

Abstract
Adoptive cell therapy with genetically modified T cells holds the promise to improve outcomes for children with recurrent/refractory solid tumors, and has the potential to reduce treatment complication for all patients. While T cells expressing chimeric antigen receptors (CARs) specific for CD19 had remarkable success for B-cell derived malignancies leading to their FDA approval, CAR T cells were less effective for solid tumors and brain tumors. Lack of efficacy is most likely multifactorial, but heterogeneous antigen expressing, limited migration of T cells to tumor sites, and the immunosuppressive, hostile tumor microenvironment have emerged as major roadblocks that need to be addressed. In this review we summarize the clinical experience with CAR T-cell therapy for pediatric solid tumors including brain tumors. In addition, we will review strategies that have and are being developed to enhance their anti-tumor activity.
INTRODUCTION
Immunotherapy for pediatric malignancies holds the promise of improving outcome and reducing treatment-related complications. Among different forms of immunotherapy that are actively being pursued, the adoptive transfer of T cells expressing chimeric antigen receptors (CARs) has garnered significant excitement due to the success of CAR T-cell therapy for CD19-positive malignancies.1–11
CARs are synthetic molecules that combine the specificity of monoclonal antibodies (MAbs) with the effector function of T cells.12–14 The prototypic CAR consists of an antigen binding domain encoded by a single chain variable fragment (scFv) derived from a MAb, a hinge and transmembrane domain, and signaling domains derived from the CD3.ζ chain, and costimulatory molecules such as CD28 and/or 41BB (Fig 1A). The majority of CAR T-cell products are generated by viral transduction using replication incompetent retro- or lentiviral vectors (Fig 1B).
Figure 1: Scheme of CAR T-cell generation.
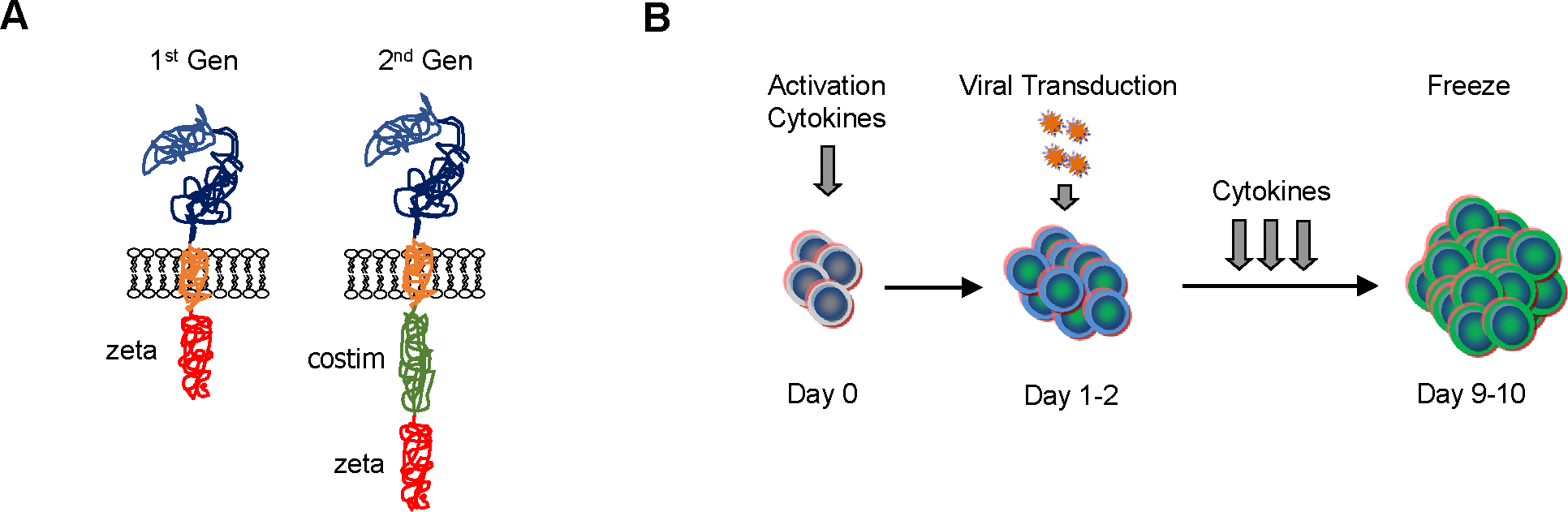
(A) CARs consist of an ectodomain, hinge and transmembrane domain, and endodomain. CARs have been designed with or without costimulatory (costim) signaling domains. As examples 1st and 2nd generation (Gen) CARs are shown. (B) CAR T cells are produced by activating T cells in the presence of cytokines. Once T-cell proliferate, they are transduced with viral vectors encoding CARs. CAR T cells are subsequently expanded with cytokines, and sufficient CAR T cells for preclinical or clinical studies are generated within 9 to 10 days of culture initiating.
