

Abstract
Clinical treatment of hepatocellular carcinoma (HCC) with 5-fluorouracil (5-FU), the primary anticancer agent, remains unsatisfactory due to the glutathione (GSH)-associated drug resistance and immunosuppressive microenvironment of HCC. To develop a facile yet robust strategy to overcome 5-FU resistance for enhanced immunotherapy treatment of HCC via all dimensional GSH exhaustion, we report in this study construction of a minimalist prodrug consisting of 5-FU linked to an indoleamine-(2,3)-dioxygenase (IDO) inhibitor (IND) via a disulfide bridge, FU-SS-IND that can further self-assemble into stabilized nanoparticles, FU-SS-IND NPs. Specifically, besides the disulfide linker-induced GSH exhaustion, IND inhibits GSH biosynthesis and enhances the effector function of T cells for turning a “cold” tumor to a “hot” one, which synergistically achieving a tumor inhibition rate (TIR) of 92.5% in a 5-FU resistant mice model. Most importantly, FU-SS-IND NPs could upregulate programmed death ligand 1 (PD-L1) expression on the surface of tumor cells, which enables facile combination with immune checkpoint blockade (ICB) for a ultimate prolonged survival lifetime of 5-FU-resistant tumors-bearing mice. Overall, the minimalist bioreducible nano-prodrug developed herein demonstrates great translatable potential for efficiently reversing drug resistance and enhancing immunotherapy of HCC.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12951-024-03027-w.
Keywords: 5-FU, IND, Nano-prodrug, Drug resistance, GSH exhaustion, PD-L1
Introduction
Chemotherapy remains the preferred clinical treatment strategy for hepatocellular carcinoma (HCC) [1, 2]. In addition to the general drawbacks of chemotherapeutic drugs such as rapid clearance, compromised targeting efficiency and systemic toxicity [1, 2], antimetabolite 5-fluorouracil (5-FU), a frontline anti-HCC drug suffered from significantly compromised therapeutic efficiency most likely attributed to its the drug resistance and complicated immunosuppressive microenvironment of HCC [3]. Therefore, the primary intent of this work is to devise a simplified and reliable approach to intensify treatment efficiency of 5-FU against HCC, which can address the Glutathione (GSH)-associated drug resistance of HCC and the immunosuppressive effect of 5-FU.
GSH-related detoxification and antioxidant defense system has been repeatedly highlighted to be a main mechanism that accounts substantially for the 5-FU treatment failure [4]. Specifically, GSH overexpression at the tumor site compromises the reactive oxygen species (ROS)-induced antitumor efficiency via direct ROS scavenging [5]. Tremendous progresses have been devoted to targeting the intracellular GSH pathway [6, 7], including construction of GSH-scavenging nanoformulation [8, 9] and development of GSH-responsive prodrugs [10, 11]. Besides the inherent high GSH level, tumor cells exert a tricky compensatory effect on intracellular GSH biosynthesis, which makes it impossible to exhaust the intracellular GSH completely for significantly compromised therapeutic efficiency of the aforementioned GSH pathway targeting strategies [12]. Therefore, an ideal means is to integrate intracellular GSH depletion and GSH biosynthesis inhibition for all dimensional GSH exhaustion. Combining the two functions in one nanoformulation exhibits more excellent performance compared to the use of a single treatment strategy for drug resistance problems [13]. A notable example of this state-of-the-art nanomedicine design is Qu’s highly potent GSH exhaustion via consuming the stored GSH and preventing SLC7A11 (a cystine/glutamate antiporter) to block the cysteine needed for GSH synthesis [14].
In addition to GSH-associated drug resistance, 5-FU has been reported to trigger immunosuppression via T lymphocytes, myelosuppression depletion and exertion of other immunosuppressive effects [15], which accounts for the clinical failure of 5-FU. To address 5-FU-induced immunosuppression, various immunotherapy-based combination strategies have been extensively explored to reshape the immunosuppressive tumor microenvironment (TME) [16]. For example, Guo et al. reported a combination treatment comprising folinic acid (FnA), oxaliplatin (OxP) and 5-FU that can significantly reverse the immunosuppressive TME by inducing immunogenic cell death (ICD) [17].
Indoleamine 2,3-dioxygenase (IDO), a regulatory enzyme shaping immune responses that can cause tumor tolerance, could be adopted as an immune checkpoint [18, 19]. IDO leads to tryptophan depletion to stimulate multiple cell-protective pathways, such as SLC7A11 expression [20]. IDO inhibitors are expected to synchronously regulate GSH and reverse the suppressive TME. Recently, small molecular inhibitors have emerged to block the IDO effector pathway and revive T cells for cancer immunotherapy, including 1-methyl-D-tryptophan (D-1MT, IND) [21, 22]. As a result, it will be a promising strategy to integrate IND and 5-FU via a nano-prodrug for combinatory therapy that can address simultaneously the GSH-associated drug resistance of HCC and the immunosuppressive TME.
The self-assembly strategy based on small molecules prodrugs can avoid the problems of complex synthesis and purification processes, poor drug encapsulation, and premature drug leakage associated with the traditional nanocarrier-based drug delivery [23, 24]. Specifically, the disulfide link-mediated small molecular self-assembly has drawn considerable attention because the incorporated disulfide link can not only promote the self-assembly process of an amphiphilic prodrug into a stabilized nanoformulation, but also enable GSH-cleavable structure to trigger particulate dissociation and promoted intracellular drug release [25–27].
Based on the above considerations, we reported in this research the development of a bio-reducible prodrug composed of 5-FU and an IDO inhibitor (IND), FU-SS-IND that can self-assembly into stabilized nanoparticles, FU-SS-IND NPs, which integrates the ICD-triggering function of 5-FU and innate immunity activation property of IND for all dimensional GSH exhaustion and overcoming 5-FU resistance. The nanoparticles (NPs) can achieve the site of tumors through an enhanced permeability and retention (EPR) effect upon intravenous injection. In 5-FU-resistant HCC cells, intracellular overexpressed of GSH can trigger the cleavage of the disulfide link for particulate dissociation and release of 5-FU and IND. The released IND can not only downregulate SLC7A11 expression for blocking cystine uptake via inhibiting the IDO signaling pathway, thereby enhancing the ICD induced by 5-FU, but also impede the entry of tryptophan (Trp) into the kynurenine (Kyn) pathway to activate the immunosuppressive T cells (Scheme 1). Most importantly, our research has identified that FU-SS-IND NPs possess the ability to reprogram immunosuppressive TME and simultaneously upregulate programmed death ligand 1 (PD-L1) expression on the surface of tumor cells. This interesting finding holds a remarkable promise for the integration of FU-SS-IND NPs with PD-L1 monoclonal antibody (PD-L1 mAb) immune checkpoint blockade therapy in vivo. Exhaustive tests in vitro and in vivo were conducted to ascertain the efficacy of this innovative nano-prodrug, and the underlying mechanism of action.
Scheme 1.

