

ABSTRACT
The treatment landscape for relapsed/refractory follicular lymphoma (RR‐FL) is marked by a pivotal debate between chimeric antigen receptor T‐cell (CAR‐T) therapy and bispecific antibodies (BsAbs). While both CAR‐T therapy and BsAbs target similar immunobiology and molecular markers, their efficacy comparisons are hindered by the lack of direct clinical trial comparisons. Key trials, such as the ZUMA‐5 study, underscore axicabtagene ciloleucel (axi‐cel)'s efficacy in treating RR‐FL, achieving a 79% complete response rate with a median duration of response exceeding 3 years. Similarly, lisocabtagene maraleucel (liso‐cel) in the TRANSCEND FL study reports a 94% complete response rate, emphasizing robust outcomes in heavily pretreated patients. Among BsAbs, mosunetuzumab showed promise in the GO29781 trial, with a 62% overall response rate in heavily pretreated RR‐FL patients. Thus, CAR‐T therapy offers potential curative benefits with a single infusion. However, its efficacy is tempered by significant adverse events such as cytokine release syndrome (CRS), neurotoxicity, and cytopenias, requiring specialized management and patient monitoring. In contrast, BsAbs provide a more tolerable treatment option counterbalancing by lower response rates and frequent dosing requirements. Personalized treatment strategies are crucial because of these distinct efficacy and safety profiles. When considering cost‐effectiveness, both therapies need to be evaluated in the context of their clinical outcomes and quality of life improvements. Cost‐effectiveness considerations are essential; while CAR‐T therapies incur higher initial costs, their potential for long‐term remission may mitigate expenses associated with repeated treatments or hospitalizations. Future research into resistance mechanisms and optimal therapeutic sequencing will further refine RR‐FL management strategies.
Keywords: BsAbs, CAR‐T, cost‐effectiveness, follicular lymphoma
1. Introduction
Follicular lymphoma (FL) is a prevalent subtype of non‐Hodgkin lymphoma (NHL), accounting for 20%–30% of cases [1]. Despite the diagnostic advancements, FL is often diagnosed at an advanced stage, with fewer than 10% of cases at Stages I and II. Approximately 70% of patients have bone marrow involvement, and less than 20% present B symptoms [1]. FL arises in germinal centers and is characterized by the t(14;18) translocation, leading to BCL‐2 overexpression. According to the fifth WHO classification, classic FL is more common, whereas follicular large B‐cell lymphoma and FL with uncommon features represent rare subtypes [2, 3].
Current FL treatment strategies include rituximab as monotherapy or watchful waiting for asymptomatic patients [4, 5], rituximab associated with chemoimmunotherapy [6, 7, 8], and maintenance therapy with rituximab [9, 10]. Lenalidomide plus rituximab‐ [11] or obinutuzumab (ZO)‐based immunochemotherapy [12] followed by maintenance have shown longer progression‐free survival (PFS) compared to rituximab‐based therapy. For relapse or refractory (RR) disease, second‐line (2L) therapies may involve re‐treatment with similar regimens or alternative combinations [13].
Nevertheless, FL remains challenging because of its tendency to relapse or become refractory to standard treatments, despite being slow‐growing and initially responsive. Overall survival (OS) following first‐line therapy for FL can extend up to 25 years; however, this survival rate declines with each subsequent line of treatment. Specifically, the median OS is reported to be 5.8 years for patients receiving third‐line therapy, which further declines to 3.6 years for those undergoing fifth‐line therapy [14]. Moreover, several observational studies have indicated that patients with FL who experience disease progression within 24 months of completing front‐line chemoimmunotherapy (designed as POD24) exhibit significantly poorer prognoses [15].
This decline underscores the need for improved therapeutic strategies. In the past 5 years, there has been a surge in targeted therapies for RR‐FL, including novel antibody‐based therapies such as tafasitamab, a CD19‐directed antibody, polatuzumab vedotin, an antibody–drug conjugate targeting CD79b, and magrolimab, which targets CD47 on macrophages rather than FL cells directly [16]. Additionally, small molecule inhibitors targeting pathways such as PI3kinase and BTK, as well as agents modulating apoptotic pathways, have demonstrated promise as new strategies for FL management. In this regard, the ROSEWOOD trial [17] has shown significant efficacy of zanubrutinib in combination with ZO for the treatment of RR‐FL patients who had received ≥ 2 lines of therapy. This study demonstrated that the combination therapy outperformed the anti‐CD20 monoclonal antibody monotherapy, achieving an overall response rate (ORR) of 69% for ZO versus 46% for O (p = 0.001). Moreover, patients treated with the ZO combination exhibited a median PFS of 28.0 months, in contrast to 10.4 months for those receiving ZO alone, corresponding to a hazard ratio (HR) of 0.50 (95% confidence interval [CI]: 0.33–0.75; p < 0.001). Nevertheless, the broader efficacy of other BTK inhibitors in this context remains less consistently established to date.
The recognition of high rates of epigenetic mutations in FL has prompted exploration into histone deacetylase inhibitors, which play a role in altering gene expression. A notable breakthrough in this area is the US Food and Drug Administration (FDA) approval of tazemetostat for RR‐FL after two prior lines of therapy. Tazemetostat specifically targets EZH2, a critical epigenetic driver involved in germinal center formation and FL pathogenesis. Both wild‐type and mutant forms of EZH2 present attractive therapeutic targets, emphasizing the importance of epigenetic modulation in FL treatment strategies [18]. Recent findings from a Phase II trial have indicated that abexinostat is both well‐tolerated and effective in RR‐FL patients. The trial reported an ORR of 61.1% (95% CI: 55.8–77.1) and a median PFS of 13.7 at a median follow‐up of 20.8 months [19].
Exciting developments in RR‐FL research are centered on immune‐based therapies like bispecific antibody (BsAb) constructs [20] and chimeric antigen receptor T cells (CAR‐Ts) [21]. These novel approaches utilize distinct mechanisms to enhance immune responses against FL cells, presenting promising avenues for therapeutic advancement.
The discussion regarding the optimal choice between CAR‐T and BsAb therapies for RR‐FL persists. Although both modalities share similarities, they diverge notably in terms of tolerability and efficacy. Thus, choosing between these therapies mandates a careful evaluation of their respective benefits, risks, and practical considerations to optimize patient outcomes effectively.
This review aims to comprehensively evaluate and compare the role of BsAbs versus CAR‐T therapy in the treatment of RR‐FL. By examining their mechanisms of action, clinical efficacy, safety profiles, and economic considerations, this review seeks to provide insights into their respective roles in clinical practice and their potential to transform the treatment landscape for FL patients.
2. Bispecific Antibodies
2.1. Structures and Mechanisms
Comprehensive reviews provided an in‐depth analysis of the structure and mechanisms of BsAbs in cancer therapy [22]. Advances in recombinant DNA technology have led to BsAbs with varied sizes, half‐lives, valencies, flexibilities, and permeabilities [22].
They are classified into two categories: IgG‐like (with Fc regions) and non–IgG‐like (without Fc regions). IgG‐like BsAbs offer enhanced solubility, stability, purification, and half‐life while avoiding antibody‐dependent cellular cytotoxicity or complement‐dependent cytotoxicity through genetic modifications [23]. Non–IgG‐like BsAbs have improved tissue penetration and faster renal clearance but shorter plasma half‐lives.
Non–IgG‐like BsAbs, produced using Fab fragments or linking variable domains, include scFv‐based BsAbs, nanobodies, and dock‐and‐lock (DNL) method antibodies [24]. Single‐chain variable fragments (scFvs) maintain specificity and antigen‐binding abilities.
ScFv‐based BsAbs offer excellent tissue permeability and reduced immunogenicity but have short half‐lives. Formats like “scFv‐HSA‐scFv” and PEGylation can extend their half‐lives [25].
BsAbs are classified into three categories depending on their targets: those targeting two tumor antigens, those targeting a tumor antigen and an immune‐related molecule, and those targeting two immune‐related molecules. Bispecific T‐cell engagers (BiTEs) belong to the second group, targeting both a CD3 molecule on T cells and a tumor antigen simultaneously redirecting T cells against tumor cells. Effective BiTE design requires suitable CD3 binding arms, differential affinity to minimize primarily cytokine release syndrome (CRS) risk, and optimal distance between binding units. High CD3 binding affinity allows continuous T‐cell activation, which can lead to depletion. An ideal affinity facilitates repeated binding and dissociation, clustering T cells for maximum tumor‐killing effect. By bridging T cells and tumor cells, BiTEs promote immunologic synapse formation, initiating antigen‐specific signal transduction and T‐cell activation. Activated T cells express CD69 and CD25, promoting proliferation [26] and effectively reactivating exhausted T cells induced by prolonged tumor antigen exposure. Immunological synapses between T cells and tumor cells facilitate T‐cell receptor (TCR) clustering and signal amplification [27], which are critical for BiTE‐mediated tumor lysis [27, 28]. Activated T cells release perforin and granzymes through these synapses, inducing cancer cell lysis by forming pores in the cancer cell membrane and endosomal membranes [27].
BiTEs targeting antigens such as CD19, CD20, CD123, CD33, CD38, and BCMA offer alternative treatments for recurrent or refractory conditions [29]. Ideal target antigens are uniquely expressed on malignant cells, avoiding on‐target/off‐cancer toxicity and reducing antigen‐loss variants [29].