Since submission of the first investigational new drug application (IND) for CD19-CAR T cells, the field as moved rapidly, culminating in the FDA approval of two CD19-CAR T-cell products in 2017.15 The interested reader is referred to recent reviews, which summarize the clinical results of CD19-CAR T cells in detail.1,2 Here, we will only highlight key findings, which need to be considered as we develop and improve current CAR T-cell therapy approaches for pediatric solid tumors.
First, lymphodepleting chemotherapy with cyclophosphamide and fludarabine is critical to allow engraftment and expansion of adoptively transferred CD19-CAR T cells. Second, CD19-CAR T cells can eradicate B-cell malignancies regardless of their underlying genetic alteration. Third, CD19-CAR T cells, can eradicate B-cell malignancies, which are refractory to chemotherapy and/or radiation, highlighting that T cells kill their target cells through different cytotoxic mechanism than conventional therapies. Fourth, CD19-CARs have to encode a costimulatory signaling domain to be effective. While CD19-CAR.CD28.ζ and CD19-CAR.41BB.ζ T cells have not been compared directly in a single clinical study, results of published studies indicate that CD19-CAR.41BB.ζ T cells persist longer.1–11 However, at present it is unclear if this translates into improved antitumor activity.1–11 Fifth, targeting a single antigen, can result in antigen loss variants, and lastly, CD19-CAR T-cell therapy can be associated with significant clinical side effects including cytokine release syndrome (CRS), and neurotoxicity.1–11
In this educational review, results of early phase clinical studies with CAR T cells for pediatric solid tumors and brain tumors are summarized (Table 1). In addition, strategies how to improve their antitumor activity, and current challenges are discussed.
Table 1:
Selected completed and future CAR T-cell therapy studies for pediatric solid tumors or brain tumors
| Disease | Target | CAR signaling domain | Vector | Cell type & delivery | Lymph depl | Comment, NCT #, or ref if published |
|---|---|---|---|---|---|---|
| Neuroblastoma | CD171 | ζ | pl | ATC, iv | − | 16 |
| CD171 | 41BB.ζ or CD28.41BB.ζ | LV | ATC, iv | + | NCT02311621 | |
| GD2 | ζ | RV | VST or ATC, iv | − | 17,18 | |
| GD2 | CD28.Ox40.ζ | RV | ATC, iv | +/− | pembrolizumab, 19 | |
| GD2 | ζ, ND | RV | ATC, iv | + | NCT03373097 | |
| GD2 | ζ, ND | RV | ATC, iv | +/− | NCT02761915 | |
| GD2 | ζ, ND | RV | iNKT, iv | + | 2nd transgene: IL15, NCT03294954 | |
| Neuroblastoma & sarcoma | GD2 | CD28.OX40.ζ | RV | ATC, iv | + | NCT02107963 |
| Sarcoma | HER2 | CD28.ζ | RV | ATC, iv | +/− | 21,22 |
| GD2 | CD28.OX40.ζ | RV | VST, iv | +/− | VZV Vaccine, NCT02932956 | |
| Hepatoblastoma & sarcoma | GPC3 | 41BB.ζ | RV | ATC, iv | + | NCT02932956 |
| Brain tumors | HER2 | CD28.ζ | RV | VST, iv | − | 24 |
| IL13Rα2 | 41BB.ζ | LV | ATC, ic | − | NCT02208362, 26 |
pl: plasmid, LV: lentivirus, RV: retrovirus, ATC: activated T cells, VST: virus-specific T cells, iNKT: invariant NK T cells, iv: intravenous, ic: intracranial, ND: not disclosed
CLINICAL STUDIES WITH CAR T CELLS FOR PEDIATRIC BRAIN AND SOLID TUMORS
Neuroblastoma
Two clinical studies have been conducted with first generation CAR T cells in neuroblastoma patients. In the first study, patients received up to 109/m2 CD8-positive CD171-specific CAR T-cell clones.16 Adoptive transfer of T cells was well tolerated, but T cells persisted only for 6 weeks, and only 1 out of 6 patients had a partial response. One strategy to improve the persistence of adoptive transferred T cells takes advantage of the specificity of the endogenous αβTCR expressed by CAR T cells. For example, virus-specific T cells receive appropriate stimulation by viral antigens presented by professional antigen-presenting cells. This concept was explored in the second clinical study, in which investigators expressed a 1st generation GD2-specific CAR in polyclonal Epstein Barr virus (EBV)-specific T cells. The in vivo fate of GD2-CAR EBV-specific T cells was directly compared to activated T cells expressing the same CAR in individual patienst.17,18 While GD2-CAR EBV-specific T cells did not expand, they persisted longer than GD2-CAR activated T cells. Five out of 11 patients with active disease showed tumor responses or necrosis. Three of them had complete responses.