Diagram outlining the preparation of small molecular self-assembly FU-SS-IND NPs and the use of FU-SS-IND NPs for robust GSH exhaustion and tumor-specific immunotherapy. Combining FU-SS-IND NPs with PD-L1 mAb further maximizes tumor inhibition
Results and discussion
Characterization of small molecular self-assembly FU-SS-IND NPs
The synthesis of small molecular prodrug FU-SS-IND was illustrated in Fig. 1a. Briefly, an esterification coupling reaction between IND and bis (2-hydroxyethyl) disulfide in the presence of DCC/DMAP afforded IND-SS-OH with a high yield of 60.0%. Following this, IND-SS-OH and 5-FU were conjugated via an ester bond, and underwent a hydrolyzed reaction to obtain the target small molecule prodrug FU-SS-IND. The constructions of the key intermediate and final product were determined using 1H NMR (Fig. S1-S5), revealing all characteristic signals attributable to 5-FU and IND. More importantly, successful preparation of FU-SS-IND with an equivalent coupling between 5-FU and IND is strongly indicated by evaluating the integrated peak intensity at 8.0 ppm assigned to 5-FU of the peak of 5.8 ppm attributed to IND. Benefiting from the inherent efficiency of disulfide-mediated small molecules in self-assembly [28, 29], we have explored the spontaneous self-assembly of FU-SS-IND into NPs in an aqueous solution. The nano-formulation, FU-SS-IND NPs, was fabricated through a classical nano-precipitation method, and the characterization of the resulting was carefully evaluated via transmission electron microscopy (TEM) and dynamic light scattering (DLS) analyses. The FU-SS-IND NPs demonstrated an extremely narrow and unimodal particle size profile, exhibiting a hydrodynamic diameter (Dh) of 186.9 nm and a polydispersity index (PDI) of 0.1 (Fig. 1b). FU-SS-IND NPs displayed a uniform spherical shape in TEM images (Fig. 1c). To investigate the stability of the FU-SS-IND NPs, these particles were observed for changes in size over a 72-hour period in saline solution. Almost unaltered diameters and PDIs indicate the excellent salt stability of FU-SS-IND NPs (Fig. 1d), which is a prerequisite for controlled release applications. These experimental results confirm that the prodrug FU-SS-IND can form stable nanoparticles. UV-vis spectrometry was used to evaluate the in vitro release characteristics of FU-SS-IND NPs in the presence of GSH concentrations of 1, 5, and 10 mM, at the physiological pH of 7.4 (Fig. 1e). The 5-FU release rate depends clearly on the GSH concentration. In the absence of GSH, FU-SS-IND NPs remained stable with only 15.1% of 5-FU release in 48 h. As GSH levels increased, the release of 5-FU accelerated, reaching 71.7% with 10 mM GSH in an identical monitoring period. A higher GSH concentration promotes the cleavage of a greater amount of disulfide links for a faster 5-FU release with a larger cumulative amount.
Fig. 1.

Synthesis and characterization of FU-SS-IND NPs. (a) The synthetic route for the designed FU-SS-IND prodrug. (b) Size and (c) TEM image of FU-SS-IND NPs. Scale bar: 200 nm. (d )The stability of FU-SS-IND NPs. (e) GSH-responsive release of FU-SS-IND NPs. n = 3
Cellular uptake of FU-SS-IND NPs
The in vitro cellular uptake efficiency of coumarin 6 (C6)-loaded nanoparticles (C6@FU-SS-IND NPs) was assessed in Hepa1-6/FU cells using flow cytometry (FCM) and fluorescence inverted microscopy (FIM) analysis. To demonstrate the cellular uptake of FU-SS-IND NPs, Hepa1-6/FU cells were incubated with either free C6 or FU-SS-IND NPs for different incubation periods of 0.5, 3, and 6 h. FU-SS-IND NPs show consistently greater cellular uptake efficiency values than free C6 at all the three time points (Fig. 2a and b). The NPs’ excellent cellular uptake efficiency is reasonably attributed to the capacity in efficiently circumventing drug resistance in Hepa1-6/FU cells. C6@FU-SS-IND NPs exhibited time-dependent endocytosis behaviors, i.e., characterized by enhanced green fluorescence intensities over extended incubation times. Subsequent FCM analysis also confirmed that the cell green fluorescence intensities gradually increased, and then reached saturation in 6 h. C6@FU-SS-IND NPs-treated Hepa1-6/FU cells displayed a mean fluorescence intensity (MFI) 7.6 times higher than that of the untreated control culture media in 6 h, revealing that the FU-SS-IND NPs had an excellent cellular uptake capacity (Fig. 2c and d).
Fig. 2.

In vitro cellular uptakes of FU-SS-IND NPs (a) In vitro cellular uptake images of Hepa1-6/FU cells treated with free C6 or C6@FU-SS-IND NPs for 0.5, 3 and 6 h using FIM (Scale bar: 200 μm), and (b) quantification of MFI by ImageJ. (c) FCM analysis of Hepa1-6/FU cells exposed to 0.5 h, 3 h, 6 h and 9 h with C6@FU-SS-IND NPs and the MFI of (d) C6 in the Hepa1-6/FU cells. n = 3, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant
In vitro antitumor effect
GSH elevation has been reported to be linked to acquired resistance of HCC cells to 5-FU [4]. 5-FU-resistant cells Hepa1-6/FU were generated by exposing parental Hepa1-6 HCC cells with 5-FU at stepwise increasing drug concentrations from 5 to 30 µM [30]. The GSH level in Hepa1-6/FU cells increased almost 3.2-fold higher than that of the parent Hepa1-6 cells (Fig. 3a). The IC50 of the resistant Hepa1-6/FU cells (IC50: 89.4 ± 8.9 µM) (Fig. 3c) to 5-FU was identified to be approximately 16.0-fold higher than that of the parental Hepa1-6 cells with an IC50 of 5.6 ± 2.9 µM at 48 h of incubation (Fig. 3b), indicating that the 5-FU-resistant cell line was successfully constructed. More importantly, FU-SS-IND NPs achieved a cytotoxic effect approximately 1.4-fold greater than that of free 5-FU on Hepa1-6/FU cells, whereas in parental cell lines FU-SS-IND NPs were much less toxic than free 5-FU, which could be possibly attributed to the cellular GSH level diversity. To verify our findings, we pretreated parental Hepa1-6 cells using varying concentrations (0, 1, 5 and 10 mM) of GSH prior to the administration of FU-SS-IND NPs (Fig. 3d). All the GSH only groups show high cell viability values around 100%, suggesting apparently an insignificant effect of GSH on the cell viability (Fig. S6). Together with the notable GSH-dependent cytotoxicity of FU-SS-IND NPs (Fig. 3d), we thus draw a conclusion that the cytotoxicity of FU-SS-IND NPs was positively correlated with the GSH levels, suggesting that a higher intracellular GSH concentration leads to a greater therapeutic efficiency of the NPs. These results provide strong corroboration to the in vitro drug release data.
Fig. 3.

In vitro anti-tumor efficacies. (a) The intracellular GSH levels were measured in parental Hepa1-6 and resistant Hepa1-6/FU cells. The cytotoxicity of 5-FU, IND, 5-FU + IND, and FU-SS-IND NPs in parental Hepa1-6 (b) and resistant Hepa1-6/FU (c) cells. (n = 5). (d) Cell viability of Hepa1-6 cells pretreated with varying concentrations (0, 1, 5 and 10 mM) of GSH for 12 h and then treated with FU-SS-IND NPs for 48 h. FCM analysis of (e) cell cycle and (f) cell apoptosis in Hepa1-6/FU cells after they had been exposed to 5-FU, IND, 5-FU + IND, and FU-SS-IND NPs for 48 h. n = 3, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant
5-FU mainly acts during the G1/S phases of the cell cycle by disrupting DNA synthesis [31]. Therefore, we further explored the effect of FU-SS-IND NPs treatment on cell cycle phases in Hepa1-6/FU cells. The experiment unveiled that Hepa1-6/FU cells treated with FU-SS-IND NPs had an obvious cell cycle arrest in the G1/S phase, along with a corresponding decrease in the G2/M phases (Fig. 3e). Next, the apoptosis of Hepa1-6/FU cells after different treatments was assessed using annexin V-FITC/propidium iodide (PI) assay kits. Contrasting with the low cytotoxicity of 5-FU, resulting in an apoptotic rate of just 25.7%, FU-SS-IND NPs showed an outstanding increase of 60.2%, indicating nearly 2.4-fold higher apoptosis when compared to 5-FU alone (Fig. 3f). This could be attributed to the synergistic action of disulfide link breakage and IND that leads to GSH exhaustion, which amplifies oxidative stress mediated by 5-FU for a synergistic antitumor effect.
