

Abstract
A series of dilanthanide benzene inverse sandwich complexes of the type (CpiPr5Ln)2(μ–η6:η6-C6H6) (1-Ln) (Ln = Y, Gd, Tb, Dy, Tm) are reported. These compounds are synthesized by reduction of the respective trivalent dimers CpiPr52Ln2I4 (Ln = Y, Gd, Tb, Dy, Tm) in diethyl ether with potassium graphite in the presence of benzene, and they feature an unusual linear coordination geometry with a highly planar benzene bridge as verified by single-crystal X-ray diffraction. The Ln–Bzcentroid distances of 1-Ln are the shortest distances observed to date, ranging from 1.943(1) Å for 1-Tm to 2.039(6) Å for 1-Gd. Structural, spectroscopic, and magnetic analyses together with density functional theory calculations support the presence of a rare, unsubstituted tetraanionic benzene in each compound, which is stabilized by strong covalent δ bonding interactions involving the filled π* orbitals of (C6H6)4– and vacant dxy and dx2–y2 orbitals of the Ln3+ ions. Notably, 1-Ln are the first examples of compounds of the later lanthanides to feature an unsubstituted tetraanionic benzene.
Introduction
Although the first reported lanthanide–arene compounds were characterized in the mid-1980s,1 they continue to be of significant interest to chemists due to their widely ranging reactivity2−5 and unusual electronic structures.6,7 Lanthanide–arene compounds are scarce compared to those of the transition metals,8 which can be ascribed to the fact that the core-like 4f valence orbitals do not participate in the covalent interactions that are responsible for the existence of stable transition metal–arene compounds.9 Structurally characterized lanthanide–arene compounds in the literature can be divided into two classes: those with formally neutral arenes and those with formally anionic arenes.8 The first of the neutral arene adducts to be reported was the chloroaluminate complex (η6-C6Me6)Sm(AlCl4)3, which features neutral hexamethylbenzene in a weak induced dipole interaction with a central Sm3+ cation.1 Compounds of the type Ln(1,3,5-tBu3C6H3)2 (Ln = Y, Gd), which feature formally zerovalent yttrium and gadolinium, have also been known since the late 1980s. These compounds were synthesized via a challenging co-condensation reaction between the corresponding metal vapor and 1,3,5-tris-tertbutylbenzene at 77 K.10
The first Ln–arene complex featuring an anionic benzene was the inverse sandwich compound [CptBu22La(μ–η6:η6-C6H6)].1,8−11 Since this initial report, the number of crystallographically characterized inverse sandwich complexes with lanthanides bound to anionic arenes has increased considerably. In the related compounds [(CpTMS2Ln)2(μ–η6:η6-C6H6)]2– (Ln = La, Ce), formed via the reaction of in situ generated [K(2,2,2-Crypt)][CpTMS3Ln(THF)] with benzene, the lanthanide ions sandwich a distorted benzene dianion, and their coordination spheres are filled by trimethylsilyl substituted cyclopentadienide anions on the periphery. The structural characteristics of [(CpTMS2Ln)2(μ–η6:η6-C6H6)]2– are typical of inverse sandwich compounds with anionic unsubstituted benzene.12,13 In particular, these compounds are frequently complex anions, with a distorted benzene subunit bridging two lanthanides coordinated by multiple bulky capping ligands in a pseudotrigonal environment. The distortion of the bound arene dianion is a consequence of its Hückel type antiaromaticity, and is virtually ubiquitous in complexes with anionic benzene.13−15 To our knowledge, the only prior examples in the literature of lanthanide compounds featuring a planar and symmetric anionic arene bridge are [Ln2(BzN6-Mes)]2– and [Ln2(BzN6-Mes)]− (Ln = Y, Gd; BzN6-Mes = 1,3,5-tris[2′,6′-(N-mesityl)dimethanamino4′-tert-butylphenyl]benzene).16 Here, the constrained ligand geometry and trigonal symmetry of the hexapodal BzN6-Mes ligand enforces a planar arene geometry, stabilizing an S = 1 benzene dianion or an S = 7/2 monoanion. The compound [(CpTMS22La)2(μ–η6:η6-C6H6)]− was previously reported to host a monoanionic benzene bridge, which exhibits an out-of-plane distortion consistent with the Jahn–Teller effect.17
In addition to mono and dianionic arenes, formally tetraanionic arene bridges have also been described in the literature.8 For example, compounds of the type [(NNTBSLn)2(μ–η6:η6-biph)]2– (Ln = Y, Gd, Dy) (NNTBS = 1,1′-fc(NSitBuMe2)2) have been isolated, all three of which feature a bridging biphenyl tetraanion.18,19 The π system of both biphenyl rings is fully conjugated in these compounds, which permits charge delocalization. As a result of the high nucleophilicity of the tetraanionic biphenyl bridge, the metal arene distances for these compounds are slightly shorter than those of previously reported dianionic arenes. Additionally, the arene bridges in [(NNTBSLn)2(μ-biph)]2– (Ln = Y, Gd, Dy) are considerably more planar than singlet arene dianions. This result is unsurprising, as the tetraanion has an aromatic 4n+2 π-electron count and therefore should not be subject to an electronically driven distortion. More recently, neutral inverse sandwich compounds supported by β-diketiminate ligands and featuring a bridging benzene tetraanion have been isolated for Y and Sm,20,21 although inverse sandwich compounds featuring a bridging benzene tetraanion are not known for the later lanthanides.
Here, we report the synthesis and characterization of a series of neutral lanthanide–benzene inverse sandwich compounds with the formula (CpiPr5Ln)2(μ–η6:η6-C6H6) 1-Ln (Ln = Y, Gd, Tb, Dy, Tm), which feature an unusual linear coordination geometry and a highly planar and symmetric unsubstituted bridging benzene anion. Single-crystal X-ray diffraction analysis, magnetometry, UV–vis spectroscopy, NMR spectroscopy and density functional theory (DFT) calculations strongly support the presence of a rare, unsubstituted benzene tetraanion in these compounds and an overall oxidation state configuration that is best described as Ln3+–Bz4––Ln3+. The planar geometry and unusual stability of the tetraanion is found to be the result of a δ-bonding interaction between the vacant dxy and dx2–y2 orbitals of the Ln3+ ions and the π* orbitals of (C6H6)4–.
Results and Discussion
Synthesis and Structural Characterization
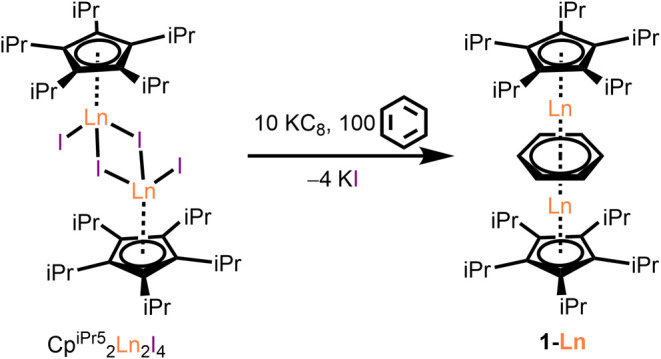
The compounds 1-Ln were synthesized from the reaction of CpiPr52Ln2I4 (Ln = Y, Gd, Tb, Dy, Tm)22 in diethyl ether (Et2O) with excess benzene followed by the addition of KC8 (Scheme 1; see the Supporting Information for details). After 4 days, the diethyl ether and excess benzene were removed from the reaction mixture under reduced pressure, and the resulting residue was extracted into boiling n-hexane. The solution was filtered to remove potassium iodide, and the filtrate was then concentrated under reduced pressure. After heating to redissolve any precipitate, the solution was allowed to cool to 25 °C overnight. Subsequent storage of these solutions at −35 °C yielded analytically pure crystals of 1-Ln suitable for X-ray diffraction analysis as dark red (1-Y), red-brown (1-Gd, 1-Tb, 1-Dy), or brown (1-Tm) prisms. Notably, in the course of this workup, it was found that n-hexane solutions of 1-Ln could be boiled at ambient pressure without evident decomposition.
