

Abstract
The thalamic reticular nucleus (TRN) is an anatomical and functional hub that modulates the flow of information between the cerebral cortex and thalamus, and its dysfunction has been linked to sensory disturbance and multiple behavioral disorders. Therefore, understanding how TRN neurons differentiate and establish connectivity is crucial to clarify the basics of TRN functions. Here, we showed that the regulatory cascade of the transcription factors Ascl1 and Isl1 promotes the fate of TRN neurons and concomitantly represses the fate of non-TRN prethalamic neurons. Furthermore, we found that this cascade is necessary for the correct development of the two main axonal connections, thalamo-cortical projections and prethalamo-thalamic projections. Notably, the disruption of prethalamo-thalamic axons can cause the pathfinding defects of thalamo-cortical axons in the thalamus. Finally, forced Isl1 expression can rescue disruption of cell fate specification and prethalamo-thalamic projections in in vitro primary cultures of Ascl1-deficient TRN neurons, indicating that Isl1 is an essential mediator of Ascl1 function in TRN development. Together, our findings provide insights into the molecular mechanisms for TRN neuron differentiation and circuit formation.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-024-05523-6.
Keywords: Diencephalon, Transcription factor, Mouse, Embryo, Differentiation
Introduction
Mounting evidence points to a disruption of functional connectivity between the cerebral cortex and thalamus in the pathogenesis of psychiatric disorders, including autism spectrum disorder, bipolar disorder, major depressive disorder, and schizophrenia [1–4]. In this context, the thalamic reticular nucleus (TRN) is particularly noteworthy because it influences cortico-thalamic and thalamo-cortical communications [5]. The TRN consists of GABAergic neurons and functions as an ideal hub, as all of the axons connecting the cortex and thalamus pass through the TRN, which receives excitatory inputs from cortico-thalamic and thalamo-cortical neurons and sends inhibitory outputs to the thalamus [6]. The TRN plays important roles in regulating sensory attention, sleep, arousal, and cognition [5, 7–10], and abnormalities in the TRN may cause behavioral deficits in individuals with autism, attention deficit hyperactivity disorder (ADHD), and schizophrenia [2, 3, 11]. Despite the functional importance of the TRN, few studies have investigated the molecular mechanisms underlying the development of the TRN with regard to neuronal differentiation and connectivity during embryogenesis.
The positioning of the TRN between the thalamus and telencephalon is crucial to an important aspect of its principal function. The TRN surrounds most of the rostrolateral surface of the thalamus and originates in proliferating progenitor cells in the alar plate of the third prosomere (p3), a diencephalic segment, which gives rise to the prethalamus [12]. Although the TRN shares a common developmental origin with other prethalamic nuclei, such as the zona incerta (ZI) and the ventral lateral geniculate nucleus (vLG), it acquires different functions and connectivity from the remainder of the prethalamus, which constitutes widespread connectivity in the forebrain, midbrain, and hindbrain [13–15]. During the patterning phase of the diencephalon, prethalamic formation is influenced by dorsoventral interactions of Wnts and Shh [16–18] and by anteroposterior interactions of Six3, Fezf, Irx, and Otx [19–22]. Inactivation of Olig2 leads to reduced prethalamus and a change in the fate of Olig2+ prethalamus to rostrally adjacent thalamic eminence [23]. Unlike well-studied brain structures such as the cortex and thalamus, little is known about how individual progenitor cells differentiate to populate distinct prethalamic nuclei, how the prethalamic nuclear identity is established, and how prethalamic nuclei undergo further functional differentiation, including axon projection.
In this study, we present evidence that the transcriptional cascade of the Ascl1 and Isl1 transcription factors orchestrates cell fate specification and axonal circuit formation in the TRN. The LIM homeobox gene Isl1 is necessary for motor neuron and interneuron differentiation in the neural tube [24] and for retinal ganglion cell differentiation [25]. Isl1 is indispensable for melanocortin neuron cell fate and for estrogen receptor-α expression [26, 27]. In the telencephalon, Isl1 plays crucial roles in the formation of the striatonigral pathway [28–30]. The proneural bHLH factor Ascl1 influences various developmental processes, including cell fate specification, differentiation and migration [31]. Ascl1 plays important roles in conferring neuronal identity in the spinal cord [32, 33], hypothalamus [34], and telencephalon [35, 36]. A previous study demonstrated that loss of Ascl1 results in defective pathfinding of thalamo-cortical axons (TCAs) accompanied by defective differentiation of brain regions that lie along the TCA pathway [37]. Herein, we focused on the TRN and examined whether the Ascl1 and Isl1 transcription factors influence prethalamus subdomain-specific cell fate decisions and axon circuitry during embryonic development.
Results
Isl1 is required for promoting TRN cell identity and repressing non-TRN prethalamic cell fates in the TRN
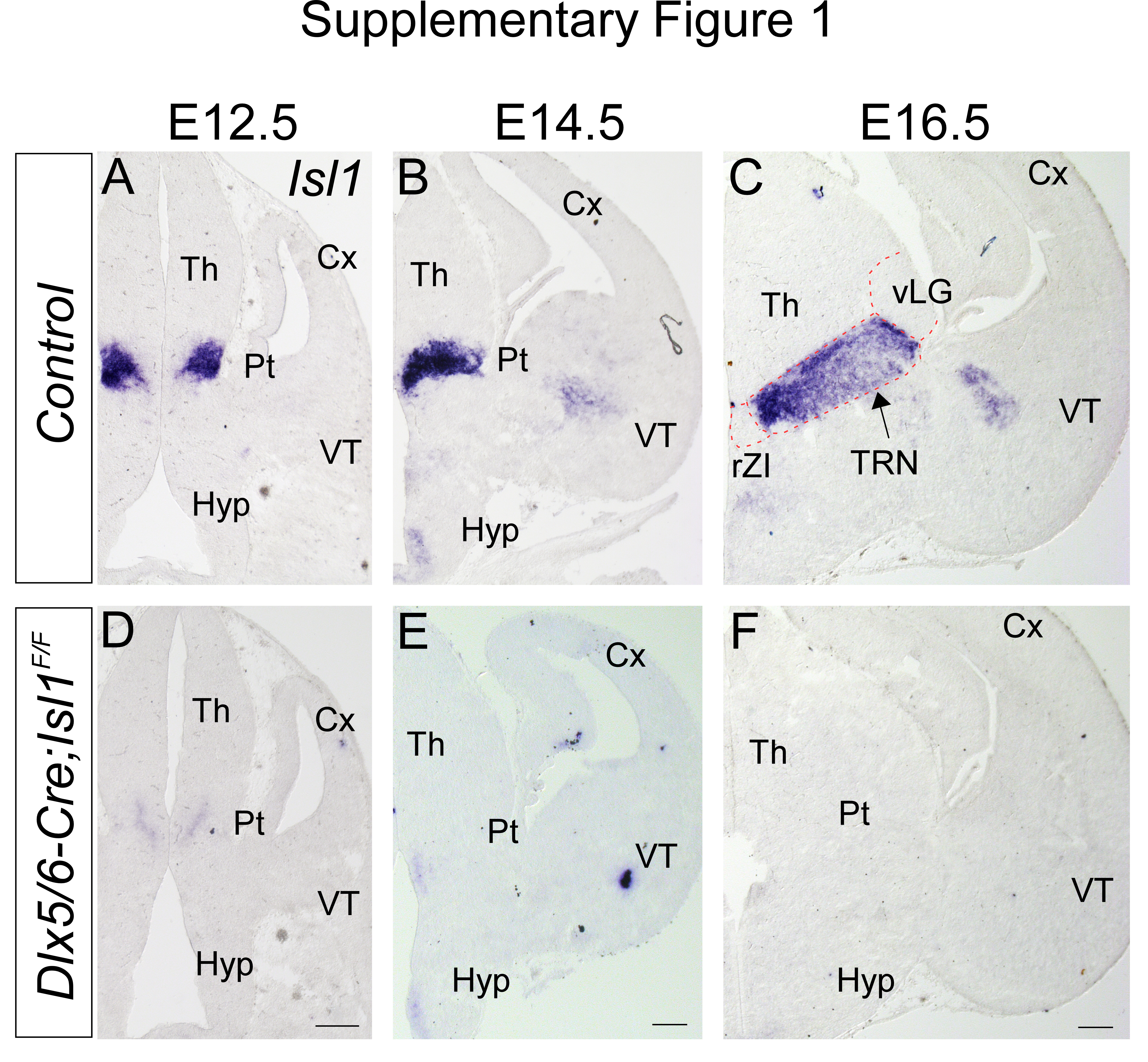
To explore whether Isl1 is required for prethalamic differentiation, we conditionally inactivated Isl1 expression by crossing an Isl1F/F mouse line, in which the homeodomain (exon 4) is flanked by loxP sites [38], with a Dlx5/6-Cre mouse line that drives Cre-mediated recombination in the ventral forebrain [39]. During embryogenesis, Isl1 is principally expressed in the TRN, a prethalamic derivative (Fig. S1A-C). In Dlx5/6-Cre; Isl1F/F embryos, Isl1 staining was almost completely lost in the prethalamus as well as in other forebrain regions (Fig. S1D-F). We first analyzed the effects of Isl1 loss on prethalamic differentiation by staining for Hes5 and Sox2, which mark proliferative progenitors, as well as for the postmitotic neuronal markers Elavl3/4 (HuC/D). At E12.5, Hes5/Sox2 and HuC/D, which exhibit mutually exclusive expression, were similarly expressed in control (Dlx5/6-Cre; Isl1F/+) and Isl1-deficient embryos (Fig. S2A-D). We next examined early patterning in the conditional mutants by analyzing the distribution of Ascl1, Gsx1/2, Helt, Olig2, and Nkx2.2, which are specifically expressed in prethalamic progenitors. None of these markers were significantly altered in Dlx5/6-Cre; Isl1F/F embryos compared to control littermates (Fig. S2E-J).
