

Abstract
Disulfidptosis is a newfound programmed cell death (PCD) mode characterized by disulfide stress. Nevertheless, the characteristics and functional significance of disulfidptosis-related genes in head and neck squamous cell carcinoma (HNSCC) are still largely unknown. In this study, several computer-aided bioinformatic analyses were performed. The Nonnegative Matrix Factorization (NMF) method classified The Cancer Genome Atlas (TCGA) patients into two clusters according to the expression of disulfidptosis-related genes. The relative compositions of cells in the tumor microenvironment (TME), mutant landscape, lasso regression analysis, and predicted clinical outcome were performed by analyzing bulk RNA-sequencing data. Besides, single-cell sequencing data (scRNA) was analyzed by Seurat, CopyKAT, and monocle2 to reveal the expression characteristics of disulfidptosis-related genes. Moreover, the spatial distribution characteristics of each cell subgroup in the section and the functional significance of cancer-associated fibroblasts (CAFs) were elucidated by STUtility, SpaCET, and SPATA2. Here, two clusters with different expression characteristics of disulfidptosis-related genes were identified. Cluster 1 (C1) patients had a worse prognosis and a higher proportion of stromal cells but lower effector T cell infiltration than cluster 2 (C2). A novel prognostic model was established and verified in our patient cohort. Additionally, diploid and inflammatory CAFs (iCAFs) showed higher disulfidptosis-related gene expression levels. Furthermore, the CCNC and CHMP1B expressions significantly changed following CAFs differentiation. Disulfidptosis-related genes exhibited extensive and differential spatial expression on tissue sections. Collectively, our study may contribute to revealing the function of disulfidptosis, and improve the expansion of knowledge of crosstalk between cancer cells and CAFs.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12672-024-01629-2.
Keywords: HNSCC, Disulfidptosis, Tumor microenvironment, CAF, Metabolism
Introduction
Head and neck squamous cell carcinomas (HNSCC) account for 95% of head and neck cancers, the eighth most common malignancy, and cause over 310,000 annual deaths worldwide [1, 2]. Although the past few decades have witnessed tremendous progress in therapeutic strategies against cancer, the long-term survival of HNSCC patients remains poor due to the highly aggressive and heterogeneous nature of HNSCC [3]. The 5‐ year survival rate is still less than 50% [4]. Surgical is the primary means of radical treatment for early HNSCC patients. However, patients with advanced-stage HNSCC often experience tumoral recurrence and systematical metastasis and show poor prognosis. Immunotherapy has displayed satisfactory effects in some patients recently, but it usually does not trigger an adequate immune response [5, 6]. It is urgent to clarify further the mechanism involved resistance of tumor cells to drugs and immunotherapy, as well as explore the new effective treatment target against HNSCC.
Programmed cell death (PCD) is essential for maintaining tissue homeostasis in multicellular organisms [7, 8]. Unlike mitosis, which promotes cell growth and proliferation, PCD eliminates damaged, mutated, or unnecessary cells, thereby ensuring the stability of the cell population [9]. By facilitating the removal of abnormal cells, PCD helps to prevent the accumulation of harmful mutations and maintain tissue integrity [10]. Thus, PCD plays a critical role in the normal development and maintenance of healthy tissues. Previously, we generally believed that the imbalance of proto-oncogene and tumor suppressor genes of cells disturbed the normal regulation of proliferation and differentiation, leading to the production of tumor cells. However, it is gradually recognized that cancer development has a significant relationship with decreased cell death [11, 12]. The disorder of necroptosis, pyroptosis, and ferroptosis has been observed in the progression of HNSCC [13–15].
Identifying specific signal pathways and developing drugs or non-coding RNA to induce tumor sensitivity to PCD have shown specific effects in some tumors [16, 17]. Cystine uptake mediated by solute carrier family seven-member 11 (SLC7A11) inhibits ferroptosis but promotes cell death under glucose starvation [18, 19]. Recent studies have found that the abnormal accumulation of intracellular disulfides in SLC7A11high cells under glucose starvation can induce a new form of cell death, disulfidptosis [19]. The SLC7A11-mediated reduction of ingested cystine to cysteine is highly dependent on the reduced nicotinamide adenine dinucleotide phosphate (NADPH), which was mainly produced by the glucose-pentose phosphate pathway [19]. NADPH in SLC7A11high cells is quickly consumed under the con starvation, and disulfides such as cystine are abnormally accumulated, leading to disulfide stress and rapid cell death [19]. Chemical proteomics and cell biology analysis show that the abnormal disulfide bonds actin cytoskeletal protein and collapse of F-actin is a feature of this new death mode [19]. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) screening and functional studies identify some disulfidptosis-related proteins [19]. However, the characteristics of these proteins still need to be discovered in HNSCC.
The application of computer-aided medicine is widely used in disease diagnosis, mechanism exploration, prognosis evaluation, and target exploration [20–22]. Computer-based bioinformatics technology has become an accurate and efficient tool for understanding various cancers [23]. Bulk-sequence data analysis can comprehensively summarize gene expression and prognostic characteristics, which are widely used to construct prognostic models and target investigation. However, gene expression characteristics usually depend on the dominant cell subgroup. single-cell sequencing data (scRNA) can accurately identify each cell subgroup, which is significant in judging cell heterogeneity and special gene differential expression. However, single-cell data lack the spatial expression characteristics of cells. The spatial transcriptome can reflect the spatial expression characteristics of cells to make up for this defect [24].
