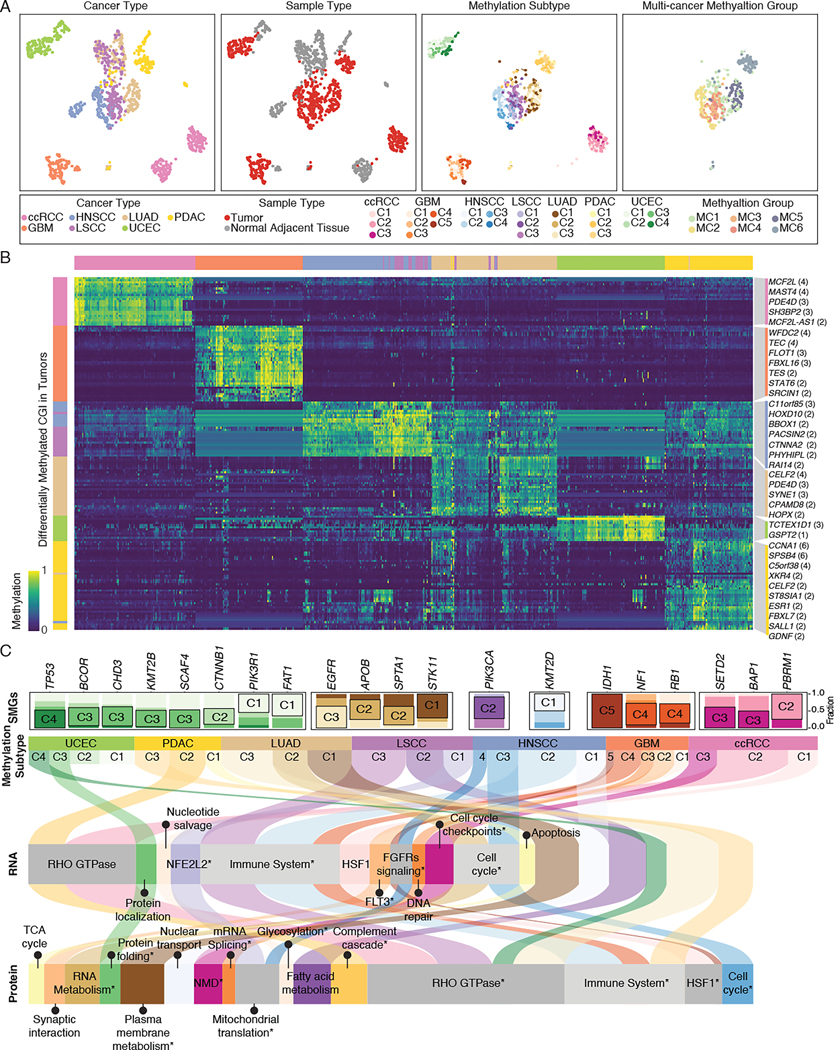
(A) Projection of the cancer methylomes. Each point is a sample and is colored based on the cancer type (first column), sample type (second column), methylation subtype (third column), or multi-cancer methylation group (fourth column).
(B) Heatmap of differentially methylated CpG sites at promoter regions in seven cancer cohorts compared to normal adjacent tissue. Selected promoters annotated with the number of differentially methylated CpG sites are shown.
(C) Alluvial plot showing the per-cancer methylation subtypes (second row), their enriched significantly mutated genes (SMGs) (first row), enriched RNA expression signature (third row), and enriched protein signature (fourth row). The curved lines across panels correspond to different methylation subtypes.
Signatures with FDR P-values < 0.05 are highlighted with *.
Next, using CGIs showing significant differences between tumors and NATs, we identified between 3 and 5 clusters from each cancer type (Figure 7A, third column, and Figure S7B). We correlated those methylation subtypes with existing RNA expression-based subtypes16,24,56,58–61 (Fisher’s exact test P < 0.05), and found that methylation subtypes captured several important genomic features and clinical characteristics (Figure S7C and Table S4). For example, the UCEC C1 to C4 methylation subtypes are enriched with POLE, CNV-low, MSI-high, and CNV-high tumors, respectively62. The LUAD C1 to C3 methylation subtypes are enriched with proximal-proliferative, proximal-inflammatory, and terminal respiratory unit tumors, respectively63,64. GBM subtypes C4 and C5 feature mesenchymal phenotype and CpG island methylator phenotype associated with IDH1 mutation, respectively22. Altogether, our results suggest that clustering cancer samples based on DNA methylation can help identify molecularly and clinically relevant subtypes.
To further explore the biological differences between methylation subtypes, we performed an over-representation pathway analysis using differentially expressed genes and proteins, revealing significantly enriched pathways related to tumorigenesis (Figure 7C and Table S5, P < 0.05). Some significant subtype-specific tumorigenic signatures were consistently observed at the transcriptomic and proteomic levels, such as enrichment of LSCC-C1 for NFE2L2 orchestrating the adaptive response to oxidative stress65,66, and enrichment of ccRCC-C1 for tumorigenic transcriptional network coordinated by HSF167. Despite comprising distinct cancer types, LSCC-C3, LUAD-C2, PDAC-C2, HNSCC-C2 and GBM-C4 were characterized by immune-related signatures at RNA and/or protein levels. This correlation suggests signaling convergence among various cancers, in line with previous studies demonstrating that some methylation subtypes are significantly associated with immune signature68–70. On the other hand, enrichment of pathway signatures was not detected in some methylation subtypes, which were thus only characterized by their distinct methylation pattern. For example, deficiency in DNA polymerase ε (POLE) proofreading generates an extensive number of somatic mutations and leads to a methylation profile, which is shown as the UCEC subtype C1 (Figure 7C and Figure S7D). Overall, these results demonstrate heterogeneity within cancer types and how distinct methylation patterns may give rise to various cancer phenotypes.
To identify the interplay between driver mutation and methylation subtypes in tumorigenesis, we correlated methylation subtypes with 299 driver mutations. We found 13 out of 25 methylation subtypes are significantly associated with cancer-specific driver mutations (Figure 7C and Figure S7D). Consistently, enrichment of seven driver mutations in particular RNA-based subtypes have been identified previously, including UCEC-C4 (CNV-high) enriched with TP53 mutations71, UCEC-C3 (MSI-high) with KMT2B mutations61, UCEC-C2 (CNV-low) with CTNNB1 mutations71, LUAD-C3 (terminal respiratory unit) with EGFR mutations72, LUAD-C1 (proximal-proliferative) with STK11 mutations64, and GBM-C4 (mesenchymal) with NF1 mutations73,74. We also identified enrichment of mutations at epigenetic modifiers that could directly affect cancer methylome. For example, IDH1 deficiency generates high levels of α-KG and leads to epigenetic reprogramming, which is shown as the GBM subtype C524. Mutations in PBRM1 (a chromatin remodeler), SETD2 (a histone methyltransferase), and BAP1 (a histone deubiquitinating enzyme) lead to distinct DNA methylation phenotypes in ccRCC75,76. The significant correlation between driver mutations and methylation subtype suggested that deregulation of driver genes induces epigenetic reprogramming, rewiring regulatory networks together during tumorigenesis.
Beyond the methylation subtyping within each cohort, we also conducted methylation profiling and signature enrichment analysis across HNSCC, LSCC, LUAD, and PDAC, which form a distinct cluster in the UMAP projection (Figure 7A, fourth column, and Figure S7E). We identified six methylation groups with various signatures, including groups enriched with immune-related signatures (MC1 and MC2), groups enriched with squamous tumors (MC3 and MC4), LUAD-dominant group (MC5), and PDAC-dominant group (MC6) (Figure S7F and Table S4, P < 0.05). MC5 and MC6 are enriched with cancer type-specific signatures such as β cell development and surfactant metabolism, respectively. Following the observation of methylation subtypes, we again observed signaling convergence among various cancers in MC1 and MC2. Notably, cancers with squamous features (MC3 and MC4) were enriched with replication stress signatures, coinciding with their high degree of genomic instability77,78. Our multi-omic integrative analysis enables the identification of common functionality arising from the same methylation profile across different cancer types.
Finally, we investigated druggable targets for sites with cis-acting dysregulated DNA methylation. We integrated cis-acting DNA methylation with the Clinical Interpretation of Variants in Cancer (CIViC)79 and analyzed target genes with outlier expressions for which pharmacological intervention might be available (Table S6). Allowing for “off-label” drug treatment, we found that 19.2% of samples (132 of 687 tumors) would likely benefit from one or more treatments targeting genes altered by DNA methylation (Figure S7G). The most frequent druggable DNA methylation events across the seven cancer types are those on MGMT (n = 45 tumors), NAPRT (n = 31 tumors), and EGFR (n = 26 tumors). These findings may have important clinical implications. For example, tumor-specific loss of NAPRT, mediated by promoter hypermethylation, is synthetically lethal with NAMPT inhibitor treatment in multiple cancer types, resulting in inactivation of nicotinic acid salvage pathways80. However, it is worth mentioning that we do not claim superiority over any other drug discovery method since it is difficult to evaluate the performance without gold standards. We claim that: (1) the high correlation among different data types provides validation to each other for the functional events in tumors; and (2) this study provides highly regulated aberrant DNA methylation events that are reflected in RNA expression and protein abundance readout, which is useful and complementary to other methods. Thus, collectively, these characterizations of cis-acting aberrant DNA methylation in cancer reveal potential new directions for treatment optimization.