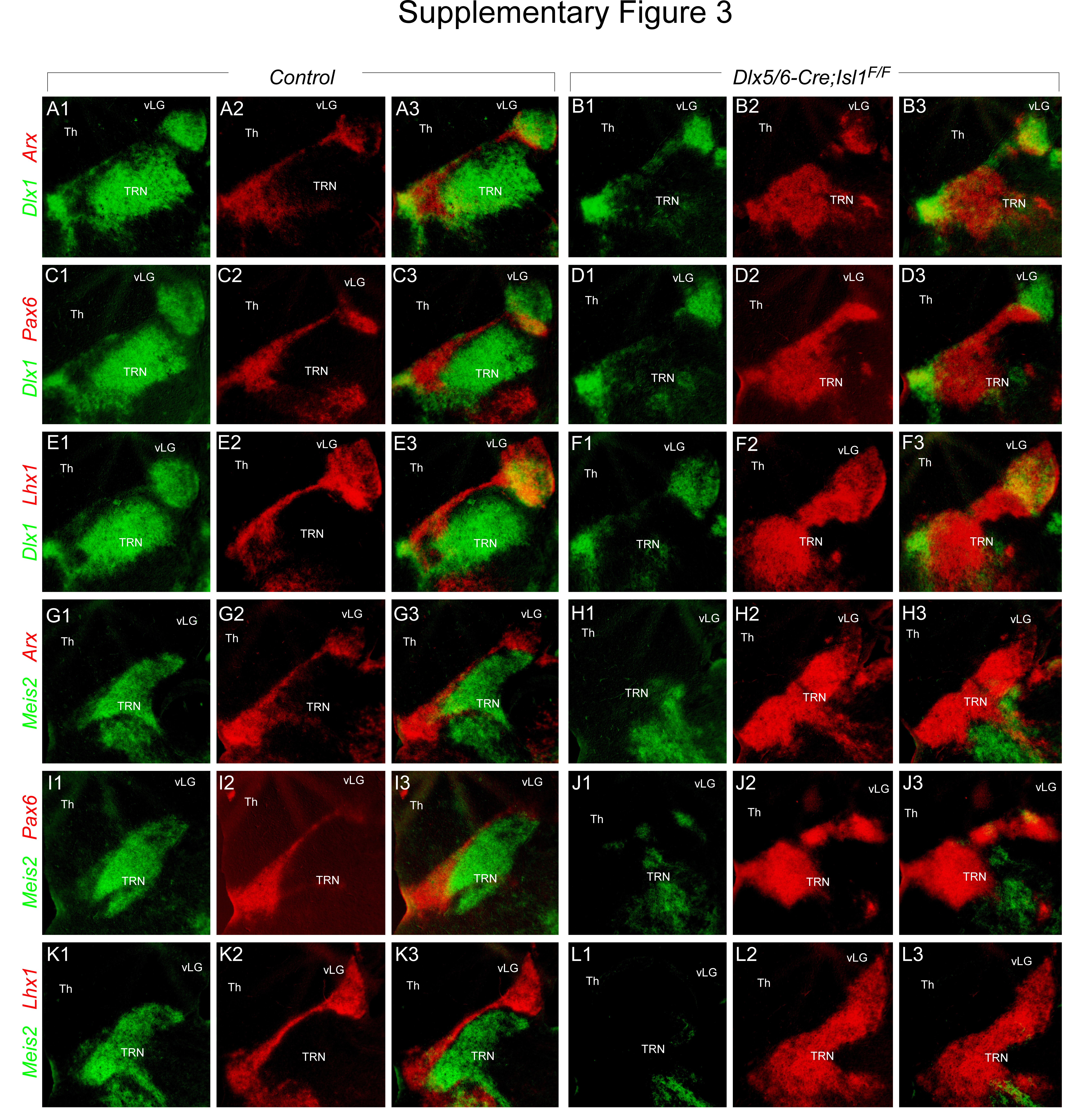
Prethalamic progenitors differentiate and populate into heterogeneous nuclear groups, including the TRN, zona incerta (ZI), and ventral lateral geniculate nucleus (vLG). At E14.5 and E16.5, these three major nuclei can be identified by postmitotic regional markers [40, 41]. The TRN consists of a sheet of cells surrounded by the rostral ZI (rZI), vLG and zona limitans intrathalamica (zli) and can be identified by staining for Dlx1 and Meis2 (Fig. 1A2,B2,C2,D2). Dlx1 was also expressed in the rZI, vLG, and zli (Fig. 1A2, B2), while Meis2 was mainly detected in the TRN (Fig. 1C2, D2). Arx, Pax6, and Lhx1/5 were expressed in the rZI and vLG at distinct levels (Fig. 1E2, F2, G2, H2, I2, J2, K2, L2). Arx and Lhx1/5 were also detected in the zli, and Pax6 expression was observed in the rostral half of the zli [40–42] (Fig. 1E2, F2, G2, H2, I2, J2, K2, L2). In contrast, the TRN does not express Arx, Pax6, or Lhx1/5 at detectable levels (Fig. 1E2, F2, G2, H2, I2, J2, K2, L2; Fig. S3). In the absence of Isl1, compared with that in controls, Dlx1 was similarly expressed in the rZI, vLG, and zli, but Dlx1 expression was undetectable in the TRN at E14.5 and E16.5 (Fig. 1A4, B4). In addition, Meis2 expression was almost completely absent in the TRN of Dlx5/6-Cre; Isl1F/F embryos (Fig. 1C4, D4). Concomitantly, the loss of Isl1 function led to the ectopic induction of Arx, Pax6 and Lhx1/5 in the TRN (Fig. 1E4, F4, G4, H4, I4, J4, K4, L4; Fig. S3), suggesting the derepression of TRN-depleted genes in the TRN in the absence of Isl1. We further examined the mRNA level of Gad1, which encodes the GABA synthetic enzyme glutamate decarboxylase. Loss of Isl1 did not result in significant changes in Gad1 expression in the prethalamic regions (Fig. 1M4, N4), indicating that the prethalamic complex maintains a GABAergic neurotransmitter phenotype in the absence of Isl1, but the neuronal subtype composition in the complex was altered. Therefore, these observations demonstrate that Isl1 is required for the acquisition of TRN identity and for suppressing non-TRN prethalamic neuronal fates in the TRN.
Fig. 1.

TRN neuronal identity is dependent on Isl1 function. (Top) Schematic showing the wild-type anatomy of the prethalamus. (A-N) In situ hybridization of prethalamic markers on control (Dlx5/6-Cre; Isl1F/+) and mutant (Dlx5/6-Cre; Isl1F/F) coronal sections. The red boxed areas are magnified as indicated. The patterns of gene expression in the prethalamus at E14.5 were similar to those at E16.5. (A-B) Dlx1 was normally expressed in all prethalamic nuclei, including the rZI, TRN, and vLG, as well as the central diencephalic organizer zli. Dlx1 expression in the TRN of control embryos was high (A2, B2), while Isl1-deficient embryos lost Dlx1 expression specifically in the TRN but not in the vLG, rZI or zli (A4, B4). (C-D) Within the prethalamus of control embryos, Meis2 expression was limited to the TRN (C2, D2). Loss of Isl1 resulted in the abrogation of Meis2 expression (C4, D4). (E-L) Arx, Pax6, and Lhx1 were normally expressed in the rZI, zli, and rostroventral parts of the vLG but were not detectable in the TRN. Lhx5 is also weakly expressed in the rZI, zli, and vLG but not in the TRN. In the absence of Isl1 function, the expression of these genes was largely unaffected in the rZI, zli, and vLG but was ectopically induced in the TRN. (M, N) A GABAergic neuronal marker Gad1 was similarly expressed in control and Isl1 mutants. (O) Quantification of in situ hybridization. The pixel intensity values of TRN or vLG were grouped and averaged from three different sections per embryo. (n = 5 embryos; Student’s t-test; **p < 0.01, ***p < 0.001, ****p < 0.0001) (P) A schematic diagram indicating that Dlx1+Meis2+ TRN cells lose their identity and instead acquire molecular features of the remainder of the prethalamus. Cx, cortex; Hyp, hypothalamus; Pt, prethalamus; rZI, rostral zona incerta; Th, thalamus; TRN, thalamic reticular nucleus; vLG, ventral lateral geniculate nucleus; zli, zona limitans intrathalamica. Scale bar, 200 μm
Isl1 is required for normal TCA navigation in the TRN
As the TRN is a diencephalic contingent that TCAs first encounter and cross, we considered potentially important factors of the TRN in normal TCA projection. Therefore, we determined the effect of Isl1 deletion on the progression and navigation of thalamic axons as they cross the TRN. To specifically label TCAs in the developing forebrain, we crossed the Dlx5/6-Cre; Isl1F allele with the Gbx2GFP allele, which expresses GFP from the Gbx2 locus [43, 44]. We examined the distribution of GFP+ TCA in coronal and sagittal sections through the forebrain in control (Gbx2GFP;Dlx5/6-Cre; Isl1F/+) and mutant (Gbx2GFP;Dlx5/6-Cre; Isl1F/F) embryos. At E14.5 ~ E16.5, in control embryos, GFP+ thalamic axons exiting the thalamus were found to extend rostrally through the prethalamus in smooth and parallel arrays of fascicles (Fig. 2A4, A5, A6, A13 and Fig. 2B1, B3, B5). In contrast, in Isl1 mutant embryos, many thalamic axons fasciculated abnormally to form much denser, thicker and more disordered bundles in the prethalamus (Fig. 2A10, A11, A12, A13, red arrows, white asterisks; B2, B4, B6, white arrows). TCAs within the thalamus are obscured by the intense GFP fluorescence expressed by cell bodies. Therefore, the TCA projection was also examined by an antibody against medium chain neurofilament (NF-M) expressed on TCAs [45]. Strikingly, abnormally thick bundles were also detected even within the thalamus, and some of the GFP+ neurofilament+ (NF+) fibers were disoriented and failed to extend rostrally through the prethalamus but instead were misrouted caudally into the pretectum (Fig. 2A9, white arrows; B2, B4, B6, white arrowheads; C4’, C4” white arrowheads). Normally, thalamic axons exiting the prethalamus make a sharp turn to avoid the hypothalamus and enter the telencephalon (Fig. 2A1-A6). In contrast, in Isl1-deficient embryos, some thalamic axons failed to cross the boundary between the diencephalon and telencephalon and instead invaded the hypothalamic region ventrally (Fig. 2A12, yellow arrowheads). Moreover, many thalamic axons that entered the ventral telencephalon were misrouted ventrally at E16.5 (Fig. 2A7-A11, red arrowheads). We also observed GFP+ axons in the subplate layer of the cortex, but the majority of them stalled at the midpoint of the cortical areas (Fig. 2A7-A11, white arrowheads).