In this study, we conducted bioinformatics analyses using transcriptomic, scRNA, and spatial transcriptomic data to investigate the expression patterns and functional roles of disulfidptosis-related genes in HNSCC (Fig. 1). We stratified 501 TCGA patients into two clusters based on the expression levels of 77 disulfidptosis-related genes. Cluster 1 (C1) patients exhibited worse prognoses, higher stromal component proportions, and lower levels of CD8 + T cell infiltration than those in Cluster 2 (C2). A prognostic model containing five risk-associated genes related to disulfidptosis was conducted. Moreover, six cell groups in tumor microenvironment (TME) were identified, and a higher disulfidptosis-score in epithelial and fibroblast cells was observed. Cyclin C (CCNC) and Charged Multivesicular Body Protein 1B (CHMP1B) were identified as vital molecules in the differentiation of CAF cells. Furthermore, this study observed a significant co-localization phenomenon among CAF, endothelium, and plasma cells. Our findings could improve our comprehension of the distinct and functional roles of disulfidptosis-related genes and discover novel therapeutic targets for treating HNSCC.
Fig. 1.

The workflow and analytical pipeline of this study
Material and methods
Data source
The gene expression data (TPM), single nucleotide polymorphism (SNP) mutation, and copy number variation data (CNV) were downloaded by using TCGAbiolinks. 501 samples were involved in downstream analysis. The single-cell transcriptome data for GSE139324, GSE173647, and GSE173964, and the space transcriptome sample were obtained from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo).
Acquisition of disulfidptosis-related genes set
The disulfidptosis-related genes set was obtained from the recent study of Junjie Chen et al. through the whole-genome CRISPR-Cas9 screening [19]. Here, 77 genes with an absolute value of NormZ score greater than 2.9 were identified as disulfidptosis-related genes (Table S1).
TCGA sample clustering, dimension reduction, and survival analysis
The TCGA expression matrix was standardized using the log2 (x + 1) method, and the expression matrix containing only 77 disulfidptosis-related genes was extracted. The samples were classified using the R package “NMF”. The parameters rank = 2:7 and method = "brunet". According to the official recommendation of the “NMF” package, we set rank = 2 to classify patients into two clusters according to the change of the copheric parameter. R package “tinyarray” was used for dimensionality reduction and visualization. R package “Survival” was employed to analyze the difference in prognosis [25].
TME analysis
CIBERSORT and ssGSEA algorithm of the “GSVA” package was performed to predict the relative proportion of immune cells infiltrated in TCGA samples. The “ESTIMATE” R package was employed to assess the stromal score, immune score, and tumor purity of the samples [26].
SNP variation analysis of disulfidptosis-related genes
R package “Maftools” was used to process TCGA-SNP mutation data and select the top 30 mutation rates genes [27].
Protein interaction analysis of disulfidptosis-related genes
Upload disulfidptosis-related genes to String database (http://string.embl.de/). The minimum required interaction score was 0.150, and the genes that did not correlate with the corresponding proteins of other genes were deleted.
Least absolute shrinkage and selection operator (LASSO) regression to build a prognosis model
R package “glmnet” was used to perform LASSO regression analysis on the expression matrix of disulfidptosis-related genes, carrying out ten cross-validations. Briefly, set family = “cox”. The cv.glmnet function was used for cross-validation, and the minimum value of λ was taken for subsequent construction. Five differentially expressed genes (DEG) were selected. The prognostic model was constructed with risk-score value calculated by the following formula (Table S2): Risk score = ∑iCoefficient (DEG mRNAi) * Expression (DEG mRNAi). The median risk-score value serves as the threshold, distinguishing high- and low-risk groups according to the median risk score. R package “Survival” and “timeROC” were conducted to analyze prognosis and draw time-dependent Receiver Operating Characteristic (ROC) curves between high and low-risk groups. TCGA patients were randomly divided into a training set and a testing set at a 1:1 ratio to examine the prognostic significance of the model.
qRT-PCR validation
A total of 52 HNSCC tissues were collected from Taizhou Central Hospital. According to the official guidance, total RNA was extracted with Trizol Reagent (Invitrogen). RNA was revers-transcribed into cDNA. qRT-PCR was conducted on SYBRH Select Master Mix (Invitrogen) with the relative expression of the five modeling genes and normalized by GAPDH. Data were quantified using the 2−ΔCt methods. High and low-risk groups were determined according to the median value of the risk score above. The primer was synthesized by Genepharma (Table S3). The research protocol was approved by the Ethics Committee of Taizhou Central Hospital (2020-KT01-E). All procedures were performed strictly in compliance with the Declaration of Helsinki 1964 or equivalent ethical principles. All patients involved in this study had given their informed consent before study.
Prediction of response to immunotherapy and chemotherapy
We processed the TPM expression matrix of TCGA sample with log2 (x + 1), subtracted the expression amount of each gene in each sample from the mean value of the gene in all samples, and uploaded the matrix to the Tumor Immune Dysfunction and Exclusion (TIDE) database (http://tide.dfci.harvard.edu/) [28]. The predicted response to immunotherapy was obtained. The sensitivity to different drugs was predicted using “pRRophic”, and the difference in drug sensitivity between high and low-risk groups was compared [29].
Quality control, cell type clustering, and identification of main cell types
Cells with higher than 20% mitochondrial gene content were removed, and 73,660 cells were obtained for downstream analysis. Here we randomly selected 1/5 cells for Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction display and downstream analysis. To standardize and normalize the single-cell data, we employed the SCTransform function available in Seurat 4.2.0 [30]. To reduce the dimensionality of the data, we utilized the RunPCA function. The parameter dims = 1:30 was set to cluster the cells. The UMAP dimension reduction diagram was optimized using “plot1cell”. Six cell groups were identified according to the instructions of CellTypist database (https://www.celltypist.org/).