2.2. Clinical Efficacy
Clinical trials evaluating BsAbs such as mosunetuzumab [30, 31] and glofitamab [32], as well as epcoritamab [33] and odronextamab [34], have demonstrated encouraging efficacy outcomes in patients with RR‐FL, both as monotherapy and potentially in combinatorial regimens (Table 1). However, common concerns in BsAbs therapy involve adverse events (AEs) related to T‐cell overactivation. Mitigating these effects involves a systematic strategy, including step‐up dosing (SUD) of BsAbs, gradually increasing from minimal to full therapeutic doses. This approach is backed by robust preclinical evidence aiming to attenuate peak cytokine release while preserving antitumor efficacy. Additional measures such as adjusting infusion rates, prophylactic corticosteroids, and specific high‐risk dose protocols are implemented to manage potential risks effectively. Furthermore, incorporating single‐dose ZO pretreatment, which depletes circulating B cells, targets T‐cell activation to minimize adverse immunological responses. Adjusting BsAbs' affinity for CD3 receptors holds promise in reducing CRS severity without compromising therapeutic efficacy, thereby enhancing treatment outcomes. This integrated approach aims to optimize safety and efficacy in BsAb therapy, supported by rigorous preclinical rationale and ongoing clinical investigation [18].
TABLE 1.
Ongoing studies investigating combinatorial regimens.
| Combinations | Clinical trials | Population |
|---|---|---|
| Mosunetuzumab + polatuzumab vedotin + obinutuzumab | NCT05169658 | Untreated indolent B‐cell lymphomas |
| Tazemetostat + mosunetuzumab | NCT05994235 | Newly diagnosed follicular lymphoma |
| Glofitamab + obinutuzumab | NCT05783596 | Untreated follicular lymphoma |
| Mosunetuzumab + lenalidomide | NCT04712097 | Relapsed follicular lymphoma |
2.2.1. Mosunetuzumab
Mosunetuzumab, a pioneering CD20 × CD3 BsAb, targets CD20 on B cells and CD3 on T cells, featuring a full‐length humanized IgG1 structure.
The Phase I dose‐escalation study involved aggressive and indolent NHL patients treated with mosunetuzumab [30]. Group A included 33 patients in eight fixed‐dose cohorts (0.05–2.8 mg), with the maximum tolerated dose (MTD) not reached. Enrollment in Group A stopped in favor of Group B, which aimed to expand the therapeutic index using a SUD schedule to mitigate CRS. Group B enrolled 197 patients across 11 dose‐escalation cohorts (0.4/1.0/2.8–1.0/2.0/60 mg). Safety and efficacy were evaluated, leading to the selection of a recommended Phase 2 dose (RP2D) of 1/2/60/60/30 mg. Group B had a median study duration of 10.1 months, with patients receiving a median of five cycles. Common AEs included neutropenia (28.4%), CRS (27.4%), and hypophosphatemia (23.4%). Grade ≥ 3 AEs occurred in 71.1% of patients. CRS was observed in 27.4% of patients, mostly of Grade 1 and 2 severity. Notably, only 2% (2 out of 90) of patients experienced Grade 3 and 4 CRS, which primarily occurred during the early treatment cycles. Additionally, there were no reported instances of Grade 3 and 4 neurotoxic AEs. Efficacy outcomes in Group B showed an ORR of 34.9% in aggressive NHL and 66.2% in indolent NHL, with complete response (CR) rates of 19.4% and 48.5%, respectively. The median duration of response was 7.6 months for aggressive NHL and 16.8 months for indolent NHL. Among 65 evaluable FL patients in Group B, the ORR was documented in 45 cases (69.2%, 95% CI: 56.6–80.1), with a CR rate detected in 33 patients (50.8%, 95% CI: 38.1–63.4). Furthermore, the data for high‐risk FL patients treated with mosunetuzumab in Group B are compelling. Among FL patients who had prior CAR‐T therapy, all four evaluable patients responded, with an impressive 100% ORR and 50% CR rate. Double refractory FL patients had a 67.6% ORR and a 55.9% CR rate, whereas those with a history of progressive disease within 24 months (POD24) showed a 75.8% ORR and 54.5% CR rate. Additionally, FL patients, refractory to PI3Ki, had the highest responses, with an 88.9% ORR and 77.8% CR rate. The median PFS across all dose levels for aggressive and indolent B‐NHL was 1.4 months (95% CI: 1.4–2.9) and 11.8 months (95% CI: 8.4–NE), respectively. These results demonstrate the substantial clinical benefit of mosunetuzumab in treating RR‐FL with an acceptable and manageable safety profile, even across challenging, high‐risk subgroups [30].
The approval of mosunetuzumab was based on results from an international, multicenter, Phase 2 trial in 90 patients with RR‐FL following at least two prior lines of systemic therapy [31]. Table 2 provides a summary of the results, highlighting the significant findings of the study. Eligibility criteria included patients who had undergone a minimum of two prior lines of therapy. Notably, over 50% of the study population exhibited disease progression within 24 months of initiating initial therapy or were doubly refractory to rituximab and an alkylating agent, and approximately 20% had a history of autologous hematopoietic stem cell transplantation. The trial results demonstrated that single‐agent mosunetuzumab achieved an overall response rate (ORR) of approximately 80%, with a notable complete remission rate of 60%. The median time to response was 1.4 months, and the median time to achieve complete remission was 3.0 months. The median duration of response was 22.8 months, with a median PFS of about 1.5 years. Importantly, mosunetuzumab exhibited significant activity in patients with early disease progression post‐initial therapy or those doubly refractory.
TABLE 2.
Evolution and advancements in CAR T‐cell generations.
| Generation | Key features | Advantages | Limitations |
|---|---|---|---|
| First | scFvs binding to the structural transmembrane domain of CD3ζ | Tumor‐specific cytotoxicity | Limited in vivo persistence; insufficient activation; poor cytokine production |
| Second | Incorporation of co‐stimulatory molecules (e.g., CD28 or 4‐1BB) along with CD3ζ | Enhanced antitumor efficacy; improved persistence and proliferation in vivo | Some safety concerns; potential for immune‐related side effects |
| Third | Inclusion of two co‐stimulatory molecules (e.g., CD28 and 4‐1BB) in addition to CD3ζ | Further improved antitumor effects; better persistence and activation | Increased complexity; higher risk of adverse immune reactions |
| Fourth | Addition of co‐stimulatory ligands (e.g., CD28, CD137, and CD134); Introduction of cell suicide genes (e.g., HSV1‐tk or iCaspase9) | Enhanced cytotoxicity; precise regulation of CAR T‐cell activity; reduced toxic side effects | More complex genetic modifications; potential for regulatory challenges |
| Fifth | Universal CARs for allogeneic therapies; knockdown of endogenous TCR and MHC molecules using gene editing (e.g., CRISPR/Cas9); knockdown of T‐cell suppressor signaling molecules (e.g., PD‐1 and CTLA4); disassembly of T‐cell signaling region for additional antigen recognition | Broader applicability; elimination of GvHD and HVGR; improved survival and cytotoxicity; potential for cost reduction in immunotherapy | Risk of severe off‐target effects; ethical and safety concerns related to gene editing; complexity in manufacturing and regulation |
Safety profiles are critical for the acceptance of novel therapies in indolent NHL‐like FL. Concerns regarding CRS and neurotoxicity, common with BsAbs, were mitigated in this study. Only 2% of patients experienced Grade 3 and 4 CRS, predominantly during early treatment cycles, and no Grade 3 and 4 neurotoxic events were reported.
The study by Budde et al. [30, 31] introduces a potent new treatment for RR‐FL, characterized by ease of administration and suitability for outpatient care because of its manageable toxicity profile. These findings have resulted in mosunetuzumab receiving a positive recommendation for third‐line treatment of FL by the Committee for Medicinal Products for Human Use of the European Medicines Agency (EMA) in April 2022, and subsequently by the FDA, marking a potential shift in treatment paradigms for later‐line therapy [35, 36]. Moreover, at the American Society of Hematology (ASH) meeting Bartlett et al. [37] provided updated results from the pivotal Phase 2 study of mosunetuzumab after a median follow‐up of 28.3 months. The 24‐month PFS, OS, and duration of CR were 48% (95% CI: 36–60), 87% (95% CI: 80–94), and 63% (95% CI: 38–88), respectively. Interestingly, whole‐exome sequencing of 51 available baseline lymphoma samples revealed that patients with common mutations, including those associated with poor prognosis such as TP53, KMT2D, EZH2, and BCL‐2, exhibited clinically meaningful response rates. No new serious AEs, Grade ≥ 3 AEs, or treatment‐related AEs were reported during the additional 10 months of follow‐up compared to the original report. CRS of all grades was observed in 44% of patients, with 26% experiencing Grade 1 CRS and 17% experiencing Grade 2 CRS. There was no correlation between the occurrence of CRS and tumor response. Mosunetuzumab‐related AEs led to treatment discontinuation in 2% of patients.