More recently, activated T cells expressing a third generation GD2-CAR with a CD28.OX40.ζ endodomain were evaluated in neuroblastoma patients.19 Three cohorts of patients were infused. The first cohort (4 patients) only received CAR T cells, the second cohort (4 patients) received lymphodepleting chemotherapy (cyclophosphamide and fludarabine) prior to CAR T-cell infusion, and the third cohort (3 patients) also received 2 doses of pembrolizumab given day-1 and day+21 in relationship to the CAR T-cell infusion. Lymphodepleting chemotherapy induced expansion of CAR T cells for up to 3 logs, which peaked at one to two weeks post infusion. Addition of pembrolizumab did not further improve T-cell expansion or persistence. There was a significant increase in the frequency of circulating myeloid cells in the peripheral blood with an immunosuppressive M2 phenotype post CAR T-cell infusion in all three cohorts. Further studies are needed to investigate the significance of this finding.
In addition to these published studies, several clinical trials are in progress including one study evaluating 2nd generation (41BB.ζ) and 3rd generation (CD28.41BB.ζ) CAR T cells targeting CD17120 in patients with neuroblastoma and ganglioneuroblastoma (NCT02311621). In addition, studies continue to explore GD2-CAR T cells for neuroblastoma including NCT02107963, NCT02761915, and NCT03373097.
Sarcoma
T cells expressing 2nd generation HER2-CARs (CD28.ζ) have been evaluated in 19 patients with refractory HER2-positive sarcoma (16 osteosarcoma, 1 Ewing sarcoma, 1 primitive neuroectodermal tumor, 1 desmoplastic small round cell tumor). HER2-CAR T-cell infusions were well tolerated with no dose limiting toxicity.21 HER2-CAR T cells persisted for at least 6 weeks in patients who received greater than 1×106/m2 HER2-CAR T cells, and were detected at tumor sites. Of 17 evaluable patients 4 had stable disease. Three of these had their tumor removed with one showing ≥90% necrosis. Subsequently, six patients received lymphodepleting chemotherapy prior to HER2-CAR T-cell infusion. Significant HER2-CAR T-cell expansion was observed without apparent toxicity, and 1 patient with refractory rhabdomyosarcoma (RMS), who only had persistent bone marrow disease, achieved a complete response for greater than 12 months.22 In addition to HER2-CAR T cells, GD2-CAR T cells are actively being explored (NCT01953900, NCT02107963) for sarcoma. In one study a third generation GD2-CAR with a CD28.OX40.ζ endodomain is expressed in varicella zoster virus (VZV)-specific T cells, and the investigators are evaluating if GD2-CAR VZV-specific T cells can be boosted with an FDA-approved VZV vaccine post infusion. Lastly, one clinical study (NCT02932956) is FDA-approved to evaluate the safety and efficacy of GPC3-CAR T cells23 in pediatric solid tumor patients including RMS. However, the study is currently not open for accrual.
Brain tumors
Clinical studies with CAR T cells targeting HER2, EGFRvIII, and IL13Rα2 have been conducted in patients with high-grade glioma.24–27 Two studies only infused adults, whereas 10 out of 17 patients were children on the HER2-CAR T-cell therapy study. T cells were either given intravenously (HER2, EGFRvIII) or directly injected into the tumor and/or ventricle (IL13Rα2). Similar to the clinical results of CAR T-cell therapy studies for solid tumors, the majority of brain tumor patients had progressive disease. However, responses were observed including one partial,24 one complete,26 and several patients had stable disease for a prolonged period of time.24
Detailed correlative studies performed post infusion of EGFRvIII-CAR T cells revealed that T cells were able to migrate to glioma sites after intravenous infusion.25 Target antigen expression was reduced in resected gliomas indicative of an ‘on target’ CAR T-cell effect. In addition, gliomas upregulated the expression of immunosuppressive molecules including indoleamine 2,3 dioxygenase (IDO) and IL-10, highlighting that gliomas have the ability to counteract infiltrating ‘pro-inflammatory’ CAR T cells. Currently, only one clinical study (NCT02208362) is actively recruiting patients with gliomas including children greater than 12 years of age. This study is evaluating the safety and efficacy of intracranial injections of IL13Rα2-CAR.41BB.ζ T cells.
In conclusion, the initial foray into the clinic with CAR T cells has demonstrated their safety for pediatric solid tumors and brain tumors. However, only subsets of patients have benefited from this approach so far. Potential strategies to increase the efficacy of CAR T cells are reviewed in the next section.
STRATEGIES TO IMPROVE CAR T-CELL THERAPY FOR SOLID TUMORS
Lack of CAR T-cell efficacy is most likely multifactorial. Major roadblocks include the i) availability of targeted antigens and their heterogeneous expression, ii) homing of T cells to tumor cells, and iii) the immunosuppressive tumor microenvironment (Fig 2).
Figure 2: Roadblocks of CAR T cells for solid tumors.