Scissor
The phenotype of single cell can be deduced from the phenotype of bulk sequencing data: Use Scissor to predict the classification or risk grouping of each cell in a single cell according to the results of NMF sample classification and the results of high and low-risk groups predicted by our prognosis model, and set the parameter family = "binary". The input bulk sequencing data uses the original TPM expression matrix.
Disulfidptosis-score
The AddModuleScore function implemented in Seurat software was utilized to quantify the expression of disulfidptosis-related genes and to assess the relative expression levels of these genes across distinct cellular populations or subgroups.
Identification of cancer cells
The epithelial cells were extracted. The SCTransform function in Seurat was used to standardize and normalize the single-cell data. The RunPCA function was employed to reduce the dimension of the data. The parameter dims = 1:30 was set to cluster the data. R package “CopyKAT” was used to predict the malignant and normal cells in the epithelial cells [31].
Trajectory analysis
Fibroblast cells were extracted and further clustering. After identifying the cell subgroups according to the differentially expressed genes, the development trajectory of cells was predicted using “monocle2”. The highly variable genes selected by “monocle2” were used to represent the developmental characteristics of cells [32].
Space transcriptome analysis
“STUtility” was used to integrate the space transcriptome data. Min.gene.count = 100 and min.spot.count = 500 was set to filter the data. The FeatureOverlay function was used to explore the expression level and location of the sections [33]. SpaCET was further used to control the data quality [34], and the SpaCET.deconvolution function was applied to explore the relative proportion of cells in each spot. The parameter cancerType = "HNSCC" was set, and SpaCET.visualize.spatialFeature function was used to visualize the inferred results. To explore the co-localization effect between cells, SpaCET.CCI.Colocalization function was employed to calculate the correlation between cells. We use SPATA2 to standardize and extract the gene expression amount in the space transcriptome for the space trajectory vector field. The AddModuleScore function scores the disulfidptosis-related gene set of each spot as the input data to represent the gene expression or the changing trend of the gene set score in the local section [35].
Statistics
Statistical data were analyzed using R. P < 0.05 was suggested as statistically significant. *P < 0.05, ** P < 0.01, *** P < 0.001 and **** P < 0.0001.
Results
Classification of patients according to the disulfidptosis-related genes
In this study, the NMF method was applied to classify a cohort of 501 samples of HNSCC (TCGA-HNSCC) based on the gene expression levels of 77 disulfidptosis-related genes (Table S1). Two patient clusters were identified (Fig. 2A, Fig. S1). We generated a heat map to visually represent the differences in gene expression between two distinct clusters (Fig. 2B). UMAP and Principal Component Analysis (PCA) dimensionality reduction analysis showed that patients in the two groups could be well distinguished (Fig. 2C, Fig. S2A). Besides, C1 patients showed unfavorable overall survival (Fig. 2D). ESTIMATE algorithm was performed, and the results showed that the stromal score of C1 was significantly higher than C2. The immune score of C1 was slightly lower than that of C2, but there was no statistical significance (Fig. 2E, Fig. S2B). We further evaluated the differences in immune cell infiltration between two clusters through CIBERSORT and ssGSEA. The results of CIRBERSORT showed that several crucial immune cell infiltration of C1 was significantly lower than C2, including CD8 + T cells. However, M2 macrophages of C1 were significantly higher than C2 (Fig. 2F, Fig. S2C). ssGSEA showed similar results, with a higher proportion of regulatory T cells but low infiltration level of CD8 + T cells in C1 than in C2 (Fig. S2D).
Fig.2.

Patients were divided into two clusters A NMF was performed, and 501 TCGA-HNSCC patients were divided into two groups according to the expression of 77 disulfidptosis-related genes. B A heat map showed the differential expression of some disulfidptosis-related genes between C1 and C2. C UMAP dimension reduction diagram. D Kaplan–Meier plots showed C1 associated with poor prognosis. E ESTIMATE results. F CIBERSORT results showed different immune cell infiltrations between C1 and C2
Genetic mutation landscape of disulfidptosis-related genes
The top 30 disulfidptosis-related genes with the highest SNP mutation rate in all samples between the two types were calculated in Fig. 3A. CNV mutation analysis was also performed. The findings of our study revealed that CNVs were observed in genes associated with disulfidptosis. Specifically, solute carrier family 3 (amino acid transporter heavy chain), member 2 (SLC3A2) showed a significant association with the copy number amplification, while serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 2 (SERPINE2) exhibited the highest frequency of CNV deletions, as depicted in Fig. 3B. The chromosomal distribution and mutations of disulfidptosis-related genes were also demonstrated in Fig. S3A. Moreover, common crosstalk among disulfidptosis-related genes was observed by String (Fig. S3B).
Fig.3.

Mutation of disulfidptosis-related risk genes A Comparison of the top 30 disulfidptosis-related genes with the highest SNP mutation rate between two clusters. B Deletion and acquisition of disulfidptosis-related genes
Establish a disulfidptosis-related prognostic model
Lasso regression analysis was utilized to select features and reduce the number of disulfidptosis-related genes in the risk model. Through optimization of the regularization parameter lambda, a total of five significant disulfidptosis-related genes were identified and subsequently incorporated into the final risk model (Fig. 4A, Table S2). TCGA patients were used to validate the prognostic significance of the model. The high-risk group exhibited a more unfavorable prognosis than the low-risk group in the train cohort and test cohort. The same pattern was also observed when combined with train cohort and test cohorts (Fig. 4B). The ROC curve of overall cohort suggested that risk score may be a valuable tool in HNSCC prognosis prediction (Fig. 4C). Besides, the number of dead patients increases following the elevation of risk score (Fig. 4D). Figure 4E showed the expression level of 5 modeling genes in high and low-risk groups. We further included 52 clinical patients in the validation of the model. As expected, high-risk patients showed poorer prognoses (Fig. 4F).