Fig. 2.

Loss of Isl1 leads to TCA pathfinding defects in the prethalamus and thalamus. (A) Immunohistochemistry for GFP on coronal sections of control (Gbx2GFP;Dlx5/6-Cre; Isl1F/+) (A1-A6) and mutant (Gbx2GFP;Dlx5/6-Cre; Isl1F/F) (A7-A12) embryos at E16.5. The red arrows in A10 and A11 indicate abnormal TCA projections in the prethalamus of Isl1 mutant embryos. White asterisks in A10, A11, and A12 indicate abnormally large bundles of TCAs crossing the prethalamus and entering the ventral telencephalon in Isl1 mutants compared to controls. GFP+ TCAs misrouted caudally to the habenula (white arrows in A9). Red asterisks in A10-A12 mark abnormal fasciculation of TCAs within the thalamus. GFP+ TCAs misrouted ventrally in the ventral telencephalon (A7-A11, red arrowheads) and hypothalamus (A12, yellow arrowhead). In the cortex, Isl1 mutant TCAs were shorter than those of control embryos (A7-A11, white arrowheads). (A13) Examples of the measurements taken for the quantification. Yellow dashed lines in A4, A5, A10, A11 indicate the position of the lines used to quantify the number of GFP+ fluorescent signals crossing the prethalamus. (n = 5 embryos; Student’s t-test; *p < 0.001). (B) Double immunofluorescence for GFP and NF-M on sagittal sections of control and Isl1 mutant embryos at E14.5. Control TCAs extended in parallel and oriented ventrolaterally, forming a thin fasciculus in the thalamus and prethalamus, including the TRN. In contrast, Isl1 mutant TCAs formed an abnormally thick fasciculus and extended randomly in the thalamus (white arrowheads) and prethalamus (white arrows). (C) Double immunofluorescence for GFP and NF-M on sagittal sections of control and Isl1 mutant embryos at E18.5. In the thalamus, GFP+ TCAs formed abnormally thick axon bundles orienting randomly and misrouting caudally into the pretectum (white arrowheads in C4’,C4”). Cx, cerebral cortex; Eth, epithalamus; Hyp, hypothalamus; Prt, pretectum; Pt, prethalamus; Th, thalamus. Scale bars, 200 μm
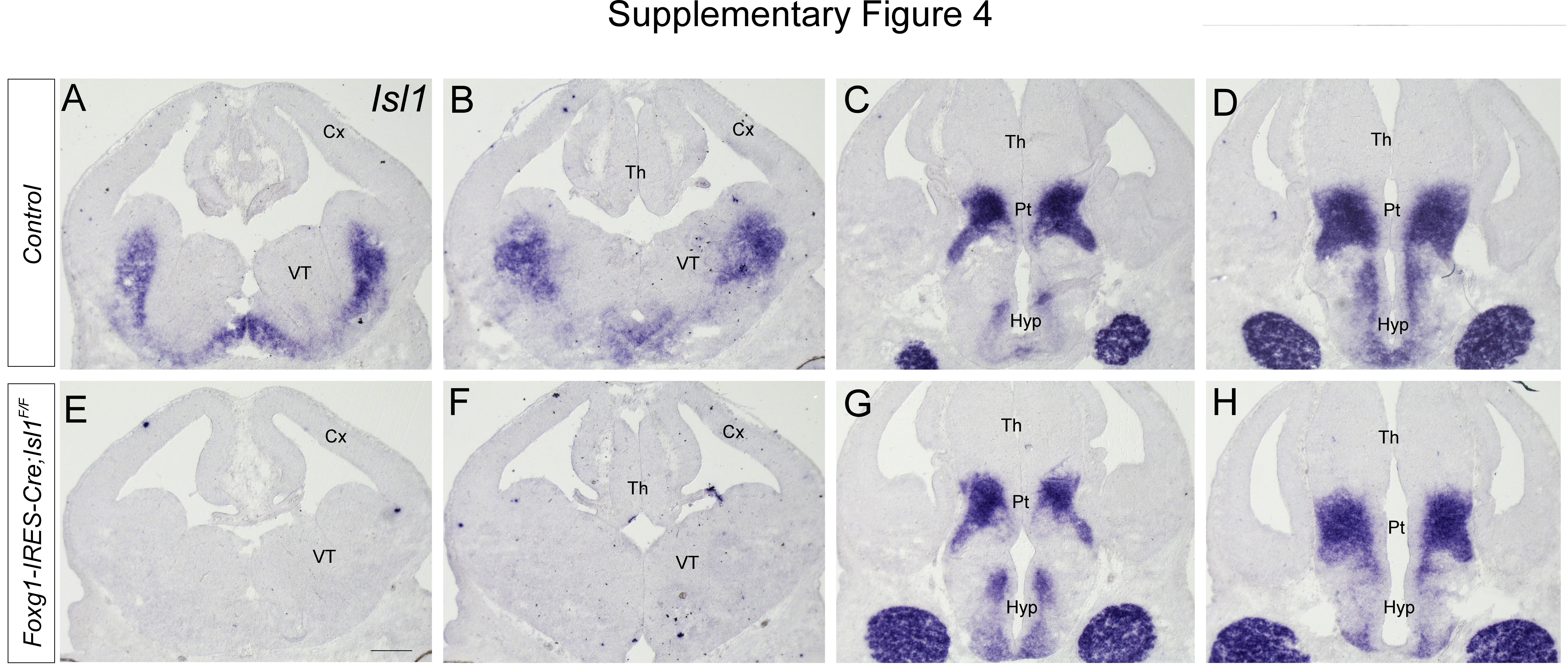
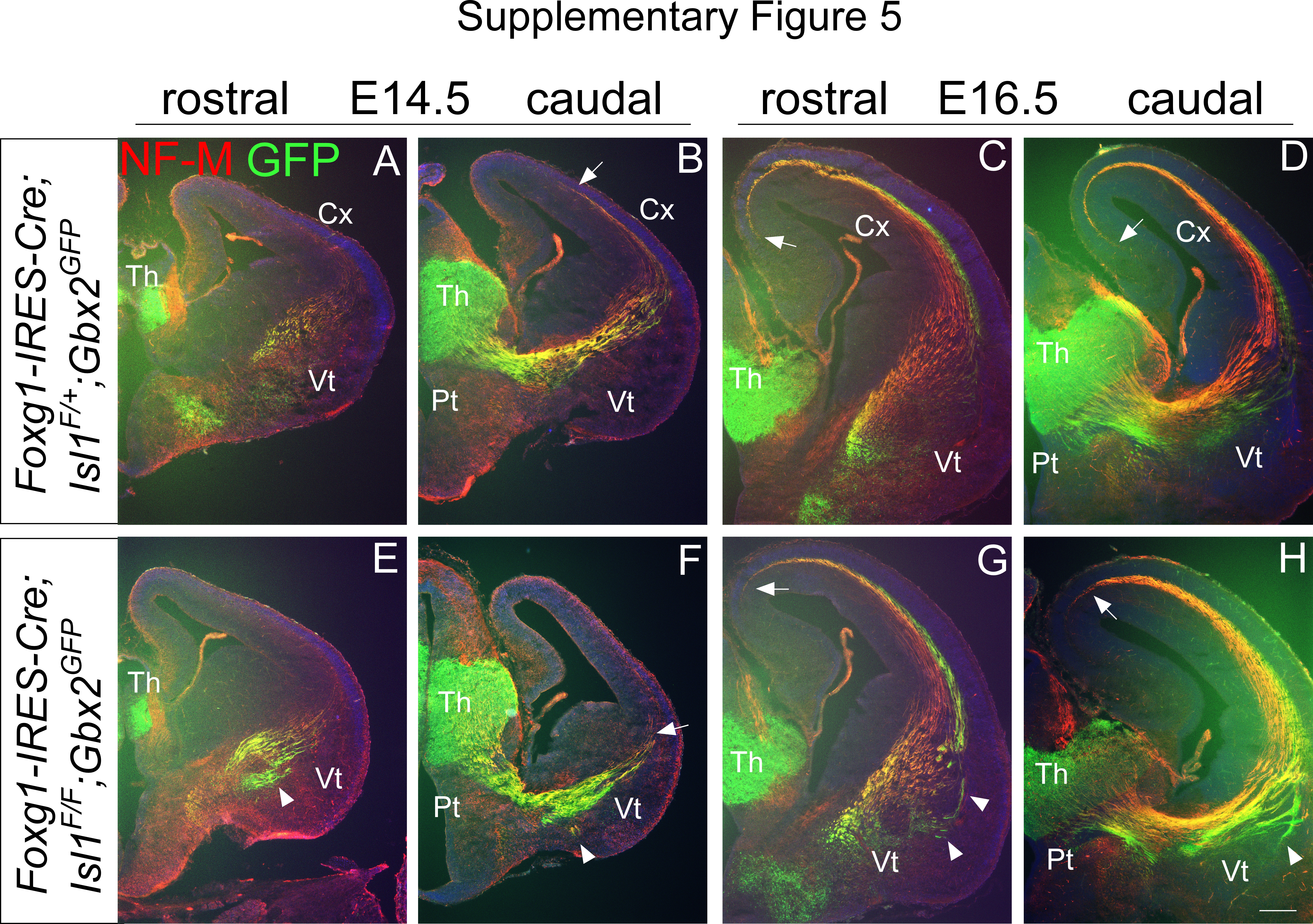
As Isl1 expression is also lost in the ventral telencephalon, we next sought to determine the developmental origin of TCA defects in Isl1-deficient mutants. We therefore generated telencephalic-specific inactivation by crossing the Isl1F/F allele with the Foxg1-IRES-Cre mouse line, in which an IRES-Cre was inserted in the 3’ UTR of the Foxg1 locus to prevent Foxg1 haploinsufficiency and ectopic Cre expression found in the widely used Foxg1-Cre mice [46]. In Foxg1-IRES-Cre; Isl1F/F embryos, Isl1 expression was completely lost in the telencephalon but remained in other brain regions, including the prethalamus (Fig. S4E-H). We then crossed the Foxg1-IRES-Cre; Isl1F/F allele with the Gbx2GFP allele. In Gbx2GFP; Foxg1-IRES-Cre; Isl1F/F embryos, GFP+ NF+ thalamic axons emerging from the thalamus converged to form normal TCA bundles as they crossed the prethalamus in a pattern similar to those in controls (Fig. S5B, D, F, H). In the ventral telencephalon, some GFP+ NF+ axons were disorganized and ran ectopically toward the ventral floor (Fig. S5E-H, arrowheads). Thalamic axons that entered the cortex exhibited delayed outgrowth compared to those of controls (Fig. S5B, C, D, F, G, H, arrows). Therefore, deletion of telencephalic Isl1 can cause TCA defects to a lesser extent than loss of Isl1 in both the diencephalon and telencephalon. Taken together, these results suggest that thalamo-cortical connectivity is dependent on the functions of Isl1 from the telencephalon and diencephalon, but diencephalic Isl1 may play a more significant role in TCA projection.
Isl1 represses the expression of Efna5, which inhibits thalamic neurite outgrowth
To further understand how defective TRN differentiation resulting from Isl1 loss leads to abnormal fasciculation and misrouting of TCAs in the prethalamus, we examined the distribution of axon guidance molecules in the developing prethalamus. Quantitative RT-PCR analysis in E13.5 prethalamus revealed that Efna5, a member of the Eph receptor-interacting ligand family was significantly upregulated in Isl1-deficient prethalamus (Fig. 3A). We then verified the differential expression of Efna5 by RNA in situ hybridization. In control embryos, Efna5 was largely undetectable in the TRN (Fig. 3B1, B7), while we observed strong upregulation of Efna5 in Dlx5/6-Cre; Isl1F/F embryos (Fig. 3B4, B10). Epha4 and Epha7, which encode the receptors for Efna5, appeared to be unchanged in the absence of Isl1 (Fig. 3B2, B3, B5, B6, B8, B9, B11, B12). To examine whether ectopic upregulation of Efna5 might affect TCA navigation into the prethalamus, thalamic explants were prepared from control (Gbx2GFP;Dlx5/6-Cre; Isl1F/+) or mutant embryos (Gbx2GFP;Dlx5/6-Cre; Isl1F/F) and cocultured with aggregates of Efna5-expressing cells. When cocultured with mock-transfected cells, thalamic explants derived from control or mutant embryos grew long neurites (Fig. 3C). In contrast, both control and mutant explants exhibited significantly shorter neurites when cocultured with Efna5-expressing cells (Fig. 3C). These results suggest that Isl1 is required to repress the expression of Efna5, which interferes with the normal growth of TCA passing through the prethalamus.
Fig. 3.