In the same ASH meeting, McGough et al. [38] reported data on an external control cohort study utilizing real‐world data from US patients with RR‐FL who received third‐line or later treatments and met the eligibility criteria of the pivotal Phase 2 trial that led to the approval of mosunetuzumab [38]. The study found a significant treatment benefit associated with mosunetuzumab, evidenced by an increased CR rate (odds ratio [OR], 3.18; 95% CI: 1.41–7.17) and improved OS (HR, 0.43; 95% CI: 0.19–0.94), thereby supporting the use of mosunetuzumab in this clinical setting.
Morschhauser et al. [39] reported a CR rate of 77% in a Phase 1b study evaluating mosunenetuzumab plus lenalidomide in patients with relapsed or refractory follicular lymphoma (R/R FL). This report paved the way for a new Phase 3 trial (NCT04712097). Olszewski et al. [40] reported a trial in progress of the same combination in front‐line use without additional AEs. Administering mosunetuzumab as a first‐line treatment could reduce reliance on chemotherapy, lowering toxicity while maintaining or enhancing efficacy. This approach also leverages the immune system's full capacity before it is weakened by multiple therapies, offering a strategic advantage. Nevertheless, any enhancement in immune activation could potentially increase the risk of immune‐related toxicities, attributable to direct T‐cell activation and on‐target off‐tumor toxicities. Additionally, preliminary results of a Phase 2 multicenter trial evaluating subcutaneous mosunetuzumab in patients with untreated high‐burden FL were recently reported [41]. Among the 43 patients enrolled, 26 were evaluated, revealing an impressive CR rate of 81%. The ORR was 96%, which remained consistent across patients with high‐risk features, including those classified as high‐risk according to FLIPI, those with Grade 3A FL, patients with bulky disease, and those exhibiting a SUVmax ≥ 13).
2.2.2. Glofitamab
A detailed analysis of the structure, mechanism of action, and pharmacokinetics of glofitamab has been provided by Minson and Dickinson [42]. The T‐cell binding domain recognizes the CD3ε chain of the TCR receptor with the consequent T‐cell activation, which occurs regardless of TCR specificity and without the need for co‐stimulation.
A Phase 1 study involved 171 adults with CD20+ B‐cell NHL (B‐NHL), including RR‐FL, exposed to a median of three prior therapies [43]. To reduce circulating B cells and mitigate the risk of CRS, patients received a 1000 mg dose of pretreatment ZO, followed by either fixed or SUD of intravenous glofitamab every 2–3 weeks. Glofitamab showed dose‐dependent clinical activity starting at 0.6 mg. SUD allowed for 25 mg escalation, resulting in fewer cases of Grade ≥ 2 CRS (28.6% vs. 47.8%), making SUD the RP2D.
At the ASH meeting, updated results of glofitamab with three different SUD regimens, as monotherapy (mono) or combined with ZO (combo), were presented [44]. Glofitamab was given intravenously in three mono cohorts and one combo cohort following SUD regimens. The ORR in the mono cohorts was 81%, with a complete metabolic response (CMR) rate of 70%. The combo cohort had a 100% ORR and 73.7% CMR rate. In the mono cohorts, 87% achieved CMR, compared to 71% in the combo cohort. Median follow‐up was insufficient to assess CMR duration and survival in terms of PFS and OS. The most frequent AE was CRS, occurring in 66% (mono) and 79% (combo). CRS events were mainly Grade 1 or 2, with no Grade 4 or 5. Tocilizumab managed CRS in 22.9% (mono) and 33.3% (combo). All CRS events were resolved by data cut‐off. Neurologic AEs were mostly low grade and not ICANS‐like. Other common AEs included infusion reactions, pyrexia, neutropenia, anemia, and thrombocytopenia.
As for the other BsAb, an increased risk of early and late infections, because of treatment‐related cytopenia, hypogammaglobulinemia, “off‐tumor on‐target” effect of B‐cell hypoplasia, and T‐cell exhaustion, has been shown. Therefore, patients should be closely monitored and supported with growth factor therapy, anti‐microbial prophylaxis (e.g., for herpes virus and Pneumocytis jiroveci), and immunoglobulin replacement, to prevent this side effect [45].
2.2.3. Epcoritamab
Epcoritamab, a subcutaneous CD3 × CD20 BsAb, addresses the need for effective treatments in patients with RR‐FL, especially those with high‐risk disease. Approved by the FDA for RR diffuse large B‐cell lymphoma (DLBCL) and high‐grade BCL after ≥ 2 lines of systemic therapy, its efficacy was investigated in the FL dose‐expansion cohort of the EPCORE NHL‐1 trial (NCT03625037; Phase 1/2). In this trial, 128 patients with CD20+ RR‐FL of systemic therapy were treated with escalating doses of epcoritamab during the first cycle, followed by 48 mg in 28‐day cycles until disease progression or unacceptable toxicity. The study enrolled 128 RR‐FL patients who had received ≥ 2 prior lines of therapy. Results showed an ORR of 82%, with a CR rate of 63%. The median PFS was 15.4 months. Common treatment‐emergent AEs (TEAEs) included CRS (66%), injection‐site reactions (57%), and COVID‐19 (40%). CRS was mainly low grade and occurred after the first full dose. No CRS events led to treatment discontinuation. Epcoritamab SC demonstrated deep, durable responses and a manageable safety profile, with no new safety signals detected [33]. Data from analysis of Arm 6 of EPCORE NHL‐2 (NCT04663347), a trial investigating the safety and effectiveness of epcoritamab as monotherapy or in association with other therapies, were recently published. The Arm 6 included treatment‐naive FL patients undergoing epcoritamab + R2 as first‐line therapy. The ORR was 90% (26/29), with 69% (20/29) achieving a CMR, and the safety profile was manageable, supporting a possible future role in earlier lines of therapy [46]. Finally, an ongoing Phase 3 trial (NCT05409066) continues to explore its efficacy in various FL treatment settings.
2.2.4. Odronextamab
Odronextamab, a human IgG4‐based CD20 × CD3 BsAb, binds CD20 on B cells and CD3 on T cells. The ELM‐2 study (Ph2, NCT03888105) [34] assessed odronextamab in patients with FL Grade 1–3a, refractory to ≥ 2 prior lines of therapy, including anti‐CD20 antibody and alkylator. Odronextamab was administered in 21‐day cycles with steroid prophylaxis and SUD to mitigate toxicity. In the earlier ELM‐1 study (Ph1, NCT02290951) [47], patients treated with ≥ 5 mg of odronextamab had an ORR of 91% and a CR rate of 72%, with a 4‐year PFS rate of 54%. In ELM‐2, the optimized step‐up regimen aimed to maintain efficacy while reducing CRS. ELM‐2 enrolled 96 patients for safety evaluation and 85 for efficacy, with a median age of 59 years. Most were refractory to their last therapy (74%) and had progression within 2 years (POD24) (48%). The ORR and CR rates by independent review were 81% and 75%, respectively, consistent across high‐risk subgroups. Responses were durable, with a median duration of response and CR of 18.2 months, and a median PFS was 20.2 months. Common TEAEs included CRS (51%), pyrexia (32%), and anemia (31%). The 0.7/4/20 step‐up regimen reduced CRS severity, with only Grade 1 CRS observed in 39% of patients, and no cell‐associated neurotoxicity syndrome (ICANS) was reported. To date, only one study reported deaths directly attributable to odronextamab, consisting of gastric perforation in a patient with gastric involvement by lymphoma, lung infection, pneumonia, and tumor‐lysis syndrome [48]. Odronextamab demonstrated significant efficacy and a manageable safety profile, showing promise for heavily pretreated, R/R FL patients [34].
Both epcoritamab and odronextamab are anticipated to be the next BsAbs approved for the treatment of FL. However, the timeline for regulatory approval and their competitive positioning relative to mosunetuzumab remains to be determined. Mosunetuzumab appears to offer advantages over odronextamab and epcoritamab because of its more favorable SUD regimen, defined treatment duration, and current recommendations for outpatient administration. Epcoritamab, in particular, currently carries a recommendation for hospitalization, further influencing its comparative utility in clinical practice.
3. CAR‐T
3.1. Structures and Mechanisms
CAR‐T therapy, a leading immunotherapy for various cancers, involves the genetic modification of T cells to express CARs, enabling these engineered cells to recognize and destroy cancer cells effectively. CAR‐T therapy has shown remarkable progress, particularly through its multiple generations, each enhancing the efficacy, persistence, and safety of the treatment. The origins of CAR‐T therapy trace back to Zelig Eshhar's introduction of the “chimeric antigen receptor (CAR)” paradigm in 1989 and Carl June's successful treatment of a refractory and relapsed patient, Emily, in 2012 [49].
The structural components of CARs, including the scFv antibody, hinge region, transmembrane domains, and intracellular signaling domain, are meticulously designed to ensure robust tumor antigen recognition and subsequent activation of T cells [50].