Scheme 1. Synthesis of 1-Ln from CpiPr52Ln2I4 (Ln = Y, Gd, Tb, Dy, Tm).
Compounds 1-Ln are isostructural (see Figure 1 for the structure of 1-Gd and Figures S1–S5 for Ln = Y, Tb, Dy, and Tm) and crystallize in the space group P2/n with similar unit cell parameters (Table S3). In each structure, the metal centers are related by a C2 rotation axis that passes through the plane of the benzene ring, and the bridging benzene exhibits in-plane rotational disorder, which was successfully resolved as two benzene positions with equivalent chemical occupancies. There are two unique Ln–benzene distances in all five structures due to this in-plane rotational disorder (see Figure S2). The average Ln–Bzcentroid distances in compounds 1-Y, 1-Gd, 1-Tb, 1-Dy, and 1-Tm are 1.989(5), 2.038(7), 2.013(6), 1.99(1), and 1.9421(7) Å, respectively (numbers in parentheses are estimated standard deviations). For compounds 1-Gd, 1-Tb, 1-Dy, and 1-Tm, the gradual decrease in Ln–Bzcentroid distance is consistent with the contraction in the lanthanide ionic radii proceeding across the series. The closest comparable Ln–Arcentroid distances in the literature are from the [(NNTBSLn)2(μ-(C6H5)2)]2– series of compounds, namely 2.053(2), 2.113(2), and 2.054(1) Å for Ln = Y, Gd, and Dy, respectively.18,19 The Ln–Ar distances in 1-Ln are all shorter than in the corresponding [(NNTBSLn)2(μ-(C6H5)2)]2– compounds. The recently reported β-diketiminate inverse sandwich compound [(BDI)Y(THF)]2(μ–η6,η6-C6H6) (BDI = HC(C(Me)N[C6H3-(3-pentyl)2-2,6])2)20 has an Y–Arcentroid distance of 2.011(1) Å, which is also longer than that in 1-Y.20 To the best of our knowledge, when comparing compounds for a given metal, the Ln–Ar distances in 1-Ln are the shortest reported for any lanthanide–arene compound in the Cambridge Structural Database (see Table 1). This suggests an unusually strong Ln–Ar interaction in 1-Ln.
Figure 1.
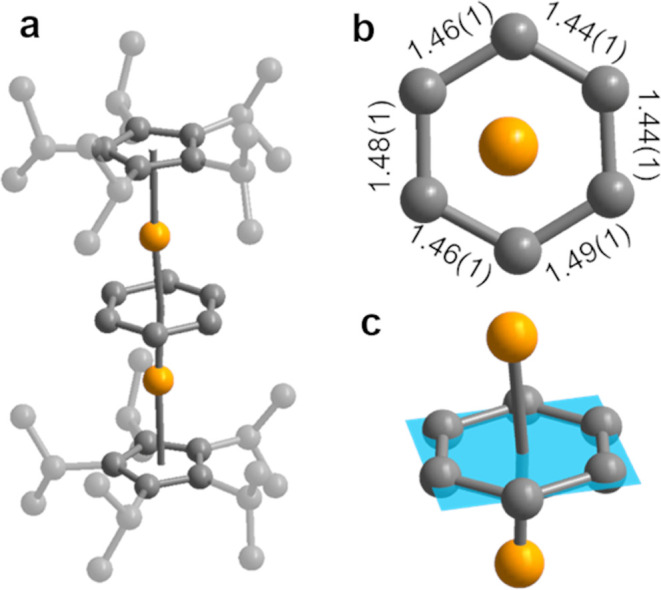
(a) X-ray crystal structure of 1-Gd. Orange and gray spheres represent Gd and C, respectively. Minor rotationally disordered positions of the benzene ring and CpiPr5 groups have been omitted for clarity. Hydrogen atom positions were also omitted for clarity. (b) Top-down representation of the 1-Gd structure highlighting the C–C distances of the benzene bridge. The CpiPr5 groups and rotationally disordered benzene position are omitted for clarity. The C–C distances are given in Å. (c) Inverse sandwich core of 1-Gd with the mean plane illustrated.
Table 1. Comparison of Ln–Arcentroid (Ln = Y, Gd, Tb, Dy, Tm) Distances in Åa.
| [(NNTBSLn)2(biph)]2– | ArLn(AlCl4)3 | [Ln2(BzN6)]2– | 1-Ln | |
|---|---|---|---|---|
| refs | (18,19) | (23−25) | (16) | |
| Y | 2.053(2) | 2.281(1) | 1.989(5) | |
| Gd | 2.113(2) | 2.309(1) | 2.038(7) | |
| Tb | 2.4882(9)b,d | 2.013(6) | ||
| Dy | 2.054(1) | 2.476(2)c | 1.990(1) | |
| Tm | 2.463(5)c,e | 1.9421(7) |
All data collected at T = 100 K unless otherwise noted.
Ar = C6Me6;
Ar = MeC6H5;
T = 143 K;
T = 163 K
The CpiPr5 groups of 1-Ln exhibit disorder, which could be resolved into one major and one minor position. The major position has a freely refined chemical occupancy of ∼0.7 in all compounds, while the minor position has a corresponding chemical occupancy value of ∼0.3. The average Ln–Cpcentroid distances reported in Table 2 are weighted averages, with the weighting coefficients equal to the CpiPr5 chemical occupancy values associated with each disordered CpiPr5 position. The average Ln–Cpcentroid distances in 1-Ln are 2.370(2), 2.421(1), 2.401(6), 2.383(2), and 2.327(1) Å for Ln = Y, Gd, Tb, Dy, and Tm, respectively. The decrease in these distances from Gd to Tm is consistent with the expected trend based on the lanthanide contraction.
Table 2. Literature Comparison of Ln–Cpcentroid (Ln = Y, Gd, Tb, Dy, Tm) Distances Measured in Åa.
| LnCpiPr52 | [LnCpiPr52]+ | CpiPr52Ln2I4b | 1-Ln | |
|---|---|---|---|---|
| refs | (20,21) | (20) | (16) | |
| Y | 2.371(6) | 2.3307(1) | 2.328(9) | 2.370(2) |
| Gd | 2.423(2) | 2.365(2) | 2.421(1) | |
| Tb | 2.4177(1) | 2.325(2) | 2.36(2) | 2.401(6) |
| Dy | 2.3848(1) | 2.321(4) | 2.34(1) | 2.383(2) |
| Tm | 2.449(3) | 2.27(2) | 2.327(1) |
All data collected at T = 100 K unless otherwise noted.
Average metal centroid distance with parenthesized standard deviations.
To gain preliminary insight into the metal oxidation state in 1-Ln, we compared the Ln–Cpcentroid distances with literature compounds featuring well-defined divalent or trivalent metal ions in similar coordination environments, namely LnCpiPr52 (Ln = Y, Gd, Tb, Dy, Tm)26,27 and [LnCpiPr52]+ (Ln = Y, Tb, Dy).26,28 In general, Ln–Cpcentroid distances in are longer than in the corresponding compounds.20,21 Further distinction can be made based on the electronic configuration of the divalent compounds: for complexes with 4fn5dz21 configurations (Ln = Gd, Tb, Dy; 4dz21 in the case of Y), the Ln–Cpcentroid distances are shorter than in complexes possessing 4fn+1 configurations (e.g., TmCpiPr52) as a result of d orbital participation in bonding. In the case of 1-Ln (Ln = Y, Gd, Tb, Dy), the Ln–Cpcentroid distances are close to the Ln–Cpcentroid distances in the corresponding LnCpiPr52 compounds and are longer than the distances in (Table 2). The Tm–Cpcentroid distance in 1-Tm is shorter than that in TmCpiPr52 (2.327(1) versus 2.449(3) Å), but longer than in the trivalent compound CpiPr52Tm2I422 (2.27(2) Å; [TmCpiPr52]+ has not been reported). Altogether, these results could suggest the possibility of d orbital occupation in all the 1-Ln compounds.