Redox imbalance
Tumor cells display a distinctive intracellular microenvironment characterized by a redox-heterogeneous state, because ROS and GSH are overproduced simultaneously in different cellular compartments [32, 33]. As far as drug resistance and tumor progression are concerned, an increase intracellular GSH level plays a crucial role [34]. The disulfide links incorporated in FU-SS-IND NPs for GSH-cleavable drugs release could consume the GSH to a certain extent 5-FU is also capable of elevating intracellular ROS levels that subsequently lead to GSH depletion [35]. The efficacy of different treatments on intracellular GSH levels in Hepa1-6/FU cells was assessed using a GSH detection kit. Interestingly, significant reductions in intracellular GSH levels were noted in the FU-SS-IND NPs group, a stark contrast to the insignificant effect on GSH levels observed in the free 5-FU group (Fig. 4a), implying the compensation of GSH synthesis for 5-FU-induced ROS production and GSH consumption. The cystine transporter SLC7A11 is required for GSH synthesis by transporting cysteine into the cell [36]. IDO can not only stimulate several cellular protective pathways (including SLC7A11 expression) [20], but also improve the immunosuppressive TME via the degradation of Trp into Kyn [37]. To confirm the function of nano-prodrug in IDO and SLC7A11 expression, we initially performed a Western blot assay to analyze the levels of these two signaling proteins. As expected, the IDO expression was downregulated in Hepa1-6/FU cells treated with FU-SS-IND NPs (Fig. 4b). The expression of SLC7A11 was also assessed by Western blot under various treatment conditions. The results revealed that IND alone insignificantly decreased SLC7A11 expression, whereas free 5-FU alone increased the SLC7A11 level (Fig. 4c). The downregulation of SLC7A11 was most pronounced in the group treated with FU-SS-IND NPs, with its expression reduced by 38% compared to that of the free 5-FU group (Fig. 4c). This result suggests that the IND released from the NPs simultaneously downregulates IDO and SLC7A11 expression for a compromised compensatory effect on intracellular GSH biosynthesis. The cysteine content showed a variation tendency almost the same as the SLC7A11 expression, with FU-SS-IND NPs inducing a significant decrease in cysteine content, which was 0.6-fold lower compared to 5-FU, demonstrating statistical significance (Fig. 4d). Intracellular GSH acts as an endogenous ROS scavenger and thus exerts a cytoprotective effect [38]. The 2,7-dichlorofluorescein diacetate (DCFH-DA) was further used to examine the effects of different treatments on intracellular ROS levels (Fig. 4e). FU-SS-IND NPs-treated Hepa1-6/FU cells showed MFI 3.5-fold higher than cells treated with 5-FU after 48 h incubation, likely due to the further enhanced ROS production through GSH depletion (Fig. 4f). Similar results were recorded by FIM, which also confirms that the ROS production capacity of FU-SS-IND NPs is stronger than that of free 5-FU (Fig. 4g). All the results collectively suggested that the FU-SS-IND NPs simultaneously achieved all dimensional GSH exhaustion via amplifying the levels of oxidative stress by depleting the existing intracellular GSH reserves and impeding GSH biosynthesis.
Fig. 4.

In vitro mechanism of FU-SS-IND NPs-induced oxidative stress. (a) Changes in GSH consumption in Hepa1-6/FU cells over time after different treatments. (b) Western blotting analysis of IDO and SLC7A11 expression. (c) The corresponding gray statistics of the expression of IDO and SLC7A11 from (b). (d) The relative intracellular cysteine content of Hepa1-6/FU cells following various treatments and incubated for 48 h. FCM analysis of (e) ROS production and the MFI of (f) DCF in Hepa1-6/FU cells after they had been treated with 5-FU, IND, 5-FU + IND, and FU-SS-IND NPs for 48 h. (g) The FIM images of Hepa1-6/FU cells with ROS production after various treatments. Scale bar: 100 μm (ROS, DCF, green). n = 3, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant
In vitro ICD induction and immune response
Reportedly, chemotherapeutic agents induce ICD, a unique form of cancer cell death and can facilitate antigen uptake by dendritic cells (DCs). The ICD occurrence is accompanied by the release of damage-associated molecular patterns (DAMPs), such as the exposure of calreticulin (CRT) and secretion of autophagy-dependent adenosine triphosphate (ATP) and high mobility group box-1 protein (HMGB1) [39, 40]. However, 5-FU as inducers of ICD remains controversial without solid detection of HMGB1 release [41]. To verify whether the nano-prodrug effectively triggers ICD in Hepa1-6/FU cells, ATP release was first detected. In contrast to the free 5-FU-treated group, FU-SS-IND NPs markedly stimulated the production of ATP from Hepa1-6/FU cells (Fig. 5a). The ATP release by FU-SS-IND NPs was 1.7-fold higher than free 5-FU, which means statistical significance. Subsequently, immunofluorescence analysis further verified the evaluation of CRT (Fig. 5b and c).and HMGB1 in Hepa1-6/FU cells (Fig. 5d). There was a significant increase in both CRT exposure and HMGB1 release in the FU-SS-IND NPs-treated group. Therefore, the ICD induction ability should be attributed to amplification of 5-FU-mediated oxidative stress via integrated GSH depletion and ROS enhancement.
Fig. 5.

In vitro ICD induction and immune activation after various stimulations. (a) The ATP release from Hepal-6/FU cells post culture with PBS (control group), free 5-FU, free IND, 5-FU + IND, and FU-SS-IND NPs for 48 h. Immunofluorescence assessment of (b) CRT exposure and (d) HMGB1 release on Hepa1-6/FU cells following various treatments for 48 h. Scale bar: 100 μm (Nuclei, DAPI, blue; CRT/HMGB1, Alexa 488, green). (c) MFI statistical results of (b). (e, f) FCM analysis of DCs maturing rate following co-incubation with various treated Hepa1-6/FU cells for 24 h. n = 3, *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001
ICD results in the DAMPs release to facilitate the cross-presentation of tumor-associated antigens to DCs, ultimately leading to the DCs activation and T lymphocyte infiltration [42]. To explore the immune response characteristics of FU-SS-IND NPs in vitro, the capability of FU-SS-IND NPs to stimulate and promote the activation of bone marrow-derived dendritic cells (BMDCs) was evaluated by co-incubation of FU-SS-IND NPs-treated Hepa1-6/FU cells with BMDCs. The quantity of mature DCs showed a significantly increase in the FU-SS-IND NPs-treated group compared to the group treated with free 5-FU, in accordance with DAMPs release data. Significantly, the DCs treated with FU-SS-IND NPs reached a maturation rate of 29.0%, outpacing the group treated with 5-FU by 1.2-fold (Fig. 5e and f). In summary, the developed nano-prodrug harnesses the benefits of both 5-FU and IND to synergistically enhance the oxidative stress within the tumor through GSH exhaustion of all dimensions to activate immunity.
In vivo distribution of nano-prodrug IR780@FU-SS-IND NPs
The hemolysis rate of FU-SS-IND NPs remains less than 5% at a concentration of 400 μg/mL, indicating that FU-SS-IND NPs exhibit favorable blood compatibility and are suitable for subsequent intravenous administration (Fig. S7). To assess the distribution of the nano-prodrug in vivo, IR780-loaded nanoparticles (IR780@FU-SS-IND NPs) were used for clear fluorescence monitoring. We first monitored the drug biodistribution properties on 5-FU-resistant tumors mice using IVIS in vivo imaging. Injected via the tail vein were the different formulations (IR780 and IR780@FU-SS-IND NPs), with fluorescence changes monitored at predetermined time intervals. For IR780@FU-SS-IND NPs, a clear fluorescence was detected in tumor tissues at 12 h, which remained stronger from 12 h to 48 h. The observed change in the fluorescence signal within the liver site was progressive over time, with the signal steadily declining from 6 h to 48 h (Fig. 6a). The drug distribution of the harvested organs was similarly monitored using fluorescence imaging at exactly 48 h post-harvesting (Fig. 6b), and consistent results were acquired, i.e., IR780@FU-SS-IND NPs exhibited significant accumulation in the tumor tissues with low fluorescence detected in the liver. It is worth pointing out that IR780@FU-SS-IND NPs mediated IR780 accumulation in tumor tissues with a percentage of 46.5% for all the examined organs and tissues. This population was 41.0% higher than that of free IR780 in 48 h (Fig. 6c). The results indicated that FU-SS-IND NPs exhibited excellent tumor targeting capability for in vivo tumor treatment.
Fig. 6.