The initial generation of CAR‐T cells demonstrated tumor‐specific cytotoxicity but had limited in vivo effectiveness because of inadequate persistence and poor cytokine production [51]. To address these issues, the second and third generations incorporated co‐stimulatory molecules, such as 4‐1BB, significantly enhancing the antitumor efficacy, persistence, and proliferation of CAR‐T cells in vivo [52]. The fourth generation further advanced these capabilities by adding co‐stimulatory ligands and cell suicide genes, allowing precise regulation and reducing toxic side effects [53]. Building on this, the fifth generation shifted focus to developing universal CARs for allogeneic therapies, aiming to eliminate GvHD and MHC‐mediated HVGR by knocking down endogenous TCR and MHC genes using gene‐editing technologies like CRISPR/Cas9 [54]. This generation also focused on enhancing survival and cytotoxicity by targeting T‐cell suppressor signaling molecules such as PD‐1 and CTLA4, thereby broadening CAR‐T applicability [48]. Table 2 summarizes the evolution and advancements in CAR‐T generations.
CAR‐Ts operate by forming immune synapses with tumor cell antigens, prompting the secretion of substances like perforin and granzyme, which create pores in the target cell membranes, leading to cell lysis. Additionally, CAR‐Ts can induce apoptosis in target cells via the Fas–FasL pathway [48].
A salient challenge tethered to universal CAR‐T therapy is the potential for immune rejection by the host, coupled with unintended cytotoxicity against host tissues. Strategies like the abrogation of the TCR and human leukocyte antigen (HLA) on CAR‐Ts have been postulated to circumvent these impediments, aiming to obliterate the immunogenicity of allogeneic T cells. Moreover, creating an optimal immune environment within the host, often via preconditioning chemotherapy regimens, is postulated as an effective approach to mitigate immune rejection [54]. However, a definitive protocol delineating the optimal chemotherapy regimen and the temporal window for CAR‐T administration post‐chemotherapy remains elusive in lymphoproliferative disorders. Recent research in a murine model showed that cyclophosphamide and fludarabine (FLU/CY) chemotherapy, followed by CD19 CAR‐T cell therapy, is more effective against B‐cell malignancies than either chemotherapeutic agent alone. The strongest anticancer effect was observed when CAR‐T cells were administered 5 days after the combined chemotherapy, likely due to disrupted mitochondrial metabolism in tumor cells [55]. However, a recent paper revealed that although both FLU/CY and bendamustine regimens created a favorable cytokine environment for T‐cell engraftment and expansion, Flu/Cy was linked to more pronounced increases in cytokines associated with CAR‐T‐related side effects [56]. This may explain the higher incidence of CRS and ICANS with FLU/CY.
3.2. Clinical Efficacy
3.2.1. Axicabtagene Ciloleucel
Axicabtagene Ciloleucel (axi‐cel) approved for RR‐FL patients after multiple lines of therapy [57] has shown impressive efficacy outcomes in clinical trials. In the pivotal ZUMA‐5 study, which enrolled 124 patients with R/R FL, 95% of efficacy‐evaluable patients achieved an overall response, with 79% achieving a CR [58]. Notably, these responses were observed in a cohort with advanced disease characteristics: 63% of patients had received three or more lines of prior therapy, and a significant proportion had high‐risk disease features such as high tumor burden and refractory disease [59]. Long‐term follow‐up data demonstrated a median duration of response of 38.6 months and a 36‐month OS rate of 75% [60]. For patients with RR‐FL, the median PFS was 40.2 months, with a 36‐month PFS rate of 54%. There were 41 progression or death events (32%) due to lymphoma or treatment, with a 36‐month cumulative incidence rate of 35%. Competing risks (non‐lymphoma deaths) occurred in 12 patients (9%), with a 36‐month cumulative incidence of 11% [60]. The median PFS for patients with POD24 was 40.2 months, whereas it was not reached for those without POD24. Prior bendamustine use was associated with a lower 36‐month PFS rate, especially if administered within 6 months of leukapheresis.
The median OS was not reached, with an estimated 36‐month OS rate of 76%. The 36‐month cumulative incidence of lymphoma‐specific death was 13%, with 12% for competing risks. Patients with lower baseline total metabolic tumor volume (TMTV) had longer PFS and DOR. The 36‐month PFS rate was 71.2% for those below the median TMTV compared to 37.3% for those above. No significant correlation was found between TMTV and ORR or CR. Finally, the results suggest that bendamustine‐based therapies administered within 6 months prior to infusion should be carefully considered for patients who are likely to need CAR‐T therapy soon, especially those at high risk. With over 3 years of follow‐up, the ZUMA‐5 study shows that axi‐cel provides durable remissions for RR‐FL, with more than half of the patients in ongoing response. These results underscore the durable efficacy of axi‐cel in this challenging patient population [60]. The safety profile of axi‐cel is characterized by manageable toxicities, primarily CRS and neurotoxicity. In ZUMA‐5, 78% of patients experienced CRS, with 6% experiencing severe CRS (Grade 3 or higher) [59]. Prompt management with tocilizumab and corticosteroids effectively mitigated these AEs in most cases [61]. Neurologic events occurred in 56% of patients, with 41% experiencing mild to moderate neurotoxicity (Grade 1 and 2) and 15% severe neurotoxicity [59]. In the ZUMA‐22 Phase 3 trial, the efficacy and safety of axi‐cel compared with standard‐of‐care therapies, that is, rituximab plus lenalidomide, rituximab plus CHOP, or rituximab plus bendamustine, is currently being assessed in patients with RR‐FL [62].
3.2.2. Tisagenlecleucel
In an earlier pilot study of tisagenlecleucel (tisa‐cel) in 14 RR‐FL patients, 71% achieved CR, with a median DOR not reached after more than 5 years. The likelihood of maintaining the response for 5 years was 60% (95% CI: 25–83) [63]. The ELARA study, involving 98 RR‐FL patients, reported a CR rate of 69.1% and an ORR of 86.2% [63]. Participants in the ELARA trial had a median of four prior therapy lines and exhibited high‐risk features, such as advanced stage and bulky disease.
Tisa‐cel showed a lower incidence of severe CRS compared to axi‐cel. In the ELARA trial, 49% of patients experienced CRS, with 34% requiring tocilizumab and 6.4% requiring corticosteroids. Neurologic events occurred in 37.1% of patients, with 4.1% experiencing severe neurotoxicity. Notably, 18% of patients received tisa‐cel in an outpatient setting, suggesting its potential for outpatient administration.
Long‐term data after a median follow‐up of 29 months showed that median PFS, DOR, and OS were not reached. The estimated 24‐month rates were 57.4% for PFS, 66.4% for DOR, and 87.7% for OS. The CR rate was 68.1%, and the ORR was 86.2%. No new safety concerns or treatment‐related deaths were reported [64].
Improved outcomes were associated with low levels of tumor‐infiltrating LAG3 + CD3+ exhausted T cells and higher baseline levels of naive CD8+ T cells. Tis‐cel demonstrated durable efficacy and a favorable safety profile over 29 months, suggesting that it may offer a favorable safety profile, particularly in managing CRS and neurotoxicity compared to other CAR‐T therapies [64].
3.2.3. Lisocabtagene Maraleucel
The TRANSCEND FL study is the largest evaluation of CAR‐T therapy for R/R FL, and the first to report outcomes in 2L R/R FL patients [65]. Participants were primarily at an advanced stage, with 82% at high or intermediate FLIPI risk, and 56% meeting the POD24 criteria, while 62% were double‐refractory, and 38% received bridging therapy. The median age was 60, consistent with Phase 2 studies of other CAR T therapies like axi‐cel and tis‐cel. The median time from leukapheresis to lisocabtagene maraleucel (liso‐cel) availability was 29 days, and the median time to infusion was 49 days. As of January 27, 2023, the median follow‐up was 18.9 months. The study achieved both primary and key secondary endpoints, showing high efficacy across different therapy lines. Liso‐cel demonstrated an ORR of 97% and a CR rate of 94% in R/R FL patients with more than three lines of therapy. These high response rates were consistent across subgroups, including those with high‐risk features and double‐refractory disease. For patients exposed to two lines of therapy who met the POD24 criteria, the ORR was 96%, with all responders achieving CR. Responses were rapid, with a median time to response of 1 month, and the median DOR and PFS were not reached at a median follow‐up of around 17 months. Efficacy was similar across subgroups, regardless of bridging therapy status. Safety profiles were manageable, with CRS occurring in 58% of patients, mostly Grades 1 and 2, and manageable with supportive care. Grade ≥ 3 infections were rare, and Grade ≥ 3 cytopenias were common but resolved by Day 90 after treatment. In conclusion, liso‐cel showed robust efficacy and manageable safety in heavily pretreated FL patients, with high and durable responses. Liso‐cel received FDA grants accelerated approval in May 2024 [66].
4. Comparative Analysis
A comparative analysis is crucial for informed decision‐making by clinicians, patients, and healthcare providers, allowing for the evaluation of different treatments' benefits, risks, and cost analyses. It helps choose the most suitable therapy based on individual patient circumstances and ensures efficient resource allocation.