The average benzene C–C bond length for all members of the series 1-Ln was found to be 1.46 Å (see Table S1 for full details), within the range of reported C–C distances for lanthanide-bound η6-arenes in the Cambridge Structural Database (1.223 to 1.517 Å).29 In general, average C–C distances similar to those in the corresponding free arene are characteristic of neutral bound arenes, whereas C–C distances that are significantly longer than in the free arene are indicative of anionic arenes, which have populated π* orbitals. The C–C distances of the C6H6 units in 1-Ln are all considerably longer than the C–C distance of free benzene (1.3946(7) Å),30 indicating that the benzene moieties are anionic. The distances are also similar to those reported for the inverse sandwich compound [NNTBSTh(THF)]2(μ-C6H6), which features a tetraanionic benzene with an average benzene C–C distance of 1.46 Å,31 and slightly longer than those reported for the inverse sandwich complexes [(BDI)Sm(THF)]2(μ–η6:η6-C6H6) (BDI = HC(C(Me)N[C6H3-(3-pentyl)2-2,6])2) and [(BDI)Sm]2(μ–η6:η6-C6H6), which feature tetraanionic benzene bridges with average benzene C–C distances of 1.44 and 1.45 Å, respectively.20,21 The expanded benzene C–C distances are therefore consistent with three possible oxidation state assignments for 1-Ln, namely Ln3+–Bz4––Ln3+, Ln2+–Bz2––Ln2+, and Ln2.5+–Bz3––Ln2.5+.
In order to quantify the degree of distortion from planarity for the bridging benzene in 1-Ln, we used the program Mercury26,32 to identify the mean plane for each benzene ligand and then calculated the so-called molecular-planarity parameter (MPP),33 which is the root-mean-square deviation of the carbon atoms in the arene ring from the mean plane (see Section 3 of the Supporting Information (SI) for details and Table S2). We also determined MPP values for Ln–(η6-Ar) complexes based on structures available in the Cambridge Structural Database29 (see Tables S3–S9). We use the MPP for benzene, which is 10–3 Å (based on a solid-state structure obtained at 100 K), as a measure of perfect planarity. Significantly larger MPP values for an anionic six-membered ring would be indicative of a distortion, and therefore an antiaromatic benzene dianion. In contrast, MPP values approaching that of benzene would indicate an aromatic tetraanionic benzene.
A representative image of the mean plane determined for 1-Gd is shown in Figure 1c. The MPP values for 1-Y, 1-Tb, and 1-Dy were found to be comparable to one another, ranging from 6 × 10–3 to 7 × 10–3 Å, and near the low end of the range of values calculated for literature compounds (≥4 × 10–3 Å). The benzene in 1-Tm exhibits the greatest amount of out-of-plane distortion, with a MPP of 2 × 10–2 Å, while the η6-C6H6 unit in 1-Gd is the most planar in the series, with a MPP of 3 × 10–3 Å that is significantly lower than for all previously reported Gd compounds (>5 × 10–3 Å). In all, the low MPP values for 1-Y, 1-Gd, 1-Tb, and 1-Dy indicate that the benzene ring is unusually planar in these complexes. Combined with the elongated average benzene C–C bond lengths, the high planarity of the benzene bridge in 1-Ln is consistent with the presence of a tetraanionic benzene, and therefore an oxidation state assignment of Ln3+–Bz4––Ln3+.
Spectroscopic Characterization
Ultraviolet–visible absorption spectra were collected for n-hexane solutions of 1-Ln from 300–800 nm. All compounds exhibit an intense absorption feature between 320 and 350 nm (Figure 2), with molar extinction coefficients (ε) ranging from ∼6 × 104 M–1 mol–1 (1-Y) to ∼0.9 × 104 M–1 mol–1 (1-Dy). A gradual decrease in the intensity of this feature occurs upon moving from 1-Gd to 1-Dy, although the intensity increases again for 1-Tm. The absorption maximum was found to shift to higher energy from 1-Gd (350 nm) to 1-Tm (320 nm). Strong visible absorption within this range, in tandem with the metal dependence of the band energy and intensity, is consistent with ligand-to-metal charge transfer from the central benzene anion π* orbitals to an unoccupied molecular orbital of mainly 5d (4d in the case of 1-Y) character.
Figure 2.
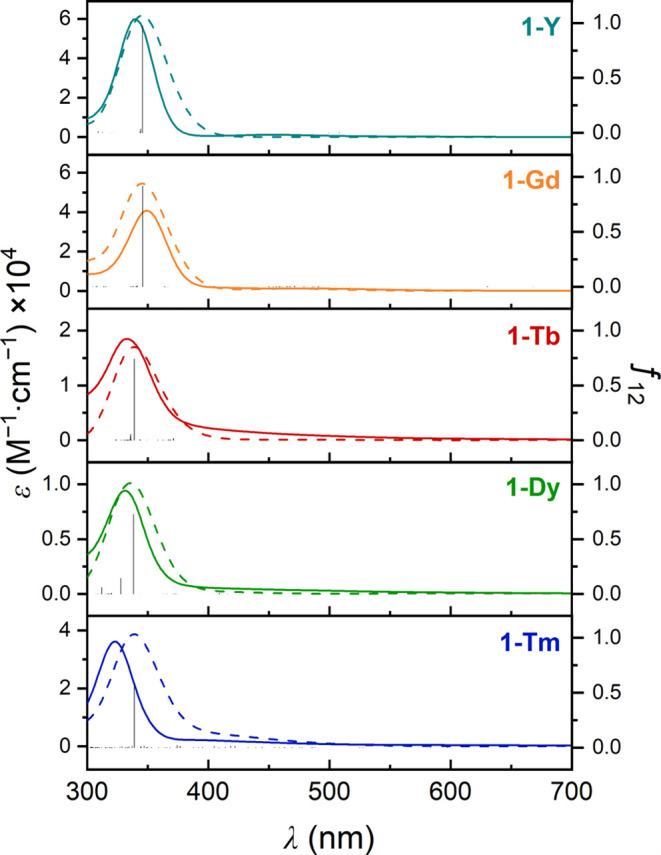
UV–visible spectra of n-hexane solutions of 1-Ln (Ln = Y, Gd, Tb, Dy, Tm) compounds. Solid lines correspond to experimental ε values (left axis). Bar plots correspond to calculated oscillator strengths (right axis) for each transition obtained from time-dependent DFT calculations. Dashed lines correspond to calculated ε values derived from the transitions obtained from time-dependent DFT. A Gaussian spectral width of 0.2 eV was used. The calculated absorption spectrum of 1-Tb was scaled by a factor of 0.33 while that of 1-Dy was scaled by a factor of 0.20.
The diamagnetic derivative 1-Y was further characterized by 1H, 13C, and 89Y NMR spectroscopy. The 1H spectra collected at 25 °C in C6D6 or toluene-d8 exhibit sharp, well resolved features (Figures S20 and S24). Interestingly, the C6H6 protons in both spectra appear as a narrow singlet, shifted upfield from the 1H resonance for free benzene (3.75 or 3.70 ppm in C6D6 or toluene-d8, respectively). Likewise, the 13C spectrum in C6D6 obtained at 25 °C features a C6H6 signal far upfield of uncoordinated benzene (59.39 ppm, see Figure S24), which appears as a triplet (J = 4.4 Hz) due to coupling with two equivalent 89Y nuclei, as observed in other reduced arene-bridged yttrium compounds.19 Taken together, these spectral signatures are consistent with a highly reduced, 6-fold symmetric η6:η6-C6H6 bridge coordinated to two equivalent Y(CpiPr5) moieties.