Fig.4.

A disulfidptosis-related prognostic model A Lasso regression was performed, and five risk disulfidptosis-related genes were selected (GYS1, STAG3, CHMP1B, GSTO1, and CCNC). B Kaplan–Meier plots showed prognosis differences in train, test, and overall cohorts. C The ROC curve of the overall cohort. D Distribution of patients' survival status. E A heat map showed the expression level of 5 modeling genes. F Verification of risk model. The representative HNSCC slice; left, 200 X magnification; right, 400 X magnification. AUC, Area Under Curve. GYS1, glycogen synthase 1; STAG3, stromal antigen 3; GSTO1, Glutathione S Transferase Omega 1
Evaluation of sensitivity to immunotherapy & chemotherapy
TIDE was used to predict the response to high/low-risk group patient immunotherapy. The results showed that the high-risk group exhibited a higher possibility of immune escape but a worse response to immunotherapy (Fig. 5). PRRophitic was used to predict the sensitivity of different drugs. Fig. S4A showed the more sensitive drugs in the high-risk group, while Fig. S4B showed the more sensitive drugs in the low-risk group. However, the sensitivity of patients in the high-risk and low-risk groups to paclitaxel and cisplatin did not show statistically significant differences (P > 0.05).
Fig.5.

Comparison of Immunotherapy Response Between Two Groups Based on TIDE Results. A Higher score on the vertical axis of TIDE indicates an increased probability of immune escape. The results show that the high-risk group has a greater probability of immune escape
scRNA profiling of disulfidptosis-related genes
UMAP and plot1cell were used to visualize the annotated clusters, and six-cell groups were obtained. Epithelial, fibroblasts, and T/NK cells constituted the main components of TME (Fig. 6A). The markers of cell group annotation were shown in Fig. 6B. We first analyze the distribution characteristics of two clusters of patients by combining them with bulk and scRNA data. A higher infiltration of B cells and T/NK cells in C2 patients was observed. Besides, a significantly different distribution of myeloid cells, epithelial cells, and fibroblasts between C1 and C2 was also observed. Heterogeneous T/NK cells and fibroblasts had a significant impact on the prognosis of patients. In contrast, the existence of the B cell group seemed to be related to the favorable prognosis (Fig. 6C). To further quantify the expression levels of disulfidptosis-related genes, the disulfidptosis-score was calculated. The results showed that the scores of epithelial and fibroblast groups were significantly higher than those for other groups (Fig. 6D, E). The cells with the highest gene expression set in the first 25% were defined as high expression, and the others were defined as low expression. High expression levels were observed mainly in the epithelial and fibroblast groups (Fig. 6F).
Fig.6.

Single-cell RNA-seq analysis A UMAP for dimensionality reduction. Six groups were identified. B Primary markers of each cell group. C The results of the Scissors. D The box chart showed the disulfidptosis-score of six groups (analysis of variance, P < 0.0001). E, F Visual disulfidptosis-related genes expression in UMAP dimensionality reduction map
Epithelial cells were extracted and classified into two subgroups by using CopyKAT (Fig. 7A). Normal epithelial cell group (diploid) showed a higher expression level of disulfidptosis-related genes, and its disulfidptosis-score was higher than cancer cells (aneuploid) (Fig. 7B, C). We then further determine the expression feature of disulfidptosis-related genes in fibroblast cells. Five CAF subgroups were identified (2 myofibroblast CAFs, two iCAFs, and smooth-muscle cells, Fig. 7D). Six genes were typically expressed in particular cell subtypes. For instance, aldo–keto reductase family 1 member C1 (AKR1C1) was found to be enriched in myofibroblast CAFs (myCAF) 1. In contrast, actin alpha cardiac muscle 1 (ACTC1) was increased in smooth muscle cells (Fig. 7E). High expression level was mainly observed in the iCAF subgroup, following the myCAFs, while the disulfidptosis-score of smooth-muscle cells was lowest (Fig. 7F, G).
Fig.7.

The expression characteristics of disulfidptosis-related genes in epithelial and fibroblast A UMAP for dimensionality reduction. Two epithelial subgroups were identified. B Difference of disulfidptosis-score between two subgroups. C A heatmap showed the differential expression of disulfidptosis-related genes. D Fibroblasts were clustered into five subgroups. E Six unique markers of 5 subgroups were identified. F, G The disulfidptosis-related gene expression difference between 5 fibroblast subgroups (analysis of variance, P < 0.0001)
The trajectory analysis revealed that CAF-differentiation regulators
The CAF cells were extracted, and conducted trajectory analysis. The pseudo time was performed, and three states of the fate trajectory of relatedness in their expression patterns were identified (Fig. 8A). Interestingly, among the five modeling genes, the CCNC and CHMP1B expression was determined to change following CAF differentiation significantly (Fig. 8B).
Fig. 8.