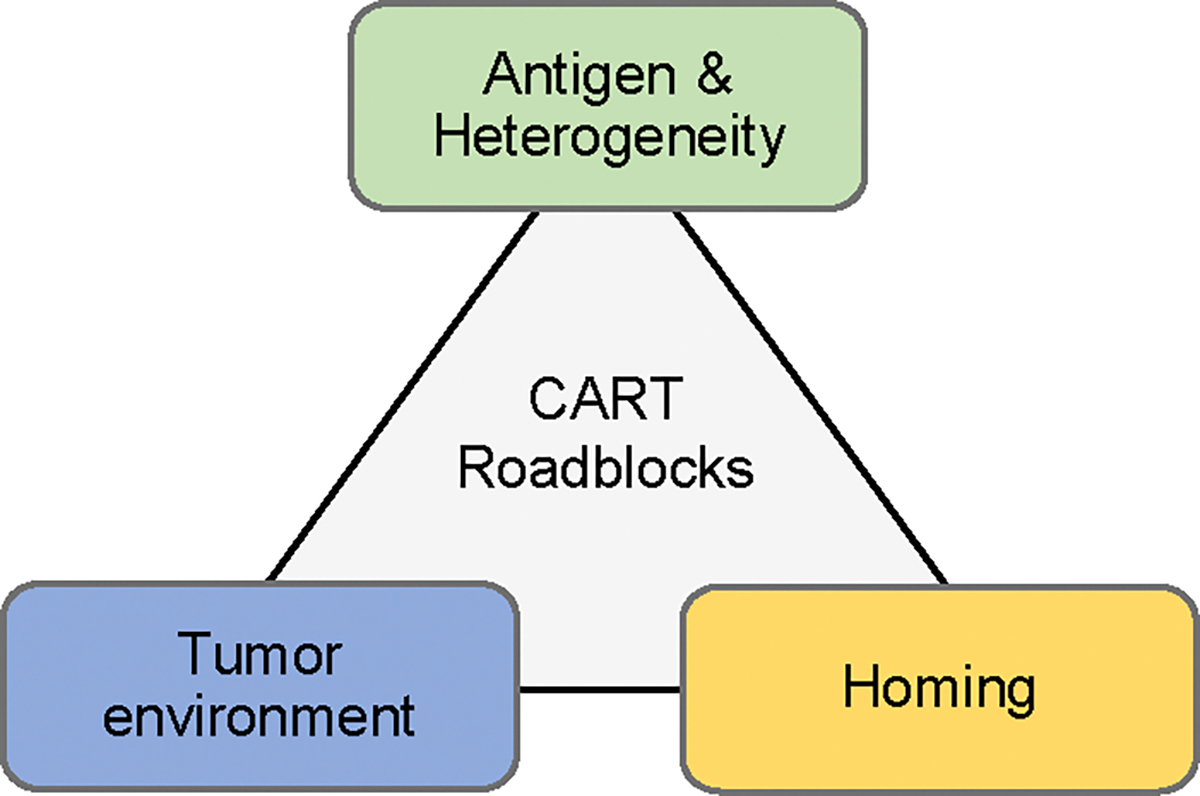
For detail see text.
Expanding the repertoire of targetable antigens
The majority of CARs developed so far recognize cell surface proteins, which were originally discovered as targets for MAbs (GD2, HER2, GPC3, and IL13Rα2). Currently, efforts are underway using gene expression array data and proteomics to identify new targetable cell surface antigens.28 The recent identification of GPC2, which is expressed at high levels on neuroblastoma, highlights the feasibility of this approach.29 However, it might be difficult to discover antigens that are not expressed at low levels in normal tissues. While private neoantigens are present albeit at low frequency in pediatric tumors,30 directly targeting these with CAR T cells is not feasible with current technology using viral vectors to generate CAR T cells. Besides the significant regulatory burden and cost, it currently takes more than 6 months to generate a clinical grade viral vector. However, optimizing CAR T cells to efficiently induce immune responses against non-targeted antigen (aka antigen spreading) would be one approach to target private neoantigens, and potentially increase the antitumor activity of CAR T cells similar to cancer vaccines.31
CAR T cells are being developed to recognize antigen patterns. Example include designing CARs that recognize two antigens or engineering T cells that express multiple CARs.32,33 T cells expressing these CARs only get fully activated in the presence of all targeted antigens. In addition, targeting multiple antigens should also offset the risk of selecting antigen loss variants. In addition, so called inhibitory chimeric antigen receptors have been developed that block potential off target T-cell responses.34 Developing CARs that recognize peptides in the context of major histocompatibility complex (MHC) class I molecules might also increase the potential repertoire of targetable antigens since 2/3 of all expressed proteins reside within the cell. This can be achieved by using a scFv derived from a T-cell receptor (TCR) mimic MAb as a CAR antigen binding domain. Examples include CARs specific for a HLA-A2-restricted peptide derived from intracellular proteins such as WT1 or Proteinase 3.35,36
Lastly, inducible expression system may also provide a potential solution to the ‘antigen dilemma’. So called synthetic notch (synNotch) signaling receptors allow the expression of CARs only once a T-cell has migrated to tumor sites, potentially enabling the targeting of antigens that are expressed in normal tissues.37,38
Enhancing migration of CAR T-cell to tumor sites and within tumors
Several preclinical studies have highlighted that there is a mismatch between chemokine secreted by solid tumors and chemokine receptors expressed by CAR T cells. Transgenic expression of chemokine receptors has been shown to overcome this roadblock.39 For example, neuroblastoma secretes high levels of CCL2, however CAR T cells lack expression of the corresponding chemokine receptor (CCR2).40 Transgenic expression of CCR2b on GD2-CAR T cells resulted in enhanced homing and antitumor activity in preclinical neuroblastoma models.40 Besides migration to tumor sites, limited migration within tumors might also contribute to the reduced antitumor activity observed in clinical studies. For example, CAR T cells are limited in their ability to degrade the extracellular matrix (ECM), resulting in poor tumor penetration. This can be improved by expressing heparanase.41 Another approach consists by directly targeting cancer associated fibroblasts (CAFs) with CAR T cells, the main producer of collagen within tumors.42
Engineering CAR T cells to resist the immunosuppressive tumor environment
Brain and solid tumors create a hostile tumor environment, which favors T-cell exhaustion and/or dysfunction induced by i) immunosuppressive cytokines (e.g. IL4, IL10, TGFβ), ii) expression of inhibitory molecules (e.g. FAS ligand, PD-L1), iii) the metabolic environment, and/or iv) recruitment of immunosuppressive cells including myeloid derived suppressor cells (MDSCs), cancer associated fibroblasts (CAFs), and/or regulatory T cells (Tregs).43–45