Proton NMR spectra for 1-Y were also obtained over a range of temperatures (25 to 75 °C in C6D6; 25 to 105 °C in toluene-d8) to probe structural dynamics in solution (Figures S23 and S27). With increasing temperature, two doublets assigned to the methyl protons of the CpiPr5 rings (−CH(CH3)2) coalesce to form a broad singlet, consistent with thermally activated rotation of the Cp–iPr bonds. In contrast, the C6H6 resonance remains a narrow singlet up to 105 °C. Notably, there was no evidence of decomposition or C6H6 substitution in C6D6 or toluene-d8 solutions, even after samples were held for several hours at 105 °C or stored for several months at ambient temperatures. The 89Y spectrum of 1-Y features a single broadened resonance at −103 ppm (C6D6 at 25 °C) that lies upfield of the 89Y resonance for the (biphenyl)4–-bridged complex (NNTBSY)2(μ-(C6H5)2)2– (189 ppm)19 and downfield of the 89Y resonance for Cp3Y(THF) at −407 ppm (THF-d8 at −45 and 25 °C, respectively).13
Magnetic Properties
In order to probe the ground-state electron configurations in the compounds 1-Gd, 1-Tb, 1-Dy, and 1-Tm, zero-field cooled dc magnetic susceptibility data were collected under applied fields of 0.1, 0.5, and 1 T at temperatures ranging from 2 to 300 K (see Figures 3 and S7 for 1-Gd and Figures S9, S11, S13, and S17 for the remaining compounds). At 300 K, the molar magnetic susceptibility-temperature product (χMT) for 1-Gd is 14.4 emu·K/mol (Figure 3). This value is consistent with the theoretical value for two uncoupled S = 7/2 GdIII centers (15.8 emu K/mol) and an S = 0 bridging arene. By contrast, the theoretical 300 K χMT value for a mixed-valence ground state (Ln2.5+–Bz3––Ln2.5+) without strong exchange coupling is higher at 16.8 emu·K/mol. Additionally, for a delocalized mixed-valence configuration with strong ferromagnetic exchange coupling, we would expect an even higher χMT value of 30.74 emu·K/mol at ambient temperature, with a similar temperature dependence to that observed for the previously reported mixed-valent compound CpiPr52Gd2I3.22 With decreasing temperature, χMT decreases monotonically to a value of 0.233 emu K/mol at 2 K, a trend that is indicative of antiferromagnetic coupling between the GdIII centers. Using the program PHI,34 the χMT versus T data could be fit using an isotropic exchange Hamiltonian for a dinuclear cluster featuring two S = 7/2 ions (eq 1) with an exchange coupling constant, Jex, of −2.94(2) cm–1. The good agreement between the experimental data and fit supports the presence of a closed shell benzene anion in 1-Gd. Together with the structural data for 1-Ln and the presence of a largely planar bridging benzene anion, this result is most consistent with the presence of a bridging benzene tetraanion, as an S = 0 benzene dianion would exhibit significant out-of-plane distortion.35 Of note, most Jex values mediated by closed shell bridging ligands are <1 cm–1 in magnitude, and therefore the exchange observed for 1-Gd is unusually strong.36 Isothermal magnetization data for 1-Gd are also consistent with strong antiferromagnetic exchange, as the magnetic moment remained unsaturated under a 7 T applied field at all measured temperatures (Figure S8). The unusually strong antiferromagnetic exchange observed for 1-Gd is likely the result of Gd–Gd superexchange enabled by the appreciable interaction between the diffuse frontier orbitals of the ligand and the 4f orbitals of each gadolinium.
Figure 3.
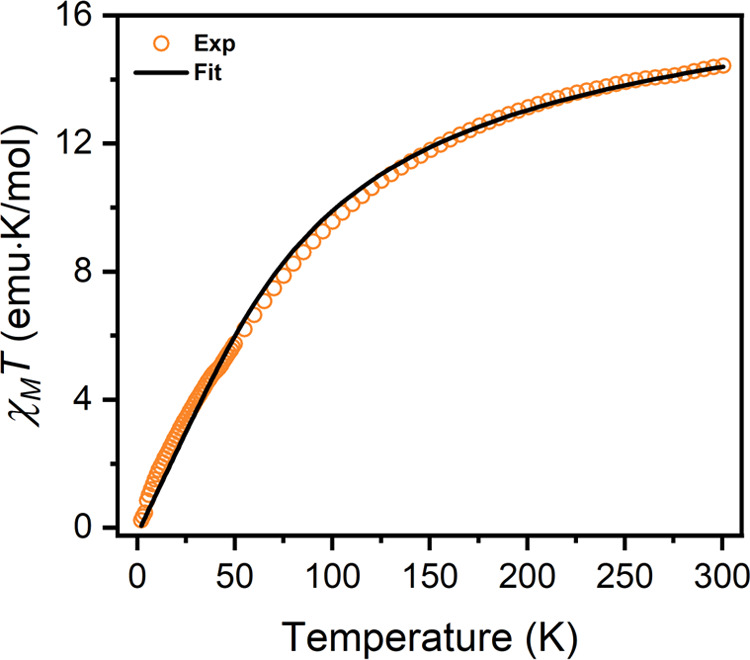
Plot of the molar magnetic susceptibility–temperature product (χMT) versus T for 1-Gd under an applied field of 0.5 T (orange circles). The black trace represents a fit using the spin-Hamiltonian given in eq 1 with two S = 7/2 GdIII centers and Jex = −2.94(2) cm–1. A TIP correction of 0.0028(1) emu·K/mol was applied.
| 1 |
At 300 K, 1-Tb, 1-Dy, and 1-Tm exhibit χMT values of 21.3, 29.3, and 14.5 emu K/mol, respectively, close to the predicted values for two TbIII (4f8, 7F6), DyIII (4f9, 6H15/2), or TmIII (3H6, 4f12) centers (22.0, 29.4, and 14.5 emu·K/mol, respectively, see Figures S10–S20) and again consistent with an Ln3+–Bz4––Ln3+ oxidation state assignment. With decreasing temperature, χMT for each compound decreased monotonically, reaching minimum values of 0.549, 0.907, and 0.606 emu K/mol for 1-Tb, 1-Dy, and 1-Tm, respectively, at 2 K. Consistent with the results for 1-Gd, the data are indicative of antiferromagnetic exchange between the lanthanide ions.
Density Functional Theory Calculations
Computational studies are often critical for elucidating the electronic structure of arene-bridged inverse sandwich compounds of the f-elements and for exploring metal–ligand interactions.13,18,19,38 Commonly, simplified models of large inverse sandwich lanthanide complexes have been investigated, instead of using the full experimental structures. However, calculations based on such simplified models can result in structural artifacts and consequently lead to inaccuracies in simulated absorption spectra or computed magnetic properties.19,20,39 Here, we treated the 1-Ln complexes in their entirety by carrying out DFT optimizations for all possible spin states for each complex, starting from the full experimental crystal structures. This thorough modeling was ultimately necessary to accurately compute the structural parameters of 1-Ln and identify their ground states. All calculations were carried out using the TURBOMOLE 7.7 quantum chemistry package.40 Geometry optimizations used the hybrid TPSSh exchange–correlation functional,41 employing def2-SVP and def2-TZVP basis sets42,43 for C and H atoms and Ln atoms, respectively. Small-core effective core potentials44 were included to model the 4f orbitals of the metals explicitly. For all-electron calculations, x2c-type basis sets45 were used (see Section 9 of the Supporting Information for details).