Trajectory analysis of CAF cells A Pseudotime analysis. A.1 showed three stages in the CAF cell differentiation process. A.2 showed the position of 5 subgroups of CAF cells. A.3 showed the time sequence. Black represented the first position. B Expression of CCNC/CHMP1B in CAF cell differentiation. C The expression of CCNC and CHMP1B in tissue section
Spatial distribution characteristics of disulfidptosis-related genes
The spatial transcriptome analysis was further performed. CCNC and CHMP1B had high expression levels and spatial differences among sections (Fig. 8C). We further clarified the distribution characteristics of each cell subgroup in the selected section. There were few stromal cells and immune cells in the area where malignant cells are concentrated. However, a significant co-localization phenomenon was observed among CAF, endothelium, and plasma cells (Fig. 9A, B). Figure 9C showed the characteristic cell references profiles, such as CAF-endothelial, CAF-plasma, and endothelial-plasma. Figure 9D showed the spatial locus vector field, indicating that the expression of disulfidptosis-related genes had significant spatial specificity.
Fig.9.

The spatial transcriptome A The spatial localization characteristic of multiple cell subgroups. B, C The characteristic cell reference profiles. D The results of spatial locus vector field. The arrow represents the gradient of expression level, and the direction of the arrow indicating higher expression level
Discussion
HNSCC has become a global health concern due to its high invasiveness and prone to lymph node metastasis [36]. The treatment needs to be improved for patients with advanced-stage, recurrent or metastatic HNSCC. Further understanding of tumor progression, anti-cell death, and therapeutic resistance, as well as pursuing new effective molecular targets, are critical to improving the prognosis of HNSCC. A significant characteristic of tumor cells is that cell death disorder under the condition of high proliferation [37]. Identifying cell death mechanisms could widen our understanding of cellular homeostasis and provide novel insights into cancer treatment. Disulfidptosis is a newly discovered form of cell death characterized by the abnormal disulfide cross-linking of actin cytoskeletal protein [19]. Although this study identified a series of disulfidptosis-related genes, the expression characteristics and functions in HNSCC are still unclear. Here, the NMF method was used to classify TCGA-HNSCC patients based on the expression of 77 disulfidptosis-related genes, and 2 clusters were identified. C1 patients showed worse prognosis and higher stromal component but lowered CD8 + T cell infiltration than C2. Common crosstalk among disulfidptosis-related genes was demonstrated. Besides, LASSO regression analysis was conducted, and a useful prognostic model containing only five risk disulfidptosis-related genes was established. High risk-group showed an unfavorable prognosis and a higher possibility of immune escape but a worse response to immunotherapy. In addition, the high-risk group was determined to be sensitive to more chemotherapy drugs. Compared with previous LASSO prognostic models, our model incorporates fewer risk genes, enhancing its potential for clinical practicality. Although its AUC value is slightly lower than previous studies [38, 39], it achieves satisfactory predictive accuracy with a streamlined gene set. This balance between model simplicity and effectiveness suggests that our model may serve as a valuable tool in clinical prognostic assessments.
Previous studies on cell heterogeneity usually rely on bulk data, immunohistochemistry, flow cytometry, etc. The development of scRNA technology and computer-aided analysis has robustly improved the accuracy and convenience of cell heterogeneity research. Because the traditional bulk analysis can only provide the average value of gene expression, mainly reflecting the phenotypic subset of dominant cellular groups [40]. Three scRNA data were included in this study. Six cell groups were identified. We set an indicator named disulfidptosis-score to evaluate the expression characteristic of 77 disulfidptosis-related genes. Epithelial and fibroblast cells had higher disulfidptosis-related gene expression than other groups. Moreover, two epithelial cell subgroups were identified by extracting epithelial cells and further clustering; the normal epithelial cell group (diploid) exhibited a higher expression of disulfidptosis-related genes. These results may indicate that several disulfidptosis-related genes were reticent in cancer cells, thus escaping disulfidptosis and maintaining cell survival. Furthermore, five CAF cell subpopulations were obtained by extracting fibroblasts and clustering. iCAF subgroup showed a higher disulfidptosis-score, followed by myCAF. Furthermore, the expression of six genes was found to be associated with specific subtypes of CAFs. Chemokine (C–C motif) ligand 2 (CCL2) could indicate iCAFs, and ACTC1 may be a marker of smooth muscle cells.
Metabolic reprogramming commonly arises in TME [41, 42]. TME alterations and tumor metabolism are closely linked through a bidirectional signal pathway involving tumor cells and CAFs [43]. CAFs represent the predominant stromal cell type in TME and exhibit significant heterogeneity and plasticity [44]. Emerging evidence indicates that CAF significantly influences tumor metabolism by dysregulating various metabolic pathways, including glucose metabolism [45] CAFs tend to produce lactic acid by glycolysis, even under aerobic conditions [46]. Metabolites such as lactic acid can provide sufficient nutrition for tumor cells. However, effector T cells can not play an anti-tumor role in an environment of high lactic acid [47]. Lactic acid has been proven to be closely related to the poor prognosis of various tumors [48, 49]. This also explains that the C1 group showed a high stromal score, while the infiltration of CD8 + T cells was significantly reduced.
Besides, the carbon use mode of proliferating cells is largely different from that of static cells. In cell proliferation, the main purpose of reducing carbon is to synthesize various biological molecules [50]. In addition, many biosynthetic reactions are reduction reactions in nature, and the proliferating cells must allocate some carbon substrates for NADPH production [51]. Tumor cells in a proliferative state have low demand for ATP and can obtain energy from lactic acid metabolism [52]. It is believed that tumor cells use glucose metabolism for NADPH production, maintain a highly proliferative state and inhibit disulfidptosis. The interaction between cancer cells and CAFs involves cross-talk that alters the metabolic pathways of cancer cells, specifically promoting the reprogramming of glycolysis. This metabolic adaptation enhances cancer cell survival and proliferation, potentially contributing to the aggressiveness and progression of cancer. Targeted CAF has become a feasible treatment strategy for increasing the response to immunotherapy, chemotherapy, and radiotherapy.