While this section is focused on engineering CAR T cells to improve their antitumor activity in the tumor microenvironment, combinatorial therapies are attractive approaches. For example, combining CAR T-cell therapy with checkpoint blockade, oncolytic viruses, chemotherapy, radiation and/or small molecules are actively being explored in preclinical studies with encouraging results.46–48 Genetic engineering approaches to enhance the antitumor activity of CAR T cells can be divided into two broad categories: i) transgenic expression of immune stimulatory molecules, and ii) silencing negative regulators.
Several preclinical studies have shown that transgenic expression of cytokines (e.g. IL12, IL15, IL18),49–51 constitutive cytokine receptors,52 41BBL,53 or CD40L54 enhance the antitumor activity of CAR T cells. One recent study has also demonstrated that transgenic expression of IL7 in combination with the chemokine CCL19 not only enhances the effector function of CAR T cells, but also enables them to induce endogenous T-cell responses against the targeted tumor indicative of antigen spreading.55
Directly blocking inhibitory cytokines or converting their signal into a T-cell stimulatory signal are other approaches that are actively being pursued. For example, expressing a dominant negative TGFβ receptor (DNR) renders T cells resistant to TGFβ in preclinical as well as clinical studies.56 In addition, DNRs have been developed to provide intrinsic protection from PD1/PD-L1 checkpoint blockade.57 Chimeric cytokine or switch receptors not only block an inhibitory signal, but convert it into a T-cell stimulatory signal. Examples include receptors that consists of the ectodomain of the IL4 receptor and the transmembrane and intracellular signaling domain of the IL2 or IL7 receptor.58,59 While siRNA approaches were initially used to silence negative regulators, more recent studies have focused on gene editing technologies such as TALENs and CRIPSR/Cas9. Pertinent examples include the silencing of FAS ligand or the knockout of PD-1 in T cells.60,61
As we enhance the effector function of CAR T cells it is advisable to insert safety switches that can be activated if side effects develop. Safety switches that have been tested clinically include the herpes simplex virus thymidine kinase (HSV-tk), which enables cell killing in the presence of ganciclovir,62 or an inducible caspase 9 (iC9),63 which can be activated by a chemical inducer of dimerization (CID). In addition, expression of cell surface molecules (EGFR, CD20),64,65 which can be targeted with FDA-approved MAbs are other suicide gene options, which have been successfully evaluated in preclinical models.
CURRENT CHALLENGES
CAR T-cell generation
The majority of clinical studies have used retroviral or lentiviral vectors to generate clinical grade CAR T-cell products. Generating clinical grade viral vectors is time consuming, costly, and their use is associated with a significant regulatory burden. In this regard reducing some of the required testing of CAR T-cell products, as recently advocated,66,67 would be a step in the right detection. These issues could be overcome with the use of non-viral DNA delivery systems that have been successfully used to generate CAR T cells.68–70 Lastly, closed cell manufacturing systems71,72 or the use of ‘off the shelf’ CAR T-cell products73 hold the promise to streamline CAR T-cell production and/or distribution.
What is the optimal T-cell subset to generate CAR T cells?
Several studies have highlighted that CAR T cells generated from central memory T cells with a defined CD4:CD8 ratio have superior effector function in comparison to CAR T cells generated from bulk T cells in preclinical models.74,75 Epigenetic profiling of T cells has provided novel insight,76 and holds the promise to further advance our ability to select the most potent T-cell subset for CAR T-cell generation. Lastly, γδ T cells and invariant natural killer T (iNKT) cells are also actively being explored as T-cell platforms for CAR T-cell therapy.77,78 In this regard a clinical study with iNKT expressing GD2-CARs and IL15 for neuroblastoma patient is FDA approved (NCT03294954), but not actively accruing patients.