The distribution of the nano-prodrug in vivo. (a) Biodistribution of free IR780 and IR780@FU-SS-IND NPs in subcutaneous Hepa1-6/FU cells tumor model at various time intervals (0.5, 6, 9, 12, 24 and 48 h) post-injection (The tumor is indicated by a red circle). (b) 48 h after dosing, tissue distribution and tumor accumulation of free IR780 and IR780@FU-SS-IND NPs from sacrificed mice. (c) After 48 h, quantification of uptake of free IR780 and IR780@FU-SS-IND NPs into tumor and organs. n = 3, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant
In vivo anticancer efficiency in a subcutaneous hepa1-6/FU Tumor model
Finally, the in vivo therapeutic potential of FU-SS-IND NPs was analyzed in a 5-FU-resistant mice model. After the tumor achieved nearly 100 mm3, mice were intravenously injected on days 1, 3, 5, and 7 with saline, 5-FU, IND, 5-FU + IND and FU-SS-IND NPs, each at a dosage equivalent to 10 mg of 5-FU/kg (Fig. 7a). After 19 days of treatment, the tumor volume growth rate of the FU-SS-IND NPs-treated group was obviously lower compared to that observed in the other treatment groups, resulting in a final mean tumor size of 110.4 ± 42.5 mm3 smaller than those of 5-FU (890.2 ± 348.9 mm3) and 5-FU + IND (343.1 ± 101.2 mm3) groups (Fig. 7b). Similar therapeutic effects were also evidenced by the tumor tissue in anatomical photos (Fig. 7d). Moreover, there was no significant body weight change in C57BL/6j mice in the FU-SS-IND NPs -treated group during the whole treatment phase, which was comparable to that observed in the saline group. However, the weight loss of the C57BL/6j mice in the free 5-FU-treated group had reached around 22.8%. Further hematoxylin-eosin (H&E) staining analyses support that the FU-SS-IND NPs -treated group had the lowest degree of cell proliferation and the greatest histological damage/apoptosis in tumor tissues with minimal impact on other primary organs. Serious liver and kidney damage from treatment with free-5-FU and 5-FU + IND accounted for the severe weight loss in mice (Fig. 7e). All the results indicated that FU-SS-IND NPs displayed excellent antitumor effect with minimized systematic side effects.
Fig. 7.

In vivo assessment of the anticancer efficiency. (a) Schematic illustration of administration. (b) Tumor volumes, (c) body weights and (d) images of tumor tissues of 5-FU-resistant tumors mice treated saline (control group), 5-FU, IND, 5-FU + IND, and FU-SS-IND NPs. (e) H&E analysis of the tumor tissues and major organs. Scale bar: 100 μm. n = 5, *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001
In vivo antitumor immune response
To further assess the immunoregulatory impact of FU-SS-IND NPs, FCM evaluated the distribution of different immune cell populations within the tumor-draining lymph nodes (TDLNs), tumor tissues, or the spleen. The release of DAMPs signaling molecules from dying tumor cells has been identified as an essential factor in promoting DCs maturation [43, 44]. This process then activates T lymphocytes and potentiates the mobilization of an adaptive immune response [45]. Firstly, the maturation of DCs in tumor tissue and TDLNs was evaluated in vivo. On one side, the percentage of mature DCs in TDLNs in the FU-SS-IND NPs-treated group reached 41.8%, which was approximately 1.6 folds that of the 5-FU + IND-treated groups (26.4%) (Fig. 8a and b). Similarly, the proportion of mature DCs in tumor tissues in the FU-SS-IND NPs-treated group achieved 56.4%, surpassing those in the free 5-FU and 5-FU + IND-treated groups by 13.3% and 9.9%, respectively (Fig. 8c). The results indicated that FU-SS-IND NPs significantly stimulated the accumulation and maturation of DCs in both TDLNs and tumor tissue. The maturation of DCs can further amplify their ability in presenting tumor antigens and stimulate antitumor immune response [46, 47]. Consequently, CD8+ T cells infiltration was evaluated in both the spleen and tumor, with the highest proportion observed in the FU-SS-IND NPs-treated groups. The percentage of CD3+CD8+ cytotoxic T cells in the spleens was 30%, which was approximately 1.5-folds and 1.9-folds that of free 5-FU and 5-FU + IND groups (Fig. 8d). In addition, a remarkable increase in CD3+CD8+ T cells was also observed in tumor tissues, with a percentage of 1.2-fold and 1.5-fold of free 5-FU and 5-FU + IND groups, respectively (Fig. 8e). The interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) as essential pro-inflammatory cytokines are predominantly released by activated T cells [48, 49]. The level of both pro-inflammatory cytokines is significantly increased in blood, suggesting that FU-SS-IND NPs may induce T-cell activation in tumor tissues (Fig. 8f and g). Meanwhile, tumors filled with regulatory T (Treg) cells are typically linked to an unfavorable prognosis, as the number of Treg cells rises in tandem with increased IDO activity, which can be reversed by IDO inhibitor IND. A substantial infiltration of intratumoral Foxp3+ Treg cells, defined as a frequency of 34.3 ± 3.1%, was observed in the saline group (Fig. 8h and i), suggesting an immunosuppressive microenvironment in Hepa1-6/FU tumors. After the treatment with FU-SS-IND NPs, the percentage of Tregs decreased dramatically to 7.9 ± 0.1% (Fig. 8h and i), demonstrating that the combination of 5-FU and IDO inhibitor significantly reduced Tregs of intratumoral. The findings of the above studies highlight that FU-SS-IND NPs, when administered intravenously, is capable of triggering an effective antitumor immune response and exhibits considerable antitumor potency by remodeling the immunosuppress microenvironment. Nonetheless, the clinical efficacy of CD8+ T cell-based tumor immunotherapy remains limited by not only insufficient T cell penetration in tumor tissue [50], but also the presence of PD-L1 on tumor cells [51, 52]. PD-L1 expression on tumor cells can prevent T cells from recognizing and attacking tumors by binding to PD-1 on T cells [53]. Unexpectedly, immunofluorescence results revealed a strong red fluorescence singling (PD-L1 expression) in the tumor tissue administered FU-SS-IND NPs (Fig. 8j), most likely due to an increased PD-L1 expression level after administration of chemotherapeutic agents [54]. Therefore, it is reasonable to postulate that combination of FU-SS-IND NPs further with PD-L1 mAb will enhance the anticancer efficacy of FU-SS-IND NPs.
Fig. 8.

In vivo assessment of the antitumor immunity efficiency. (a, b) FCM profiles and quantitative statistics of mature DCs in TDLNs with different therapies. FCM quantitative statistics indicating the maturity rate of (c) DCs in tumors following various treatments. FCM evaluation of the proportions of CD8+ T cells in the (d) spleens and (e) tumors following various treatments. Immunological parameters: enzyme-linked immunosorbent assay (ELISA) was employed to quantify serum levels of cytokines, including TNF-α (f), and IFN-γ (g). (h, i) FCM profiles and quantitative analyses of Tregs in the tumors following various treatments. (j) Immunofluorescence staining of PD-L1 levels in Hepa1-6/FU tumors after treatment with saline, 5-FU, IND, 5-FU + IND and FU-SS-IND NPs. Scale bar: 100 μm. n = 3, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant
FU-SS-IND NPs combined with PD-L1 mAb could synergistically improve anticancer effect and immunotherapy in a Hepa1-6/FU tumor model
Given that the FU-SS-IND NPs group has been observed to induce a robust upregulation of PD-L1 expression on murine tumor cells, we explored the potential synergy between FU-SS-IND NPs and the administration of PD-L1 mAb (10 mg kg− 1) in the treatment of 5-FU-resistant tumor mice models (Fig. 9a). First, the results showed that FU-SS-IND NPs in combination with PD-L1 mAb led to significantly superior tumor growth inhibition compared to treatment with either FU-SS-IND NPs or PD-L1 mAb separately (Fig. 9b). Specifically, the tumor volume of 5-FU-resistant tumors mice administered saline, PD-L1 mAb, and FU-SS-IND NPs + PD-L1 mAb was 1738.0 mm3, 1142.3 mm3, 187.4 mm3, and 54.2 mm3, respectively, indicating that FU-SS-IND NPs + PD-L1 mAb had optimal antitumor effect. Second, the association of body weight changes among the different drug treatment groups revealed that statistically, these differences were negligible (Fig. 9c). Furthermore, the survival rate in the combination of FU-SS-IND NPs and PD-L1 mAb treatment group was significantly increased compared to the FU-SS-IND NPs alone group. The survival rate of mice in the FU-SS-IND NPs + PD-L1 mAb in combination of reached 60% within the next 60 days, while all mice in the FU-SS-IND NPs alone group died (Fig. 9d). To verify whether FU-SS-IND NPs + PD-L1 mAb could further enhance T cell immunity, we subsequently assessed the infiltration of CD8+ T cells and memory T cells in the tumor tissues. In the tumor tissues of mice treated with FU-SS-IND NPs + PD-L1 mAb, the percentage of CD3+CD8+ T cells was 59.4%, which was 1.3-fold and 1.7-fold that of FU-SS-IND NPs or PD-L1 mAb alone (Fig. 9e and f). To conduct a further examination of the immunological memory response, central memory T cells (Tcm, CD3+CD8+CD44+CD62L+) in the spleen were performed using FCM (Fig. 9g and h). The percentage of Tcm in FU-SS-IND NPs + PD-L1 mAb group was 11.8%, which was 1.6-fold and 2.3-fold that of FU-SS-IND NPs or PD-L1 mAb administered separately. Finally, the expression of PD-L1 in the tumor tissues of 5-FU is resistant mice treated with different therapeutic interventions has been examined by FCM (Fig. 9i and j). The results indicated that the FU-SS-IND NPs treatment group significantly enhanced the PD-L1 expression, with downregulating the PD-L1 levels in the tumor cells of mice treated with FU-SS-IND NPs + PD-L1 mAb group.