Through trajectory analysis, two potential targeted proteins against CAFs were identified. CCNC is a member of the cyclin family of proteins, which interacts with cyclin-dependent kinase eight and induces phosphorylation of the carboxy-terminal domain of the large subunit of RNA polymerase II [53]. Overexpression of CCNC in tumor and tumor-adjacent tissues compared with that in normal tissues was observed and infer serve as promising targets for managing breast cancer [54]. Some studies indicate that CCNC could be a response to chemosensitivity in gastric cancer [55]. CHMP1B belongs to the chromatin-modifying protein/charged multivesicular body protein (CHMP) family, which is related to protein domain-specific binding [56]. CHMP1B has been proven to be related to esophageal squamous cell carcinoma and colorectal cancer [57, 58]. The spatial transcriptome analysis was conducted to offset the loss of spatial information of scRNA. CCNC and CHMP1B had high expression levels and spatial differences among sections. Furthermore, a significant co-localization phenomenon was observed among CAF, endothelium, and plasma cells. Cadamuro et al. proved that fibroblasts promote the expansion of the lymphatic vasculature and tumor cell intravasation. This critical process could be blocked by inducing CAF apoptosis [59]. Targeted CAF has become a feasible treatment strategy for increasing the effectiveness of clinical treatment.
Although this study has comprehensively clarified the expression characteristics and functions of disulfidptosis-related genes in HNSCC, some limitations still exist. This study mainly relies on computer-aided analysis, and the limited sample size introduces potential biases in clustering and risks of overfitting in LASSO regression models. Besides, our model has a slightly lower AUC value, necessitating extensive validation across large sample sizes to determine its clinical applicability. Moreover, additional in vitro and in vivo experiments are required to clarify the biological function of specific disulfidptosis-related proteins. The spatial locus vector field analysis suggested that disulfidptosis-related genes have significant spatial specificity. Given that computer-aided results cannot fully capture the complexities of the TME, further investigations into effect of disulfidptosis by crosstalk between cancer cells and TME are needed.