Need for preclinical testing in range of animal models
The majority of preclinical studies have relied on xenograft models, which do not reliably recapitulate the complex tumor microenvironment. Immune competent animal models have been adapted for CAR T-cell therapies and these models will be invaluable as combinatorial therapies are being developed.79–81 In addition patient-derived xenograft (pdx) models should enable preclinical testing of CAR T cells against a panel of human tumors that more closely mimic patient tumors than tumor cell lines that have been propagated in vitro. Lastly, large animal models hold the promise to evaluate CAR T cells in spontaneous tumor models.82
Correlative studies, in vivo tracking of infused T cells, and clinical response criteria
There is currently only one published CAR T-cell therapy (see Brain tumor section) that has systematically studied tumor biopsies post infusion.25 These type of studies will be critical to understand current therapeutic failure and devise evidence-based approaches to overcome them. In addition, our ability to track infused CAR T cells in patients is limited unless they are genetically modified with a reporter gene such HSV-tk.83 Even being able to routinely rack CAR T cells for 48 to 72 hours post infusion would be a major advance, giving us invaluable insight into their initial biodistribution and ability to migrate to tumor sites. While diagnostic imaging immune response criteria have been implemented for the assessment of immunotherapies for solid tumors and brain tumors,84,85 these were developed in the ‘cancer vaccine era’ and might require further fine tuning for cell-based immunotherapies including CAR T cells.
CONCLUSIONS
The initial foray of CAR T cells into the clinic for pediatric solid tumor and brain tumors demonstrated their safety, but also has highlighted their limited antitumor activity. Additional genetic modification of CAR T cells has greatly enhanced their anti-tumor activity in preclinical studies, and we are hopeful that some of the devised strategies will translate into improved anti-tumor activity in humans. To advance the field there is an urgent need to discover novel antigens that can be targeted with CAR T cells, and to improve our ability to evaluate CAR T-cell therapies in preclinical models. Lastly, being able to track CAR T cells noninvasively and preform detailed studies on patients enrolled on CAR T-cell therapy should enable us to advance the field. We remain hopeful that within the next 5 to 10 years solid tumor patients will benefit from CAR T-cell therapies to the same degree as patients with B-cell derived malignancies today.
PRACTICAL APPLICATIONS BULLET POINTS.
CAR T cells for pediatric solid tumors are safe, but have limited antitumor activity in early phase clinical studies
Carefully designed correlative studies will be critical to understand current failures of CAR T-cell therapies and devise strategies to improve them
Additional genetic modification have improved the antitumor activity of CAR T cells in preclinical models, but ‘improved’ CAR T cells have not been tested in the clinic
Costs and regulatory requirements associated with clinical testing of CAR T cells have the potential to impede progress
ACKNOWLEDGEMENTS
This work was supported by NIH grants 5T32HL092332 and 1R01CA173750, and CRPIT grant RP101335.
REFERENCES
- 1.Ruella M, June CH: Chimeric Antigen Receptor T cells for B Cell Neoplasms: Choose the Right CAR for You. Curr Hematol Malig Rep 11:368–84, 2016 [DOI] [PubMed] [Google Scholar]
- 2.Park JH, Geyer MB, Brentjens RJ: CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood 127:3312–20, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kochenderfer JN, Dudley ME, Feldman SA, et al. : B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119:2709–2720, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kochenderfer JN, Dudley ME, Kassim SH, et al. : Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 33:540–9, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, Frey N, Shaw PA, et al. : Chimeric antigen receptor T cells for sustained remissions in leukemia. N.Engl.J.Med. 371:1507–1517, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. : T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385:517–28, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turtle CJ, Hanafi LA, Berger C, et al. : Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 8:355ra116, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turtle CJ, Riddell SR, Maloney DG: CD19-Targeted chimeric antigen receptor-modified T-cell immunotherapy for B-cell malignancies. Clin Pharmacol Ther 100:252–8, 2016 [DOI] [PubMed] [Google Scholar]
- 9.Gardner RA, Finney O, Annesley C, et al. : Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 129:3322–3331, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brentjens RJ, Davila ML, Riviere I, et al. : CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci.Transl.Med. 5:177ra38, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park JH, Riviere I, Gonen M, et al. : Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med 378:449–459, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eshhar Z, Waks T, Gross G, et al. : Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc.Natl.Acad.Sci.U.S.A 90:720–724, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dotti G, Gottschalk S, Savoldo B, et al. : Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 257:107–126, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sadelain M: Chimeric antigen receptors: driving immunology towards synthetic biology. Curr Opin Immunol 41:68–76, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadelain M: CD19 CAR T Cells. Cell 171:1471, 2017 [DOI] [PubMed] [Google Scholar]
- 16.Park JR, Digiusto DL, Slovak M, et al. : Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol.Ther. 15:825–833, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Pule MA, Savoldo B, Myers GD, et al. : Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14:1264–1270, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louis CU, Savoldo B, Dotti G, et al. : Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118:6050–6056, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heczey A, Louis CU, Savoldo B, et al. : CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol Ther 25:2214–2224, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunkele A, Taraseviciute A, Finn LS, et al. : Preclinical Assessment of CD171-Directed CAR T-cell Adoptive Therapy for Childhood Neuroblastoma: CE7 Epitope Target Safety and Product Manufacturing Feasibility. Clin Cancer Res 23:466–477, 2017 [DOI] [PubMed] [Google Scholar]