Fig. 9.

Evaluation of the in vivo anticancer and immunological efficacy of FU-SS-IND NPs plus PD-L1 mAb. (a) Schematic illustration of administration. (b) Volumes of tumors, (c) body weights and (d) survival curves of 5-FU-resistant tumors mice treated with saline, FU-SS-IND NPs, PD-L1 mAb and FU-SS-IND NPs + PD-L1 mAb. (e, f) FCM profiles and quantification of CD8+ T cells in tumors with various treatments. (g, h) FCM profiles and quantification of Tcm in spleens with different treatments. (i, j) FCM analysis and quantification of PD-L1 expression in tumors of mice following various treatment groups. n = 3–5, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant
Conclusions
In summary, a minimalist bio-reducible nano-prodrug based on 5-FU and IND, FU-SS-IND was constructed to enhance the treatment efficiency of 5-FU for HCC. The disulfide links not only participated in the self-assembly process of an amphiphilic prodrug into stabilized NPs, but also enabled GSH-triggered particulate dissociation for an accelerated intracellular drug release toward enhanced therapeutic efficiency. Specifically, GSH consumption via disulfide bonds in Hepa1-6/FU cells with an elevated GSH level can further increase the intracellular ROS production for inducing oxidative stress. Meanwhile, the released IND served as an IDO inhibitor to reverse the immunosuppressive microenvironment and downregulated SLC7A11 expression for compromised GSH synthesis and amplified oxidative stress. Therefore, FU-SS-IND NPs reshaped the immunosuppressive TME by stimulating CD8+ T cell activation and inhibiting Treg. More importantly, FU-SS-IND NPs significantly enhanced the expression of PD-L1 on the tumor cell surface and effectively increased the PD-L1 mAb response rate in tumor tissues, leading to synergistic chemotherapy with immunotherapy to extend the survival lifetime of 5-FU-resistant HCC mice. However, there remains several limitations that should be given further consideration. Firstly, our initial assessment focused on vital organs like the liver and kidney, necessitating further comprehensive evaluation of the nanoprodrugs’ toxicity to fully understand its potential effects. Secondly, while laboratory-cultured tumor cells typically exhibit a clear phenotype, naturally occurring tumors show different characteristics, thus further utilization of organoids or primary tumor cells provides a more accurate indication of the practical therapeutic index of FU-SS-IND NPs. Last but not least, this study is limited to subcutaneous tumor models, and in situ models or tumor models induced by risk factors may have more potential ranslational prospects. Overall, the minimalist bio-reducible nano-prodrug developed herein demonstrates great translatable potential for efficient overcoming 5-FU-resistant HCC, which could be facile combined with immune checkpoint blockade (ICB) therapy for ultimately promising clinical outcomes.
Methods
Materials
Dichloromethane (DCM), Tetrahydrofuran (THF), Ethyl acetate, Methanol (MeOH) were supplied by Hengyang Hongjin Chemical Company. Sodium sulfate (anhydrous) was obtained from Tianjin Beilian Fine Chemicals Development Co., Ltd. 5-FU, IND, Trifluoroacetic acid (TFA), Dicyclohexylcarbodiimide (DCC), Bromoacetic acid, 4-dimethylaminopyridine (DMAP), 2,2’-Dithiodiethanol, Di-tert-butyl dicarbonate ((BOC)2O), Potassium hydroxide (KOH) and C6 were obtained from Macklin Co., Ltd. GSH (Reduced) and Sodium bicarbonate (NaHCO3) were supplied by Shanghai Aladdin Biochemical Technology Co., Ltd. 0.25% trypsin-EDTA, penicillin-streptomycin, RIPA Lysis Buffered Solution, Fetal bovine serum (FBS) and DMEM medium were obtained from MeilunBio Co., Ltd. MTT was supplied by Coolaber. GSH and oxidized glutathione (GSSG) Assay Kit, Annexin V-FITC Assay Kit, ROS Assay Kit and Hoechst 33,342 were obtained from Beyotime Biotechnology Co., Ltd.
C57BL/6j mice were supplied from Hunan Slack Jingda Experimental Animal Co., Ltd. (Hunan, China). All animals were kept in the Laboratory Animal Centre in a specific pathogen-free (SPF) condition.
Synthesis of FUA
First, the 5-FU derivative FUA was synthesized according to previous report. Briefly, KOH (1.32 g) was mixed with deionized water (5 mL), followed by mixing with 5-FU (1.046 g). Stirring at 60 °C continued until a transparent solution was formed. At 60℃, bromoacetic acid (1.146 g) was introduced and stirred for 6 h. After the solution was cooled, the pH was adjusted to 5, and impurities were removed by suction filtration after refrigeration for 6–8 h. The pH was then lowered to 2, and the mixture was refrigerated until the product separated out. The crystallized compound FUA was separated and dried under vacuum.
Synthesis of bOC-IND
A mixture of equal volumes of THF and deionized water (35 mL) was dissolved with NaHCO3 (740 mg), (BOC)2O (630 mg), and IND (520 mg). The solution was stirred with ice cooling for 10 min, then transferred to room temperature, where it was allowed to react for 24 h. Following this, THF was removed, and the remaining liquid was neutralized to pH 1.0 using 1 M hydrochloric acid, followed by overnight refrigeration. Next, the residue was extracted with ethyl acetate, and after evaporating the ethyl acetate, a yellow solid was obtained. The final products were characterized using 1H NMR.
Synthesis of bOC-IND-SS-OH
BOC-IND (300 mg), DCC (466.7 mg) and 2,2′Dithiobisethanol were combined in DCM (10 mL) and kept reaction at 30℃ for 24 h. Following filtration and rotary evaporation, the target compound was separated and purified using chromatographic separation, eluting with MeOH/DCM (v/v, 1:200-1:100). The final products were characterized using 1H NMR.
Synthesis of FU-SS-IND-BOC
The BOC-IND-SS-OH was stirred in DCM (10 mL) at 25℃, and DMAP, DCC, FUA were added to kept reaction for 24 h. Following filtration and rotary evaporation, the target compound was separated and purified using chromatographic separation, eluting with MeOH/DCM (v/v, 1:80 − 1:30). The final products were characterized using 1H NMR.
Deprotection of bOC groups of FU-SS-IND
FU-SS-IND-BOC was combined with a mixture of DCM and TFA for 2 h at 25℃. Following filtration and rotary evaporation, the target compound was separated and purified using chromatographic separation, eluting with MeOH/DCM (v/v, 1:80 − 1:25). The final products were characterized using 1H NMR.