A study by Ray et al. [67] aimed to compare the effectiveness and safety of axi‐cel (ZUMA‐5 trial) and mosunetuzumab (GO29781 trial, NCT02500407) in treating patients with RR‐FL who had received at least two prior systemic treatments. Because no direct head‐to‐head trials exist, the authors utilized unanchored matching‐adjusted indirect comparisons to assess their relative efficacy. This comparative analysis suggests that axi‐cel generally leads to superior efficacy compared to mosunetuzumab, with 12‐month PFS rates of 81.7% (95% CI: 74.8–89.2) for axi‐cel, and 55.9% (95% CI: 46.0–67.9) for mosunetuzumab. A greater difference was observed in terms of 2‐year PFS, with 71.1% (95% CI: 63.0–80.4) for axi‐cel, and 29.5% (95% CI: 15.5–56.3) for mosunetuzumab. Notably, axi‐cel requires a single infusion, unlike mosunetuzumab, which is administered in multiple cycles. Safety analysis revealed higher rates of all‐grade AEs with axi‐cel, though rates of Grade 3 and 4 CRS and treatment‐related AEs were statistically similar to those of mosunetuzumab. Although axi‐cel showed promising efficacy advantages, the comparison for OS was limited due to less mature data from the mosunetuzumab trial.
It is important to note that a primary limitation of the MAIC analysis is the relatively short median follow‐up period of 24 months, which may not adequately capture long‐term risks such as nonrelapse mortality (NRM). This limited follow‐up duration may overlook late‐onset toxicities and treatment‐related mortalities that can emerge over time. This concern is particularly pertinent for therapies such as CAR‐T, where late complications, including NRM, may manifest only after prolonged follow‐up. The wider CIs observed for toxicities in this preliminary data may reflect the constraints imposed by the limited sample size and follow‐up duration. Therefore, longer term data will be essential for a more comprehensive evaluation of the long‐term safety profile, especially regarding the risks associated with NRM.
Finally, logistic challenges and treatment availability differences were also noted between the two therapies, with fewer centers offering CAR‐T therapies compared to BsAbs like mosunetuzumab.
Similarly, Nastoupil et al. [68] conducted an unanchored MAIC comparing liso‐cel and mosunetuzumab for third‐line or later RR‐FL. Using patient data from TRANSCEND FL (NCT04245839; median follow‐up of 19.3 months) and GO29781 (median follow‐up of 18.3 months), they demonstrated that liso‐cel exhibited superior efficacy and a favorable safety profile compared to mosunetuzumab. Liso‐cel showed lower rates of severe CRS and serious infections while demonstrating higher incidences of any‐grade CRS and neurological events. These findings suggest a positive benefit–risk profile for the treatment of R/R FL with liso‐cel. Similar to the prior comparative study [69], the MAIC analysis comparing mosunetuzumab and liso‐cel is constrained by a follow‐up period of less than 24 months. Given the nature of these therapies, an extended follow‐up duration is essential to adequately evaluate durable responses, late‐onset toxicities, and OS outcomes. A follow‐up period of under 24 months may not sufficiently capture the long‐term efficacy or safety of these treatments. Consequently, the conclusions drawn from this comparison should be interpreted with caution. Further studies incorporating extended follow‐up are necessary to gain a more comprehensive understanding of the long‐term performance of these therapies.
Cost‐effectiveness analysis compares clinical and cost outcomes between treatment options, producing an incremental cost‐effectiveness ratio (ICER) that measures costs per health benefit, typically in quality‐adjusted life years (QALYs). Such analyses can guide treatment decisions in RR‐FL. A study initially presented at the ASH meeting [58] and later published in full [58] found that axi‐cel may be cost‐effective compared to mosunetuzumab at a US willingness‐to‐pay threshold of $150 000 per QALY. The base case ICER for axi‐cel was $108 307 per QALY, with 64% of probabilistic analyses falling below this threshold. Key factors affecting results were patient age and quality of life utility values, highlighting the importance of early axi‐cel treatment to maximize survival benefits. Axi‐cel extends PFS, improving quality of life and potentially offsetting costs over a lifetime. The ICER would decrease to $102 695 per QALY based on 48‐month data [58].
These findings contrast with recent studies on the cost‐effectiveness of mosunetuzumab for third‐line and beyond RR‐FL in the United States. Two studies, published in abstract form [70, 71], found that mosunetuzumab is either cost‐effective or superior to axi‐cel and other comparator treatments, except for the combination of rituximab and lenalidomide. In the Lin study [70], mosunetuzumab was shown to be more cost‐effective than both axi‐cel and tisa‐cel at 1, 2, 5, and 10 years for treating RR‐FL. Even though axi‐cel had better QALYs at 10 years due to improved long‐term PFS, mosunetuzumab remained cost‐effective. Similarly, Matasar's study [71] compared mosunetuzumab with seven treatments, including axi‐cel and tisa‐cel, using efficacy data from the GO29781 trial and real‐world cohort data. The cost calculations, adjusted to 2022 US dollars, included treatment costs, AEs, and routine and terminal care. Mosunetuzumab showed dominance over some treatments and cost‐effectiveness against others, with ICERs ranging from $21 521 to $78 604 per QALY.
The comparison of different analyses reveals that each study employs distinct methodologies and assumptions, which significantly impact their outcomes and conclusions. Matasar et al. [71] used a three‐state partitioned survival model and an MAIC, but key details like matching methods and hazard ratios were not fully disclosed, making it difficult to thoroughly assess their approach. Their assumption of no cure effect for treatments like mosunetuzumab and CAR‐T therapies might underestimate the benefits of CAR‐T, despite evidence supporting cure effects. In contrast, the study cited by Lin et al. [70] lacks clarity on how treatment efficacy was adjusted for different trial populations and does not provide insights into assumptions for long‐term extrapolation. This is evident in their estimated 5‐year PFS rates, which differ significantly from those observed in long‐term ZUMA‐5 follow‐up data, potentially underestimating the value of axi‐cel in their analysis.
In the Oluwole study [72], assumptions were more closely aligned with published evidence by considering both the potential cure effects of CAR‐T therapies and the shorter PFS associated with mosunetuzumab. This approach offers a nuanced understanding of treatment benefits, using comprehensive data and published findings. The analysis was based on inpatient treatment for axi‐cel patients, consistent with the ZUMA‐5 trial, but the findings should be viewed in light of evolving treatment patterns, such as the increasing outpatient administration of CAR‐T therapies [72, 73] and monoclonal antibodies [74]. CAR‐T trials were primarily conducted in inpatient settings to manage AEs like CRS and ICANS. Future cost‐effectiveness analyses should incorporate real‐world evidence on costs and AEs in outpatient CAR‐T therapy [75].
Further research, including head‐to‐head trials and real‐world studies, is crucial to refine treatment strategies and optimize patient outcomes in RR‐FL.
5. Conclusion
Until recently, a definitive standard of care for RR‐FL had not been established, as evidenced by the findings of the SCHOLAR‐5 trial. However, the advent of CAR‐T therapy and BsAbs has shown significant efficacy in the setting of this hard‐to‐treat population [69]. Nevertheless, the ongoing debate over the selection and sequencing of CAR‐T therapy versus BsAbs for RR‐FL underscores the complexity of treatment decisions in this rapidly evolving field.
Figure 1 summarizes the key differences of several critical aspects that influence the choice between BsAbs and CAR‐T therapy in treating RR‐FL.
FIGURE 1.
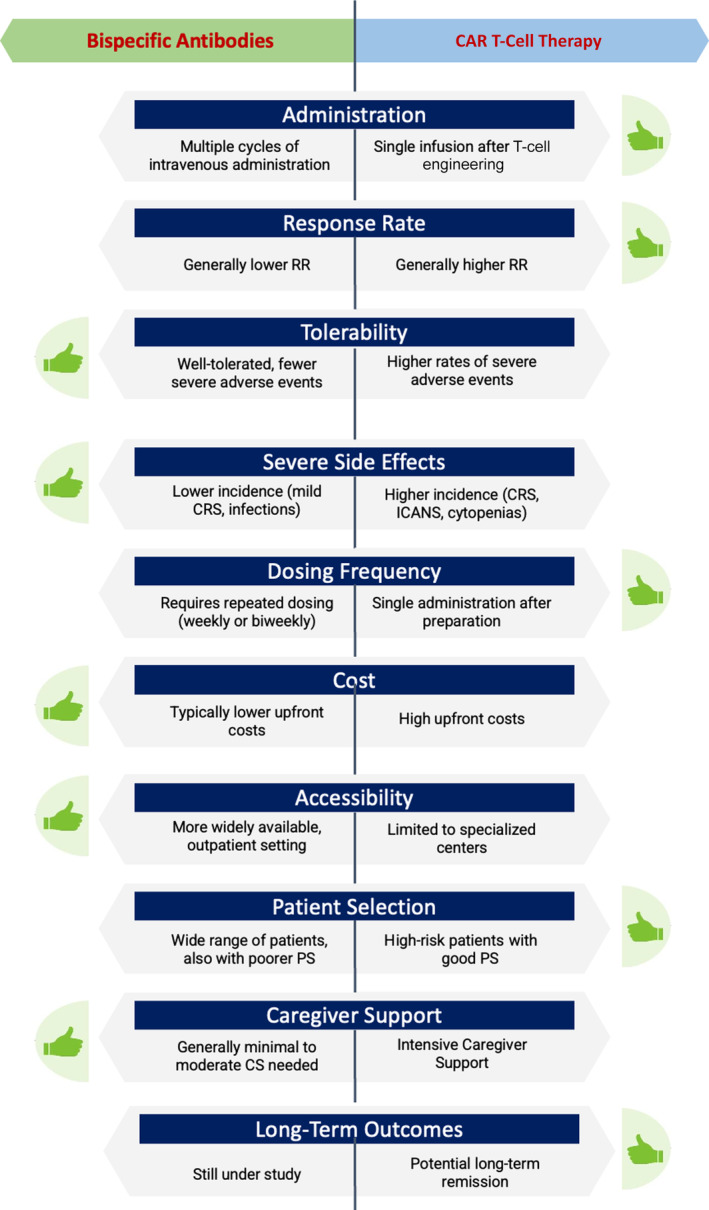
Key differences between bispecific antibodies and CAR T‐cell therapy in the treatment of relapsed/refractory follicular lymphoma.