Starting with the 1-Ln crystal structures, we optimized the geometries of the complexes for all possible spin states. This included examining scenarios in which the lanthanide ions were trivalent or divalent, consistent with 4fn, 4fn+1 or 4fn5d1 configurations. We also considered both ferromagnetic and antiferromagnetic coupling between the metal ions. Additionally, singlet and triplet states of the benzene were also explored (see Section 9 of the Supporting Information for details). The DFT results indicated that the structural parameters of the optimized complexes, particularly the Ln···Ln distances and the C–C of the C6H6 ligand, are highly sensitive to their electronic configurations.
Among the optimized 1-Ln (Ln = Y, Gd, Tb, Dy) structures, those with a singlet ground state (S = 0) were found to most accurately reproduce the experimental structures obtained from single-crystal X-ray diffraction analysis, with all metrical parameters differing from the experimental values by only ∼1–2%, within the error margin of the methodology used39 (see Table 3). For example, the calculations predicted a Ln–Bzcentroid distance of 2.019 Å for the 1-Gd singlet structure, compared with the experimental distance of 2.038(7) Å from X-ray diffraction analysis. The singlet ground-state assignment is also consistent with antiferromagnetic coupling of paramagnetic lanthanides (Gd, Tb, and Dy). Interestingly, 1-Tm was found to be an outlier: in particular a quintet ground state was computed to be energetically similar to the singlet state, and the structural parameters are identical for optimized 1-Tm compounds with quintet and singlet states and agree well with experimental data. As a result, based on these data it remains unclear if the Tm ions in 1-Tm are coupled ferromagnetically or antiferromagnetically, and our results suggest the possibility of a dynamic equilibrium between the two configurations.
Table 3. Comparison of the Ln···Ln Distances and Average Benzene C–C Distances for the 1-Ln Singlet Structures with the Values Determined Experimentally from Single-Crystal X-ray Diffractiona.
| Y | Gd | Tb | Dy | Tm | |
|---|---|---|---|---|---|
| Ln···Lnb | 3.9707(4) | 4.0710(3) | 4.0177(3) | 3.9810(4) | 3.8748(3) |
| Ln···Lnc | 3.963 | 4.038 | 3.986 | 3.963 | 3.995 |
| C–Cb | 1.46(1) | 1.46(2) | 1.46(1) | 1.46(1) | 1.46(1) |
| C–Cc | 1.472 | 1.468 | 1.469 | 1.467 | 1.464 |
All Values are Given in Å.
Structural parameters from crystallographic data.
Structural parameters from DFT models.
DFT computations showed that the benzene rings in the optimized structures of singlet 1-Y, 1-Gd, and 1-Dy are planar, while a slight distortion is apparent for 1-Tb, and 1-Tm. The average C–C bond lengths for all 1-Ln structures are consistent with those determined experimentally (see Table 3). In particular, the average C–C lengths in the benzene ring are longer by 0.08–0.07 Å than the length of ∼1.39 Å typically seen in neutral benzene, consistent with previous observations in benzene-bridged inverse sandwich complexes12,13 and indicative of charge localization on the arene bridge. Additionally, DFT predicts the six C–C bonds of benzene in each 1-Ln are equally elongated, consistent with a reduced, aromatic benzene ligand.
We briefly note that, in the cases of 1-Ln (Ln = Gd, Tb, Dy), the singlet state was not the absolute lowest energy state (see Tables S15, S19, and S23), although it produced structural parameters most consistent with the experimental results. DFT predictions of the relative ordering of spin state energetics in open-shell transition metal and lanthanide organometallics may not be as reliable as its description of most ground-state properties,46 and this is particularly true for lanthanide complexes,47 where the contracted nature of the 4f orbitals may lead to several low-lying electronic states with near-degeneracies.
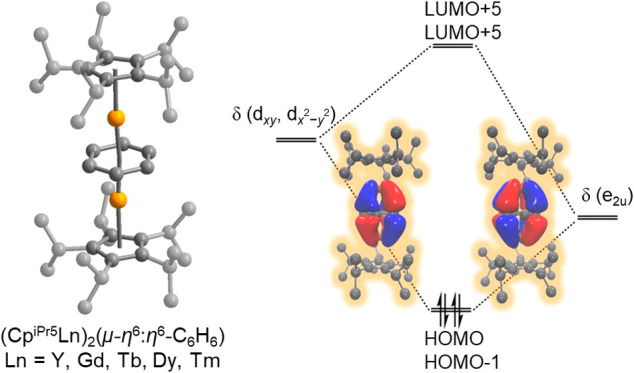
Mulliken population analysis48 of the HOMO and HOMO–1 for the 1-Ln complexes indicated significant mixing between metal d orbitals and the π* orbitals of the reduced C6H6 (see Figure 4a for 1-Gd and Figures S48, S53, and S55 for 1-Y, 1-Tb, and 1-Dy, respectively). For example, in the singlet structure of 1-Y, the HOMO and HOMO–1 exhibit a 62% contribution from the benzene π* orbitals and 38% from 4dxy and 4dx2–y2 orbitals of the Y ions (see Table S12; the molecular orbital (MO) energies are also summarized in this table). The orbital mixing in the HOMO and HOMO–1 are indicative of δ-symmetric bonding, characterized by two nodal planes perpendicular to the benzene ring, suggesting a strong metal–arene interaction in all 1-Ln congeners. In the 1-Gd complex, as illustrated in Figure 4a, the frontier orbitals clearly illustrate the formation of a unique δ covalent bond between the Gd ions and the benzene ligand in a linear geometry, which helps to rationalize the very short Gd–Arcentroid distance, relative to reported inverse sandwich arene-bridged lanthanide complexes (Table 1).
Figure 4.
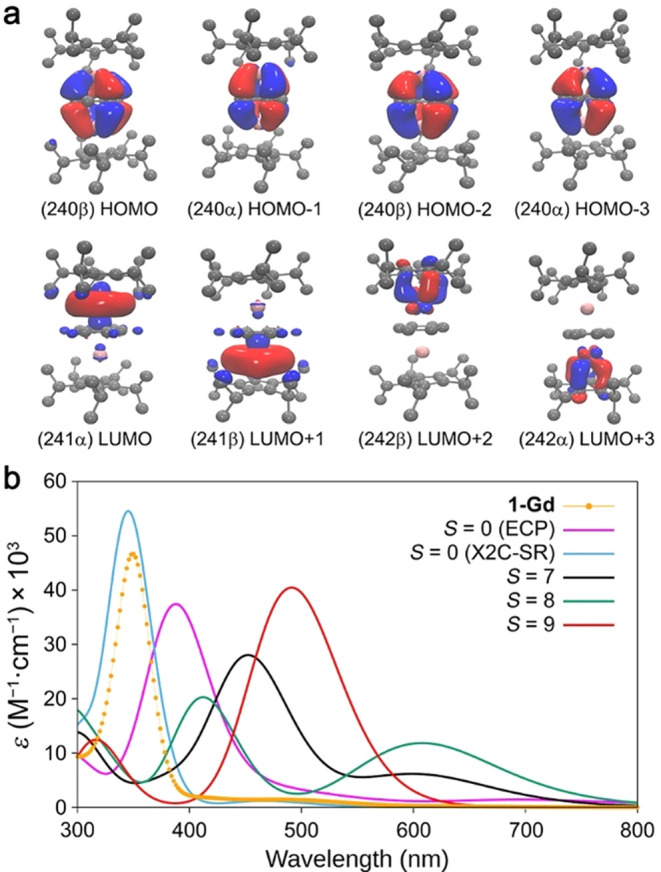
(a) Frontier molecular orbitals of the 1-Gd singlet ground state with a contour value of 0.03. H atoms are omitted for clarity. (b) Comparison of the experimental UV–vis spectrum for 1-Gd (orange data) with calculated spectra using time-dependent DFT (solid purple, light blue, black, turquoise and red traces) for different possible ground states of 1-Gd. The time-dependent DFT calculations included hexane solvent effects with the COSMO37 model and used the same choice of functional and basis sets as ground-state computations.