Loss of Isl1 derepresses the expression of Efna5, a repellent for TCAs, in the TRN. (A) Differential expression of Efna5 in the prethalamus of E13.5 mutant embryos (Dlx5/6-Cre; Isl1F/F) compared with control embryos (Dlx5/6-Cre; Isl1F/+) based on qRT-PCR. Prethalamic Efna5 expression was significantly elevated in Isl1 mutant embryos (two-way ANOVA with Sidak’s multiple comparison test; ****p ≤ 0.001). (B) RNA in situ hybridization for Efna5, Epha4, and Epha7 on coronal sections of control and Isl1 mutants at E13.5 and E14.5. Loss of Isl1 resulted in significant upregulation of Efna5 in the TRN (red dashed areas), despite similar staining in other brain regions. Scale bar, 200 μm. (B13) Quantification of in situ hybridization. The pixel intensity values of TRN were grouped and averaged from three different sections per embryo. (n = 3 embryos; Student’s t-test; **p < 0.01) (C) Thalamic explants from control (Gbx2GFP;Dlx5/6-Cre; Isl1F/+) and mutant (Gbx2GFP; Dlx5/6-Cre; Isl1F/F) embryos were cocultured with Efna5-expressing cells (outlined by a white dashed line). Explants were photographed, and outgrowth from the distal and proximal sides of the explants was quantified by GFP fluorescence-occupied area. The values were averaged from two explants per embryo, and the averaged values from three independent experiments were plotted. A significant increase in the distal-proximal (D:P) fluorescence ratio was observed in the presence of Efna5-expressing cells (two-way ANOVA with Sidak’s multiple comparison test; *p < 0.05). Pt, prethalamus; Th, thalamus; TRN, thalamic reticular nucleus
Prethalamo-thalamic axons (PTAs) precede TCAs, and loss of PTAs by Isl1 inactivation causes the pathfinding defects of TCAs in the thalamus
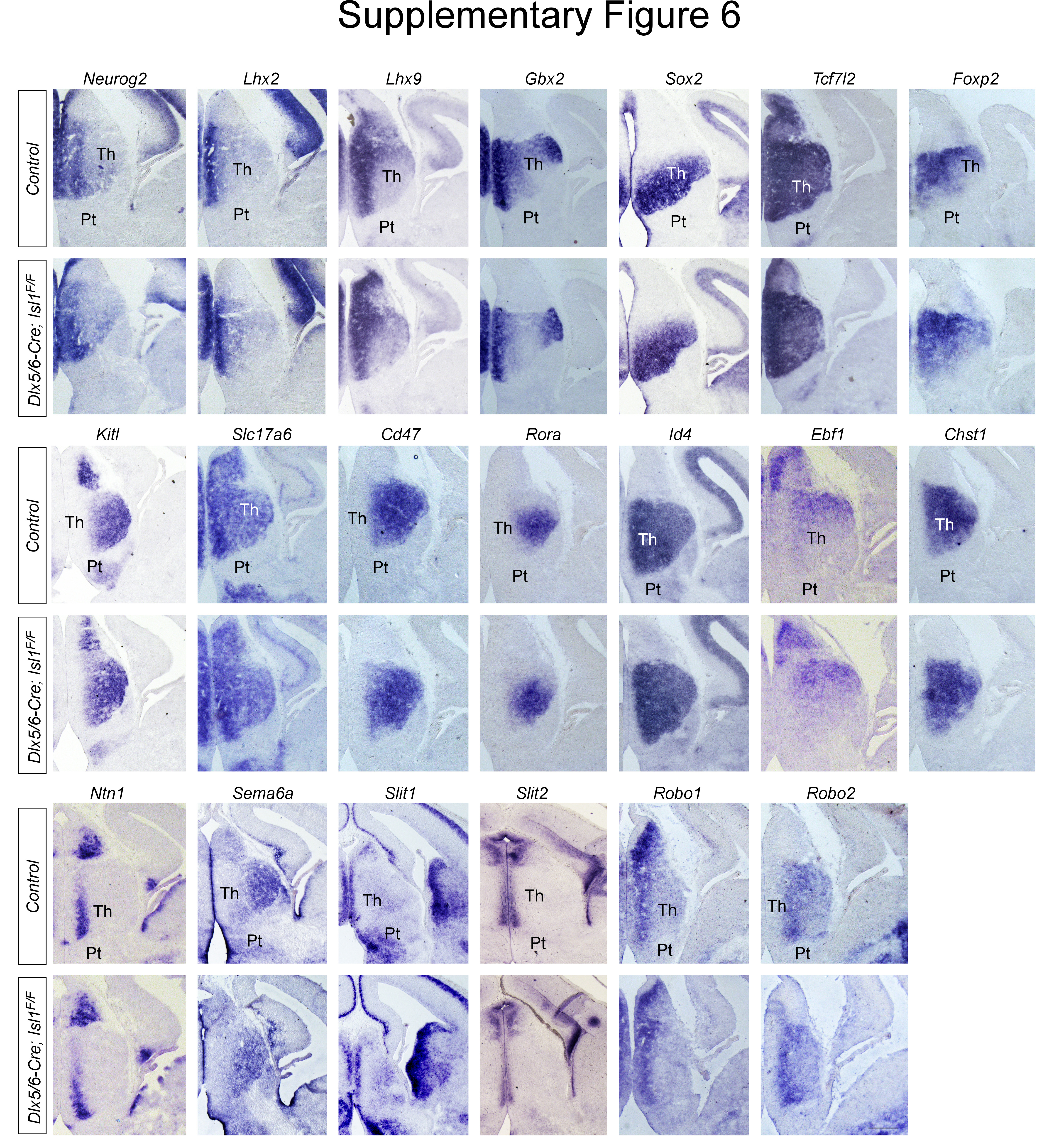
Our findings that abnormal fasciculation and misrouting of TCA were also evident within the thalamus itself led us to investigate whether Isl1 deletion affected patterning or regionalization of the thalamus, although the expression pattern of Isl1 does not suggest a role in thalamic development. We first analyzed the effects of Isl1 loss on early thalamic differentiation. Hes5, Sox2, Elavl3/4, and Olig2 were similarly expressed in control and Isl1-deficient thalamus (Fig. S2A-D, I). We next examined the expression profiles of a panel of transcription factors whose functions influence TCA development. Loss of Neurog2 or Lhx2 disrupts normal TCA growth and pathfinding [47, 48]. Previous genetic studies demonstrated crucial functions of Gbx2 and Tcf7l2 in regulating correct TCA guidance [44, 49]. During development, we did not observe significant alterations in the distribution of these factors in Isl1-deficient thalamus (Fig. S6). We also examined the distribution of additional postmitotic neuronal markers or axon guidance molecules, and none of these markers expression were altered in the thalamus of Dlx5/6-Cre; Isl1F/F embryos (Fig. S6). Overall, our gene expression analysis indicated that Isl1 deletion does not significantly perturb the molecular regionalization of the thalamus. Therefore, TCA abnormalities are unlikely to result from obvious defects in thalamic development.
Neurons within the prethalamus extend axons to the thalamus. These prethalamo-thalamic axons (PTAs) have been suggested to act as pioneer axons for TCA navigation [50–52]. However, the developmental and spatiotemporal relationships between these two axonal projections remain largely unknown. We thus investigated whether PTAs are involved in correct TCA formation within the thalamus. To mark PTAs, we crossed the Dlx5/6-Cre allele with the R26-tdTomato mouse line. At E13.5 and E16.5, we observed tdTomato+ axons that emerged from the prethalamus and extended into the thalamus of Dlx5/6-Cre; R26-tdTomato embryos (Fig. 4A). To address the temporal relationships between PTAs and TCAs, we simultaneously labeled these two axonal pathways by crossing the Gbx2GFP reporter line onto the Dlx5/6-Cre; R26-tdTomato mouse line. At E11.5, a significant portion of tdTomato+ PTAs emerged from the marginal zone of the developing prethalamus and extended caudally through the thalamus (Fig. 4B1, arrowheads), while we did not observe TCAs entering the prethalamus, indicating that PTAs precede TCAs. To assess the spatial relationships between these axonal projections within the thalamus, we visualized TCAs with NF-M immunofluorescence because after E11.5, GFP+ TCA fibers are masked by the bright GFP staining expressed by cell bodies. We found that tdTomato+ prethalamic fibers and NF-M+ thalamic fibers exhibited close associations in the thalamus (Fig. 4C3, C9). To address whether Isl1 is required for the formation of PTAs, we crossed mice carrying the floxed Isl1 allele and R26-tdTomato with the Dlx5/6-Cre line. Isl1 deletion in the prethalamus caused a profound reduction in the number of tdTomato+ axons projecting from the prethalamus to the thalamus in E13.5 and E14.5 embryos (Fig. 4C4, C10, C13). In the thalamus of control embryos, we observed tdTomato+ axons and NF-M+ TCAs in ordered and parallel arrays (Fig. 4C2, C3, C8, C9), while NF-M+ TCAs exhibited erroneous and disorganized trajectories in the thalamus of Isl1-deficient embryos (Fig. 4C5, C6, C11, C12), suggesting a potential pioneering role of PTAs in guiding thalamic axons.
Fig. 4.

Isl1 is required for the formation of prethalamo-thalamic pioneer axons. (A) Immunohistochemistry for tdTomato on coronal sections of Dlx5/6-Cre; R26-tdTomato embryos at E13.5 and E16.5. Prethalamo-thalamic axons (PTAs) labeled by tdTomato extended into the thalamus, forming ordered and parallel projections. (B) Costaining for tdTomato and GFP on coronal sections of Dlx5/6-Cre; R26-TdTomato; Gbx2GFP embryos at E11.5. PTAs labeled with tdTomato extended into the thalamus, while GFP+ thalamic axons had yet to enter the prethalamus. (C) Costaining for tdTomato and NF-M on coronal sections of control (Dlx5/6-Cre; Isl1F/+:R26-tdTomato) and mutant (Dlx5/6-Cre; Isl1F/F:R26-tdTomato) embryos at E13.5 and E14.5. Loss of Isl1 caused a visible reduction in the number of tdTomato+ axons projecting from the prethalamus to the thalamus in E13.5 and E14.5 mutant embryos. Control NF-M+ thalamic axons formed ordered and parallel projections, but mutant NF-M+ thalamic axons aggregated into thick bundles that ran laterally. (C13) Examples of the measurements taken for the quantification. Yellow dashed lines in C1, C4, C7, C10 indicate the position of the lines used to quantify the number of axon bundles crossing the thalamus. (two-way ANOVA with Sidak’s multiple comparison test; *p < 0.05, **p < 0.01, ***p < 0.001). (D) Implanting the chemical tracer Neurovue into the prethalamus of control (Dlx5/6-Cre; Isl1F/+) at E13.5 visualized PTA projections across the thalamus. Coronal sections from five independent experiments revealed a defective pattern in the outgrowth of PTAs in mutant embryos (Dlx5/6-Cre; Isl1F/F) similar to that shown by tdTomato immunohistochemistry in Dlx5/6-Cre; Isl1F/F; R26-TdTomato embryos. Pt, prethalamus; Th, thalamus. Scale bars: 200 μm
Isl1 is also expressed in the ventral telencephalon, and neurons in the ventral telencephalon project to the thalamus [53]. To confirm whether the tdTomato+ axons that were lost in Dlx5/6-Cre; Isl1F/F;R26-tdTomato embryos originated from the prethalamus, we injected the neuronal tracer Neurovue into the prethalamus of E13.5 control and Dlx5/6-Cre; Isl1F/F embryos. Neurovue placed in the prethalamus of control embryos successfully labeled axons projecting to the thalamus, while we did not observe Neurovue+ axons passing through the thalamus in the absence of Isl1 (Fig. 4D). Taken together, these results suggest that Isl1 regulates correct TCA projections within the thalamus by promoting proper formation of PTAs.
Prolonged Isl1 function is required to maintain TRN neuron fate
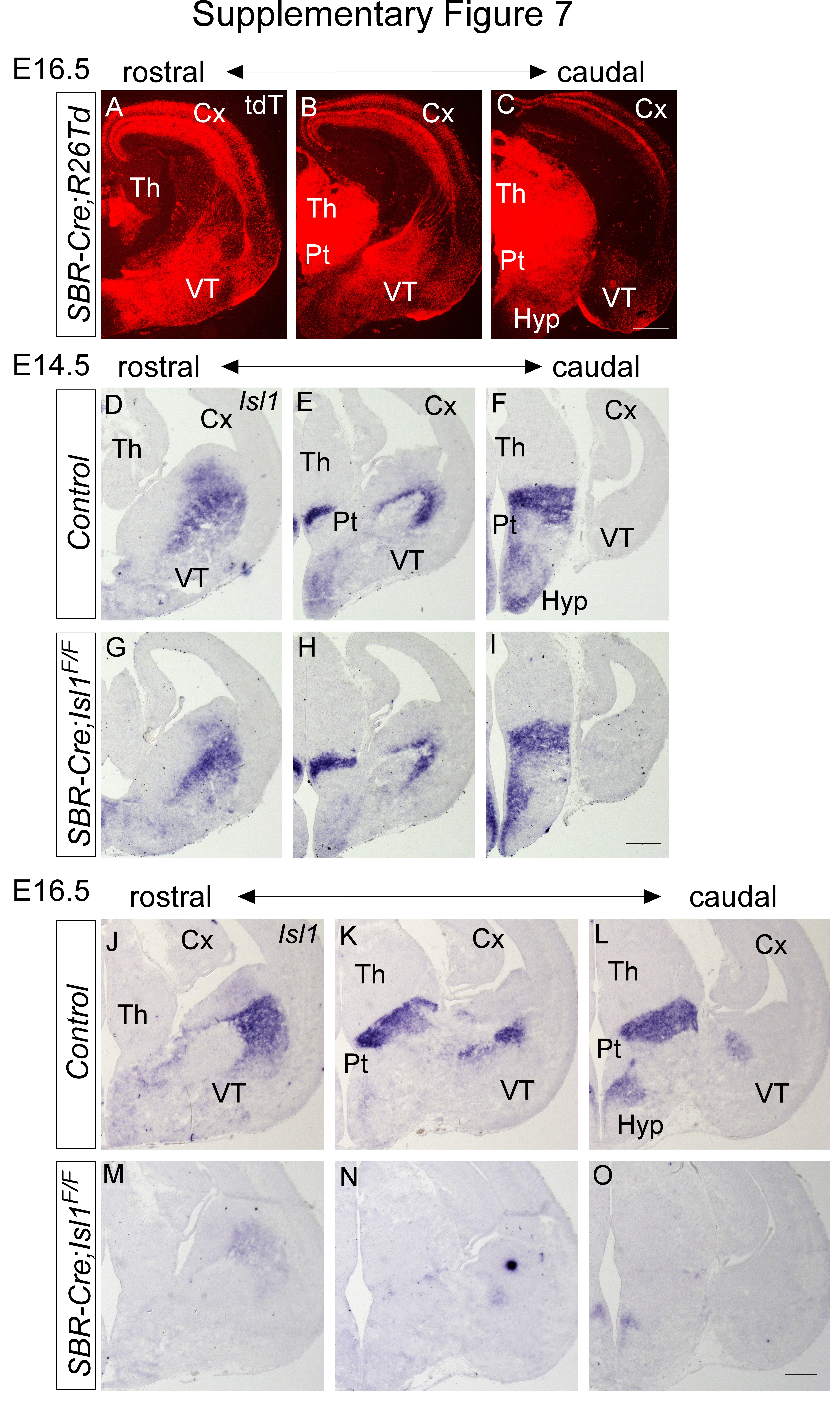
Given that Isl1 expression persists in the TRN during late embryogenesis (Fig. S1C), our observations raise the intriguing possibility that Isl1 has a continued role in prethalamic development. To address whether the dependency of TRN development on Isl1 is temporally regulated, we took advantage of a newly generated mouse line that drives Cre-mediated recombination in the forebrain. This driver relies on a Sox2-bound regulatory element (SBR), which was previously shown to direct the transcription of a reporter gene to the telencephalon and diencephalon at late developmental stages [54]. To determine the efficiency of Cre-mediated recombination, SBR-Cre transgenic mice were generated and crossed with a R26-tdTomato reporter strain. At E16.5, strong tdTomato staining was detected in the developing forebrain (Fig. S7A, B, C). This SBR-Cre allele was then crossed with Isl1F/F mice to generate conditional mutant progeny. Isl1 staining was almost completely lost in the prethalamus of SBR-Cre; Isl1F/F embryos at E16.5 (Fig. S7J-O). Like in the prethalamus of Dlx5/6-Cre; Isl1F/F mutants, Dlx1 and Meis2 expression was significantly downregulated in the TRN of SBR-Cre; Isl1F/F embryos (Fig. 5G, H, M). Moreover, we observed ectopic activation of Arx, Pax6, and Lhx1/5 in the TRN of SBR-Cre; Isl1F/F mutants in a pattern similar to that of Dlx5/6-Cre; Isl1F/F mutants (Fig. 5I-L, M), indicating that Isl1 continues to control TRN identity during embryogenesis.