Both modalities share similar immunobiology and molecular targets, positioning them as crucial components and viable alternatives. BsAbs generally offer improved tolerability with a lower incidence of severe treatment‐related AEs. This favorable safety profile suggests a possible utilization before CAR‐Ts, particularly given their high response rates and reduced occurrence of immune‐related toxicities, despite the requirement for more frequent dosing. In this regard, glofitamab and mosunetuzumab have been investigated in fixed duration schedules, demonstrating benefits in terms of patient quality of life. In contrast, epcoritamab and odronextamab are administered continuously until progression or the emergence of unacceptable toxicity.
CAR‐T therapy is known for achieving higher response rates and offers the potential for improved outcomes with a single infusion; however, extended follow‐up is warranted to confirm its benefits in terms of OS. Currently, there is limited information available concerning NRM following CAR‐T therapy. A recent meta‐analysis investigated the incidence of NRM in patients with lymphoproliferative disorders who received CAR‐T therapy. The analysis revealed that the incidence of NRM in indolent lymphomas is approximately 6%, with the primary causes being infections, secondary malignancies, and cardiovascular events [76]. Given these considerations, early initiation of CAR‐T therapy is recommended for high‐risk patients, especially those experiencing early relapse following 2L therapy (POD24), as well as for young patients with minimal comorbidities.
Conversely, the CAR‐T approach is associated with more frequent and severe side effects such as CRS, ICANS, and cytopenias. In this respect, patients with better performance status may benefit more from axi‐cel due to its CD28 co‐stimulatory domain, while those with poorer status may find tisa‐cel more suitable with its 4‐1BB co‐stimulatory domain.
Additionally, the intensive caregiver support required for CAR‐T therapy is a notable drawback, adding psychosocial stress and potential caregiver burden, including burnout risks. Accessibility is further limited to certified academic treatment centers, and the high cost of a single infusion heavily influences treatment decisions.
With methodological differences across studies, attempts for cost‐effectiveness analyses seem to favor axi‐cel at certain thresholds, although some studies find mosunetuzumab more cost‐effective in specific scenarios [58, 68, 70, 71]. Overall, these findings emphasize the importance of integrating clinical and economic considerations in treatment decisions for RR‐FL and suggest further research to refine therapeutic strategies and optimize patient outcomes.
Two critical factors in selecting and sequencing CAR‐T therapies and BsAbs are T‐cell exhaustion and antigen loss, both actively investigated in current research. Antigen loss variants necessitate ongoing monitoring and potentially sequential or combination therapies to address resistant clones and maintain treatment efficacy [77].
Future research should focus on optimizing CAR design to improve efficacy, reduce toxicity, and maximize durable remissions by enhancing in vivo durability and minimizing antigen escape. Integrating functional genomics with bioengineering, including novel armoring systems, is crucial for advancing CAR‐T therapy. Future advancements in understanding the genetics and immunobiology of RR‐FL, particularly regarding mechanisms of resistance, are expected to drive rational therapeutic developments, potentially leading to improved patient outcomes in the years ahead.
Author Contributions
All authors contributed to the manuscript and were involved in revisions and proofreading. All authors approved the submitted version.
Conflicts of Interest
The authors declare no conflicts of interest.
Fortunato Morabito and Enrica Antonia Martino contributed equally to the first name.
Funding: The authors received no specific funding for this work.
Considering the new therapeutic frontiers of relapsed/refractory follicular lymphoma, we have conducted an accurate review of the literature to provide a comparison in terms of efficacy, toxicity, and cost‐effectiveness between BsAbs and CAR‐T therapy.
Contributor Information
Fortunato Morabito, Email: f.morabito53@grade.it.
Massimo Gentile, Email: massim.gentile@tiscali.it.
Data Availability Statement
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.
References
- 1. Freedman A., “Follicular Lymphoma: 2018 Update on Diagnosis and Management,” American Journal of Hematology 93 (2018): 296–305. [DOI] [PubMed] [Google Scholar]
- 2. Alaggio R., Amador C., Anagnostopoulos I., et al., “The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms,” Leukemia 36, no. 7 (July 2022): 1720–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ardeshna K. M., Qian W., Smith P., et al., “Rituximab Versus a Watch‐And‐Wait Approach in Patients With Advanced‐Stage, Asymptomatic, Non Bulky Follicular Lymphoma: An Open‐Label Randomised Phase 3 Trial,” Lancet Oncology 15 (2014): 424–435. [DOI] [PubMed] [Google Scholar]
- 4. Solal‐Céligny P., Bellei M., Marcheselli L., et al., “Watchful Waiting in Low‐Tumor Burden Follicular Lymphoma in the Rituximab Era: Results of an F2‐Study Database,” Journal of Clinical Oncology 30 (2012): 3848–3853. [DOI] [PubMed] [Google Scholar]
- 5. Nastoupil L. J., Sinha R., Byrtek M., et al., “Outcomes Following Watchful Waiting for Stage II–IV Follicular Lymphoma Patients in the Modern Era,” British Journal of Haematology 172 (2016): 724–734. [DOI] [PubMed] [Google Scholar]
- 6. Luminari S., Ferrari A., Manni M., et al., “Long‐Term Results of the FOLL05 Trial Comparing R‐CVP Versus R‐CHOP Versus R‐FM for the Initial Treatment of Patients With Advanced‐Stage Symptomatic Follicular Lymphoma,” Journal of Clinical Oncology 36 (2018): 689–696. [DOI] [PubMed] [Google Scholar]
- 7. Rummel M. J., Niederle N., Maschmeyer G., et al., “Bendamustine Plus Rituximab Versus CHOP Plus Rituximab as First‐Line Treatment for Patients With Indolent and Mantle‐Cell Lymphomas: An Open‐Label, Multicentre, Randomised, Phase 3 Non‐inferiority Trial,” Lancet 381 (2013): 1203–1210. [DOI] [PubMed] [Google Scholar]
- 8. Flinn I. W., van der Jagt R., Kahl B. S., et al., “Randomized Trial of Bendamustine‐Rituximab or R‐CHOP/R‐CVP in First‐Line Treatment of Indolent NHL or MCL: The BRIGHT Study,” Blood 123 (2014): 2944–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bachy E., Seymour J. F., Feugier P., et al., “Sustained Progression‐Free Survival Benefit of Rituximab Maintenance in Patients With Follicular Lymphoma: Long‐Term Results of the PRIMA Study,” Journal of Clinical Oncology 37 (2019): 2815–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hill B. T., Nastoupil L., Winter A. M., et al., “Maintenance Rituximab or Observation After Frontline Treatment With Bendamustine‐Rituximab for Follicular Lymphoma,” British Journal of Haematology 184 (2019): 524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morschhauser F., Nastoupil L., Feugier P., et al., “Six‐Year Results From RELEVANCE: Lenalidomide Plus Rituximab (R2) Versus Rituximab‐Chemotherapy Followed by Rituximab Maintenance in Untreated Advanced Follicular Lymphoma,” Journal of Clinical Oncology 40 (2022): 3239–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marcus R., Davies A., Ando K., et al., “Obinutuzumab for the First‐Line Treatment of Follicular Lymphoma,” New England Journal of Medicine 377, no. 14 (October 2017): 1331–1344. [DOI] [PubMed] [Google Scholar]
- 13. Matasar M. J., Luminari S., Barr P. M., et al., “Follicular Lymphoma: Recent and Emerging Therapies, Treatment Strategies, and Remaining Unmet Needs,” Oncologist 24 (2019): e1236–e1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ghione P., Palomba M. L., Ghesquieres H., et al., “Treatment Patterns and Outcomes in Relapsed/Refractory Follicular Lymphoma: Results From the International SCHOLAR‐5 Study,” Haematologica 108, no. 3 (March 2023): 822–832, 10.3324/haematol.2022.281421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Casulo C., Dixon J. G., Le‐Rademacher J., et al., “Validation of POD24 as a Robust Early Clinical End Point of Poor Survival in FL From 5225 Patients on 13 Clinical Trials,” Blood 139, no. 11 (March 2022): 1684–1693, 10.1182/blood.2020010263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gordon M. J., Smith M. R., and Nastoupil L. J., “Follicular Lymphoma: The Long and Winding Road Leading to Your Cure?,” Blood Reviews 57 (January 2023): 100992. [DOI] [PubMed] [Google Scholar]
- 17. Zinzani P. L., Mayer J., Flowers C. R., et al., “ROSEWOOD: A Phase II Randomized Study of Zanubrutinib Plus Obinutuzumab Versus Obinutuzumab Monotherapy in Patients With Relapsed or Refractory Follicular Lymphoma,” Journal of Clinical Oncology 41, no. 33 (November 2023): 5107–5117, 10.1200/JCO.23.00775. [DOI] [PubMed] [Google Scholar]
- 18. Ma J., Mo Y., Tang M., et al., “Bispecific Antibodies: From Research to Clinical Application,” Frontiers in Immunology 12 (2021): 626616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi Y., Gui L., Cheng Y., et al., “A Phase II Multicenter Study of Abexinostat, an Oral Histone Deacetylase Inhibitor, in Patients With Relapsed/Refractory Follicular Lymphoma,” Journal of Clinical Oncology 42 (2024): 7059, 10.1200/JCO.2024.42.16_suppl.7059. [DOI] [Google Scholar]