To complement the DFT analysis, time-dependent DFT calculations were performed to simulate the UV–vis spectra of 1-Ln complexes with all the different candidate spin multiplicities. The computed electronic spectra for 1-Ln with singlet states are in excellent agreement with the experimental spectra for all compounds (see Figure 2 and Table S33), corroborating the spin state assignment made using DFT. In contrast, for optimized structures with higher spin states, the simulated absorption spectra exhibit features at longer wavelengths than those in the experimental spectra (see Figures 4b and S54 for 1-Gd and Figures S56, S58, and S60 for 1-Tb, 1-Dy, and 1-Tm, respectively). In the case of 1-Tm, the quintet state yielded an electronic spectrum identical to that for the singlet state, both of which are consistent with the experimental data (Figure S60). For singlet 1-Y, transitions from the HOMO and HOMO–1 to LUMO+2 and LUMO+3, respectively, give rise to the intense band located at 345 nm in the simulated spectrum, while a very weak and broad absorption around 500 nm arises due to transitions from HOMO and HOMO–1 to LUMO and LUMO+1, respectively (Figure 5). Similarly, strong electronic transitions between molecular orbitals with similar characters give rise to the intense absorption features in the higher energy region (300–400 nm) for the other 1-Ln compounds and are consistent with strong Ln–C6H6 δ bonding. These results collectively show that the singlet states are the best model for the 1-Ln (Ln = Y, Gd, Tb, Dy) complexes, demonstrating the usefulness of DFT in understanding the electronic structures of triple-decker lanthanide molecular complexes.
Figure 5.
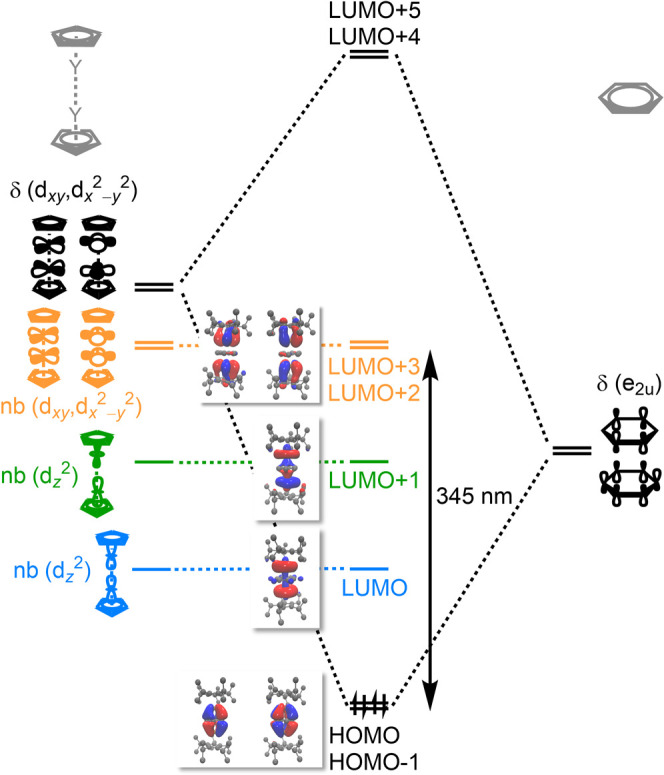
Qualitative diagram illustrating the metal–benzene interaction in 1-Y. The LMCT transition between HOMO/HOMO–1 and LUMO+2/LUMO+3 is indicated with a black arrow. Surface plots of the HOMOs and four LUMOs are pictured. A contour value of 0.03 was used in the orbital depictions.
The unusually short Ln–Bzcentroid distances as well as the elongated Ln–Cpcentroid distances observed in the experimental structures for all five 1-Ln compounds are readily explained within this covalent bonding picture. Indeed, formal population of δ bonding orbitals would necessarily yield a relatively short Ln–C6H6 distance and an elongated Ln–CpiPr5 distance, as the HOMOs are bonding with respect to the metal ions and benzene and are weakly antibonding with respect to the metal ions and CpiPr5. Finally, the benzene ring in this configuration would not be susceptible to an electronically driven distortion, given that its e2u set is fully occupied. Ultimately, our experimental and computational data strongly support an oxidation state assignment for all five compounds of Ln3+–Bz4––Ln3+, in which the bridging tetraanion is stabilized through a highly covalent interaction with the neighboring metal ions.
Conclusions
In summary, we have synthesized and characterized a series of lanthanide–benzene inverse sandwich compounds of the type (CpiPr5Ln)2(μ–η6:η6-C6H6) (1-Ln; Ln = Y, Gd, Tb, Dy, Tm), through the reduction of CpiPr52Ln2I4 with KC8 in the presence of benzene. The compounds feature an unusual linear coordination geometry and overall neutral charge state, and the structural features and magnetic properties of 1-Ln, supported by computational analysis, are consistent with the presence of an S = 0 tetraanionic benzene bridge, the first example of such a bridge for any compound of the later lanthanides. Further, computations revealed that the frontier orbitals of the benzene tetraanion in 1-Ln, rather than being fully localized, are δ-bonding combinations of the 5dxy and 5dx2–y2 (4dxy and 4dx2–y2 for 1-Y) orbitals of the two Ln ions and the π* orbitals of benzene. Ultimately, our data support an oxidation state assignment for all five compounds of Ln3+–Bz4––Ln3+, in which the bridging benzene tetraanion is stabilized through a highly covalent interaction with the neighboring metal ions. Notably, the one-electron oxidation of these complexes can be anticipated to result in species featuring an unprecedented benzene trianion radical with, in the cases of Ln = Gd, Tb, and Dy, strongly coupled high-magnetic moment ground states.
Acknowledgments
We thank the U.S. National Science Foundation for support of the experimental research under NSF grant CHE-2102603 (A.H.V. and J.R.L.) and the theoretical research under NSF grant CHE-2102568 (A.R. and F.F.). This work was additionally funded by the Naval Air Warfare Center Weapons Division ILIR program (K.R.M. and B.G.H.). A.R. acknowledges support from an Eddleman Quantum Institute Fellowship and Dmitrij Rappoport for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c12278.
Synthesis and structural characterization details and detailed magnetic, spectroscopic, and computational results for all compounds (PDF)
Author Contributions
∇ K.R.M., A.H.V., and A.R. are cofirst authors, each contributing equally to the work.
The authors declare the following competing financial interest(s): Principal investigator Filipp Furche has an equity interest in TURBOMOLE GmbH. The terms of this arrangement have been reviewed and approved by the University of California, Irvine, in accordance with its conflict of interest policies.