Discussion
The pan-cancer multi-omic analysis revealed driver gene regulation via DNA methylation, providing insights into methylation-based stratification of cancer patients. We identified and characterized methylation subtypes enriched with various RNA and protein signatures that have potential therapeutic and prognostic implications. Of interest, we observed that subsets of tumors in different organs may share a convergent immune-related signature. For these, the cancer methylome may offer opportunities for patient stratification to increase the efficacy of immune-based therapies. Moreover, we observed clinically relevant alterations with important therapeutic potential in 132 out of 687 tumors. Targeting those common aberrant methylation events could enhance the therapeutic reach of existing drugs by broadening the treatable patient and tumor populations. To maximize this benefit, future studies may look toward optimizing epigenetic therapies.
We uncovered several bona fide DNA methylation drivers showing functional consequences, including hypomethylated FGFR2 and EGFR, which could be informative in expanding patient eligibility of conventional genotype-directed clinical trials. In addition, the apparent co-occurrence of FGFR2 hypomethylation and amplification suggests that epigenetic enhancement of FGFR2 expression may offer selective advantage for developing a second FGFR2 alteration, perhaps from enhanced FGFR2 signaling in these cells. The converse relationship is also possible: genomic alterations to FGFR2 disrupt the reading, writing, or maintaining of DNA methylation machinery in tumor cells, subsequently leading to aberrantly reduced methylation within the FGFR2 promoter. Regardless of the direction, this relationship takes, since UCEC tumors harboring epigenetic and genetic alterations are significantly associated with FGFR2 upregulation, it is likely that FGFR2 hypomethylation works in concert with amplification at FGFR2 to promote tumorigenesis81.
Studies have shown that HNSCC tumors evade the host immune system by manipulating their own immunogenicity53. Our findings reinforce the critical nature of STAT5A as a signaling hub in modulating tumor immunogenicity across squamous cancers. Together, these findings suggest opportunities for therapeutic intervention by targeting epigenetic alterations within the STAT5A promoter. DNMT inhibitors, such as 5’-azacytidine, have been shown to reduce methylation of the STAT5A promoter in cell lines52 and are FDA-approved for treating myelodysplastic syndrome82. Furthermore, activation of STAT5A signaling may transform an immunologically cold, inactive tumor into a hot, inflamed one and thus increase the anti-tumor immune response. Additional investigation is required to uncover the mechanisms mediating STAT5A hypermethylation and downstream immune-related signaling pathways, including the interaction between them, and the impact on therapeutic sensitivity.
There are several limitations to this study. First, recent investigations have shown that not only promoters, but also intragenic and intergenic regions, are widely modulated during disease progression83. Here, we only focus on cis-acting DNA methylation at promoter regions, while trans-acting DNA methylation (i.e. DNA methylation acting upon other target genes) and other regulatory elements are not discussed. Second, previous studies have shown that DNA methylation and gene expression are not as frequently correlated as previously thought84. Therefore, DNA methylation may have critical functions other than gene expression regulation. One possibility is that DNA methylation changes influence transcriptional potential rather than actual transcription status and could therefore be involved in the epigenetic plasticity of tumor cells85,86. Another possibility is that aberrant intra- or intergenic DNA methylation in cancer cells may lead to increased non-synonymous mutation rate. Finally, although our integrative multi-omic analysis has limitation in differentiating DNA methylation changes specifically in cancer epithelial cells or tumor-infiltrating lymphocytes, we provide a reliable estimate across a wide range of tumors, laying the groundwork for future single-cell analyses and spatial omics investigations. We emphasize the importance of isolating cancer epithelial cells and employing DNA methylome sequencing for future study. Altogether, future studies at single cell resolution may help reveal additional mechanistic details underlying the contribution of aberrant DNA methylation to tumor development.
Overall, our results help identify the contribution of DNA methylation in tumorigenesis and delineate its role in initiating and maintaining malignancies. This thorough account of cis-acting events and characterization of the cancer methylome will inform systematic explorations of aberrant DNA methylation and associated functional consequences, ultimately revealing potential new disease mechanisms and therapeutic opportunities.
Consortia
The members of the National Cancer Institute Clinical Proteomic Tumor Analysis Consortium for Pan-Cancer are François Aguet, Yo Akiyama, Eunkyung An, Shankara Anand, Meenakshi Anurag, Özgün Babur, Jasmin Bavarva, Chet Birger, Michael J. Birrer, Anna Calinawan, Lewis C. Cantley, Song Cao, Steven A. Carr, Michele Ceccarelli, Daniel W. Chan, Arul M. Chinnaiyan, Hanbyul Cho, Shrabanti Chowdhury, Marcin P. Cieslik, Karl R. Clauser, Antonio Colaprico, Daniel Cui Zhou, Felipe da Veiga Leprevost, Corbin Day, Saravana M. Dhanasekaran, Li Ding, Marcin J. Domagalski, Yongchao Dou, Brian J. Druker, Nathan Edwards, Matthew J. Ellis, Myvizhi Esai Selvan, David Fenyö, Steven M. Foltz, Alicia Francis, Yifat Geffen, Gad Getz, Michael A. Gillette, Tania J. Gonzalez Robles, Sara J. C. Gosline, Zeynep H. Gümüş, David I. Heiman, Tara Hiltke, Runyu Hong, Galen Hostetter, Yingwei Hu, Chen Huang, Emily Huntsman, Antonio Iavarone, Eric J. Jaehnig, Scott D. Jewell, Jiayi Ji, Wen Jiang, Jared L. Johnson, Lizabeth Katsnelson, Karen A. Ketchum, Iga Kolodziejczak, Karsten Krug, Chandan Kumar-Sinha, Alexander J. Lazar, Jonathan T. Lei, Yize Li, Wen-Wei Liang, Yuxing Liao, Caleb M. Lindgren, Tao Liu, Wenke Liu, Weiping Ma, D R. Mani, Fernanda Martins Rodrigues, Wilson McKerrow, Mehdi Mesri, Alexey I. Nesvizhskii, Chelsea J. Newton, Robert Oldroyd, Gilbert S. Omenn, Amanda G. Paulovich, Samuel H. Payne, Francesca Petralia, Pietro Pugliese, Boris Reva, Ana I. Robles, Karin D. Rodland, Henry Rodriguez, Kelly V. Ruggles, Dmitry Rykunov, Shankha Satpathy, Sara R. Savage, Eric E. Schadt, Michael Schnaubelt, Tobias Schraink, Stephan Schürer, Zhiao Shi, Richard D. Smith, Xiaoyu Song, Yizhe Song, Vasileios Stathias, Erik P. Storrs, Jimin Tan, Nadezhda V. Terekhanova, Ratna R. Thangudu, Mathangi Thiagarajan, Nicole Tignor, Joshua M. Wang, Liang-Bo Wang, Pei Wang, Ying Wang, Bo Wen, Maciej Wiznerowicz, Yige Wu, Matthew A. Wyczalkowski, Lijun Yao, Tomer M. Yaron, Xinpei Yi, Bing Zhang, Hui Zhang, Qing Zhang, Xu Zhang, Zhen Zhang.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Li Ding (lding@wustl.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Raw and processed proteomics as well as open-access genomic data, can be obtained via Proteomic Data Commons (PDC) at https://pdc.cancer.gov/pdc/cptac-pancancer. Raw genomic and transcriptomic data files can be accessed via the Genomic Data Commons (GDC) Data Portal at https://portal.gdc.cancer.gov with dbGaP Study Accession: phs001287.v16.p6. Complete CPTAC Pan-Cancer controlled and processed data can be accessed via the Cancer Data Service (CDS, https://dataservice.datacommons.cancer.gov/). The CPTAC Pan-Cancer data hosted in CDS is controlled data and can be accessed through the NCI DAC approved, dbGaP compiled whitelists. Users can access the data for analysis through the Seven Bridges Cancer Genomics Cloud (SB-CGC) which is one of the NCI-funded Cloud Resource/platform for compute intensive analysis. Instructions to access data: 1. Create an account on CGC, Seven Bridges (https://cgc-accounts.sbgenomics.com/auth/register 2. Get approval from dbGaP to access the controlled study (https://www.ncbi.nlm.nih.gov/projects/gap/cgibin/study.cgi?study_id=phs001287. v16.p6 ) 3. Log into CGC to access Cancer Data Service (CDS) File Explore 4. Copy data into your own space and start analysis and exploration 5. Visit the CDS page on CGC to see what studies are available and instructions and guides to use the resources. (https://docs.cancergenomicscloud.org/page/cds-data).