- 20. Falchi L., Vardhana S. A., and Salles G. A., “Bispecific Antibodies for the Treatment of B‐Cell Lymphoma: Promises, Unknowns, and Opportunities,” Blood 141, no. 5 (Feb 2023): 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hirayama A. V., Gauthier J., Hay K. A., et al., “High Rate of Durable Complete Remission in Follicular Lymphoma After CD19 CAR‐T Cell Immunotherapy,” Blood 134, no. 7 (August 2019): 636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tian Z., Liu M., Zhang Y., and Wang X., “Bispecific T Cell Engagers: An Emerging Therapy for Management of Hematologic Malignancies,” Journal of Hematology & Oncology 14, no. 1 (May 2021): 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hosseini S. S., Khalili S., Baradaran B., et al., “Bispecific Monoclonal Antibodies for Targeted Immunotherapy of Solid Tumors: Recent Advances and Clinical Trials,” International Journal of Biological Macromolecules 167 (2021): 1030–1047. [DOI] [PubMed] [Google Scholar]
- 24. Kontermann R. E. and Brinkmann U., “Bispecific Antibodies,” Drug Discovery Today 20, no. 7 (2015): 838–847. [DOI] [PubMed] [Google Scholar]
- 25. Kontermann R. E., “Strategies for Extended Serum Half‐Life of Protein Therapeutics,” Current Opinion in Biotechnology 22, no. 6 (2011): 868–876. [DOI] [PubMed] [Google Scholar]
- 26. Huehls A. M., Coupet T. A., and Sentman C. L., “Bispecific T‐Cell Engagers for Cancer Immunotherapy,” Immunology and Cell Biology 93, no. 3 (2015): 290–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dustin M. L., “The Immunological Synapse,” Cancer Immunology Research 2, no. 11 (2014): 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Velasquez M. P., Bonifant C. L., and Gottschalk S., “Redirecting T Cells to Hematological Malignancies With Bispecific Antibodies,” Blood 131, no. 1 (2018): 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Braig F., Brandt A., Goebeler M., et al., “Resistance to Anti‐CD19/CD3 BiTE in Acute Lymphoblastic Leukemia May Be Mediated by Disrupted CD19 Membrane Trafficking,” Blood 129, no. 1 (2017): 100–104. [DOI] [PubMed] [Google Scholar]
- 30. Budde L. E., Assouline S., Sehn L. H., et al., “Single‐Agent Mosunetuzumab Shows Durable Complete Responses in Patients With Relapsed or Refractory B‐Cell Lymphomas: Phase I Dose‐Escalation Study,” Journal of Clinical Oncology 40, no. 5 (2022): 481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Budde L. E., Sehn L. H., Matasar M., et al., “Safety and Efficacy of Mosunetuzumab, a Bispecific Antibody, in Patients With Relapsed or Refractory Follicular Lymphoma: A Single‐Arm, Multicentre, Phase 2 Study,” Lancet Oncology 23, no. 8 (2022): 1055–1065. [DOI] [PubMed] [Google Scholar]
- 32. Morschhauser F., Carlo‐Stella C., Dickinson M., et al., “Glofitamab as Monotherapy and in Combination With Obinutuzumab Induces High Complete Response Rates in Patients (Pts) With Multiple Relapsed or Refractory (R/R) Follicular Lymphoma (FL),” Blood 138, no. S1 (2021): 128. [Google Scholar]
- 33. Linton K. M., Vitolo U., Jurczak W., et al., “Epcoritamab Monotherapy in Patients With Relapsed or Refractory Follicular Lymphoma (EPCORE NHL‐1): A Phase 2 Cohort of a Single‐Arm, Multicentre Study,” Lancet Haematology 11, no. 8 (August 2024): e593–e605. [DOI] [PubMed] [Google Scholar]
- 34. Kim T. M., Taszner M., Cho S.‐G., et al., “Odronextamab in Patients With Relapsed/Refractory (R/R) Follicular Lymphoma (FL) Grade 1–3a: Results From a Prespecified Analysis of the Pivotal Phase II Study ELM‐2,” Blood 140, no. S1 (2022): 2280–2282. [Google Scholar]
- 35. Kang C., “Mosunetuzumab: First Approval,” Drugs 82, no. 11 (July 2022): 1229–1234. [DOI] [PubMed] [Google Scholar]
- 36. “FDA Grants Accelerated Approval to Mosunetuzumab‐axgb for Relapsed or Refractory Follicular Lymphoma,” News release, US Food and Drug Administration, accessed March 21, 2023, 10.1182/bloodadvances.2022009260. [DOI]
- 37. Bartlett N. L., Sehn L. H., Matasar M. J., et al., “Mosunetuzumab Monotherapy Demonstrates Durable Efficacy With a Manageable Safety Profile in Patients With Relapsed/Refractory Follicular Lymphoma Who Received ≥2 Prior Therapies: Updated Results From a Pivotal Phase II Study,” Blood 140, no. S1 (2022): 1467–1470. [Google Scholar]
- 38. McGough S. F., Shamas N., Wang J., et al., “An External Control for Mosunetuzumab Using Real‐World Data in Follicular Lymphoma in the Third or Subsequent Lines of Systemic Therapy,” Blood 140, no. S1 (2022): 3658–3660. [Google Scholar]
- 39. Morschhauser F., Bishton M., Eyre T. A., et al., “Mosunetuzumab in Combination With Lenalidomide Has a Manageable Safety Profle and Encouraging Activity in Patients With Relapsed/Refractory Follicular Lymphoma: Initial Results From a Phase Ib Study,” Blood 138, no. S1 (2021): 129. [Google Scholar]
- 40. Olszewski A. J., Huntington S. F., Bannerji R., Ollila T. A., McMahon J., and Dubielecka P. M., “Mosunetuzumab With Lenalidomide Augmentation as First‐Line Therapy for Follicular (FL) and Marginal Zone Lymphoma (MZL),” Blood 140, no. S1 (2022): 6492–6493. [Google Scholar]
- 41. Falchi L., Okwali M., Ghione P., et al., “Subcutaneous (SC) Mosunetuzumab (Mosun) as First‐Line Therapy for Patients (Pts) With High Tumor‐Burden Follicular Lymphoma (FL): First Results of a Multicenter Phase 2 Study,” Blood 142, no. S1 (2023): 604, 10.1182/blood-2023-179906. [DOI] [Google Scholar]
- 42. Minson A. and Dickinson M., “Glofitamab CD20‐TCB Bispecific Antibody,” Leukemia & Lymphoma 62, no. 13 (2021): 3098–3108. [DOI] [PubMed] [Google Scholar]
- 43. Hutchings M., Morschhauser F., Iacoboni G., et al., “Glofitamab, a Novel, Bivalent CD20‐Targeting T‐Cell‐Engaging Bispecific Antibody, Induces Durable Complete Remissions in Relapsed or Refractory B‐Cell Lymphoma: A Phase I Trial,” Journal of Clinical Oncology 39, no. 18 (June 2021): 1959–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Morschhauser F., Carlo‐Stella C., Dickinson M., et al., “Glofitamab as Monotherapy and in Combination With Obinutuzumab Induces High Complete Response Rates in Patients With Multiple Relapsed or Refractory Follicular Lymphoma,” Blood 138, no. S1 (2021): 128. [Google Scholar]
- 45. Rafei H. and Rezvani K., “Mitigating Infection Risks: The Promise and Challenge of Bispecific Antibodies in Haematological Malignancies,” British Journal of Haematology 205 (July 2024): 764–766, 10.1111/bjh.19668. [DOI] [PubMed] [Google Scholar]
- 46. Falchi L., Leslie L. A., Belada D., et al., “Subcutaneous Epcoritamab in Combination With Rituximab + Lenalidomide (R2) for First‐Line Treatment of Follicular Lymphoma: Initial Results From Phase 1/2 Trial,” Blood 140, no. S1 (2022): 1471–1473, 10.1182/blood-2022-158232. [DOI] [Google Scholar]
- 47. Bannerji R., Arnason J. E., Advani R. H., et al., “Odronextamab, a Human CD20 × CD3 Bispecific Antibody in Patients With CD20‐Positive B‐Cell Malignancies (ELM‐1): Results From the Relapsed or Refractory Non‐Hodgkin Lymphoma Cohort in a Single‐Arm, Multicentre, Phase 1 Trial,” Lancet Haematology 9, no. 5 (May 2022): e327–e339, 10.1016/S2352-3026(22)00072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Turtle C. J., Hanafi L. A., Berger C., et al., “Immunotherapy of Non‐Hodgkin's Lymphoma With a Defined Ratio of CD8(+) And CD4(+) CD19‐Specific Chimeric Antigen Receptor‐Modified T Cells,” Science Translational Medicine 8, no. 355 (2016): 355ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Luo J. and Zhang X., “Challenges and Innovations in CAR‐T Cell Therapy: A Comprehensive Analysis,” Frontiers in Oncology 11, no. 14 (June 2024): 1399544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mitra A., Barua A., Huang L., Ganguly S., Feng Q., and He B., “From Bench to Bedside: The History and Progress of CAR T Cell Therapy,” Frontiers in Immunology 15, no. 14 (2023): 118–129, 10.3389/fimmu.2023.1188049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sadelain M., Rivière I., and Riddell S., “Therapeutic T Cell Engineering,” Nature 545 (2017): 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Eshhar Z., Waks T., Gross G., and Schindler D. G., “Specific Activation and Targeting of Cytotoxic Lymphocytes Through Chimeric Single Chains Consisting of Antibody‐Binding Domains and the Gamma or Zeta Subunits of the Immunoglobulin and T‐Cell Receptors,” Proceedings of the National Academy of Sciences of the United States of America 90 (1993): 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mihara K., Andreansky M., Nicholson I. C., et al., “Chimeric Receptors With 4‐1BB Signaling Capacity Provoke Potent Cytotoxicity Against Acute Lymphoblastic Leukemia,” Leukemia 18 (2004): 676–684. [DOI] [PubMed] [Google Scholar]
- 54. Park A. K., Fong Y., Kim S. I., et al., “Effective Combination Immunotherapy Using Oncolytic Viruses to Deliver CAR Targets to Solid Tumors,” Science Translational Medicine 12, no. 559 (2020): eaaz1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Martínez Bedoya D., Dutoit V., and Migliorini D., “Allogeneic CAR T Cells: An Alternative to Overcome Challenges of CAR T Cell Therapy in Glioblastoma,” Frontiers in Immunology 3, no. 12 (2021): 640–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xia Z., Tian M., Cheng Y., et al., “Preclinical Evaluation of Cyclophosphamide and Fludarabine Combined With CD19 CAR‐T in the Treatment of B‐Cell Hematologic Malignancies In Vivo,” Oncology Research 32, no. 6 (May 2024): 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Food and Drug Administration , “FDA Approves Axi‐Cel for Relapsed/Refractory Indolent Follicular Lymphoma,” 2021, https://www.onclive.com/view/fda‐approves‐axi‐cel‐for‐relapsed‐refractory‐indolent‐follicular‐lymphoma.