Supplementary Material
References
- Cotton F. A.; Schwotzer W. Sm(η6-C6Me6)(η2-AlCl4)3: The First Structure of a Rare Earth Complex with a Neutral π-Ligand. J. Am. Chem. Soc. 1986, 108 (15), 4657–4658. 10.1021/ja00275a070. [DOI] [Google Scholar]
- Dzudza A.; Marks T. J. Lanthanide Triflate-Catalyzed Arene Acylation. Relation to Classical Friedel–Crafts Acylation. J. Org. Chem. 2008, 73 (11), 4004–4016. 10.1021/jo800158k. [DOI] [PubMed] [Google Scholar]
- Huang W.; Diaconescu P. L. Aromatic C–F Bond Activation by Rare-Earth-Metal Complexes. Organometallics 2017, 36 (1), 89–96. 10.1021/acs.organomet.6b00661. [DOI] [Google Scholar]
- Halter D. P.; Palumbo C. T.; Ziller J. W.; Gembicky M.; Rheingold A. L.; Evans W. J.; Meyer K. Electrocatalytic H2O Reduction with f-Elements: Mechanistic Insight and Overpotential Tuning in a Series of Lanthanide Complexes. J. Am. Chem. Soc. 2018, 140 (7), 2587–2594. 10.1021/jacs.7b11532. [DOI] [PubMed] [Google Scholar]
- Zhu M.; Chai Z.; Li T.; Wei J.; Lv Z.-J.; Zhang W.-X. Selective C–C Coupling of Two Nitriles Affording Rare-Earth Diazametallacyclopentadienes: Synthesis, Cooperative Reactivity and Mechanistic Studies. Inorg. Chem. Front. 2023, 10 (15), 4569–4577. 10.1039/D3QI00946G. [DOI] [Google Scholar]
- Huang W.; L Diaconescu P. Rare-Earth Metal π-Complexes of Reduced Arenes, Alkenes, and Alkynes: Bonding, Electronic Structure, and Comparison with Actinides and Other Electropositive Metals. Dalton Trans. 2015, 44 (35), 15360–15371. 10.1039/C5DT02198G. [DOI] [PubMed] [Google Scholar]
- Xin T.; Wang X.; Yang K.; Liang J.; Huang W. Rare Earth Metal Complexes Supported by a Tripodal Tris(Amido) Ligand System Featuring an Arene Anchor. Inorg. Chem. 2021, 60 (20), 15321–15329. 10.1021/acs.inorgchem.1c01922. [DOI] [PubMed] [Google Scholar]
- Bochkarev M. N. Synthesis, Arrangement, and Reactivity of Arene–Lanthanide Compounds. Chem. Rev. 2002, 102 (6), 2089–2118. 10.1021/cr010329i. [DOI] [PubMed] [Google Scholar]
- Cotton S.Lanthanide and Actinide Chemistry; John Wiley & Sons, 2013. [Google Scholar]
- Brennan J. G.; Cloke F. G. N.; Sameh A. A.; Zalkin A. Synthesis of Bis(η-1,3,5-Tri-t-Butylbenzene) Sandwich Complexes of Yttrium(0) and Gadolinium(0); the X-Ray Crystal Structure of the First Authentic Lanthanide(0) Complex, [Gd(η-But3C6H3)2]. J. Chem. Soc. Chem. Commun. 1987, (21), 1668–1669. 10.1039/C39870001668. [DOI] [Google Scholar]
- Cassani M. C.; Duncalf D. J.; Lappert M. F. The First Example of a Crystalline Subvalent Organolanthanum Complex: [K([18]Crown-6)-(η2-C6H6)2][(LaCptt)2(μ-η6:η6-C6H6)]·2C6H6 (Cptt = η5-C5H3Bu2t-1,3). J. Am. Chem. Soc. 1998, 120 (49), 12958–12959. 10.1021/ja980377t. [DOI] [Google Scholar]
- Fieser M. E.; Ferrier M. G.; Su J.; Batista E.; Cary S. K.; Engle J. W.; Evans W. J.; Lezama Pacheco J. S.; Kozimor S. A.; Olson A. C.; Ryan A. J.; Stein B. W.; Wagner G. L.; Woen D. H.; Vitova T.; Yang P. Evaluating the Electronic Structure of Formal LnII Ions in LnII (C5H4SiMe3)31– Using XANES Spectroscopy and DFT Calculations. Chem. Sci. 2017, 8 (9), 6076–6091. 10.1039/C7SC00825B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotyk C. M.; Fieser M. E.; Palumbo C. T.; Ziller J. W.; Darago L. E.; Long J. R.; Furche F.; Evans W. J. Isolation Of +2 Rare Earth Metal Ions with Three Anionic Carbocyclic Rings: Bimetallic Bis (Cyclopentadienyl) Reduced Arene Complexes of La2+ and Ce2+ Are Four Electron Reductants. Chem. Sci. 2015, 6 (12), 7267–7273. 10.1039/C5SC02486B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentner T. X.; Rösch B.; Ballmann G.; Langer J.; Elsen H.; Harder S. Low Valent Magnesium Chemistry with a Super Bulky β-Diketiminate Ligand. Angew. Chem., Int. Ed. 2019, 58 (2), 607–611. 10.1002/anie.201812051. [DOI] [PubMed] [Google Scholar]
- Reinfandt N.; Michenfelder N.; Schoo C.; Yadav R.; Reichl S.; Konchenko S. N.; Unterreiner A. N.; Scheer M.; Roesky P. W. d/f-Polypnictides Derived by Non-Classical Ln2+ Compounds: Synthesis, Small Molecule Activation and Optical Properties. Chem. Eur. J. 2021, 27 (29), 7862–7871. 10.1002/chem.202100605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould C. A.; Marbey J.; Vieru V.; Marchiori D. A.; David Britt R.; Chibotaru L. F.; Hill S.; Long J. R. Isolation of a Triplet Benzene Dianion. Nat. Chem. 2021, 13 (10), 1001–1005. 10.1038/s41557-021-00737-8. [DOI] [PubMed] [Google Scholar]
- Palumbo C. T.; Darago L. E.; Dumas M. T.; Ziller J. W.; Long J. R.; Evans W. J. Structure, Magnetism, and Multi-Electron Reduction Reactivity of the Inverse Sandwich Reduced Arene La2+ Complex [{[C5H3(SiMe3)2]2La}2(μ-η6:η6-C6H6)]1–. Organometallics 2018, 37 (19), 3322–3331. 10.1021/acs.organomet.8b00523. [DOI] [Google Scholar]
- Huang W.; Le Roy J. J.; Khan S. I.; Ungur L.; Murugesu M.; Diaconescu P. L. Tetraanionic Biphenyl Lanthanide Complexes as Single-Molecule Magnets. Inorg. Chem. 2015, 54 (5), 2374–2382. 10.1021/ic5029788. [DOI] [PubMed] [Google Scholar]
- Huang W.; Dulong F.; Wu T.; Khan S. I.; Miller J. T.; Cantat T.; Diaconescu P. L. A Six-Carbon 10π-Electron Aromatic System Supported by Group 3 Metals. Nat. Commun. 2013, 4 (1), 1448 10.1038/ncomms2473. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang Y.; Liang J.; Tan B.; Deng C.; Huang W. Neutral Inverse-Sandwich Rare-Earth Metal Complexes of the Benzene Tetraanion. Chem. Sci. 2024, 15 (23), 8740–8749. 10.1039/D4SC02491E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur S. K.; Roig N.; Monreal-Corona R.; Langer J.; Alonso M.; Harder S. Similarities and Differences in Benzene Reduction with Ca, Sr, Yb and Sm: Strong Evidence for Tetra-Anionic Benzene. Angew. Chem., Int. Ed. 2024, 63 (25), e202405229 10.1002/anie.202405229. [DOI] [PubMed] [Google Scholar]
- Gould C. A.; McClain K. R.; Reta D.; Kragskow J. G. C.; Marchiori D. A.; Lachman E.; Choi E.-S.; Analytis J. G.; Britt R. D.; Chilton N. F.; Harvey B. G.; Long J. R. Ultrahard Magnetism from Mixed-Valence Dilanthanide Complexes with Metal-Metal Bonding. Science 2022, 375 (6577), 198–202. 10.1126/science.abl5470. [DOI] [PubMed] [Google Scholar]
- Fagin A. A.; Bochkarev M. N.; Kozimor S. A.; Ziller J. W.; Evans W. J. Comparative Reductive Reactivity of SmI2 with TmI2 in the Synthesis of Lanthanide Arene Complexes. Z. Anorg. Allg. Chem. 2005, 631 (13–14), 2848–2853. 10.1002/zaac.200500173. [DOI] [Google Scholar]