Preparation of the FU-SS-IND NPs
The FU-SS-IND NPs were synthesized using the nanoprecipitation method. Briefly, 50 µL methanol with dissolved FU-SS-IND (10 mg mL− 1) was gradually introduced into 1.2 mL water, followed by stirring for half an hour. FU-SS-IND NPs loaded with C6 or IR780 were prepared employing a comparable nanoprecipitation technique. The morphology and particle size of nano-prodrug were assessed by TEM and Malvern ZS90 DLS instrument, respectively.
Drug release profile of nano-prodrug in vitro
By employing a dialysis method and UV-visible spectrophotometer, the release characteristics of FU-SS-IND NPs in vitro were investigated. In this investigation, FU-SS-IND NPs (1 mg mL− 1, 0.5 mL) were enclosed in a dialysis bag (MWCO = 3500 Da) and placed into a 30 mL aqueous solution with varying GSH concentrations (0, 1, 5, 10 mM). The system was then kept at 37℃ for 48 h with agitation. Samples were collected at specific time points (0, 0.5, 1, 2, 4, 6, 8, 12, 24, 36 and 48 h), the release of 5-FU in FU-SS-IND NPs was determined using UV-vis spectroscopy.
Cell culture
Hepa1-6 (a mouse HCC cell line) was supplied from the Cell Bank of the Chinese Academy of Sciences. At 37℃ in a 5% CO2 atmosphere, the cells were cultured in DMEM high glucose medium containing 10% FBS and 1% penicillin/streptomycin.
To establish 5-FU resistance in Hepal-6 cells, Hepal-6 cells were continuously cultured in escalating doses of 5-FU (5–30 µM) for at least 6 months, until the cells could be expanded in medium containing 30 µM 5-FU.
In vitro cellular uptake of FU-SS-IND NPs
We added C6 to construct C6@FU-SS-IND NPs to evaluate Hepa1-6/FU uptake behavior. Hepa1-6/FU cells were cultured in 12-well plates or 24-well plates. Subsequently, free C6 or C6@FU-SS-IND NPs were introduced into the culture medium. After 0 h, 0.5 h, 3 h, 6 h incubation, 12-well plates cells were harvested and suspended in 500 µL PBS for detecting by FCM. For 24-well plates, cells were immobilized with 4% paraformaldehyde, subsequently stained with Hoechst 33,342 for 15 min. The resulting cells were imaged using a FIM to obtain fluorescent images.
In vitro cytotoxicity assessment
The cytotoxicity assessment used 96-well plates and the seeding number of Hepa1-6/FU cells was 3 × 105 cells per well, followed by treated with the drug/nano-prodrug for 48 h. Furthermore, to assess the cytotoxicity of FU-SS-IND NPs after GSH treatment, Hepa1-6 cells were exposed to varying concentrations of GSH (0, 1, 5 and 10 mM) for 12 h prior to dosing, followed by 48 h with FU-SS-IND NPs, with the separate GSH group serving as a control. Then, MTT solution (5 mg/mL) was applied and treated for 4 h. The crystals that resulted were dissolved in 100 µL SDS solution, and a microplate read recorded their absorbance at OD 562 nm. Cell viability was calculated as:
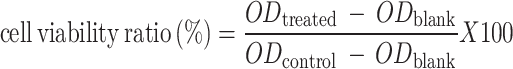 |
In vitro apoptosis analysis
The apoptosis experiment used 12-well plates and the seeding number of Hepa1-6/FU cells was 1 × 105 cells per well. Then, PBS, 5-FU, IND, 5-FU + IND and nano-prodrug FU-SS-IND NPs were diluted with DMEM medium containing 5% FBS to treat the cells respectively. After 48 h, cells were treated by the prescribed procedure of the Annexin V-FITC/PI Assay Kit. Quantitative detection of apoptotic rate in Hepa1-6/FU cells was performed using FCM.
Cysteine content detection assay
To exploit cysteine levels, Hepa1-6/FU cells were exposed to either PBS, 5-FU, IND, 5-FU + IND or FU-SS-IND NPs. 48 h post treatment, cells were harvested. The levels of cysteine were then measured by the Cysteine Assay Kits.
ICD induction
The HMGB1 release and CRT exposure were evaluated using FIM. The seeding number of Hepa1-6/FU cells was 8 × 104 cells/well on 24-well plates. Following 24 h incubation, the cells were then exposed to either 5-FU, IND, 5-FU + IND or FU-SS-IND NPs. 48 h post incubation, cells were fixed with ice-cold methanol or 4% paraformaldehyde, blocked by 1% bovine serum albumin. Following 1 h treatment, then the cells were treated with either anti-CRT antibody or anti-HMGB1 antibody overnight at 4℃. After PBS washing, secondary antibodies were treated with cells for 2 h at room temperature. Afterward, cells were stained for 15 min with Hoechst 33,342. The resulting cells were imaged using a FIM to obtain fluorescent images.
The ATP assay Kit was employed to assess the released ATP levels. Hepa1-6/FU cells were treated with either 5-FU, IND, 5-FU + IND or FU-SS-IND NPs. The culture media was gathered for analysis after 48 h of incubation.
Intracellular GSH and ROS content
The intracellular GSH levels between Hepa1-6 and Hepa1-6/FU cells and the intracellular GSH content of Hepa1-6/FU incubated with various preparations were assessed by GSH and GSSG Assay Kit. The cells were incubated with PBS, 5-FU, IND, 5-FU + IND, FU-SS-IND NPs. After 48 h treatment, cells were harvested by centrifugation. The levels of GSH were then assessed by the GSH and GSSG Assay Kits.
The intracellular ROS levels in Hepa1-6/FU exposed to various conditions were investigated using the ROS fluorescence probe (DCFH-DA). In brief, Hepa1-6/FU cells were incubated in 24-well plates. After 24 h incubation, Hepa1-6/FU cells were expose to 5-FU, IND, 5-FU + IND and FU-SS-IND NPs for 48 h. Subsequently, serum-free medium supplemented with DCFH-DA was utilized to replace the medium and treated for a further 30 min at 37℃. The cellular ROS generation was imaged by FIM and quantitatively analyzed by FCM.
Hemolysis evaluation
Hemolysis test was used to evaluate the blood biocompatibility and feasibility of intravenous administration of FU-SS-IND NPs. Fresh Sprague-Dawley rat blood was centrifuged at 3000 rpm for 5 min, the pellet was collected, and red blood cells were extracted by washing 3 times with PBS. Add PBS (pH7.4) to prepare a 2% (v/v) red blood cell suspension. Mix different concentrations of FU-SS-IND NPs solution (80, 400, and 800 μg/mL) with an equal volume of red blood cell suspension and add to the centrifuge tube while incubating at 37℃ for 3 h. PBS and 0.1% Triton X-100 were used as negative and positive controls, respectively. Then, the mixture was centrifuged at 1200×g for 10 min at 4℃, the supernatant was transferred to a new 96-well plate, and absorbance was measured at OD 560 nm using a microscope. The hemolysis rate was calculated according to the following equation:
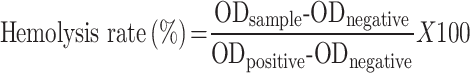 |
In vivo biodistribution
In the right lower buttocks of the mice, tumor-bearing mouse models were established by inoculating a total of 5 × 106 Hepa1-6/FU cells/mouse. The mice were partitioned into two groups at random once their tumor volume reached nearly 400 mm³. The one group was injected with IR780@FU-SS-IND NPs intravenously, the other one injected with free IR780 was used as its control. All the mice were imaged at 0.5, 6, 9, 12, 24, 36, 48 h after injection, using a small animal optical molecular imaging system for acquiring the body fluorescence images.
In vivo antitumor performance
The mice were allocated into 5 groups (n = 5) after their tumor volume reached nearly 100 mm³.The mice received treatments of saline, 5-FU, IND, 5-FU + IND, FU-SS-IND NPs once every 2 days for 4 doses, at a dose of 10 mg kg− 1 5-FU. The formula for calculating tumor volume: volume = width2 × length × 0.5.