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-STAT5A antibody | Atlas Antibodies | Catalog: HPA042128, RRID: AB_2677864 |
| Biological samples | ||
| Primary tumor and normal adjacent tissue samples | CANCER-CELL-D- 22–00603 companion Pan-Cancer resource manuscript47 | https://pdc.cancer.gov/pdc/cptac-pancancer |
| Chemicals, peptides, and recombinant proteins | ||
| Dako Protein Block, Serum-free blocking solution | Agilent Technologies Inc | Catalog: X090930–2 |
| Dako Taget Retrieval Solution, pH=6 | Agilent Technologies Inc | Catalog: S236984–2 |
| Dako Wash Buffer 10X | Agilent Technologies Inc | Catalog: S3006 |
| Critical commercial assays | ||
| TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold | Illumina | Catalog: RS-122–2301 |
| Infinium MethylationEPIC Kit | Illumina | Catalog: WG-317–1003 |
| Nextera DNA Exosome Kit | Illumina | Catalog: 20020617 |
| KAPA Hyper Prep Kit, PCR-free | Roche | Catalog: 07962371001 |
| TMT-11 Reagent Kit | ThermoFisher Scientific | Catalog: A34808 |
| BCA Protein Assay Kit | ThermoFisher Scientific | Catalog: 23225 |
| PTMScan® Acetyl-Lysine Motif [Ac-K] Kit | Cell Signaling | Catalog: 13416 |
| EnVision FLEX Visualizing Kit | Agilent Technologies Inc | Catalog: K800221–2 |
| Deposited data | ||
| CIViC nightly, 062220 | Griffith et al.79 | https://civicdb.org/home |
| FANTOM5 | FANTOM Consortium et al.87 | http://fantom.gsc.riken.jp/5/ |
| TARGET Methylation data | Pugh et al.88 | https://ocg.cancer.gov/programs/target/data-matrix |
| TCGA Methylation data and RNA-seq data | TCGA et al.64,71,78,89–92 | https://gdac.broadinstitute.org/ |
| JASPAR | Khan et al.93 | https://jaspar.genereg.net/ |
| InfiniumAnnotation | Zhou et al.94 | https://zwdzwd.github.io/InfiniumAnnotation |
| OmniPath | Türei et al.95 | http://omnipathdb.org/ |
| ccRCC scRNA-seq data | Li et al.25 | https://portal.gdc.cancer.gov/projects/CPTAC-3 |
| GBM scRNA-seq data | Wang et al.24 | https://portal.gdc.cancer.gov/projects/CPTAC-3 |
| PDAC scRNA-seq data | Cui Zhou et al.26 | https://data.humantumoratlas.org/ |
| Lungs scRNA-seq data | Travaglini et al.27 | https://www.synapse.org/#!Synapse:syn21041850 |
| CPTAC clinical and proteomic data | CANCER-CELL-D- 22–00603 companion Pan-Cancer resource manuscript47 | https://pdc.cancer.gov/pdc/cptac-pancancer |
| CPTAC genomic and transcriptomic data | CANCER-CELL-D- 22–00603 companion Pan-Cancer resource manuscript47 | https://pdc.cancer.gov/pdc/cptac-pancancer and Cancer Data Service (CDS) |
| CPTAC DNA methylation data | This study | https://pdc.cancer.gov/pdc/cptac-pancancer |
| CPTAC acetylation data | CANCER-CELL-D- 22–00603 companion Pan-Cancer resource manuscript47 | https://pdc.cancer.gov/pdc/cptac-pancancer |
| Software and algorithms | ||
| RESET | Saghafinia et al.9 | http://ciriellolab.org/reset/reset.html |
| ChAMP | Morris et al.96 | https://www.bioconductor.org/packages/release/bioc/vignettes/ChAMP/inst/doc/ChAMP.html |
| Methylation array analysis pipeline for CPTAC | This study | https://github.com/ding-lab/cptac_methylation |
| methylationArrayAnalysis v3.9 | Maksimovic et al.97 | https://master.bioconductor.org/packages/release/workflows/html/methylationArrayAnalysis.html |
| Illumina EPIC methylation array v0.6 | See link | https://bioconductor.org/packages/release/data/annotation/html/IlluminaHumanMethylationEPICanno.ilm10b2.hg19.html |
| ConsensusClusterPlus v1.48.0 | Wilkerson et al.98 | https://bioconductor.org/packages/ConsensusClusterPlus/ |
| xCell v1.2 | Aran et al.45 | http://xcell.ucsf.edu/ |
| SomaticWrapper | Ding Lab | https://github.com/ding-lab/somaticwrapper |
| Strelka2 | Saunders et al.99 | https://github.com/Illumina/strelka |
| MUTECT v1.1.7 | Cibulskis et al.100 | https://software.broadinstitute.org/gatk/download/archive |
| VarScan v2.3.8 | Koboldt et al.101 | http://varscan.sourceforge.net |
| Pindel v0.2.5 | Ye et al.102 | http://gmt.genome.wustl.edu/packages/pindel/ |
| Fusion calling pipeline for CPTAC | Gao et al.103 | https://github.com/cuidaniel/Fusion_hg38 |
| STAR-Fusion v1.5.0 | Haas et al.104 | https://github.com/STAR-Fusion/STAR-Fusion/wiki |
| EricScript v0.5.5 | Benelli et al.105 | https://sites.google.com/site/bioericscript |
| Integrate v0.2.6 | Zhang et al.106 | https://sourceforge.net/p/integrate-fusion/wiki/Home/ |
| Copy Number Variant Calling | Ding Lab | https://github.com/ding-lab/BICSEQ2 |
| BIC-seq2 | Xi et al.107 | http://compbio.med.harvard.edu/BIC-seq/ |
| SomaticSV | Ding Lab | https://github.com/ding-lab/SomaticSV |
| Manta v1.6.0 | Chen et al.108 | https://github.com/Illumina/manta |
| MSFragger v3.4 | Kong et al.109 | https://msfragger.nesvilab.org/ |
| Philosopher toolkit v4.0.1 | da Veiga Leprevost et al.110 | https://philosopher.nesvilab.org/ |
| TMT-Integrator | Djomehri et al.111 | http://tmt-integrator.nesvilab.org/ |
| HTSeq v0.11.2 | Anders et al.117 | https://htseq.readthedocs.io/en/master/ |
| DreamAI | Ma et al.112 | https://github.com/WangLab-MSSM/DreamAI |
| ProteintPaint Lollipop | Zhou Lab | https://viz.stjude.cloud/zhou-lab/visualization/proteintpaint-lollipop-example~57 |
| LIMMA v3.36 (R Package) | Ritchie et al.113 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| HOMER | Heinz et al.114 | http://homer.salk.edu/homer/ |
| QuPath v0.3.2 | Bankhead et al.115 | https://qupath.github.io/ |
| Reactome | Fabregat et al.116 | https://reactome.org/ |
| Scanpy v1.7.0 | Wolf et al.117 | https://github.com/scverse/scanpy |
| Python v3.7 | Python Software Foundation | https://www.python.org/ |
| R v3.6 | R Development Core Team | https://www.R-project.org/ |
| Bioconda | Grüning et al.118 | https://bioconda.github.io/ |
| Bioconductor v3.9 | Huber et al.119 | https://bioconductor.org/ |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Human participants
A total of 687 participants were included in strict accordance with the CPTAC-3 protocol with informed consent from the patients. Prospective biospecimen collection (tumor and adjacent normal samples where feasible) followed a tumor type specific protocol and standard operating procedures, where sample collection, qualification and processing were optimized for both genomics and proteomics16,24,56,58–61. CPTAC samples were collected by 30+ tissue source sites from both domestic and international locations and processed by a central biospecimen core resource. The samples were pathology qualified by a general pathologist and later reconfirmed by a disease-specific expert pathologist through histopathology image review and immunohistochemistry assays where applicable.
Clinical data annotation
Clinical data were obtained from TSS and aggregated by the Biospecimen Core Resource (BCR, Van Andel Research Institute (Grand Rapids, MI)). Data forms were stored as Microsoft Excel files (.xls). Clinical data can be accessed and downloaded from the CPTAC Data Portal and https://pdc.cancer.gov/pdc/cptac-pancancer as described in [CANCER-CELL-D-22–00603 companion Pan-Cancer resource manuscript]47.
METHODS DETAILS
CPTAC datasets description
We aggregated somatic variants, copy number variations, transcriptomic, proteomic, and clinical data generated by the National Cancer Institute CPTAC from CPTAC data portal, Genomic Data Commons (GDC), and published studies47 (See Data and Code Availability). The datasets include CPTAC Clear Cell Renal Cell Carcinoma (ccRCC) Discovery Study56, CPTAC Glioblastoma (GBM) Discovery Study24, CPTAC Lung Adenocarcinoma (LUAD) Discovery Study60, CPTAC Lung Squamous Cell Carcinoma (LSCC) Discovery Study59, CPTAC Head and Neck Cancer (HNSCC) Discovery Study58, CPTAC Pancreatic Ductal Adenocarcinoma (PDAC) Discovery Study16, and CPTAC Uterine Corpus Endometrial Carcinoma (UCEC) Discovery Study61. Of note, previous CPTAC cohorts (Breast Invasive Carcinoma, Colon Adenocarcinoma, and Ovarian Serous Cystadenocarcinoma) did not obtain DNA methylation measurement, therefore we could not include those CPTAC cohorts in this study.