- 21.Ahmed N, Brawley VS, Hegde M, et al. : Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol 33:1688–96, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hegde M, DeRenzo CC, Zhang H, et al. : Expansion of HER2-CAR T cells after lymphodepletion and clinical responses in patients with advanced sarcoma. Journal of Clinical Oncology 35:10508–10508, 2017 [Google Scholar]
- 23.Li W, Guo L, Rathi P, et al. : Redirecting T Cells to Glypican-3 with 4–1BB Zeta Chimeric Antigen Receptors Results in Th1 Polarization and Potent Antitumor Activity. Hum Gene Ther 28:437–448, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmed N, Brawley V, Hegde M, et al. : HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol 3:1094–1101, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Rourke DM, Nasrallah MP, Desai A, et al. : A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 9, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown CE, Alizadeh D, Starr R, et al. : Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med 375:2561–9, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown CE, Badie B, Barish ME, et al. : Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin Cancer Res 21:4062–72, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orentas RJ, Yang JJ, Wen X, et al. : Identification of cell surface proteins as potential immunotherapy targets in 12 pediatric cancers. Front.Oncol. 2:194, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bosse KR, Raman P, Zhu Z, et al. : Identification of GPC2 as an Oncoprotein and Candidate Immunotherapeutic Target in High-Risk Neuroblastoma. Cancer Cell 32:295–309 e12, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grobner SN, Worst BC, Weischenfeldt J, et al. : The landscape of genomic alterations across childhood cancers. Nature, 2018 [DOI] [PubMed] [Google Scholar]
- 31.Butterfield LH, Ribas A, Dissette VB, et al. : Determinant spreading associated with clinical response in dendritic cell-based immunotherapy for malignant melanoma. Clin.Cancer Res. 9:998–1008, 2003 [PubMed] [Google Scholar]
- 32.Hegde M, Mukherjee M, Grada Z, et al. : Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest 126:3036–52, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bielamowicz K, Fousek K, Byrd TT, et al. : Trivalent CAR T-cells Overcome Interpatient Antigenic Variability in Glioblastoma. Neuro Oncol, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fedorov VD, Themeli M, Sadelain M: PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci.Transl.Med. 5:215ra172, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rafiq S, Purdon TJ, Daniyan AF, et al. : Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia 31:1788–1797, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma Q, Garber HR, Lu S, et al. : A novel TCR-like CAR with specificity for PR1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy 18:985–94, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roybal KT, Lim WA: Synthetic Immunology: Hacking Immune Cells to Expand Their Therapeutic Capabilities. Annu Rev Immunol 35:229–253, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morsut L, Roybal KT, Xiong X, et al. : Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 164:780–91, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kershaw MH, Wang G, Westwood JA, et al. : Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum.Gene Ther. 13:1971–1980, 2002 [DOI] [PubMed] [Google Scholar]
- 40.Craddock JA, Lu A, Bear A, et al. : Enhanced Tumor Trafficking of GD2 Chimeric Antigen Receptor T Cells by Expression of the Chemokine Receptor CCR2b. J Immunother, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caruana I, Savoldo B, Hoyos V, et al. : Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med 21:524–9, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kakarla S, Chow KK, Mata M, et al. : Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther 21:1611–1620, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kesarwani P, Kant S, Prabhu A, et al. : The interplay between metabolic remodeling and immune regulation in glioblastoma. Neuro Oncol 19:1308–1315, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamran N, Chandran M, Lowenstein PR, et al. : Immature myeloid cells in the tumor microenvironment: Implications for immunotherapy. Clin Immunol, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou W: Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat.Rev.Cancer 5:263–274, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Nishio N, Diaconu I, Liu H, et al. : Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 74:5195–5205, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanoue K, Rosewell Shaw A, Watanabe N, et al. : Armed Oncolytic Adenovirus-Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res 77:2040–2051, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.John LB, Devaud C, Duong CP, et al. : Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin.Cancer Res. 19:5636–5646, 2013 [DOI] [PubMed] [Google Scholar]
- 49.Krenciute G, Prinzing BL, Yi Z, et al. : Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Ralpha2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol Res 5:571–581, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koneru M, Purdon TJ, Spriggs D, et al. : IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 4:e994446, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu B, Ren J, Luo Y, et al. : Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep 20:3025–3033, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shum T, Omer B, Tashiro H, et al. : Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov 7:1238–1247, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao Z, Condomines M, van der Stegen SJC, et al. : Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 28:415–428, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Curran KJ, Seinstra BA, Nikhamin Y, et al. : Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther 23:769–78, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adachi K, Kano Y, Nagai T, et al. : IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol, 2018 [DOI] [PubMed] [Google Scholar]