The mice were allocated into 4 groups (n = 5) after their tumor volume reached nearly 100 mm³. The mice received treatments of saline, FU-SS-IND NPs, PD-L1 mAb and FU-SS-IND NPs + PD-L1 mAb once every 2 days for 4 doses (FU-SS-IND NPs were injected intravenously on days 1, 3, 5, and 7, respectively; intraperitoneal injection of 10 mg kg− 1 of PD-L1 mAb on days 2, 4, 6, and 8, respectively). The mice’s body weight and tumor volume were recorded bidaily, and the survival rate of the mice in each group was monitored within 60 days.
Tumors and main organs of the mice were harvested the day the treatment experiment was concluded, immobilized with 4% (v/v) paraformaldehyde, processed for paraffin embedding for subsequent H&E staining or immunofluorescence assays.
In vivo immune response experiments
Tumor-infiltrating lymphocytes or spleen cells with various treatments were assessed through FCM. Lymph nodes, tumors and spleens from each group of tumor-bearing mice were harvested on day 9 after treatment, dispersed through a filter screen into cell suspension, stained with the fitting antibodies, and analyzed by FCM. Tcm cells were detected by CD3, CD8a, CD62L, and CD44 antibodies, CD8+ T cells were assessed with CD3, CD8a, and CD4 antibodies, Treg cells were identified using CD3, CD4, CD25 and Foxp3 antibodies, the maturation of DCs were assessed with CD11c, CD86 and CD80 antibodies, and PD-L1 level in tumor cells was detected by PD-L1 antibody.
Whole blood was harvested from mice to extract serum on the day the experiment ended, and the levels of IFN-γ and TNF-α were determined through ELISA kit.
Statistical analysis
The results were presented as mean ± standard deviation (SD). The statistical analysis utilized one-way analysis of variance (ANOVA) and Student’s t-test in GraphPad Prism version 8 software. Statistical significance between the datasets was defined as follows based on the p-values: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This study received financial support from several sources, including the National Natural Science Foundation of China (82373826), the Hunan Science and Technology Innovation Leading Talent Project (Project No. 2022RC3080), the Natural Science Foundation of Hunan province (2022JJ40381, 2023JJ50138), and the Key R & D Program of Hunan province (2021SK2036, 2023SK2043).
Author contributions
T.Z., Y.H., T.T.Z., and S.Y.H. are responsible for all phases of the research,including Conceptualization, Data curation, Formal analysis, Investigation, Validation, Writing-Original Draft & Revision. J.L., Y.L.C., Z.H.L. and M.X.L. are responsible for Data curation. H.W. and C.Y.Y. are responsible for Conceptualization, Funding acquisition, Project administration, Supervision, Writing-Original Draft, Review, Editing & Revision, Writing-original draft.
Data availability
Data is provided within the manuscript or supplementary information files.
Declarations
Ethics approval and consent to participate
All experimental involving animals were approved by the Experimental Animal Ethics Committee of the University of South China, Hunan, People’s Republic of China (approval 4304079008946).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ting Zou, Yun Huang, Zongtao Zhou and Shuangyan He contributed equally to this work.
Contributor Information
Hua Wei, Email: weih@usc.edu.cn.
CuiYun Yu, Email: yucuiyunusc@hotmail.com.
References
- 1.Qin S, Bai Y, Lim HY, Thongprasert S, Chao Y, Fan J, Yang TS, Bhudhisawasdi V, Kang WK, Zhou Y, et al. Randomized, multicenter, open-label study of oxaliplatin plus fluorouracil/leucovorin versus doxorubicin as palliative chemotherapy in patients with advanced hepatocellular carcinoma from Asia. J Clin Oncol. 2013;31:3501–8. [DOI] [PubMed] [Google Scholar]
- 2.Hayashi H, Higashi T, Yokoyama N, Kaida T, Sakamoto K, Fukushima Y, Ishimoto T, Kuroki H, Nitta H, Hashimoto D, et al. An Imbalance in TAZ and YAP expression in Hepatocellular Carcinoma confers Cancer Stem Cell-like behaviors contributing to Disease Progression. Cancer Res. 2015;75:4985–97. [DOI] [PubMed] [Google Scholar]
- 3.Wei L, Wang X, Lv L, Liu J, Xing H, Song Y, Xie M, Lei T, Zhang N, Yang M. The emerging role of microRNAs and long noncoding RNAs in drug resistance of hepatocellular carcinoma. Mol Cancer. 2019;18:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin J, Huang M, Wei HL, Liu GT. Mechanism of 5-fluorouracil required resistance in human hepatocellular carcinoma cell line Bel(7402). World J Gastroenterol. 2002;8:1029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiong Y, Xiao C, Li Z, Yang X. Engineering nanomedicine for glutathione depletion-augmented cancer therapy. Chem Soc Rev. 2021;50:6013–41. [DOI] [PubMed] [Google Scholar]
- 6.Xiao X, Wang K, Zong Q, Tu Y, Dong Y, Yuan Y. Polyprodrug with glutathione depletion and cascade drug activation for multi-drug resistance reversal. Biomaterials. 2021;270:120649. [DOI] [PubMed] [Google Scholar]
- 7.Yang YX, Sun BJ, Zuo SY, Li XM, Zhou S, Li LX, Luo C, Liu HZ, Cheng MS, Wang YJ, et al. Trisulfide bond-mediated doxorubicin dimeric prodrug nanoassemblies with high drug loading, high self-assembly stability, and high tumor selectivity. Sci Adv. 2020;6:eabc1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Wang D, Liu H, Liu L, Li S, Zhou Z, Lu L, Liu X, He L, He D, et al. A clinically translatable Ternary Platinum(IV) Prodrug for synergistically reversing Drug Resistance. J Med Chem. 2023;66:4045–58. [DOI] [PubMed] [Google Scholar]
- 9.Gao F, Yang X, Luo X, Xue X, Qian C, Sun M. Photoactivated Nanosheets accelerate Nucleus Access of Cisplatin for Drug-Resistant Cancer Therapy. Adv Funct Mater. 2020;30:2001546. [Google Scholar]
- 10.Zeng X, Wang Y, Han J, Sun W, Butt HJ, Liang XJ, Wu S. Fighting against drug-resistant tumors using a dual-responsive pt(IV)/Ru(II) Bimetallic Polymer. Adv Mater. 2020;32:e2004766. [DOI] [PubMed] [Google Scholar]
- 11.Ling X, Tu J, Wang J, Shajii A, Kong N, Feng C, Zhang Y, Yu M, Xie T, Bharwani Z, et al. Glutathione-responsive Prodrug nanoparticles for Effective Drug Delivery and Cancer Therapy. ACS Nano. 2019;13:357–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niu B, Liao K, Zhou Y, Wen T, Quan G, Pan X, Wu C. Application of glutathione depletion in cancer therapy: enhanced ROS-based therapy, ferroptosis, and chemotherapy. Biomaterials. 2021;277:121110. [DOI] [PubMed] [Google Scholar]
- 13.Gao Y, Li Y, Cao H, Jia H, Wang D, Ren C, Wang Z, Yang C, Liu J. Hypertoxic self-assembled peptide with dual functions of glutathione depletion and biosynthesis inhibition for selective tumor ferroptosis and pyroptosis. J Nanobiotechnol. 2022;20:390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y, Wu S, Zhang L, Deng Q, Ren J, Qu X. A metabolic multistage glutathione depletion used for Tumor-Specific Chemodynamic Therapy. ACS Nano. 2022;16:4228–38. [DOI] [PubMed] [Google Scholar]
- 15.Cho YH, Ro EJ, Yoon JS, Mizutani T, Kang DW, Park JC, Il Kim T, Clevers H, Choi KY. 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/β-catenin pathway activation. Nat Commun. 2020;11:5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shurin MR, Naiditch H, Gutkin DW, Umansky V, Shurin GV. ChemoImmunoModulation: immune regulation by the antineoplastic chemotherapeutic agents. Curr Med Chem. 2012;19:1792–803. [DOI] [PubMed] [Google Scholar]
- 17.Guo J, Yu Z, Das M, Huang L. Nano Codelivery of Oxaliplatin and Folinic Acid achieves synergistic chemo-immunotherapy with 5-Fluorouracil for Colorectal Cancer and Liver Metastasis. ACS Nano. 2020;14:5075–89. [DOI] [PubMed] [Google Scholar]