Collectively, a new PCD mode characterized by the abnormal disulfide bonds actin cytoskeletal protein and collapse of F-actin was defined, and its related genes have recently been identified. Based on transcriptome, scRNA, and spatial transcriptome data, our study has comprehensively revealed the expression characteristics and functions of disulfidptosis-related genes in HNSCC using a series of computer-assisted bioinformatics analyses. A useful prognostic model was successfully constructed and verified in our patient cohort; high-risk patients showed poor prognosis and worse response to immunotherapy but sensitivity to more chemotherapy drugs. In addition, the results of scRNA suggested that disulfidptosis-related genes were highly expressed in epithelial and fibroblasts. Among them, the expression of normal epithelial cells was higher than cancer cells, and the expression of iCAF group was higher than that of myCAF and smooth muscle cells. The spatial transcriptome analysis comprehensively showed the interaction between tumor cells and TME and the spatial distribution characteristics of disulfidptosis-related genes. The results showed that CAFs might participate in metabolic reprogramming, inhibit disulfidptosis, restrain immunity, and promote angiogenesis. CCNC and CHMP1B were identified as potential regulatory genes for CAF differentiation. Our study focuses on disulfidptosis, a novel PCD mode, and preliminarily reveals the role and mechanism of disulfidptosis-related genes in HNSCC.
Supplementary Information
Supplementary material1 Table S1. Disulfidptosis-related genes. (XLS 22 KB)
Supplementary material2 Table S2. Five risk genes. (DOC 15 KB)
Supplementary material3 Table S3. The Primers. (DOC 17 KB)
{kind=link}
Supplementary material4 Fig. S1 NMF method. (A) Parameter changes. (B) Classification of patients with different ranks. (JPG 1465 KB)
{kind=link}
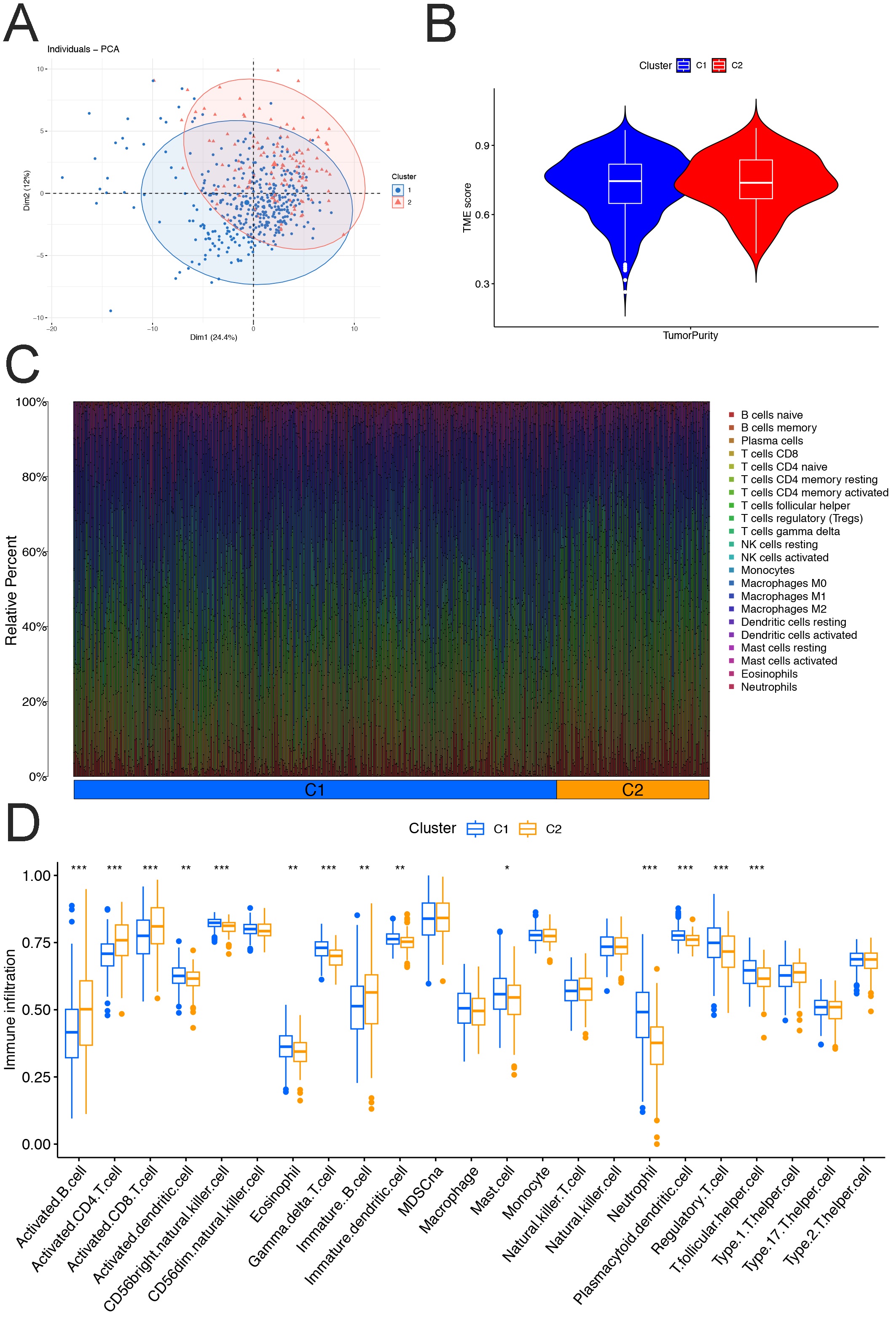
Supplementary material5 Fig. S2 Characteristic of disulfidptosis-related genes. (A) The results of PCA. (B) ESTIMATE results. (C) CIRBERSORT results. (D) ssGSEA results. (JPG 1264 KB)
{kind=link}
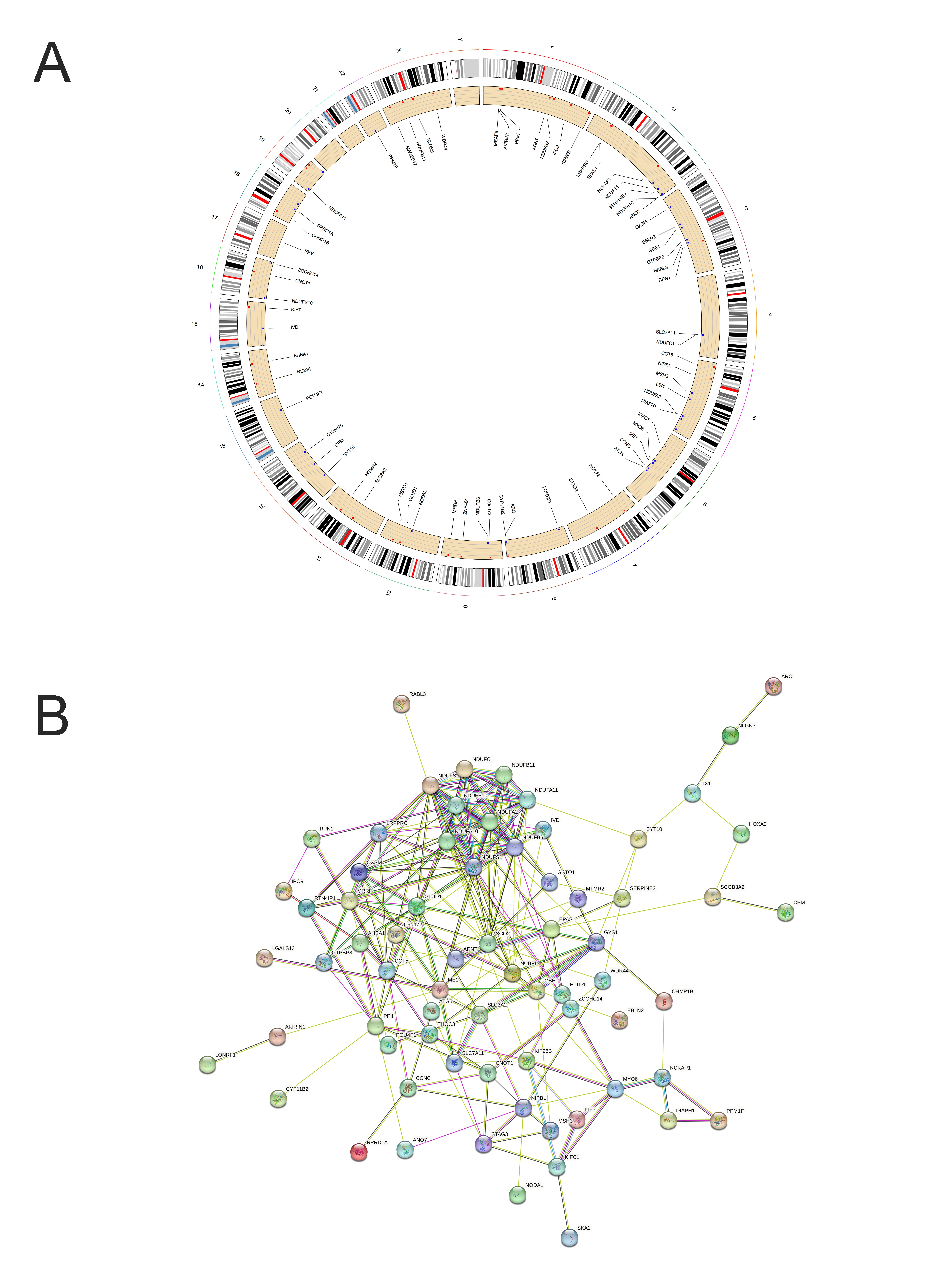
Supplementary material6 Fig. S3 Mutation of disulfidptosis-related risk genes. (A) The circle chart showed the location and CNV of some disulfidptosis-related genes. Red represents the increase in copy number, and blue represents the decrease. (B) PPI network. (JPG 3402 KB)
{kind=link}
Supplementary material7 Fig. S4 Comparison responses to chemotherapy between two groups. PRRophitic results showed sensitivity to drug treatment. (A) The drugs to which the high-risk group was more sensitive, (B) those to which the low-risk group was more sensitive. (JPG 763 KB)
Acknowledgements
The authors thank the contributions from the TCGA and GEO network.