- 58. Oluwole O. O., Ray M. D., Zur R., et al., “Cost‐Effectiveness of Axicabtagene Ciloleucel Versus Mosunetuzumab in Relapsed/Refractory Follicular Lymphoma in the US,” Blood 142, no. S1 (2023): 5082. [Google Scholar]
- 59. Jacobson C. A., Chavez J. C., Sehgal A. R., et al., “Axicabtagene Ciloleucel in Relapsed or Refractory Indolent Non‐Hodgkin Lymphoma (ZUMA‐5): A Single‐Arm, Multicentre, Phase 2 Trial,” Lancet Oncology 23, no. 1 (January 2022): 91–103, 10.1016/S1470-2045(21)00591-X. [DOI] [PubMed] [Google Scholar]
- 60. Neelapu S. S., Chavez J. C., Sehgal A. R., et al., “Three‐Year Follow‐Up Analysis of Axicabtagene Ciloleucel in Relapsed/Refractory Indolent Non‐Hodgkin Lymphoma (ZUMA‐5),” Blood 143, no. 6 (February 2024): 496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chong E. A., Ruella M., and Schuster S. J., “Lymphoma Program Investigators at the University of Pennsylvania. Five‐Year Outcomes for Refractory B‐Cell Lymphomas With CAR T‐Cell Therapy,” New England Journal of Medicine 384 (2021): 673–674. [DOI] [PubMed] [Google Scholar]
- 62. Flinn I. W., Jacobson C., Nastoupil L. J., et al., “ZUMA‐22: A Phase 3, Randomized Controlled Study of Axicabtagene Ciloleucel (axi‐cel) Versus Standard‐of‐Care Therapy in Patients With Relapsed or Refractory (R/R) Follicular Lymphoma (FL),” Journal of Clinical Oncology 41 (2023): TPS7579, 10.1200/JCO.2023.41.16_suppl.TPS7579. [DOI] [Google Scholar]
- 63. Fowler N. H., Dickinson M., Dreyling M., et al., “Tisagenlecleucel in Adult Relapsed or Refractory Follicular Lymphoma: The Phase 2 ELARA Trial,” Nature Medicine 28 (2022): 325–332. [DOI] [PubMed] [Google Scholar]
- 64. Dreyling M., Fowler N. H., Dickinson M., et al., “Durable Response After Tisagenlecleucel in Adults With Relapsed/Refractory Follicular Lymphoma: ELARA Trial Update,” Blood 143, no. 17 (April 2024): 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Morschhauser F., Dahiya S., Palomba M. L., et al., “Lisocabtagene Maraleucel in Follicular Lymphoma: The Phase 2 TRANSCEND FL Study,” Nature Medicine 30 (June 2024): 2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. “FDA Grants Accelerated Approval to Lisocabtagene Maraleucel for Follicular Lymphoma,” FDA, accessed May 15, 2024, https://www.fda.gov/drugs/resources‐information‐approved‐drugs/fda‐grants‐accelerated‐approval‐lisocabtagene‐maraleucel‐follicular‐lymphoma.
- 67. Ray M. D., Kanters S., Beygi S., et al., “Matching‐Adjusted Indirect Comparisons of Axicabtagene Ciloleucel to Mosunetuzumab for the Treatment of Relapsed/Refractory Follicular Lymphoma,” Transplantation and Cell Therapy 30, no. 9 (September 2024): 885.e1–885.e11. [DOI] [PubMed] [Google Scholar]
- 68. Nastoupil L. J., Bonner A., Wang P., et al., “Matching‐Adjusted Indirect Comparison (MAIC) of Efficacy and Safety of Lisocabtagene Maraleucel (Liso‐Cel) and Mosunetuzumab for the Treatment (Tx) of Third Line or Later (3L+) Relapsed or Refractory (R/R) Follicular Lymphoma (FL),” Blood 142 (2023): 2338. [Google Scholar]
- 69. Gribben J. G., Ghione P., Palomba M. L., et al., “An Updated Comparison of Clinical Outcomes From 4‐Year Follow‐Up of Zuma‐5 (Axicabtagene Ciloleucel) and the International Scholar‐5 External Control Cohort in Relapsed/Refractory Follicular Lymphoma,” Blood 142, no. S1 (2023): 4869, 10.1182/blood-2023-186842. [DOI] [Google Scholar]
- 70. Lin M., Weiss J., Phillips T. J., et al., “Cost Effectiveness of Mosunetuzumab and CAR‐T Cell Therapy in Relapsed/Refractory Follicular Lymphoma,” Blood 142 (2023): 256, 10.1182/blood-2023-182244. [DOI] [Google Scholar]
- 71. Matasar M., Sanchez Alvarez J., Parise H., et al., “EE514 A Cost‐Effectiveness Analysis of Mosunetuzumab for Treatment of Third‐ Or Higher‐Line Relapsed or Refractory (R/R) Follicular Lymphoma (FL) in the United States (US),” Value in Health 26 (2023): S153. [Google Scholar]
- 72. Oluwole O. O., Dholaria B., Knight T. E., et al., “Chimeric Antigen Receptor T‐Cell Therapy in the Outpatient Setting: An Expert Panel Opinion From the American Society for Transplantation and Cellular Therapy,” Transplantation and Cell Therapy 30, no. 2 (February 2024): 131–142. [DOI] [PubMed] [Google Scholar]
- 73. Perez A., Al Sagheer T., Nahas G. R., and Linhares Y. P. L., “Outpatient Administration of CAR T‐Cell Therapy: A Focused Review With Recommendations for Implementation in Community Based Centers,” Frontiers in Immunology 8, no. 15 (May 2024): 1412002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Crombie J. L., Graff T., Falchi L., et al., “Consensus Recommendations on the Management of Toxicity Associated With CD3 × CD20 Bispecific Antibody Therapy,” Blood 143, no. 16 (April 2024): 1565–1575. [DOI] [PubMed] [Google Scholar]
- 75. Seyedin R., Hasegawa K., Rajagopalan K., and Wade S. W., “Chimeric Antigen Receptor T‐Cell Therapy Setting of Care: A Retrospective Cohort Analysis of MCL and FL Patients in the US,” Blood 140 (2022): 3642–3643. [Google Scholar]
- 76. Cordas Dos Santos D. M., Tix T., Shouval R., et al., “A Systematic Review and Meta‐Analysis of Nonrelapse Mortality After CAR T Cell Therapy,” Nature Medicine 30, no. 9 (September 2024): 2667–2678, 10.1038/s41591-024-03084-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Duell J., Leipold A. M., Appenzeller S., et al., “Sequential Antigen Loss and Branching Evolution in Lymphoma After CD19‐ and CD20‐Targeted T‐Cell‐Redirecting Therapy,” Blood 143, no. 8 (February 2024): 685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.