Fig. 5.

Isl1 continues to regulate TRN identity during late embryogenesis (A-L) In situ hybridization for prethalamic markers on control (SBR-Cre; Isl1F/+) and mutant (SBR-Cre; Isl1F/F) coronal sections at E16.5. Dlx1 expression in the TRN but not in other prethalamic nuclei was lost in Isl1 mutant embryos (G). Meis2 expression, which was restricted to the TRN of control embryos, was abrogated in mutant embryos (H). Loss of Isl1 resulted in ectopic induction of Arx, Pax6, and Lhx1/5 in the TRN (I-L). (M) Quantification of in situ hybridization. The pixel intensity values of TRN or vLG were grouped and averaged from three different sections per embryo (n = 3 embryos; Student’s t-test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Scale bars, 200 μm
Ascl1 is required for promoting TRN neuron fate and PTA outgrowth and regulates Isl1 expression in the prethalamus
The TCA pathfinding defects resulting from the loss of Ascl1 [37] closely resemble the defects in TCAs caused by Isl1 deficiency. Ascl1 mutant embryos exhibit abnormal growth and pathfinding of TCAs in the developing forebrain [37]. To explore whether these errors are due to the disruption of prethalamic factors, we analyzed the Ascl1KI allele, in which the entire coding region of Ascl1 was replaced with GFP [55]. Similar to the phenotype described for Isl1- deficient mutants, the loss of Ascl1 caused severe downregulation of Dlx1 in the TRN of the prethalamus (Fig. 6A6, A16). In contrast to the Isl1- deficient mutants, Dlx1 expression was also lost in the vLG and zli in the absence of Ascl1 (Fig. 6A6, A16). Meis2 expression was greatly reduced in the TRN of Ascl1 mutants in a pattern similar to that of Isl1- deficient mutants (Fig. 6A7, A17). The narrow band corresponding to Arx, Pax6, or Lhx1 expression along the zli was mostly missing, and the expression of these genes was also lost in the vLG in the absence of Ascl1 (Fig. 6A8, A9, A10, A18, A19, A20). In contrast, Arx, Pax6, and Lhx1 expression was ectopically induced in the TRN of Ascl1KI/KI mutants (Fig. 6A8, A9, A10, A18, A19, A20). These data suggest that, similar to Isl1, Ascl1 is required to promote TRN identity and repress non-TRN prethalamic cell fates within the TRN and that unlike Isl1, Ascl1 is also important for the differentiation of other prethalamic nuclei.
We next analyzed circuit formation between the prethalamus and thalamus. Consistent with previous observations [37], NF-M+ thalamic axons formed abnormally thick fascicles and extended randomly in the prethalamus (Fig. 6B3, B4). Axonal arrangement was also severely disrupted within the thalamus of Ascl1KI/KI mutants. NF-M+ thalamic axons formed thin fascicles and were arranged in parallel in the control thalamus, whereas those in Ascl1 mutants formed thick bundles and crossed each other with abnormal overlapping course of axons (Fig. 6B3, B4, arrows). Although defective TCAs within the thalamus could be a secondary consequence of a total failure in morphological maturation of the prethalamus, we assessed the potential importance of PTAs in the establishment of thalamocortical connectivity. We therefore crossed mice carrying the Ascl1 null allele and R26-tdTomato with the Dlx5/6-Cre line. Loss of Ascl1 resulted in a marked reduction in the number of tdTomato+ axons projecting from the prethalamus to the thalamus in E13.5 and E14.5 embryos (Fig. 6C). When the Neurovue tracer was injected into prethalamic regions, Neurovue labeling accumulated in the prethalamus of both control and mutant embryos, but Neurovue+ axons were not apparent in the thalamus of Ascl1 mutants (Fig. 6D), confirming that Ascl1 deletion disrupts the formation of axons that project from the prethalamus to the thalamus. These results demonstrate that Ascl1 and Isl1 have a significant degree of overlapping regulatory function in prethalamic differentiation and circuit formation.
Fig. 6.

Ascl1 controls TRN cell fate and PTA outgrowth and is essential for Isl1 expression in the prethalamus. (A) In situ hybridization for prethalamic markers on control (Ascl1KI/+) and mutant (Ascl1KI/KI) coronal sections at E14.5 and E16.5. In Ascl1 mutants, Dlx1 expression was lost in the TRN as well as in the rZI, vLG, and zli (A6, A16). Loss of Ascl1 resulted in severe downregulation of Meis2 expression in the TRN (A7, A17). Arx, Pax6, or Lhx1 expression in the vLG and zli of Ascl1KI/KI mutants was downregulated but ectopically induced in the TRN (A8-A10, A18-A20). (A21) Quantification of in situ hybridization. The pixel intensity values of TRN or vLG were grouped and averaged from three different sections per embryo (n = 5 embryos; Student’s t-test; **p < 0.01, ***p < 0.001, ****p < 0.0001). (B) Immunohistochemistry for NF-M on coronal sections of control (Ascl1KI/+) and mutant (Ascl1KI/KI) embryos at E14.5 and E16.5. In control embryos, NF-M+ thalamic axons extended in ordered and parallel arrays that were less fasciculated in the thalamus and prethalamus (B1,B2) than in mutant embryos (B3,B4). Moreover, in Ascl1 mutants, thick NF-M+ bundles crossed each other with abnormal overlapping course of axons (arrows in B3,B4). (C) Immunohistochemistry for tdTomato on coronal sections of control (Dlx5/6-Cre; R26-TdTomato; Ascl1KI/+) and mutant (Dlx5/6-Cre; R26-TdTomato; Ascl1KI/KI) embryos at E13.5 and E14.5. The control prethalamus formed ordered and parallel tdTomato+ projections to the thalamus (C1,C3), while very few PTAs were detected in the mutant embryos (C2,C4). (C5) Examples of the measurements taken for the quantification. Yellow dashed lines in C1-C4 indicate the position of the lines used to quantify the number of axon bundles crossing the thalamus. (n = 3 embryos; two-way ANOVA with Sidak’s multiple comparison test; **p < 0.01, ***p < 0.001, ****p < 0.0001). (D) Implanting the chemical tracer Neurovue into the prethalamus of control (Ascl1KI/+) and mutant embryos (Ascl1KI/KI) at E13.5. Compared to those in controls (D1), no Neurovue+ PTAs were labeled in the thalamus in the absence of Ascl1 (D2). (E) In situ hybridization for Isl1 and Shh on control (Ascl1KI/+) and mutant (Ascl1KI/KI) coronal sections at E12.5. Shh is a specific marker for the zli. Loss of Ascl1 abrogated Isl1 expression in the TRN. (E5) Quantification of in situ hybridization. The pixel intensity values of TRN were grouped and averaged from four different sections per embryo (n = 4 embryos; Student’s t-test; ****p < 0.0001).Pt, prethalamus: Hyp, hypothalamus; Pt, prethalamus; rZI, rostral zona incerta; Th, thalamus; TRN, thalamic reticular nucleus; vLG, ventral lateral geniculate; zli, zona limitans intrathalamica. Scale bars, 200 μm
We then investigated the gene regulatory hierarchy between Isl1 and Ascl1. In the developing prethalamus, Ascl1 expression is restricted to ventricular zone progenitors (Fig. S2E), whereas Isl1 is expressed both in the progenitor and postmitotic regions (Fig. 6E1, E2). When in situ hybridization for Isl1 on coronal sections was performed with Shh, a zli-specific marker, to locate the affected area more precisely, we found that Isl1 expression was lost in the prethalamus of Ascl1KI/KI mutant embryos (Fig. 6E3, E4). In contrast, Ascl1 expression was largely unaffected in the prethalamus of Dlx5/6-Cre; Isl1F/F mutants (Fig. S2E). These observations revealed a crucial role for Ascl1 in the regulation of Isl1 expression in the prethalamus.
Isl1 is necessary for Ascl1-mediated fate specification and neurite outgrowth in in vitro cultured prethalamic neurons
The above-described observations led us to ask whether Isl1 is an essential component of Ascl1-mediated genetic programs that support neuronal fate specification and connectivity of the TRN. To address this question, we established primary cell cultures from the prethalamus of E13.5 Ascl1KI/+ and Ascl1KI/KI embryonic brains. To verify whether cultured cells were indeed derived from TRN tissues, cells that had been cultured for 2 days in vitro (DIV) were immunostained with Isl1, Dlx1, and Pax6 antibodies. The majority of primary cultured cells from control TRN tissues were Isl1+, Dlx1+ and Pax6−, while Ascl1-deficient cells exhibited Isl1−, Dlx1− and Pax6+ immunoreactivity (Fig. 7A, B), indicating TRN cell identity. We next examined neurite outgrowth of neurons in primary cultures from Ascl1KI/+ and Ascl1KI/KI embryos using Tuj1, a neuron-specific class III beta-tubulin that is widely used to visualize neuronal extensions. We measured the number of neurites, the length of the longest neurite, and the total length of neurites to assess neurite outgrowth in primary cultures. Consistent with our embryo experiments, Ascl1KI/KI primary cultured neurons developed fewer neurites than did Ascl1KI/+ neurons at DIV2 and DIV6 (Fig. 7C, D). The longest neurites and the total length of neurites in Ascl1KI/KI neurons were shorter than those in Ascl1KI/+ neurons (Fig. 7C, D). To assess the ability of Isl1 to rescue the TRN phenotypes caused by Ascl1 deficiency, an Isl1 expression construct was transfected into Ascl1KI/KI TRN cells that had been cultured for 1 DIV together with a tdTomato reporter expression vector to identify the transfected cells. After 6 additional days of culture, the tdTomato+ cells were evaluated for neuron identity and neuritogenesis. Compared with that in mock-transfected Ascl1KI/KI cells, the number of Dlx1+ cells in primary cultures of Ascl1KI/KI cells transfected with the Isl1 expression construct increased, while the number of Pax6+ cells in Isl1-transfected cells decreased (Fig. 7E, F). For each Tuj1+ cell, the number of neurites was greater in Ascl1-deficient cells transfected with the Isl1 expression construct than in mock-transfected cells (Fig. 7G, H). The longest neurites and the total length of neurites in Isl1-transfected Ascl1KI/KI cells were longer than those in mock-transfected cells (Fig. 7G, H), indicating that Isl1 significantly rescued defective neuritogenesis caused by Ascl1 deficiency. Taken together, these results suggest that Isl1 is an important mediator of Ascl1 function in the fate specification and axon formation of in vitro-derived TRN neurons.
Fig. 7.