- 18.Terness P, Bauer TM, Röse L, Dufter C, Watzlik A, Simon H, Opelz G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196:447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boasso A, Herbeuval JP, Hardy AW, Anderson SA, Dolan MJ, Fuchs D, Shearer GM. HIV inhibits CD4 + T-cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood. 2007;109:3351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiore A, Zeitler L, Russier M, Groß A, Hiller MK, Parker JL, Stier L, Köcher T, Newstead S, Murray PJ. Kynurenine importation by SLC7A11 propagates anti-ferroptotic signaling. Mol Cell. 2022;82:920–e932927. [DOI] [PubMed] [Google Scholar]
- 21.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, Mellor AL, Prendergast GC, Munn DH. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67:792–801. [DOI] [PubMed] [Google Scholar]
- 22.Yang W, Zhang F, Deng H, Lin L, Wang S, Kang F, Yu G, Lau J, Tian R, Zhang M, et al. Smart nanovesicle-mediated immunogenic cell death through Tumor Microenvironment Modulation for effective photodynamic immunotherapy. ACS Nano. 2020;14:620–31. [DOI] [PubMed] [Google Scholar]
- 23.Feng B, Zhou F, Hou B, Wang D, Wang T, Fu Y, Ma Y, Yu H, Li Y. Binary Cooperative Prodrug nanoparticles improve immunotherapy by synergistically modulating Immune Tumor Microenvironment. Adv Mater. 2018;30:e1803001. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Zhang J, Tu Y, Zhu L. Potential-independent Intracellular Drug Delivery and mitochondrial targeting. ACS Nano. 2022;16:1409–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo C, Sun J, Liu D, Sun B, Miao L, Musetti S, Li J, Han X, Du Y, Li L, et al. Self-assembled Redox Dual-Responsive Prodrug-Nanosystem formed by single thioether-bridged paclitaxel-fatty acid conjugate for Cancer Chemotherapy. Nano Lett. 2016;16:5401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun B, Luo C, Yu H, Zhang X, Chen Q, Yang W, Wang M, Kan Q, Zhang H, Wang Y, et al. Disulfide bond-driven oxidation- and reduction-responsive Prodrug nanoassemblies for Cancer Therapy. Nano Lett. 2018;18:3643–50. [DOI] [PubMed] [Google Scholar]
- 27.Luo C, Sun J, Sun B, Liu D, Miao L, Goodwin TJ, Huang L, He Z. Facile fabrication of Tumor Redox-Sensitive nanoassemblies of small-molecule oleate Prodrug as Potent Chemotherapeutic Nanomedicine. Small. 2016;12:6353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu C, Zhang F, Li B, Li Z, Xie X, Huang Y, Yao Z, Chen Y, Ping Y, Pan W. A Self-Assembly Nano-Prodrug for Combination Therapy in Triple-negative breast Cancer stem cells. Small. 2023;19:e2301600. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Liu D, Zheng Q, Zhao Q, Zhang H, Ma Y, Fallon JK, Fu Q, Haynes MT, Lin G, et al. Disulfide bond bridge insertion turns hydrophobic anticancer prodrugs into self-assembled nanomedicines. Nano Lett. 2014;14:5577–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pardee TS, Gomes E, Jennings-Gee J, Caudell D, Gmeiner WH. Unique dual targeting of thymidylate synthase and topoisomerase1 by FdUMP[10] results in high efficacy against AML and low toxicity. Blood. 2012;119:3561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin H, Xie F, Zhang J, Yang Y, Deng B, Sun J, Wang Q, Qu X, Mao H. Combination of interferon-α and 5-fluorouracil induces apoptosis through mitochondrial pathway in hepatocellular carcinoma in vitro. Cancer Lett. 2011;306:34–42. [DOI] [PubMed] [Google Scholar]
- 32.Fang J, Seki T, Maeda H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv Drug Deliv Rev. 2009;61:290–302. [DOI] [PubMed] [Google Scholar]
- 33.Sun B, Luo C, Zhang X, Guo M, Sun M, Yu H, Chen Q, Yang W, Wang M, Zuo S, et al. Probing the impact of sulfur/selenium/carbon linkages on prodrug nanoassemblies for cancer therapy. Nat Commun. 2019;10:3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun HJ, Lee WT, Leng B, Wu ZY, Yang Y, Bian JS. Nitroxyl as a potential Theranostic in the Cancer Arena. Antioxid Redox Signal. 2020;32:331–49. [DOI] [PubMed] [Google Scholar]
- 35.Fu Y, Yang G, Zhu F, Peng C, Li W, Li H, Kim HG, Bode AM, Dong Z, Dong Z. Antioxidants decrease the apoptotic effect of 5-Fu in colon cancer by regulating src-dependent caspase-7 phosphorylation. Cell Death Dis. 2014;5:e983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagasaki T, Schuyler AJ, Zhao J, Samovich SN, Yamada K, Deng Y, Ginebaugh SP, Christenson SA, Woodruff PG, Fahy JV, et al. 15LO1 dictates glutathione redox changes in asthmatic airway epithelium to worsen type 2 inflammation. J Clin Invest. 2022;132:e151685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 inhibitors: from bench to Bedside. Cancer Res. 2017;77:6795–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drain CM, Varotto A, Radivojevic I. Self-organized porphyrinic materials. Chem Rev. 2009;109:1630–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Zhao Z, Qiu N, Zhou Q, Wang G, Jiang H, Piao Y, Zhou Z, Tang J, Shen Y. Co-delivery of IOX1 and doxorubicin for antibody-independent cancer chemo-immunotherapy. Nat Commun. 2021;12:2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petersen SH, Kua LF, Nakajima S, Yong WP, Kono K. Chemoradiation induces upregulation of immunogenic cell death-related molecules together with increased expression of PD-L1 and galectin-9 in gastric cancer. Sci Rep. 2021;11:12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rébé C, Ghiringhelli F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–61. [DOI] [PubMed] [Google Scholar]
- 42.Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired single-cell analyses. Cell. 2017;169:750–e765717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. 2021;21:298–312. [DOI] [PubMed] [Google Scholar]
- 44.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, Krummel MF. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for Tumor Antigen Trafficking and priming of T cell immunity in Melanoma. Cancer Cell. 2016;30:324–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Somarribas Patterson LF, Vardhana SA. Metabolic regulation of the cancer-immunity cycle. Trends Immunol. 2021;42:975–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heras-Murillo I, Adán-Barrientos I, Galán M, Wculek SK, Sancho D. Dendritic cells as orchestrators of anticancer immunity and immunotherapy. Nat Rev Clin Oncol. 2024;21:257–77. [DOI] [PubMed] [Google Scholar]
- 47.Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24. [DOI] [PubMed] [Google Scholar]
- 48.Au-Yeung BB, Shah NH, Shen L, Weiss A. ZAP-70 in Signaling, Biology, and Disease. Annu Rev Immunol. 2018;36:127–56. [DOI] [PubMed] [Google Scholar]
- 49.Schreiber H, Rowley DA. Cancer. Awakening immunity. Science. 2010;330:761–2. [DOI] [PubMed] [Google Scholar]
- 50.Zhang T, Yu W, Cheng X, Yeung J, Ahumada V, Norris PC, Pearson MJ, Yang X, van Deursen W, Halcovich C, et al. Up-regulated PLA2G10 in cancer impairs T cell infiltration to dampen immunity. Sci Immunol. 2024;9:eadh2334. [DOI] [PubMed] [Google Scholar]
- 51.Lei X, Lei Y, Li JK, Du WX, Li RG, Yang J, Li J, Li F, Tan HB. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020;470:126–33. [DOI] [PubMed] [Google Scholar]
- 52.Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol. 2016;27:1492–504. [DOI] [PubMed] [Google Scholar]
- 53.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheng J, Fang W, Yu J, Chen N, Zhan J, Ma Y, Yang Y, Huang Y, Zhao H, Zhang L. Expression of programmed death ligand-1 on tumor cells varies pre and post chemotherapy in non-small cell lung cancer. Sci Rep. 2016;6:20090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is provided within the manuscript or supplementary information files.