All the data were harmonized by CPTAC pipelines47, which included alignment to the GDC hg38 human reference genome (GRCh38.d1.vd1), annotation with GENCODE v22 (RNA expression quantification) or v34 (the others), and thorough quality checks. Briefly, somatic mutations were called by the SomaticWrapper pipeline from Washington University in St Louis, which includes four different callers: Strelka299, MUTECT v1.1.7100, VarScan v.2.3.8101, and Pindel v.0.2.5102. Copy number variation and structure variants were identified by BIC-seq2107 and Manta v.1.6.0108, respectively (CPTAC pipeline from Washington University in St. Louis). Gene fusions in RNA-Seq samples were identified using three callers: STAR-Fusion104, EricScript105, and Integrate106, with fusions reported by at least 2 callers or reported by STAR-Fusion being retained (CPTAC pipeline from Washington University in St Louis)103. For transcriptomic data, gene-level stranded read counts were obtained using HTSeq v0.11.2120 and then converted to Fragments Per Kilobase of transcript per Million mapped reads Upper Quartile (FPKM-UQ) values by following the GDC’s RNA-Seq pipeline, except running the quantification tools in the stranded mode (CPTAC pipeline from Washington University in St Louis). Tandem mass tags-based global proteomic and phosphoproteomic data were searched using the MSFragger search engine v3.4109 against a GENCODE v34 protein FASTA database, processed by Philosopher toolkit v4.0.1110, and quantified by TMT-Integrator111. All the cohorts were uniformly processed and harmonized using the Philosopher toolkit. Missing values for the proteins or phosphosites that appeared in at least 50% of samples were imputed using DreamAI112 on each cohort separately. The acetylation data for six cancer types (BRCA, GBM, LUAD, LSCC and UCEC) were generated using MS/MS spectra. These spectra were analyzed with Spectrum Mill (SM) v7.08 (proteomics.broadinstitute.org) to identify and quantify the shared acetylation sites among the cohorts.
DNA methylation data preprocessing
Raw methylation image files generated by Illumina Infinium EPIC BeadChip were downloaded from the CPTAC GDC (See Data and Code Availability). We calculated methylated (M) and unmethylated (U) intensities for tumor and normal adjacent tissue samples as described in the methylation processing pipeline on GitHub121 (See Key Resources Table). Generally, we flagged a locus as NA if probes did not meet a detection P-value of 0.01. Probes with a minor allele frequency more than 0.1 were removed. Probes located on the sex chromosomes and samples with more than 85% NA values were removed from subsequent analysis. Infinium EPIC probes annotated as poor performing were filtered out, leaving 832,749 unique probes94. To map EPIC arrays to GRCh38 assembly, all probes are reannotated by InfiniumAnnotation GENCODE v36, which was downloaded from the InfiniumAnnotation website (https://zwdzwd.github.io/InfiniumAnnotation)94.
The raw methylation image files of TARGET-NBL88, TCGA-GBM89, and TCGA-UCEC71 datasets generated by Illumina Infinium HumanMethylation450 BeadChip assays, used here for normal adjacent tissue approximation, were downloaded from TCGA FireHose or TARGET data matrix and processed as described above. We combined the data derived from the two platforms, but only used data on common probes (GBM: 364518 probes, UCEC: 364518 probes) (Table S1).
Batch correction of DNA methylation value
To account for batch effects introduced by different data sources, we performed batch-effect correction on the DNA methylation data using the ChAMP Bioconductor package96 with default parameters. The methylation data matrix was first imputed using the champ.impute() function and then batch-corrected using champ.runCombat() function. Singular value decomposition (SVD) was conducted before and after batch correction for each cohort using champ.SVD() function. The choice of variable for batch correction was determined based on SVD diagnostic plots, where most cancer types were corrected using the “batch” variable, while for GBM and UCEC samples, the “source” variable was utilized. TCGA GBM and TCGA UCEC tumor samples were included solely for the purpose of batch correction and subsequently removed (Table S1). No batch correction was applied to HNSCC samples due to the lack of improvement in SVD diagnostic plots.
Multi-omic data correlation analysis
To investigate the correlation between mean promoter methylation, RNA expression, and protein abundance for 12,934 genes, we utilized linear regression models to fit three different relationships (methylation versus RNA expression, methylation versus protein expression, and RNA expression versus protein expression) and evaluated the significance of each model using adjusted R-squared values, regression coefficients, and P-values of coefficients. Specifically, we used the following model:
where Y is a (n x 1) vector representing either protein or RNA abundance of a given gene, M is a vector indicating the methylation status for that particular gene (g) in a tumor sample, and the tumor purity (P) calculated by ESTIMATE122 was also included as a covariate. Cancer type was one-hot encoded and was included as an additional covariate (C). Lastly, the error 𝜖 is assumed to be normally distributed with a constant variance 𝜎.
Additionally, we accounted for multiple comparisons and applied an FDR correction to determine the adjusted P-values and assess the significance of the associations with a threshold of P-value < 0.05. This comprehensive approach enabled us to identify which pairs were significantly associated.
Defining aberrant DNA methylation using RESET
We mapped CpG probes to canonical transcriptional start sites and unconventional exonic TSS as defined by FANTOM5 consortium87, and then applied the RESET algorithm9 separately on each tumor type using the corresponding adjacent normal tissue samples. For integrating HM450 (adjacent normal tissue) and EPIC (tumor) datasets for GBM and UCEC, the arrays were intersected to a HM450 probeset to ensure comparability. To identify aberrant DNA methylation events associated with transcriptional or translational changes, aggregated mRNA expression and global proteomic data were used as an input, respectively. We considered the association as significant if the methylation event had FDR < 10%.
A parallel RESET run was conducted using purity-adjusted beta values. Briefly, a linear regression model is fitted to each tumor cohort using the original methylation estimate as the dependent and “1-purity” as the independent variable to obtain a linear fit, where the resultant intercept is the pure tumor methylation state123. After normalizing values between 0 to 1 to satisfy the beta distribution of methylation values, the purity-adjusted beta value at individual CpG sites were obtained. We then repeated our association analysis with purity-adjusted beta values, and provided the mean FDR value in the “FDR_mean|RNA(Purity) “ and “FDR_mean|Protein(Purity)” columns of Table S2 as an additional annotation to the reported events.
For Figure 2C, the methylation difference is derived from the mean beta values differences between samples with or without aberrant methylation. For RNA and protein differences, the values are derived from the mean differences of scaled RNA sequencing data or proteomic data between samples with or without aberrant methylation.
Validation in TCGA cohorts
For aberrant DNA methylation events with concurrent RNA and protein changes where DNA methylation (HM450 array) and RNA-seq data were available from TCGA, we downloaded and uniformly processed the raw DNA methylation data from 2,011 TCGA tumors across 7 cancer types, including ccRCC (N of tumors = 273), GBM (N = 58), HNSCC (N = 514), LSCC (N = 363), LUAD (N = 452), PDAC (N = 177), and UCEC (N = 174). The harmonized RNA expression, protein abundance data (reverse phase protein array, RPPA), mutation profiles, and copy number variation files were downloaded from GDAC. We then identified the aberrant methylation associated with RNA expression or protein abundance changes using the same RESET analysis aforementioned. Since the EPIC array used in this study represents a bigger genomic coverage when compared to the one from TCGA (HM450), we performed a direct comparison of genes where their promoters have been covered in both studies. At least FDR < 10% was required to determine if the event was validated. Due to the sparsity of protein measurements included in the RPPA dataset from TCGA (the mean number of protein measurements in each cohort is 171.1), we calculated the validation rate at RNA level only. The validation results were shown in the “FDR_mean|RNA(TCGA)”, “FDR_mean|Protein(TCGA)”, “No.Methylation.Events_mean|RNA(TCGA)”, and “ProbID|RNA(TCGA)” columns of Table S2.
DEG analysis using scRNA-seq dataset
We used published single-cell RNA-seq (scRNA-seq) datasets24–27 to annotate whether the reported aberrant DNA methylation event was likely to be identified in cancer cells, immune cells, or stromal cells. Briefly, we separated the cells into three main categories including tumor, immune, and stromal. Differentially expressed genes (DEGs) corresponding to each category were identified by the FindMarkers function in Seurat124 using the Wilcoxon rank sum test. DEGs were further filtered using the criteria of FDR < 0.05. DEGs identified in the same cancer type with a reported DNA methylation event at the corresponding promoters were annotated in the “scRNA-seq|cluster_matched_cancertype” column of Table S2. DEGs that were uniquely identified in cancer cells, immune cells, or stromal cells of the other cancer types were annotated in the “scRNA-seq|cluster_cross_cancertype” column of Table S2.
Defining cancer-associated genes
Cancer-associated genes were compiled from a 299 driver gene list defined by Bailey et al29 and cancer-associated genes listed in Mertins et al125 and adapted from Vogelstein et al126.
TFBS enrichment analysis
The frequency distribution of transcription factor bind sites (TFBS) of hypermethylation and hypomethylation probes was conducted at locations of these probes and human hg38 TFBS locations in the JASPAR database93. We further defined hyper and hypomethylation significant probes with driver genes and adopted the HOMER software114 to explore motifs enriched in these significant probes.
Profiling genetic alterations at driver genes
We obtained the somatic mutations in 299 driver genes29 harboring aberrant DNA methylation as described in “Identification of significantly mutated genes in methylation subtypes”. We further collected gene fusions involved in the driver genes. Aberrant DNA methylation was obtained as described in “Defining aberrant DNA methylation using RESET”. For the CPTAC samples, we defined the log2 copy ratios of each gene larger than 0.3 or smaller than −0.3 as amplification or deletion, respectively. For the TCGA samples, we defined the GISTIC value of −2, −1, 1, 2 as deep deletion, shallow deletion, shallow amplification, and deep amplification, respectively. We separated all the tumors into “No Genomic Alteration”, “Mutation”, and “CNV” categories based on the genomic alteration profiles of the locus being tested. For tumors harboring mutation, CNV, and gene fusions, we categorized them as “Mutation”. The missense mutations at FGFR2 locus were visualized using ProteintPaint Lollipop.