Ascl1 requires Isl1 to promote TRN neuronal specification and neurite outgrowth in cultured neurons. (A, B) Isolation and primary culture of TRN cells from E13.5 control (Ascl1KI/+) and Ascl1KI/KI mutant embryonic brains. TRN cells were cultured on coverslips coated with L-ornithine and laminin in 24-well plates. TRN cell identity was verified by immunocytochemistry of DIV2 cells using antibodies against Isl1, Dlx1 and Pax6. Consistent with the gene expression profiles of the embryos, most of the control cultured cells derived from Ascl1KI/+ TRN were Isl1+, Dlx1+, and Pax6−, while many of the Ascl1KI/KI cells were Isl1−, Dlx1−, and Pax6+. A total of 100 DAPI+ cells were quantified for each field, and three measurements were averaged for each coverslip. The average values from six coverslips prepared from three embryos were plotted for each genotype (Student’s t-test; **p ≤ 0.01). (C, D) Tuj1 immunostaining was performed at DIV2 and DIV6 to determine the number of neurites, the length of the longest neurite, and the total length of neurites. Quantification of the number of neurites, length of the longest neurite, and total length of neurites were achieved from 20 neurons for each field, and three measurements were averaged for each coverslip. The average values from ten coverslips prepared from five embryos were plotted for each genotype (two-way ANOVA with Sidak’s multiple comparison test; **p < 0.01, ****p < 0.0001). (E-H) TRN cells isolated from Ascl1KI/KI mutant E13.5 brains were cultured for 1 DIV and transfected with an Isl1 expression construct (pCMV-Isl1) or an empty vector (pCMV). The reporter expression vector (pCMV-tdTomato) was cotransfected to identify the transfected cells. (E, F) Quantification was achieved from transfected Ascl1KI/KI mutant cells cultured for 5 additional DIV by counting Dlx1+ or Pax6+ cells among 100 tdTomato+ cells for each field. Three measurements were averaged for each coverslip, and the average values from eight coverslips prepared from four embryos were plotted for each genotype (Student’s t-test; **p < 0.01). (G, H) The number of neurites, length of the longest neurite, and total length of neurites were measured for each tdTomato+ neuron cultured for 5 additional DIV following transfection. Quantification was achieved from 20 tdTomato+- transfected cells for each field. Three measurements were averaged for each coverslip, and the average values from ten coverslips prepared from five embryos were plotted for each genotype (Student’s t-test; *p < 0.05, **p ≤ 0.01)
Discussion
Our study demonstrated the key roles of the Ascl1-Isl1 regulatory cascade in orchestrating TRN neuronal fate specification and connectivity. A set of TRN-enriched genes, such as Dlx1 and Meis2, was specifically downregulated in Isl1-deficient TRN, while the loss of Isl1 did not influence Dlx1 expression in other prethalamic nuclei. The importance of Isl1 in the determination of TRN neuronal identity was further evidenced by the concomitant induction of a set of TRN-depleted genes, such as Arx, Pax6, and Lhx1/5, in Isl1-deficient TRN. This coordinated regulation by Isl1 implicates a central role in determining the cell fate of the TRN. Given that the Gad1 expression pattern was unaltered in the entire prethalamus in the absence of Isl1, the downregulation of TRN-enriched genes and upregulation of TRN-depleted genes were not due to deprivation of TRN tissue. Therefore, these findings indicate that the role of Isl1 in the developing prethalamus is likely to specify the fate of TRN cells by promoting TRN-enriched genes and suppressing TRN-depleted genes. Conditional deletion of Isl1 at the late developmental stage caused similar phenotypes, indicating that TRN cells continue to depend on Isl1 for their normal differentiation during late gestation. Embryonic inactivation of Isl1 causes abnormalities in the formation of TRN at adult stages, as demonstrated by reduced Meis2 expression [28]. Previous studies demonstrated critical roles for Isl1 in specifying striatal neurons in the ventral telencephalon [28, 29]. Loss of Isl1 led to the downregulation of striatonigral genes and concomitant upregulation of striatopallidal genes, while forced expression of Isl1 repressed the striatopallidal genetic program, indicating that Isl1 plays dual roles by promoting striatonigral neuron identity and repressing striatopallidal neuron identity. With respect to the molecular processes by which Isl1 regulates cell types, it has been reported that Isl1 controls motor neuron fate specification by cooperating with Lhx3 and striatal neuron identity by interacting with Lhx8 [56, 57]. Whether the differentiation of TRN neurons is dependent on the combined activity of Isl1 and other LIM homeodomain proteins requires further investigation.
In our study, the loss of Isl1 resulted in abnormal fasciculation and pathfinding of TCAs by disrupting the development of structures located on the path of thalamic axons. The TRN is thought to be a crucial terrain for axon projections because all cortico-thalamic and thalamo-cortical axons must cross the TRN. How does proper formation of the TRN regulated by Isl1 control the normal extension of TCAs? It is possible that in the absence of Isl1, TCAs entering the prethalamus undergo an abrupt change in the expression levels of TRN-enriched/-depleted genes and encounter an abnormal attractive/repulsive environment. Previously, similar defects in the pathfinding of TCAs were observed in Dlx1/2−/− mutant embryos [58]. Our results showed that disruption of TRN development by Isl1 inactivation was accompanied by dysregulated expression of the axon guidance molecule Efna5, inhibiting the growth of TCAs passing through the prethalamus. Efna5, which encodes one of the membrane-tethered ligands for EphA receptors, was not expressed at detectable levels in the wild-type TRN, while loss of Isl1 led to strong upregulation of this gene in the TRN. Efna ligands transduce the signaling of EphA receptors, leading primarily to cell repulsion [59]. Previous studies have demonstrated that Efna5 functions as an inhibitor of thalamic axonal growth [60] or serves as a repellent cue of thalamic axons [61]. Our observations and those of others indicate that normal thalamo-cortical connectivity requires the correct expression pattern of Efna5, while the failure to regulate Efna5 leads to misprojection of TCAs. It is therefore likely that another crucial role of Isl1 is to ensure the correct extension of TCAs by promoting appropriate regionalization of the prethalamus and preventing inappropriate expression of the axon guidance molecule.
The current study provides intriguing new perspectives on the growth and significance of PTAs extending into the thalamus. Another important new finding is that the loss of Isl1 resulted in abnormal fasciculation and random orientation of thalamic axons within the thalamus despite that Isl1 deletion had no impact on the formation of the thalamus. Disrupted neuronal specification in the TRN and ectopic expression of the repellent Efna5 in the prethalamus do not account for all TCA defects within the thalamus because many disordered thick bundles were also detected in the caudal thalamus distant from the prethalamus, and some of the misrouted fibers extended more caudally into the pretectum. It is, therefore, likely that in the absence of Isl1, TCAs might have undergone abnormal fasciculation and pathfinding errors to a certain degree before leaving the thalamus. These results suggest that there must be additional factors underlying the phenotypes. It has been suggested that prethalamic nuclei, including the TRN, contain a population of cells that extend axons to the thalamus and function to guide TCA navigation [37, 50–52]. First, we found that proper formation of PTAs relies on Isl1 function. Second, we determined the relative developmental timing between TCAs and PTAs because if PTAs pioneer TCAs, PTA outgrowth should precede the extension of TCAs into the prethalamus. By simultaneous visualization of thalamic and prethalamic axons, we showed that PTAs begin to penetrate thalamic areas before TCAs invade the prethalamus. Third, Isl1 mutant mice phenocopy the TCA pathfinding defects observed in the prethalmus and thalamus of Ascl1 mutant embryos [37]. We found that PTAs require Ascl1 for their normal development and that Isl1 functions downstream of Ascl1. Therefore, our data and those of others corroborate a role for PTAs in guiding thalamo-cortical axons. However, further work will be required to dissociate the impact of PTAs on the pathfinding of TCAs from that of axon guidance molecules.
Our study uncovered striking similarities in the aberrant phenotypes of TRN neuron specification between Isl1 and Ascl1 mutants. Furthermore, the phenotypes of defective prethalamo-thalamic and thalamo-cortical projections in Isl1 mutant brains were similar to those observed in Ascl1 mutant embryos. However, there are some differences in the phenotypes between Ascl1 mutant mice and Isl1 mutant mice. Loss of Ascl1 disrupted neuronal differentiation in all of the prethalamic nuclei, while Isl1 inactivation affected the development of the TRN but not the remainder of the prethalamus. This finding suggested that TRN neurons are very different from other prethalamic neurons. As Ascl1 is expressed in the progenitor region along the anteroposterior axis of the prethalamus and has the ability to specify neuronal progenitors, it is likely that loss of Ascl1 could block the generation of committed neuronal precursors in the prethalamus. Previously, in Ascl1 mutant mice, multiple neurogenesis defects, including defects in the proliferation and neuronal specification of progenitor cells, have been observed in the telencephalon, and midbrain [36, 62, 63]. Notably, we found that Isl1 is functionally required for Ascl1-mediated neuronal specification and axonal projections in in vitro cultured TRN cells. These findings suggest that Ascl1 may regulate the distribution of Isl1, which may, in turn, affect TRN differentiation. However, the impairments in gene expression and neurite outgrowth in in vitro cultured Ascl1-deficient cells could be partially restored by forced expression of Isl1, suggesting that the ability of Ascl1 to promote TRN differentiation is unlikely to be mediated entirely by Isl1. The question of whether parallel action by other transcription factors participates in the regulation of TRN development by Ascl1 needs further investigation.
Materials and methods
Mouse lines
The generation of Isl1loxp, Ascl1KI, Dlx5/6-Cre, Foxg1-IRES-Cre, and R26-tdTomato mice obtained from The Jackson Laboratory was described previously [38, 39, 46, 64]. All animals used for these experiments were maintained on mixed backgrounds. To confirm mating, vaginal plugs were checked each morning, and plug dates were considered as E0.5.
Production of transgenic mice
The SBR sequence was amplified by PCR with the primers 5′-ATTAGCGGCCGCCCTTTCTTTCTGTCTGCTGATTGCC-3′ and 5′-ATTAGCGGCCGCATGTTCAACTCCTTGGAACTTACC-3’ and cloned into the NotI site of an expression construct containing a β-globin minimal promoter, the Cre gene, and an SV40 poly(A) signal. The resulting plasmid transgene was linearized with SalI for microinjection as previously described [54]. Stable transgenic mouse lines were generated by pronuclear injection into fertilized eggs derived from the FVBN mouse strain.
Section in situ hybridization
The pairs of PCR primer sequences used to generate template DNA for in vitro transcription by T7 RNA polymerase were obtained from the Allen Brain Atlas or designed against unique regions of transcripts to avoid cross-reactivity (Supplementary Table 1). Embryonic brains were collected from timed pregnant females. Section in situ hybridization were performed as described previously [49]. The brains were fixed overnight in 4% paraformaldehyde at 4 °C, washed three times in PBS, pH7.4, submerged in 30% sucrose in PBS, pH7.4, at 4 °C, embedded in optimal cutting temperature embedding medium (OCT), quickly frozen on dry ice and cryosectioned at 25 μm. Experiments for any given riboprobe were performed in at least three independent biological replicates.
To quantify the signal intensity of in situ hybridization, after the stained sections were photographed under the same illumination conditions, all images were converted to 8-bit grayscale using Fiji software [65]. The mean pixel intensity of the staining signal was obtained by subtracting the mean intensity value of the background region from that of the stained region.
Immunostaining
For immunohistochemistry, brains were fixed in 4% paraformaldehyde in PBS, pH7.4, for 2 h at 4 °C, immersed in 30% sucrose, embedded in OCT compound, and sectioned using a cryostat at 25 μm. The cryosections were washed with PBS, pH7.4, containing 0.4% Triton X-100 and incubated in blocking buffer containing 0.4% Triton X-100, 10% sheep serum and 5% Mouse on Mouse (MOM) blocking reagent (VectorLabs, MKB-2213-1) in PBS, pH7.4, for 2 h at 4 °C. For immunocytochemistry, coverslips containing prethalamic neurons obtained from cell culture were washed with PBS, pH7.4, containing 0.1% Triton X-100 and incubated in blocking buffer containing 0.4% Triton X-100, 10% sheep serum and 5% MOM blocking reagent in PBS, pH7.4, for 1 h at 4 °C. Sections or coverslips were then incubated overnight at 4 °C with the following primary antibodies: anti-Tubulin beta III isoform (Millipore, MAB1637, 1:600), anti-NF-M (DSHB, 2H3, 1:100), anti-GFP (Abcam ab6673, 1:500), anti-Isl1 (DSHB, 39.3F7, 1:100), anti-tdTomato (SICGEN, AB8181–200, 1:500), anti-Dlx1 (Chemicon, AB5724, 1:400) and anti-Pax6 (DSHB, 1:100). Following incubation with primary antibodies, sections or coverslips were incubated with species-specific secondary antibodies conjugated to Alexa Fluor (Molecular Probes, A11034, A11003, A11010, A11029, A11055, A11056; 1:200).