- 56.Bollard CM, Tripic T, Cruz CR, et al. : Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients With Relapsed Hodgkin Lymphoma. J Clin Oncol:JCO2017743179, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cherkassky L, Morello A, Villena-Vargas J, et al. : Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 126:3130–44, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Levy R: A perspective on monoclonal antibody therapy: where we have been and where we are going. Semin.Hematol.2000.Oct.;37.(4.Suppl.7.):43.−6. 37:43–46 [DOI] [PubMed] [Google Scholar]
- 59.Leen AM, Sukumaran S, Watanabe N, et al. : Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol.Ther. 22:1211–1220, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dotti G, Savoldo B, Pule M, et al. : Human cytotoxic T lymphocytes with reduced sensitivity to Fas-induced apoptosis. Blood 105:4677–4684, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rupp LJ, Schumann K, Roybal KT, et al. : CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep 7:737, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lupo-Stanghellini MT, Provasi E, Bondanza A, et al. : Clinical impact of suicide gene therapy in allogeneic hematopoietic stem cell transplantation. Hum.Gene Ther. 21:241–250, 2010 [DOI] [PubMed] [Google Scholar]
- 63.Di Stasi A, Tey SK, Dotti G, et al. : Inducible apoptosis as a safety switch for adoptive cell therapy. N.Engl.J.Med. 365:1673–1683, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vogler I, Newrzela S, Hartmann S, et al. : An improved bicistronic CD20/tCD34 vector for efficient purification and in vivo depletion of gene-modified T cells for adoptive immunotherapy. Mol.Ther. 18:1330–1338, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang X, Chang WC, Wong CW, et al. : A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 118:1255–1263, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cornetta K, Duffy L, Turtle CJ, et al. : Absence of Replication-Competent Lentivirus in the Clinic: Analysis of Infused T Cell Products. Mol Ther 26:280–288, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heslop HE, Brenner MK: Seek and You Will Not Find: Ending the Hunt for Replication-Competent Retroviruses during Human Gene Therapy. Mol Ther 26:1–2, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kebriaei P, Singh H, Huls MH, et al. : Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest 126:3363–76, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manuri PV, Wilson MH, Maiti SN, et al. : piggyBac transposon/transposase system to generate CD19-specific T cells for the treatment of B-lineage malignancies. Hum.Gene Ther. 21:427–437, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakazawa Y, Huye LE, Salsman VS, et al. : PiggyBac-mediated Cancer Immunotherapy Using EBV-specific Cytotoxic T-cells Expressing HER2-specific Chimeric Antigen Receptor. Mol.Ther. 19:2133–2143, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu F, Shah N, Xu H, et al. : Closed-system manufacturing of CD19 and dual-targeted CD20/19 chimeric antigen receptor T cells using the CliniMACS Prodigy device at an academic medical center. Cytotherapy, 2017 [DOI] [PubMed] [Google Scholar]
- 72.Lock D, Mockel-Tenbrinck N, Drechsel K, et al. : Automated Manufacturing of Potent CD20-Directed Chimeric Antigen Receptor T Cells for Clinical Use. Hum Gene Ther 28:914–925, 2017 [DOI] [PubMed] [Google Scholar]
- 73.Poirot L, Philip B, Schiffer-Mannioui C, et al. : Multiplex Genome Editing of TCRa/CD52 Genes as a Platform for “Off the Shelf” Adoptive T-Cell Immunotherapies. Mol Ther 22:S201–S202, 2014 [Google Scholar]
- 74.Greenberg P, Klarnet J, Kern D, et al. : Requirements for T cell recognition and elimination of retrovirally- transformed cells. Princess Takamatsu Symp. 19:287–301, 1988 [PubMed] [Google Scholar]
- 75.Sommermeyer D, Hudecek M, Kosasih PL, et al. : Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30:492–500, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abdelsamed HA, Moustaki A, Fan Y, et al. : Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med 214:1593–1606, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heczey A, Liu D, Tian G, et al. : Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Capsomidis A, Benthall G, Van Acker HH, et al. : Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol Ther 26:354–365, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnson LA, Sanchez-Perez L, Suryadevara CM, et al. : Chimeric antigen receptor engineered T cells can eliminate brain tumors and initiate long-term protection against recurrence. Oncoimmunology 3:e944059, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barber A, Rynda A, Sentman CL: Chimeric NKG2D expressing T cells eliminate immunosuppression and activate immunity within the ovarian tumor microenvironment. J Immunol 183:6939–47, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pituch KC, Miska J, Krenciute G, et al. : Adoptive Transfer of IL13Ralpha2-Specific Chimeric Antigen Receptor T Cells Creates a Pro-inflammatory Environment in Glioblastoma. Mol Ther, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Panjwani MK, Smith JB, Schutsky K, et al. : Feasibility and Safety of RNA-transfected CD20-specific Chimeric Antigen Receptor T Cells in Dogs with Spontaneous B Cell Lymphoma. Mol Ther 24:1602–14, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Keu KV, Witney TH, Yaghoubi S, et al. : Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci Transl Med 9, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Okada H, Weller M, Huang R, et al. : Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol 16:e534–42, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wolchok JD, Hoos A, O’Day S, et al. : Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin.Cancer Res. 15:7412–7420, 2009 [DOI] [PubMed] [Google Scholar]