- Liu S.-S.; Yan B.; Meng Z.-S.; Gao C.; Wang B.-W.; Gao S. Two Half-Sandwich Organometallic Single-Ion Magnets with Toluene Coordinated to the Dy(III) Ion: The [(C7H8)Dy(AlCl4)3] and [(C7H8)Dy(AlBr4)3] Complexes. Inorg. Chem. Commun. 2017, 86, 312–314. 10.1016/j.inoche.2017.05.006. [DOI] [Google Scholar]
- Liu S.-S.; Ziller J. W.; Zhang Y.-Q.; Wang B.-W.; Evans W. J.; Gao S. A Half-Sandwich Organometallic Single-Ion Magnet with Hexamethylbenzene Coordinated to the Dy(III) Ion. Chem. Commun. 2014, 50 (77), 11418–11420. 10.1039/C4CC04262J. [DOI] [PubMed] [Google Scholar]
- Gould C. A.; McClain K. R.; Yu J. M.; Groshens T. J.; Furche F.; Harvey B. G.; Long J. R. Synthesis and Magnetism of Neutral, Linear Metallocene Complexes of Terbium(II) and Dysprosium(II). J. Am. Chem. Soc. 2019, 141 (33), 12967–12973. 10.1021/jacs.9b05816. [DOI] [PubMed] [Google Scholar]
- McClain K. R.; Gould C. A.; Marchiori D. A.; Kwon H.; Nguyen T. T.; Rosenkoetter K. E.; Kuzmina D.; Tuna F.; Britt R. D.; Long J. R.; Harvey B. G. Divalent Lanthanide Metallocene Complexes with a Linear Coordination Geometry and Pronounced 6s–5d Orbital Mixing. J. Am. Chem. Soc. 2022, 144 (48), 22193–22201. 10.1021/jacs.2c09880. [DOI] [PubMed] [Google Scholar]
- Randall McClain K.; Gould C. A.; Chakarawet K.; Teat S. J.; Groshens T. J.; Long J. R.; Harvey B. G. High-Temperature Magnetic Blocking and Magneto-Structural Correlations in a Series of Dysprosium(III) Metallocenium Single-Molecule Magnets. Chem. Sci. 2018, 9 (45), 8492–8503. 10.1039/C8SC03907K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom C. R.; Bruno I. J.; Lightfoot M. P.; Ward S. C. The Cambridge Structural Database. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016, 72 (2), 171–179. 10.1107/S2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woińska M.; Grabowsky S.; Dominiak P. M.; Woźniak K.; Jayatilaka D. Hydrogen Atoms Can Be Located Accurately and Precisely by X-Ray Crystallography. Sci. Adv. 2016, 2 (5), e1600192 10.1126/sciadv.1600192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C.; Liang J.; Deng C.; Lefèvre G.; Cantat T.; Diaconescu P. L.; Huang W. Arene-Bridged Dithorium Complexes: Inverse Sandwiches Supported by a δ Bonding Interaction. J. Am. Chem. Soc. 2020, 142 (51), 21292–21297. 10.1021/jacs.0c11215. [DOI] [PubMed] [Google Scholar]
- Macrae C. F.; Sovago I.; Cottrell S. J.; Galek P. T. A.; McCabe P.; Pidcock E.; Platings M.; Shields G. P.; Stevens J. S.; Towler M.; Wood P. A. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53 (1), 226–235. 10.1107/S1600576719014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T. Simple, Reliable, and Universal Metrics of Molecular Planarity. J. Mol. Model. 2021, 27 (9), 263 10.1007/s00894-021-04884-0. [DOI] [PubMed] [Google Scholar]
- Chilton N. F.; Anderson R. P.; Turner L. D.; Soncini A.; Murray K. S. PHI: A Powerful New Program for the Analysis of Anisotropic Monomeric and Exchange-Coupled Polynuclear d- and f-Block Complexes. J. Comput. Chem. 2013, 34 (13), 1164–1175. 10.1002/jcc.23234. [DOI] [PubMed] [Google Scholar]
- Falceto A.; Casanova D.; Alemany P.; Alvarez S. Distortions of π-Coordinated Arenes with Anionic Character. Chem. - Eur. J. 2014, 20 (45), 14674–14689. 10.1002/chem.201403889. [DOI] [PubMed] [Google Scholar]
- Demir S.; Jeon I.-R.; Long J. R.; Harris T. D. Radical Ligand-Containing Single-Molecule Magnets. Coord. Chem. Rev. 2015, 289–290, 149–176. 10.1016/j.ccr.2014.10.012. [DOI] [Google Scholar]
- Klamt A.; Schüürmann G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc., Perkin Trans. 1993, 2 (5), 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Vlaisavljevich B.; Diaconescu P. L.; Lukens W. L. Jr; Gagliardi L.; Cummins C. C. Investigations of the Electronic Structure of Arene-Bridged Diuranium Complexes. Organometallics 2013, 32 (5), 1341–1352. 10.1021/om3010367. [DOI] [Google Scholar]
- Eisenstein O.; Maron L. DFT Studies of Some Structures and Reactions of Lanthanides Complexes. J. Organomet. Chem. 2002, 647 (1), 190–197. 10.1016/S0022-328X(01)01407-3. [DOI] [Google Scholar]
- Franzke Y. J.; Holzer C.; Andersen J. H.; Begušić T.; Bruder F.; Coriani S.; Della Sala F.; Fabiano E.; Fedotov D. A.; Fürst S.; et al. Turbomole: Today and Tomorrow. J. Chem. Theory Comput. 2023, 19 (20), 6859–6890. 10.1021/acs.jctc.3c00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staroverov V. N.; Scuseria G. E.; Tao J.; Perdew J. P. Comparative Assessment of a New Nonempirical Density Functional: Molecules and Hydrogen-Bonded Complexes. J. Chem. Phys. 2003, 119 (23), 12129–12137. 10.1063/1.1626543. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7 (18), 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Gulde R.; Pollak P.; Weigend F. Error-Balanced Segmented Contracted Basis Sets of Double-ζ to Quadruple-ζ Valence Quality for the Lanthanides. J. Chem. Theory Comput. 2012, 8 (11), 4062–4068. 10.1021/ct300302u. [DOI] [PubMed] [Google Scholar]
- Dolg M.; Stoll H.; Preuss H. Energy-Adjusted Abinitio Pseudopotentials for the Rare Earth Elements. J. Chem. Phys. 1989, 90 (3), 1730–1734. 10.1063/1.456066. [DOI] [Google Scholar]
- Pollak P.; Weigend F. Segmented Contracted Error-Consistent Basis Sets of Double-and Triple-ζ Valence Quality for One-and Two-Component Relativistic All-Electron Calculations. J. Chem. Theory Comput. 2017, 13 (8), 3696–3705. 10.1021/acs.jctc.7b00593. [DOI] [PubMed] [Google Scholar]
- Hostaš J.; Pérez-Becerra K. O.; Calaminici P.; Barrios-Herrera L.; Lourenço M. P.; Tchagang A.; Salahub D. R.; Köster A. M. How Important Is the Amount of Exact Exchange for Spin-State Energy Ordering in DFT? Case Study of Molybdenum Carbide Cluster, Mo4C2. J. Chem. Phys. 2023, 159 (18), 184301 10.1063/5.0169409. [DOI] [PubMed] [Google Scholar]
- Rajabi A.; Grotjahn R.; Rappoport D.; Furche F. A DFT Perspective on Organometallic Lanthanide Chemistry. Dalton Trans. 2024, 53 (2), 410–417. 10.1039/D3DT03221C. [DOI] [PubMed] [Google Scholar]
- Mulliken R. S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23 (10), 1833–1840. 10.1063/1.1740588. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.