Histone acetylation analysis
To investigate the impact of IDH1/2 somatic alterations and aberrant methylations on the correlations between histone acetylation levels and methylation levels at IDH2 or α-KG target genes, we categorized a total of 393 patients with available acetylation and methylation data into four groups based on their IDH1/2 mutation and methylation status: IDH1/IDH2 wild-type (n=374), IDH1 mutants (n=11), IDH2 hypomethylated samples (n=5), and IDH2 mutants (n=3). To assess the levels of histone acetylation and DNA methylation at target genes in each group, we calculated Pearson correlation coefficients and determined the significance levels (P-values) of the correlations using the R function cor.test(). We focused on the acetylation-methylation pairs that showed significant correlations for further analysis (Pearson’s r > 0.2, P < 0.05, or Pearson’s r < −0.2, P < 0.05). To examine the differences in correlation coefficients among the groups, we utilized the R package rstatix (version 0.7.2). Specifically, we compared the group differences separately for positively and negatively correlated acetylation-methylation pairs.
We also compared the impact of IDH1 mutation in GBM and IDH2 hypomethylation in LSCC on histone acetylation levels. By examining 46 specific sites on histones H1, H2, H3, and H4, we compared the abundance of histone acetylation between IDH1 mutant (n=7) and wild-type (n=87) groups in GBM, as well as between IDH2 hypomethylation (n=5) and normal methylation (n=99) groups in LSCC using the Wilcoxon signed-rank test.
Frequency of gene hyper- or hypomethylation
To determine the frequency of hyper- or hypomethylation in each tumor, we calculated the proportion by aggregating the counts of CGI sites exhibiting aberrant methylation and dividing it by the total number of CGI sites in the tumor sample. This analysis was performed using the RESET output.
Regulon association analysis
We collected transcription factor-interacting genes from publicly available databases OmniPath95, and grouped the samples into regulon-high (>0) and regulon-low (<0) based on the sum of transcription factor-interacting genes using RNA expression and protein abundance data. To test the association of the regulons with the DNA methylation status of transcription factors, we used Fisher’s exact test to test for overrepresentation of samples with aberrantly methylated transcription factors in the set of samples defining the regulon activity as described above. To account for multiple comparisons in Fisher’s exact test, we applied the Benjamini-Hochberg (BH) method to adjust the P-values for false discovery rate (FDR) control. We narrowed the list of transcription factors down to STAT5A and STAT5A-interacting genes based on FDR P-value <= 0.1 and the number of interacting genes >=5. We then performed unsupervised clustering of the STAT5A regulon using the Ward.D2 linkage method to generate heatmaps.
Cell type enrichment deconvolution
We inferred the abundance of each cell type using the xCell web tool45 using the FPKM-UQ expression matrix as input. xCell is a gene signature-based method learned from thousands of pure cell types from various sources, which performed the cell type enrichment analysis from gene expression data for 64 immune and stromal cell types (default xCell signature). xCell generates an immune score per sample that integrates the enrichment scores of immune cells (B cells, CD4+ T-cells, CD8+ T-cells, DC, eosinophils, macrophages, monocytes, mast cells, neutrophils, and NK cells), a stroma score, and a micro-environment score which is the sum of the immune score and stroma score28,47.
Immunohistochemistry analysis
Immunohistochemistry (IHC) staining was performed on 4-micron formalin-fixed, paraffin-embedded (FFPE) tissue sections. Prior to staining, antigen retrieval was performed using the heat-induced epitope retrieval method at pH 6. Staining employed the Dako Autostainer Link 48 with EnVision FLEX visualizing kit (K800221-2; Dako, Agilent Technologies Inc.) and rabbit polyclonal antibody against human STAT5A (HPA042128, Atlas Antibodies, 1:150 dilution). Appropriate known positive and negative control tissue were run in each assay batch.
A semi-quantitative product score for tumor-infiltrating lymphocytes (TILs) and peritumoral lymphocytes were scored by the study pathologists. Percentage of lymphocytes represents the positive lymphocytes relative to the total number of cells (tumors and lymphocytes) in the stained slide, while the intensity represents staining intensity of STAT5A (none, 0; weak, 1; moderate, 2; strong, 3). An independent H-score of STAT5A abundance in the stained slide is calculated using QuPath115.
Methylation-driven subtyping
To identify subtype based on cancer methylome, we took CGIs showing significant differences between tumors and NATs and performed unsupervised classification of tumors using consensus clustering on the most variable 8,000 CGIs by the R package ConsensusClusterPlus (parameters: reps = 2000, pItem = 0.9, pFeature = 0.9, clusterAlg = “kmdist”, distance = “spearman”)98. Samples were assigned to the optimal number of clusters.
Pathway over-representation analysis
To designate the representative pathways of methylation subtypes from transcriptomic and proteomic data, we used the Wilcoxon rank sum test to select the top 250 differentially expressed features (RNA expression and protein abundance) for each subtype or methylation group. We then performed hierarchical clustering on these features. Each set of clustered features underwent pathway enrichment analysis using Reactome116. Pathways with P-value smaller than 0.05 were manually reviewed and selected based on the following rules: (1) if similar signatures showed up repeatedly in the significant list, (2) if the significant pathway is supported by literature, and (3) if the signature had been supported by FDR test with P-value smaller than 0.05, (4) if the signatures were consistently observed at the transcriptomic and proteomic levels. Those manually reviewed pathways are highlighted in Figure 7C and Figure S7F.
Identification of significantly mutated genes
A list of 299 driver genes was downloaded from Bailey et al.29 and results of somatic mutations were downloaded from the CPTAC discovery studies16,24,56,58–61. We compiled a genomic alteration profile for each sample (file name: PanCan_Union_Maf_Broad_WashU_v1.1.maf), including frameshift deletion, frameshift insertion, inframe deletion, inframe insertion, missense mutation, nonsense mutation, nonstop mutation, splice site mutation, and intron mutation in 299 driver genes. We then categorized samples with driver mutations based on their methylation subtypes, and conducted Fisher’s exact test to test for overrepresentation of any key driver somatic alterations in the methylation subtypes.
RNA expression-based subtypes
The classifications of RNA expression-based subtypes of ccRCC, GBM, HNSCC, LSCC, LUAD, PDAC, and UCEC samples were downloaded from the CPTAC discovery studies16,24,58–61. We then compared the association of RNA expression-based subtype and methylation subtypes using Fisher’s exact test.
Druggable genes with aberrant methylation
CIViC is a curated list of druggable variants describing their therapeutic, prognostic, diagnostic and predisposing relevance. We downloaded the list and intersected the list of genes with aberrant methylation. The potential druggability of each gene was manually reviewed to see if the altered expression of such gene is associated with therapeutic relevance supported by literature.
ADDITIONAL RESOURCES
The CPTAC program website, which includes details about program initiatives, investigators, and datasets, can be accessed at https://proteomics.cancer.gov/programs/cptac.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
All statistical analyses were performed using R or Python unless explained otherwise. Multiple comparisons were adjusted by the Benjamini-Hochberg127. Statistical parameters for each experiment are reported in the respective figure legend.
Supplementary Material
Table S1. Dataset overview and summary of linear model results, related to Figure 1.
Table S3. Summary of STAT5A regulon analysis and metadata associated with squamous tumors, Related to Figures 5 and 6.
Table S4. Summary of cancer-specific promoters, per-cancer methylation subtypes, and multi-cancer methylation groups, related to Figure 7.
Table S5. Summary of Reactome analysis of per-cancer methylation subtypes, related to Figure 7.
Table S6. Summary of druggable DNA methylation events, related to Figure 7.
Highlights.
Pan-cancer epigenetic aberrations and their transcriptional and translational changes
FGFR2 and EGFR hypomethylation are bona fide driver DNA methylation events.
STAT5A methylation is a potential switch for immunosuppression in squamous tumors.
Methylation subtypes illuminate cell origin, tumor heterogeneity, and tumor phenotype.