For quantification of number of TCAs, we positioned a line across the prethalamus, and generated a GFP intensity profile using Fiji software. Intensity profiles were then processed by tracing and quantifying the number of GFP+ fluorescent signals crossing the line. For quantification of number of PTAs, we positioned three lines across the thalamus (1: at the thalamic-prethalamic border; 2: at a caudal thalamic region; 3: at the midpoint position between the two other lines), and generated a tdTomato intensity profile.
Quantitative reverse transcription PCR
Brains were dissected from Dlx5/6-Cre; Isl1F/+;R26-tdTomato or Dlx5/6-Cre; Isl1F/F;R26-tdTomato embryos at E13.5 and then cut in half along the dorsal and ventral midline. Fluorescence by tdTomato expression allowed us to distinguish prethalamic tissues from surrounding tissues. After prethalamic tissues from the two halves of the same embryo were harvested in TRIzol Reagent (Invitrogen), total RNA was isolated using an RNeasy Mini kit (Qiagen) and used to prepare cDNA using a SuperScriptIII first-strand synthesis system (Invitrogen). Quantitative PCR was carried out in duplicate for each RNA sample using QuantiTect SYBR Green Supermix (Qiagen) following the manufacturers’ instructions. The reactions were monitored using an Mx4000 multiplex quantitative PCR system (Stratagene, La Jolla, CA). The housekeeping gene β-actin was used for normalization, and relative quantification was calculated using comparative threshold cycle values for three biological replicates. All primers used are listed in Supplementary Table 1.
Explant culture
Dissection and explant culture were carried out as described previously [44, 66]. Thalamic explants were isolated from E13.5 Dlx5/6-Cre; Gbx2GFP; Isl1F/+ or Dlx5/6-Cre; Gbx2GFP; Isl1F/F embryos. Hanging drops of 293T cells transfected with pCMV alone or pCMV-Efna5 were embedded in basement membrane matrices (Corning Matrigel). Thalamic explants were positioned approximately 200–400 μm from the transfected 293T cell aggregates and cocultured in Neurobasal medium (Thermofisher) containing B-27 supplements (Invitrogen) and 1% penicillin/streptomycin. After 48 h, the cultures were fixed in 4% paraformaldehyde in PBS, pH7.4, for 30 min at 4 °C. Assays for axon outgrowth were quantified by measuring the area of fluorescence associated with GFP+-axons extending from the distal or proximal sides of the explant with respect to the cell aggregate as described previously [66].
Primary neuron cultures and transfection
Embryonic brains were dissected from the Ascl1KI mouse line at E13.5 in 3 cm sterile Petri dish containing ice cold dissection medium, consisting of Hanks’ balanced salt solution (HBSS) (Gibco), 20 mM D-glucose (Sigma), 200 µM ascorbic acid (Sigma), and 100 units/ml penicillin/streptomycin (Gibco) and then cut in half along the dorsal and ventral midline. GFP fluorescence, which labels Ascl1-expressing cells, allowed us to locate and dissect TRN tissues. TRN tissues were collected in a 1.5 ml sterile tube, and digested with StemPro Accutase Cell Dissociation reagent (Thermo Fisher Scientific) for 10–20 min at 37 °C. The digestion was stopped by removing the supernatant and washing the tissue with dissociation medium consisting of Neurobasal medium (Invitrogen), 2% B-27 supplement (Thermo Fisher Scientific), 2% B-27 supplement Plus (Thermo Fisher Scientific), 1% FBS (Gibco), 200 µM L-glutamine (Thermo Fisher Scientific) and 200 µM ascorbic acid. The tissue was resuspended in 1 ml of dissociation medium and triturated to obtain single cell suspension. After the cells were spun at 200 × g, the cell pellet was resuspended in the dissociation medium. The number of dissociated cells and the ratio of viable to damaged cells were determined by Trypan blue staining using a hemocytometer. The cell suspension was transferred on top of 12 mm coverslips coated with 20 µg/ml poly-L-ornithine (Sigma) and 10 µg/ml laminin (Thermo Fisher Scientific) (100,000 cells per coverslip). After 1 h of incubation in a humidified tissue culture incubator at 37 °C with 5% CO2, the coverslips were carefully transferred into 24-well plates containing the dissociation medium. Transient transfection was performed at DIV1. The transfected plasmids (0.3 µg) consisted of a mixture of 0.1 µg of pCMV-tdTomato (a gift from Davidson M, Shaner N, and Tsien R; Addgene plasmid #54642) [67] + 0.2 µg of pCMV-Isl1 (Sino Biological plasmid #MG56983-UT). Mock transfection (0.3 µg) consisted of 0.1 µg of pCMV-tdTomato + 0.2 µg of pCMV empty vector. For individual wells in 24-well plates, a total of 0.3 µg of DNA was diluted in 25 µl of Opti-MEM reduced serum medium (Gibco), and 2.5 µl of Lipofectamine 2000 (Invitrogen) was diluted in 25 µl of Opti-MEM in a separate tube. The diluted DNA-Lipofectamine mixture was incubated at RT for 20 min and applied to Ascl1KI/KI cells.
Measurement of neurite outgrowth
For quantitation of neurite outgrowth, images of stained cultures were captured using a Leica DMI6000B digital camera (Leica Microsystems), and the number of neurites, the length of the longest neurite and the total length of neurites were scored from the images captured by using the neurite tracer plugin in ImageJ [68]. Images are optimized to reduce artifacts, subtract background staining, and enhance contrast. Following optimization, images were thresholded and skeletonized, and then neurite length was measured by tracing down Tuj1 staining from the edge of the cell body to the furthest point. Such measurements were carried out for every neurite of each neuron.
Statistical analysis
All the mutant phenotypes we observed in the Dlx5/6-Cre; Isl1F/F, Foxg1-IRES-Cre; Isl1F/F, SBR-Cre; Isl1F/F, and Ascl1KI/KI mice were fully penetrant. The number of independent values in each experiment is as follows: Fig. 1, five independent biological replicates (n = 5) were used for RNA in situ hybridization; Fig. 2, n = 5 for immunohistochemistry; Fig. 3, n = 3 for real-time RT-PCR, n = 3 for ISH, n = 3 for explant culture; Fig. 4, n = 3 for immunohistochemistry, n = 5 for Neurovue tracing; Fig. 5, n = 3 for RNA in situ hybridization; Fig. 6, n = 5 for RNA in situ hybridization (A), n = 4 for RNA in situ hybridization (E), n = 3 for immunohistochemistry, n = 5 for Neurovue tracing; Fig. 7, n = 3 for Isl1, Dlx1, and Pax6 immunostaining in control and Ascl1KI/KI cells, n = 5 for neurite outgrowth in control and Ascl1KI/KI cells, n = 4 for Dlx1 and Pax6 immunostaining in transfected Ascl1KI/KI cells, n = 5 for neurite outgrowth in transfected Ascl1KI/KI cells. Relevant information for each experiment, including statistical tests and p values, is included in the legend corresponding to each figure. In all cases, p ≤ 0.05 was considered to indicate statistical significance.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Not applicable.
Author contributions
Conceived the project: YJ. Designed the experiments: QN, YJ. Performed experiments: QN, HT. Analyzed the data: QN, HT, YJ. Wrote the paper: YJ. Reviewed the paper: QN, HT, YJ.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grants funded by the Korea Ministry of Science and ICT (No. 2022R1F1A1064555).
Data availability
All data generated or analyzed during this study are included in the manuscript and supporting files.
Declarations
Competing interests
There are no competing interests.
Ethics approval
All animal procedures were carried out in accordance with the guidelines and protocols approved by the Kyung Hee University Institutional Animal Care and Use Committee.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nair A, Treiber JM, Shukla DK et al (2013) Impaired thalamocortical connectivity in autism spectrum disorder: a study of functional and anatomical connectivity. Brain 136:1942–1955. 10.1093/brain/awt079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krol A, Wimmer RD, Halassa MM, Feng G (2018) Thalamic reticular dysfunction as a Circuit Endophenotype in Neurodevelopmental disorders. Neuron 98:282–295. 10.1016/j.neuron.2018.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steullet P, Cabungcal JH, Bukhari SA et al (2018) The thalamic reticular nucleus in schizophrenia and bipolar disorder: role of parvalbumin-expressing neuron networks and oxidative stress. Mol Psychiatry 23:2057–2065. 10.1038/mp.2017.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheffield JM, Huang AS, Rogers BP et al (2020) Thalamocortical anatomical connectivity in schizophrenia and psychotic bipolar disorder. Schizophr Bull 46:1062–1071. 10.1093/schbul/sbaa022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinault D (2004) The thalamic reticular nucleus: structure, function and concept. Brain Res Rev 46:1–31. 10.1016/j.brainresrev.2004.04.008 [DOI] [PubMed] [Google Scholar]
- 6.Jones E (2007) The Thalamus, 2nd edn. Cambridge University Press, Cambridge [Google Scholar]
- 7.Sherman SM, Guillery RW (2002) The role of the thalamus in the flow of information to the cortex. Philos Trans R Soc B Biol Sci 357:1695–1708. 10.1098/rstb.2002.1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McAlonan K, Cavanaugh J, Wurtz RH (2006) Attentional modulation of thalamic reticular neurons. J Neurosci 26:4444–4450. 10.1523/JNEUROSCI.5602-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halassa MM, Chen Z, Wimmer RD et al (2014) State-dependent architecture of thalamic reticular subnetworks. Cell 158:808–821. 10.1016/j.cell.2014.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong P, Wang H, Shen XF et al (2019) A novel cortico-intrathalamic circuit for flight behavior. Nat Neurosci 22:941–949. 10.1038/s41593-019-0391-6 [DOI] [PubMed] [Google Scholar]
- 11.Saletin JM, Coon WG, Carskadon MA (2017) Stage 2 Sleep EEG Sigma Activity and Motor Learning in Childhood ADHD: a pilot study. J Clin Child Adolesc Psychol 46:188–197. 10.1080/15374416.2016.1157756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puelles L, Rubenstein JLR (2003) Forebrain gene expression domains and the evolving prosomeric model. Trends Neurosci 26:469–476. 10.1016/S0166-2236(03)00234-0 [DOI] [PubMed] [Google Scholar]
- 13.Halassa MM, Acsády L (2016) Thalamic inhibition: diverse sources, diverse scales. Trends Neurosci 39:680–693. 10.1016/j.tins.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crabtree JW (2018) Functional diversity of thalamic reticular subnetworks. Front Syst Neurosci 12:1–18. 10.3389/fnsys.2018.00041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Lopez-Huerta VG, Adiconis X et al (2020) Distinct subnetworks of the thalamic reticular nucleus. Nature 583:819–824. 10.1038/s41586-020-2504-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiecker C, Lumsden A (2004) Hedgehog signaling from the ZLI regulates diencephalic regional identity. Nat Neurosci 7:1242–1249. 10.1038/nn1338 [DOI] [PubMed] [Google Scholar]
- 17.Bluske KK, Vue TY, Kawakami Y et al (2012) β-Catenin signaling specifies progenitor cell identity in parallel with shh signaling in the developing mammalian thalamus. Development 139:2692–2702. 10.1242/dev.072314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hagemann AIH, Scholpp S (2012) The tale of the three brothers - shh, wnt, and Fgf during development of the thalamus. Front Neurosci 6:1–9. 10.3389/fnins.2012.00076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi D, Kobayashi M, Matsumoto K et al (2002) Early subdivisions in the neural plate define distinct competence for inductive signals. Development 129:83–93. 10.1242/dev.129.1.83 [DOI] [PubMed] [Google Scholar]