Acknowledgments
We thank InPrint for the editing, Matthew A. Wyczalkowski for feedback on figures, and BioRender for diagrams. The Clinical Proteomic Tumor Analysis Consortium (CPTAC) is supported by the National Cancer Institute of the National Institutes of Health under award numbers U24CA210955, U24CA210985, U24CA210986, U24CA210954, U24CA210967, U24CA210972, U24CA210979, U24CA210993, U01CA214114, U01CA214116, and U01CA214125 as U24CA210972 to D.F., L.D., and S.P., and Contract GR0012005 to L.D. This project has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261201500003I, Task Order HHSN26100064. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work the author(s) used ChatGPT to enhance its readability. After using this tool/service, the author(s) reviewed and edited the content as needed and take(s) full responsibility for the content of the publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Feinberg AP, Ohlsson R, and Henikoff S. (2006). The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 7, 21–33. [DOI] [PubMed] [Google Scholar]
- 2.Baylin SB, and Jones PA (2011). A decade of exploring the cancer epigenome - biological and translational implications. Nat. Rev. Cancer 11, 726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen H, and Laird PW (2013). Interplay between the cancer genome and epigenome. Cell 153, 38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ehrlich M. (2002). DNA methylation in cancer: too much, but also too little. Oncogene 21, 5400–5413. [DOI] [PubMed] [Google Scholar]
- 5.Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CPE, van Dijk CM, Tollenaar RA, et al. (2012). Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina--associated domains. Nat. Genet. 44, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Easwaran H, Tsai H-C, and Baylin SB (2014). Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 54, 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahuja N, Sharma AR, and Baylin SB (2016). Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 67, 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalari S, and Pfeifer GP (2010). Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv. Genet. 70, 277–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saghafinia S, Mina M, Riggi N, Hanahan D, and Ciriello G. (2018). Pan-Cancer Landscape of Aberrant DNA Methylation across Human Tumors. Cell Rep. 25, 1066–1080.e8. [DOI] [PubMed] [Google Scholar]
- 10.Fan S, Tang J, Li N, Zhao Y, Ai R, Zhang K, Wang M, Du W, and Wang W. (2019). Integrative analysis with expanded DNA methylation data reveals common key regulators and pathways in cancers. NPJ Genom Med 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez H, Zenklusen JC, Staudt LM, Doroshow JH, and Lowy DR (2021). The next horizon in precision oncology: Proteogenomics to inform cancer diagnosis and treatment. Cell 184, 1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Beyer A, and Aebersold R. (2016). On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165, 535–550. [DOI] [PubMed] [Google Scholar]
- 13.Buccitelli C, and Selbach M. (2020). mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 10.1038/s41576-020-0258-4. [DOI] [PubMed] [Google Scholar]
- 14.Tang Z, Wang L, Bajinka O, Wu G, and Tan Y. (2022). Abnormal Gene Expression Regulation Mechanism of Myeloid Cell Nuclear Differentiation Antigen in Lung Adenocarcinoma. Biology 11. 10.3390/biology11071047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ziller MJ, Gu H, Müller F, Donaghey J, Tsai LT-Y, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al. (2013). Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao L, Huang C, Cui Zhou D, Hu Y, Lih TM, Savage SR, Krug K, Clark DJ, Schnaubelt M, Chen L, et al. (2021). Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 184, 5031–5052.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vidal E, Sayols S, Moran S, Guillaumet-Adkins A, Schroeder MP, Royo R, Orozco M, Gut M, Gut I, Lopez-Bigas N, et al. (2017). A DNA methylation map of human cancer at single base-pair resolution. Oncogene 36, 5648–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simpkins SB, Bocker T, Swisher EM, Mutch DG, Gersell DJ, Kovatich AJ, Palazzo JP, Fishel R, and Goodfellow PJ (1999). MLH1 promoter methylation and gene silencing is the primary cause of microsatellite instability in sporadic endometrial cancers. Hum. Mol. Genet. 8, 661–666. [DOI] [PubMed] [Google Scholar]
- 19.Hegi ME, Diserens A-C, Gorlia T, Hamou M-F, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352, 997–1003. [DOI] [PubMed] [Google Scholar]
- 20.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, et al. (2011). Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 43, 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Witte T, Plass C, and Gerhauser C. (2014). Pan-cancer patterns of DNA methylation. Genome Med. 6, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. (2010). Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang F, Liu D, Deng Y, Wang J, Mei S, Ge S, Li H, Zhang C, and Zhang T. (2020). Frequent promoter methylation of HOXD10 in endometrial carcinoma and its pathological significance. Oncol. Lett. 19, 3602–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L-B, Karpova A, Gritsenko MA, Kyle JE, Cao S, Li Y, Rykunov D, Colaprico A, Rothstein JH, Hong R, et al. (2021). Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell. 10.1016/j.ccell.2021.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Lih T-SM, Dhanasekaran SM, Mannan R, Chen L, Cieslik M, Wu Y, Lu RJ-H, Clark DJ, Kołodziejczak I, et al. (2022). Histopathologic and proteogenomic heterogeneity reveals features of clear cell renal cell carcinoma aggressiveness. Cancer Cell. 10.1016/j.ccell.2022.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cui Zhou D, Jayasinghe RG, Chen S, Herndon JM, Iglesia MD, Navale P, Wendl MC, Caravan W, Sato K, Storrs E, et al. (2022). Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat. Genet. 54, 1390–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J, et al. (2020). A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.CPTAC (2023). Companion Pan-Cancer Driver paper. Cell. [Google Scholar]
- 29.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu H, Wang G, and Qian J. (2016). Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 17, 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iotti G, Longobardi E, Masella S, Dardaei L, De Santis F, Micali N, and Blasi F. (2011). Homeodomain transcription factor and tumor suppressor Prep1 is required to maintain genomic stability. Proc. Natl. Acad. Sci. U. S. A. 108, E314–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, He Y, Tan Z, Lu J, Li L, Song X, Shi F, Xie L, You S, Luo X, et al. (2018). Wild-type IDH2 promotes the Warburg effect and tumor growth through HIF1α in lung cancer. Theranostics 8, 4050–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergaggio E, and Piva R. (2019). Wild-Type IDH Enzymes as Actionable Targets for Cancer Therapy. Cancers 11. 10.3390/cancers11040563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu K, Herr AB, Waksman G, and Ornitz DM (2000). Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc. Natl. Acad. Sci. U. S. A. 97, 14536–14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pollock PM, Gartside MG, Dejeza LC, Powell MA, Mallon MA, Davies H, Mohammadi M, Futreal PA, Stratton MR, Trent JM, et al. (2007). Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 26, 7158–7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gartside MG, Chen H, Ibrahimi OA, Byron SA, Curtis AV, Wellens CL, Bengston A, Yudt LM, Eliseenkova AV, Ma J, et al. (2009). Loss-of-function fibroblast growth factor receptor-2 mutations in melanoma. Mol. Cancer Res. 7, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Byron SA, Chen H, Wortmann A, Loch D, Gartside MG, Dehkhoda F, Blais SP, Neubert TA, Mohammadi M, and Pollock PM (2013). The N550K/H mutations in FGFR2 confer differential resistance to PD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neoplasia 15, 975–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao RG, Jung J, Tchaicha J, Wilkerson MD, Sivachenko A, Beauchamp EM, Liu Q, Pugh TJ, Pedamallu CS, Hayes DN, et al. (2013). Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res. 73, 5195–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng PK-S, Li J, Jeong KJ, Shao S, Chen H, Tsang YH, Sengupta S, Wang Z, Bhavana VH, Tran R, et al. (2018). Systematic Functional Annotation of Somatic Mutations in Cancer. Cancer Cell 33, 450–462.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saito Y, Koya J, Araki M, Kogure Y, Shingaki S, Tabata M, McClure MB, Yoshifuji K, Matsumoto S, Isaka Y, et al. (2020). Landscape and function of multiple mutations within individual oncogenes. Nature 582, 95–99. [DOI] [PubMed] [Google Scholar]
- 41.Robertson SC, Tynan J, and Donoghue DJ (2000). RTK mutations and human syndromes: when good receptors turn bad. Trends Genet. 16, 368. [DOI] [PubMed] [Google Scholar]
- 42.Rani A, and Murphy JJ (2016). STAT5 in Cancer and Immunity. J. Interferon Cytokine Res. 36, 226–237. [DOI] [PubMed] [Google Scholar]
- 43.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. (2017). Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 19, 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Platanias LC (2005). Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386. [DOI] [PubMed] [Google Scholar]
- 45.Aran D, Hu Z, and Butte AJ (2017). xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.CPTAC (2023). Companion PTM paper. [Google Scholar]
- 47.CPTAC (2023). Companion Pan-Cancer Resource paper. [Google Scholar]
- 48.Karmodiya K, Krebs AR, Oulad-Abdelghani M, Kimura H, and Tora L. (2012). H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics 13, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang S, Fukuda S, Lee Y, Hangoc G, Cooper S, Spolski R, Leonard WJ, and Broxmeyer HE (2000). Essential role of signal transducer and activator of transcription (Stat)5a but not Stat5b for Flt3-dependent signaling. J. Exp. Med. 192, 719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, Li D, Durum SK, Jiang Q, Bhandoola A, et al. (2006). Stat5a/b are essential for normal lymphoid development and differentiation. Proc. Natl. Acad. Sci. U. S. A. 103, 1000–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park J-H, Adoro S, Guinter T, Erman B, Alag AS, Catalfamo M, Kimura MY, Cui Y, Lucas PJ, Gress RE, et al. (2010). Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxic-lineage T cells. Nat. Immunol. 11, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Q, Wang HY, Liu X, and Wasik MA (2007). STAT5A is epigenetically silenced by the tyrosine kinase NPM1-ALK and acts as a tumor suppressor by reciprocally inhibiting NPM1-ALK expression. Nat. Med. 13, 1341–1348. [DOI] [PubMed] [Google Scholar]
- 53.Ferris RL (2015). Immunology and Immunotherapy of Head and Neck Cancer. J. Clin. Oncol. 33, 3293–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Villarino AV, Kanno Y, and O’Shea JJ (2017). Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 18, 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McInnes L, Healy J, Saul N, and Großberger L. (2018). UMAP: Uniform Manifold Approximation and Projection. Journal of Open Source Software 3, 861. 10.21105/joss.00861. [DOI] [Google Scholar]
- 56.Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, da Veiga Leprevost F, Reva B, Lih T-SM, Chang H-Y, et al. (2019). Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 179, 964–983.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC, et al. (2014). The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 26, 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang C, Chen L, Savage SR, Eguez RV, Dou Y, Li Y, da Veiga Leprevost F, Jaehnig EJ, Lei JT, Wen B, et al. (2021). Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell 39, 361–379.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Satpathy S, Krug K, Jean Beltran PM, Savage SR, Petralia F, Kumar-Sinha C, Dou Y, Reva B, Kane MH, Avanessian SC, et al. (2021). A proteogenomic portrait of lung squamous cell carcinoma. Cell 184, 4348–4371.e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, Petralia F, Li Y, Liang W-W, Reva B, et al. (2020). Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 182, 200–225.e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dou Y, Kawaler EA, Cui Zhou D, Gritsenko MA, Huang C, Blumenberg L, Karpova A, Petyuk VA, Savage SR, Satpathy S, et al. (2020). Proteogenomic Characterization of Endometrial Carcinoma. Cell 180, 729–748.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. (2018). Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilkerson MD, Yin X, Walter V, Zhao N, Cabanski CR, Hayward MC, Miller CR, Socinski MA, Parsons AM, Thorne LB, et al. (2012). Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PLoS One 7, e36530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cancer Genome Atlas Research Network (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jeong Y, Hoang NT, Lovejoy A, Stehr H, Newman AM, Gentles AJ, Kong W, Truong D, Martin S, Chaudhuri A, et al. (2017). Role of KEAP1/NRF2 and TP53 Mutations in Lung Squamous Cell Carcinoma Development and Radiation Resistance. Cancer Discov. 7, 86–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ryoo I-G, and Kwak M-K (2018). Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 359, 24–33. [DOI] [PubMed] [Google Scholar]
- 67.Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, and Lindquist S. (2012). HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150, 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wauters E, Janssens W, Vansteenkiste J, Decaluwé H, Heulens N, Thienpont B, Zhao H, Smeets D, Sagaert X, Coolen J, et al. (2015). DNA methylation profiling of non-small cell lung cancer reveals a COPD-driven immune-related signature. Thorax 70, 1113–1122. [DOI] [PubMed] [Google Scholar]
- 69.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V, et al. (2018). Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173, 291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pereira B, Billaud M, and Almeida R. (2017). RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer Res. 3, 506–528. [DOI] [PubMed] [Google Scholar]
- 71.Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al. (2013). Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yatabe Y, Kosaka T, Takahashi T, and Mitsudomi T. (2005). EGFR mutation is specific for terminal respiratory unit type adenocarcinoma. Am. J. Surg. Pathol. 29, 633–639. [DOI] [PubMed] [Google Scholar]
- 73.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, et al. (2006). Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173. [DOI] [PubMed] [Google Scholar]
- 75.Tiedemann RL, Hlady RA, Hanavan PD, Lake DF, Tibes R, Lee J-H, Choi J-H, Ho TH, and Robertson KD (2016). Dynamic reprogramming of DNA methylation in SETD2-deregulated renal cell carcinoma. Oncotarget 7, 1927–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen Y-C, Gotea V, Margolin G, and Elnitski L. (2017). Significant associations between driver gene mutations and DNA methylation alterations across many cancer types. PLoS Comput. Biol. 13, e1005840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Papalouka C, Adamaki M, Batsaki P, Zoumpourlis P, Tsintarakis A, Goulielmaki M, Fortis SP, Baxevanis CN, and Zoumpourlis V. (2023). DNA Damage Response Mechanisms in Head and Neck Cancer: Significant Implications for Therapy and Survival. Int. J. Mol. Sci. 24. 10.3390/ijms24032760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cancer Genome Atlas Research Network (2012). Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Griffith M, Spies NC, Krysiak K, McMichael JF, Coffman AC, Danos AM, Ainscough BJ, Ramirez CA, Rieke DT, Kujan L, et al. (2017). CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 49, 170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shames DS, Elkins K, Walter K, Holcomb T, Du P, Mohl D, Xiao Y, Pham T, Haverty PM, Liederer B, et al. (2013). Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin. Cancer Res. 19, 6912–6923. [DOI] [PubMed] [Google Scholar]
- 81.Saito Y, Koya J, and Kataoka K. (2021). Multiple mutations within individual oncogenes. Cancer Sci. 112, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tsai H-C, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, et al. (2012). Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 21, 430–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shenker N, and Flanagan JM (2012). Intragenic DNA methylation: implications of this epigenetic mechanism for cancer research. Br. J. Cancer 106, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Spainhour JC, Lim HS, Yi SV, and Qiu P. (2019). Correlation Patterns Between DNA Methylation and Gene Expression in The Cancer Genome Atlas. Cancer Inform. 18, 1176935119828776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raynal NJ-M, Si J, Taby RF, Gharibyan V, Ahmed S, Jelinek J, Estécio MRH, and Issa J-PJ (2012). DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res. 72, 1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Flavahan WA, Gaskell E, and Bernstein BE (2017). Epigenetic plasticity and the hallmarks of cancer. Science 357. 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.FANTOM Consortium and the RIKEN PMI and CLST (DGT), Forrest, A.R.R., Kawaji H, Rehli M, Baillie JK, de Hoon MJL, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, et al. (2014). A promoter-level mammalian expression atlas. Nature 507, 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, et al. (2013). The genetic landscape of high-risk neuroblastoma. Nat. Genet. 45, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cancer Genome Atlas Research Network (2013). Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cancer Genome Atlas Research Network. Electronic address: andrew_aguirre@dfci.harvard.edu, and Cancer Genome Atlas Research Network (2017). Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 32, 185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cancer Genome Atlas Network (2015). Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Chèneby J, Kulkarni SR, Tan G, et al. (2018). JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou W, Laird PW, and Shen H. (2017). Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 45, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Türei D, Korcsmáros T, and Saez-Rodriguez J. (2016). OmniPath: guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 13, 966–967. [DOI] [PubMed] [Google Scholar]
- 96.Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, and Beck S. (2014). ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 30, 428–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maksimovic J, Phipson B, and Oshlack A. (2016). A cross-package Bioconductor workflow for analysing methylation array data. F1000Res. 5, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wilkerson MD, and Hayes DN (2010). ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, and Cheetham RK (2012). Strelka: accurate somatic small-variant calling from sequenced - sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- 100.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G. (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ye K, Schulz MH, Long Q, Apweiler R, and Ning Z. (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gao Q, Liang W-W, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, Liao W-W, Reynolds SM, Wyczalkowski MA, Yao L, et al. (2018). Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 23, 227–238.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Haas BJ, Dobin A, Li B, Stransky N, Pochet N, and Regev A. (2019). Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 20, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Benelli M, Pescucci C, Marseglia G, Severgnini M, Torricelli F, and Magi A. (2012). Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics 28, 3232–3239. [DOI] [PubMed] [Google Scholar]
- 106.Zhang J, White NM, Schmidt HK, Fulton RS, Tomlinson C, Warren WC, Wilson RK, and Maher CA (2016). INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res. 26, 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xi R, Lee S, Xia Y, Kim T-M, and Park PJ (2016). Copy number analysis of whole-genome data using BIC-seq2 and its application to detection of cancer susceptibility variants. Nucleic Acids Res. 44, 6274–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. [DOI] [PubMed] [Google Scholar]
- 109.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, and Nesvizhskii AI (2017). MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 14, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.da Veiga Leprevost F, Haynes SE, Avtonomov DM, Chang H-Y, Shanmugam AK, Mellacheruvu D, Kong AT, and Nesvizhskii AI (2020). Philosopher: a versatile toolkit for shotgun proteomics data analysis. Nat. Methods 17, 869–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Djomehri SI, Gonzalez ME, da Veiga Leprevost F, Tekula SR, Chang H-Y, White MJ, Cimino-Mathews A, Burman B, Basrur V, Argani P, et al. (2020). Quantitative proteomic landscape of metaplastic breast carcinoma pathological subtypes and their relationship to triple-negative tumors. Nat. Commun. 11, 1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ma W, Kim S, Chowdhury S, Li Z, Yang M, Yoo S, Petralia F, Jacobsen J, Li JJ, Ge X, et al. (2020). DreamAI: algorithm for the imputation of proteomics data. bioRxiv, 2020.07.21.214205. 10.1101/2020.07.21.214205. [DOI] [Google Scholar]
- 113.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci. Rep. 7, 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fabregat A, Sidiropoulos K, Viteri G, Forner O, Marin-Garcia P, Arnau V, D’Eustachio P, Stein L, and Hermjakob H. (2017). Reactome pathway analysis: a high-performance in-memory approach. BMC Bioinformatics 18, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Grüning B, Dale R, Sjödin A, Chapman BA, Rowe J, Tomkins-Tinch CH, Valieris R, Köster J, and Bioconda Team (2018). Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat. Methods 15, 475–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Anders S, Pyl PT, and Huber W. (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fortin J-P, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, Greenwood CM, and Hansen KD (2014). Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 15, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Staaf J, and Aine M. (2022). Tumor purity adjusted beta values improve biological interpretability of high-dimensional DNA methylation data. PLoS One 17, e0265557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, et al. (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, and Kinzler KW (2013). Cancer genome landscapes. Science 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Benjamini Y, and Hochberg Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Dataset overview and summary of linear model results, related to Figure 1.