- 20.Hirata T, Nakazawa M, Muraoka O et al (2006) Zinc-finger genes Fez and Fez-like function in the establishment of diencephalon subdivisions. Development 133:3993–4004. 10.1242/dev.02585 [DOI] [PubMed] [Google Scholar]
- 21.Scholpp S, Foucher I, Staudt N et al (2007) Otx1l, Otx2 and Irx1b establish and position the ZLI in the diencephalon. Development 134:3167–3176. 10.1242/dev.001461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scholpp S, Lumsden A (2010) Building a bridal chamber: development of the thalamus. Trends Neurosci 33:373–380. 10.1016/j.tins.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ono K, Clavairoly A, Nomura T et al (2014) Development of the prethalamus is crucial for thalamocortical projection formation and is regulated by Olig2. Dev 141:2075–2084. 10.1242/dev.097790 [DOI] [PubMed] [Google Scholar]
- 24.Pfaff SL, Mendelsohn M, Stewart CL et al (1996) Requirement for LIM homeobox gene Isl1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell 84:309–320. 10.1016/S0092-8674(00)80985-X [DOI] [PubMed] [Google Scholar]
- 25.Elshatory Y, Everhart D, Deng M et al (2007) Islet-1 controls the differentiation of retinal bipolar and cholinergic amacrine cells. J Neurosci 27:12707–12720. 10.1523/JNEUROSCI.3951-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gay F, Anglade I, Gong Z, Salbert G (2000) The LIM/Homeodomain Protein Islet-1 modulates estrogen receptor functions. Mol Endocrinol 14:1627–1648. 10.1210/mend.14.10.0538 [DOI] [PubMed] [Google Scholar]
- 27.Nasif S, De Souza FSJ, González LE et al (2015) Islet 1 specifies the identity of hypothalamic melanocortin neurons and is critical for normal food intake and adiposity in adulthood. Proc Natl Acad Sci U S A 112:E1861–E1870. 10.1073/pnas.1500672112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ehrman LA, Mu X, Waclaw RR et al (2013) The LIM homeobox gene Isl1 is required for the correct development of the striatonigral pathway in the mouse. Proc Natl Acad Sci U S A 110. 10.1073/pnas.1308275110 [DOI] [PMC free article] [PubMed]
- 29.Lu K-M, Evans SM, Hirano S, Liu F-C (2013) Dual role for Islet-1 in promoting striatonigral and repressing striatopallidal genetic programs to specify striatonigral cell identity. Proc Natl Acad Sci 111. 10.1073/pnas.1319138111 [DOI] [PMC free article] [PubMed]
- 30.Ehrman JM, Merchan-Sala P, Ehrman LA et al (2022) Formation of the Mouse Internal Capsule and Cerebral Peduncle: a pioneering role for Striatonigral axons as revealed in Isl1 conditional mutants. J Neurosci 42:3344–3364. 10.1523/JNEUROSCI.2291-21.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guillemot F, Hassan BA (2017) Beyond proneural: emerging functions and regulations of proneural proteins. Curr Opin Neurobiol 42:93–101. 10.1016/j.conb.2016.11.011 [DOI] [PubMed] [Google Scholar]
- 32.Parras CM, Schuurmans C, Scardigli R et al (2002) Divergent functions of the proneural genes Mash1 and Ngn2 in the specification of neuronal subtype identity. Genes Dev 16:324–338. 10.1101/gad.940902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S, Misra K, Matise MP, Xiang M (2005) Foxn4 acts synergistically with Mash1 to specify subtype identity of V2 interneurons in the spinal cord. Proc Natl Acad Sci U S A 102:10688–10693. 10.1073/pnas.0504799102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aslanpour S, Rosin JM, Balakrishnan A et al (2020) Ascl1 is required to specify a subset of ventromedial hypothalamic neurons. Development 147:1–15. 10.1242/dev.180067 [DOI] [PubMed] [Google Scholar]
- 35.Horton S, Meredith A, Richardson JA, Johnson JE (1999) Correct coordination of neuronal differentiation events in ventral forebrain requires the bHLH factor MASH1. Mol Cell Neurosci 14:355–369. 10.1006/mcne.1999.0791 [DOI] [PubMed] [Google Scholar]
- 36.Casarosa S, Fode C, Guillemot F (1999) Mash1 regulates neurogenesis in the ventral telencephalon. Development 126:525–534. 10.1242/dev.126.3.525 [DOI] [PubMed] [Google Scholar]
- 37.Tuttle R, Nakagawa Y, Johnson JE, O’Leary DDM (1999) Defects in thalamocortical axon pathfinding correlate with altered cell domains in Mash-1-deficient mice. Development 126:1903–1916. 10.1242/dev.126.9.1903 [DOI] [PubMed] [Google Scholar]
- 38.Sun Y, Dykes IM, Liang X et al (2008) A central role for Islet1 in sensory neuron development linking sensory and spinal gene regulatory programs. Nat Neurosci 11:1283–1293. 10.1038/nn.2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monory K, Massa F, Egertová M et al (2006) The Endocannabinoid System Controls Key Epileptogenic circuits in the Hippocampus. Neuron 51:455–466. 10.1016/j.neuron.2006.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kitamura K, Miura H, Yanazawa M et al (1997) Expression patterns of Brx1 (Rieg gene), sonic hedgehog, Nkx2.2, Dlx1 and Arx during zona limitans intrathalamica and embryonic ventral lateral geniculate nuclear formation. Mech Dev 67:83–96. 10.1016/S0925-4773(97)00110-X [DOI] [PubMed] [Google Scholar]
- 41.Nakagawa Y, O’Leary DDM (2001) Combinatorial expression patterns of LIM-homeodomain and other regulatory genes parcellate developing thalamus. J Neurosci 21:2711–2725. 10.1523/jneurosci.21-08-02711.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caballero IM, Manuel MN, Molinek M et al (2014) Cell-Autonomous repression of shh by transcription factor Pax6 regulates Diencephalic Patterning by Controlling the Central Diencephalic Organizer. Cell Rep 8:1405–1418. 10.1016/j.celrep.2014.07.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen L, Guo Q, Li JYH (2009) Transcription factor Gbx2 acts cell-nonautonomously to regulate the formation of lineage-restriction boundaries ofthe thalamus. Development 136:1317–1326. 10.1242/dev.030510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chatterjee M, Li K, Chen L et al (2012) Gbx2 regulates thalamocortical axon guidance by modifying the LIM and Robo codes. Dev 139:4633–4643. 10.1242/dev.086991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deck M, Lokmane L, Chauvet S et al (2013) Pathfinding of Corticothalamic Axons Relies on a rendezvous with thalamic projections. Neuron 77:472–484. 10.1016/j.neuron.2012.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawaguchi D, Sahara S, Zembrzycki A, O’Leary DDM (2016) Generation and analysis of an improved Foxg1-IRES-Cre driver mouse line. Dev Biol 412:139–147. 10.1016/j.ydbio.2016.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marcos-Mondéjar P, Peregrín S, Li JY et al (2012) The Lhx2 transcription factor controls thalamocortical axonal guidance by specific regulation of Robo1 and Robo2 receptors. J Neurosci 32:4372–4385. 10.1523/JNEUROSCI.5851-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seibt J, Schuurmans C, Gradwhol G et al (2003) Neurogenin2 specifies the connectivity of thalamic neurons by controlling axon responsiveness to intermediate target cues. Neuron 39:439–452. 10.1016/S0896-6273(03)00435-5 [DOI] [PubMed] [Google Scholar]
- 49.Lee M, Yoon J, Song H et al (2017) Tcf7l2 plays crucial roles in forebrain development through regulation of thalamic and habenular neuron identity and connectivity. Dev Biol 424:62–76. 10.1016/j.ydbio.2017.02.010 [DOI] [PubMed] [Google Scholar]
- 50.Quintana-Urzainqui I, Hernández-Malmierca P, Clegg JM et al (2020) The role of the diencephalon in the guidance of thalamocortical axons in mice. Dev 147. 10.1242/dev.184523 [DOI] [PMC free article] [PubMed]
- 51.Mitrofanis J, Baker GE (1993) Development of the thalamic reticular and perireticular nuclei in rats and their relationship to the course of growing corticofugal and corticopetal axons. J Comp Neurol 338:575–587. 10.1002/cne.903380407 [DOI] [PubMed] [Google Scholar]
- 52.Mitrofanis J, Guillery RW (1993) New views of the thalamic reticular nucleus in the adult and the developing brain. Trends Neurosci 16:240–245. 10.1016/0166-2236(93)90163-G [DOI] [PubMed] [Google Scholar]
- 53.López-Bendito G, Molnár Z (2003) Thalamocortical development: how are we going to get there? Nat Rev Neurosci 4:276–289. 10.1038/nrn1075 [DOI] [PubMed] [Google Scholar]
- 54.Lee B, Song H, Rizzoti K et al (2013) Genomic code for Sox2 binding uncovers its regulatory role in Six3 activation in the forebrain. Dev Biol 381:491–501. 10.1016/j.ydbio.2013.06.016 [DOI] [PubMed] [Google Scholar]
- 55.Leung CT, Coulombe PA, Reed RR (2007) Contribution of olfactory neural stem cells to tissue maintenance and regeneration. Nat Neurosci 10:720–726. 10.1038/nn1882 [DOI] [PubMed] [Google Scholar]
- 56.Thaler JP, Lee SK, Jurata LW et al (2002) LIM factor Lhx3 contributes to the specification of motor neuron and interneuron identity through cell-type-specific protein-protein interactions. Cell 110:237–249. 10.1016/S0092-8674(02)00823-1 [DOI] [PubMed] [Google Scholar]
- 57.Cho H-H, Cargnin F, Kim Y et al (2014) Isl1 directly controls a cholinergic neuronal identity in the developing forebrain and spinal cord by forming cell type-specific complexes. PLoS Genet 10:e1004280. 10.1371/journal.pgen.1004280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garel S, Yun K, Grosschedl R, Rubenstein JLR (2002) Th early topography of thalamocortical projections is shifted in Ebf1 and Dlx/2 mutant mice. Development 129:5621–5634. 10.1242/dev.00166 [DOI] [PubMed] [Google Scholar]
- 59.Kania A, Klein R (2016) Mechanisms of ephrin-eph signalling in development, physiology and disease. Nat Rev Mol Cell Biol 17:240–256. 10.1038/nrm.2015.16 [DOI] [PubMed] [Google Scholar]
- 60.Gao PP, Yue Y, Zhang JH et al (1998) Regulation of thalamic neurite outgrowth by the eph ligand ephrin-A5: implications in the development of thalamocortical projections. Proc Natl Acad Sci U S A 95:5329–5334. 10.1073/pnas.95.9.5329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vanderhaeghen P, Lu Q, Prakash N et al (2000) A mapping label required for normal scale of body representation in the cortex. Nat Neurosci 3:358–365. 10.1038/73929 [DOI] [PubMed] [Google Scholar]
- 62.Vasconcelos FF, Castro DS (2014) Transcriptional control of vertebrate neurogenesis by the proneural factor ascll. Front Cell Neurosci 8:1–6. 10.3389/fncel.2014.00412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peltopuro P, Kala K, Partanen J (2010) Distinct requirements for Ascl1 in subpopulations of midbrain GABAergic neurons. Dev Biol 343:63–70. 10.1016/j.ydbio.2010.04.015 [DOI] [PubMed] [Google Scholar]
- 64.Madisen L, Zwingman TA, Sunkin SM et al (2010) A robust and high-throughput cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schindelin J, Arganda-Carreras I, Frise E et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brose K, Bland KS, Wang KH et al (1999) Slit Proteins Bind Robo Receptors and have an evolutionarily conserved Role in Repulsive Axon Guidance. Cell 96:795–806. 10.1016/S0092-8674(00)80590-5 [DOI] [PubMed] [Google Scholar]
- 67.Shaner NC, Campbell RE, Steinbach PA et al (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22:1567–1572. 10.1038/nbt1037 [DOI] [PubMed] [Google Scholar]
- 68.Pool M, Thiemann J, Bar-Or A, Fournier AE (2008) NeuriteTracer: a novel ImageJ plugin for automated quantification of neurite outgrowth. J Neurosci Methods 168:134–139. 10.1016/j.jneumeth.2007.08.029 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in the manuscript and supporting files.