Table S3. Summary of STAT5A regulon analysis and metadata associated with squamous tumors, Related to Figures 5 and 6.
Table S4. Summary of cancer-specific promoters, per-cancer methylation subtypes, and multi-cancer methylation groups, related to Figure 7.
Table S5. Summary of Reactome analysis of per-cancer methylation subtypes, related to Figure 7.
Table S6. Summary of druggable DNA methylation events, related to Figure 7.
Data Availability Statement
Raw and processed proteomics as well as open-access genomic data, can be obtained via Proteomic Data Commons (PDC) at https://pdc.cancer.gov/pdc/cptac-pancancer. Raw genomic and transcriptomic data files can be accessed via the Genomic Data Commons (GDC) Data Portal at https://portal.gdc.cancer.gov with dbGaP Study Accession: phs001287.v16.p6. Complete CPTAC Pan-Cancer controlled and processed data can be accessed via the Cancer Data Service (CDS, https://dataservice.datacommons.cancer.gov/). The CPTAC Pan-Cancer data hosted in CDS is controlled data and can be accessed through the NCI DAC approved, dbGaP compiled whitelists. Users can access the data for analysis through the Seven Bridges Cancer Genomics Cloud (SB-CGC) which is one of the NCI-funded Cloud Resource/platform for compute intensive analysis. Instructions to access data: 1. Create an account on CGC, Seven Bridges (https://cgc-accounts.sbgenomics.com/auth/register 2. Get approval from dbGaP to access the controlled study (https://www.ncbi.nlm.nih.gov/projects/gap/cgibin/study.cgi?study_id=phs001287. v16.p6 ) 3. Log into CGC to access Cancer Data Service (CDS) File Explore 4. Copy data into your own space and start analysis and exploration 5. Visit the CDS page on CGC to see what studies are available and instructions and guides to use the resources. (https://docs.cancergenomicscloud.org/page/cds-data).
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-STAT5A antibody | Atlas Antibodies | Catalog: HPA042128, RRID: AB_2677864 |
| Biological samples | ||
| Primary tumor and normal adjacent tissue samples | CANCER-CELL-D- 22–00603 companion Pan-Cancer resource manuscript47 | https://pdc.cancer.gov/pdc/cptac-pancancer |
| Chemicals, peptides, and recombinant proteins | ||
| Dako Protein Block, Serum-free blocking solution | Agilent Technologies Inc | Catalog: X090930–2 |
| Dako Taget Retrieval Solution, pH=6 | Agilent Technologies Inc | Catalog: S236984–2 |
| Dako Wash Buffer 10X | Agilent Technologies Inc | Catalog: S3006 |
| Critical commercial assays | ||
| TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold | Illumina | Catalog: RS-122–2301 |
| Infinium MethylationEPIC Kit | Illumina | Catalog: WG-317–1003 |
| Nextera DNA Exosome Kit | Illumina | Catalog: 20020617 |
| KAPA Hyper Prep Kit, PCR-free | Roche | Catalog: 07962371001 |
| TMT-11 Reagent Kit | ThermoFisher Scientific | Catalog: A34808 |
| BCA Protein Assay Kit | ThermoFisher Scientific | Catalog: 23225 |
| PTMScan® Acetyl-Lysine Motif [Ac-K] Kit | Cell Signaling | Catalog: 13416 |
| EnVision FLEX Visualizing Kit | Agilent Technologies Inc | Catalog: K800221–2 |
| Deposited data | ||
| CIViC nightly, 062220 | Griffith et al.79 | https://civicdb.org/home |
| FANTOM5 | FANTOM Consortium et al.87 | http://fantom.gsc.riken.jp/5/ |
| TARGET Methylation data | Pugh et al.88 | https://ocg.cancer.gov/programs/target/data-matrix |
| TCGA Methylation data and RNA-seq data | TCGA et al.64,71,78,89–92 | https://gdac.broadinstitute.org/ |
| JASPAR | Khan et al.93 | https://jaspar.genereg.net/ |
| InfiniumAnnotation | Zhou et al.94 | https://zwdzwd.github.io/InfiniumAnnotation |
| OmniPath | Türei et al.95 | http://omnipathdb.org/ |
| ccRCC scRNA-seq data | Li et al.25 | https://portal.gdc.cancer.gov/projects/CPTAC-3 |
| GBM scRNA-seq data | Wang et al.24 | https://portal.gdc.cancer.gov/projects/CPTAC-3 |
| PDAC scRNA-seq data | Cui Zhou et al.26 | https://data.humantumoratlas.org/ |
| Lungs scRNA-seq data | Travaglini et al.27 | https://www.synapse.org/#!Synapse:syn21041850 |
| CPTAC clinical and proteomic data | CANCER-CELL-D- 22–00603 companion Pan-Cancer resource manuscript47 | https://pdc.cancer.gov/pdc/cptac-pancancer |
| CPTAC genomic and transcriptomic data | CANCER-CELL-D- 22–00603 companion Pan-Cancer resource manuscript47 | https://pdc.cancer.gov/pdc/cptac-pancancer and Cancer Data Service (CDS) |
| CPTAC DNA methylation data | This study | https://pdc.cancer.gov/pdc/cptac-pancancer |
| CPTAC acetylation data | CANCER-CELL-D- 22–00603 companion Pan-Cancer resource manuscript47 | https://pdc.cancer.gov/pdc/cptac-pancancer |
| Software and algorithms | ||
| RESET | Saghafinia et al.9 | http://ciriellolab.org/reset/reset.html |
| ChAMP | Morris et al.96 | https://www.bioconductor.org/packages/release/bioc/vignettes/ChAMP/inst/doc/ChAMP.html |
| Methylation array analysis pipeline for CPTAC | This study | https://github.com/ding-lab/cptac_methylation |
| methylationArrayAnalysis v3.9 | Maksimovic et al.97 | https://master.bioconductor.org/packages/release/workflows/html/methylationArrayAnalysis.html |
| Illumina EPIC methylation array v0.6 | See link | https://bioconductor.org/packages/release/data/annotation/html/IlluminaHumanMethylationEPICanno.ilm10b2.hg19.html |
| ConsensusClusterPlus v1.48.0 | Wilkerson et al.98 | https://bioconductor.org/packages/ConsensusClusterPlus/ |
| xCell v1.2 | Aran et al.45 | http://xcell.ucsf.edu/ |
| SomaticWrapper | Ding Lab | https://github.com/ding-lab/somaticwrapper |
| Strelka2 | Saunders et al.99 | https://github.com/Illumina/strelka |
| MUTECT v1.1.7 | Cibulskis et al.100 | https://software.broadinstitute.org/gatk/download/archive |
| VarScan v2.3.8 | Koboldt et al.101 | http://varscan.sourceforge.net |
| Pindel v0.2.5 | Ye et al.102 | http://gmt.genome.wustl.edu/packages/pindel/ |
| Fusion calling pipeline for CPTAC | Gao et al.103 | https://github.com/cuidaniel/Fusion_hg38 |
| STAR-Fusion v1.5.0 | Haas et al.104 | https://github.com/STAR-Fusion/STAR-Fusion/wiki |
| EricScript v0.5.5 | Benelli et al.105 | https://sites.google.com/site/bioericscript |
| Integrate v0.2.6 | Zhang et al.106 | https://sourceforge.net/p/integrate-fusion/wiki/Home/ |
| Copy Number Variant Calling | Ding Lab | https://github.com/ding-lab/BICSEQ2 |
| BIC-seq2 | Xi et al.107 | http://compbio.med.harvard.edu/BIC-seq/ |
| SomaticSV | Ding Lab | https://github.com/ding-lab/SomaticSV |
| Manta v1.6.0 | Chen et al.108 | https://github.com/Illumina/manta |
| MSFragger v3.4 | Kong et al.109 | https://msfragger.nesvilab.org/ |
| Philosopher toolkit v4.0.1 | da Veiga Leprevost et al.110 | https://philosopher.nesvilab.org/ |
| TMT-Integrator | Djomehri et al.111 | http://tmt-integrator.nesvilab.org/ |
| HTSeq v0.11.2 | Anders et al.117 | https://htseq.readthedocs.io/en/master/ |
| DreamAI | Ma et al.112 | https://github.com/WangLab-MSSM/DreamAI |
| ProteintPaint Lollipop | Zhou Lab | https://viz.stjude.cloud/zhou-lab/visualization/proteintpaint-lollipop-example~57 |
| LIMMA v3.36 (R Package) | Ritchie et al.113 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| HOMER | Heinz et al.114 | http://homer.salk.edu/homer/ |
| QuPath v0.3.2 | Bankhead et al.115 | https://qupath.github.io/ |
| Reactome | Fabregat et al.116 | https://reactome.org/ |
| Scanpy v1.7.0 | Wolf et al.117 | https://github.com/scverse/scanpy |
| Python v3.7 | Python Software Foundation | https://www.python.org/ |
| R v3.6 | R Development Core Team | https://www.R-project.org/ |
| Bioconda | Grüning et al.118 | https://bioconda.github.io/ |
| Bioconductor v3.9 | Huber et al.119 | https://bioconductor.org/ |