Abbreviations
- PCD
Programmed cell death
- HNSCC
Head and neck squamous cell carcinoma
- NMF
Nonnegative Matrix Factorization
- TCGA
The Cancer Genome Atlas
- TME
Tumor microenvironment
- scRNA
Single-cell sequencing
- CAFs
Cancer-associated fibroblasts
- C1
Cluster 1
- C2
Cluster 2
- iCAFs
Inflammatory cancer-associated fibroblasts
- SLC7A11
Solute carrier family seven-member 11
- NADPH
Nicotinamide adenine dinucleotide phosphate
- CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats
- TME
Tumor microenvironment
- CCNC
Cyclin C
- CHMP1B
Charged Multivesicular Body Protein 1B
- TPM
The gene expression data
- SNP
Single nucleotide polymorphism
- CNV
Copy number variation
- LASSO
Least absolute shrinkage and selection operator
- DEG
Differentially expressed genes
- ROC
Receiver Operating Characteristic
- TIDE
Tumor Immune Dysfunction and Exclusion
- UMAP
Uniform Manifold Approximation and Projection
- PCA
Principal Component Analysis
- SLC3A2
Solute carrier family 3 (amino acid transporter heavy chain), member 2
- SERPINE2
Serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 2
- AUC
Area Under Curve
- GYS1
Glycogen synthase 1
- STAG3
Stromal antigen 3
- GSTO1
Glutathione S Transferase Omega 1
- myCAFs
Myofibroblast cancer-associated fibroblasts
- AKR1C1
Aldo–keto reductase family 1 member C1
- ACTC1
Actin alpha cardiac muscle 1
- CCL2
Chemokine (C–C motif) ligand 2
Author contributions
Conceptualization, Chifeng Zhao, Haoran Zhu and Zhenxing Zhang; Formal analysis, Chifeng Zhao, Haiqian Zhu, Xuhui Xu and Conglin Hu; Funding acquisition, Zhenxing Zhang; Project administration, Haiqian Zhu and Zhenxing Zhang; Supervision, Zhenxing Zhang; Writing—original draft, Chifeng Zhao, Haoran Zhu and Haiqian Zhu; Writing—review and editing, Haiqian Zhu and Zhenxing Zhang.
Funding
This study was supported by the Taizhou Social Development Science and Technology Project (No.23ywa20).
Data availability
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Code availability
R software was used in this study. The relevant R package was explained in the Materials and Methods.
Declarations
Ethics approval and consent to participate
The research protocol was approved by the Ethics Committee of Taizhou Central Hospital (2020-KT01-E). All procedures were performed strictly in compliance with the Declaration of Helsinki 1964 or equivalent ethical principles. All patients involved in this study had given their informed consent before study.
Consent for publication
All authors approved the final manuscript and the submission to this journal.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Haiqian Zhu, Chifeng Zhao and Haoran Zhu have contributed equally to this work and share first authorship.
References
- 1.Cramer JD, Burtness B, Le QT, Ferris RL. The changing therapeutic landscape of head and neck cancer. Nat Rev Clin Oncol. 2019;16(11):669–83. [DOI] [PubMed] [Google Scholar]
- 2.Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2020;6(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leemans CR, Snijders PJF, Brakenhoff RH. The molecular landscape of head and neck cancer. Nat Rev Cancer. 2018;18(5):269–82. [DOI] [PubMed] [Google Scholar]
- 4.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- 5.Cohen EEW, Bell RB, Bifulco CB, Burtness B, Gillison ML, Harrington KJ, et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J Immunother Cancer. 2019;7(1):184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diao P, Jiang Y, Li Y, Wu X, Li J, Zhou C, et al. Immune landscape and subtypes in primary resectable oral squamous cell carcinoma: prognostic significance and predictive of therapeutic response. J Immunother Cancer. 2021;9(6): e002434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moujalled D, Strasser A, Liddell JR. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021;28(7):2029–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gibellini L, Moro L. Programmed cell death in health and disease. Cells. 2021;10(7):1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, Hong M, Li Y, Chen D, Wu Y, Hu Y. Programmed cell death tunes tumor immunity. Front Immunol. 2022;13: 847345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strasser A, Vaux DL. Cell death in the origin and treatment of cancer. Mol Cell. 2020;78(6):1045–54. [DOI] [PubMed] [Google Scholar]
- 11.Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L, et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct Target Ther. 2022;7(1):286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Messmer MN, Snyder AG, Oberst A. Comparing the effects of different cell death programs in tumor progression and immunotherapy. Cell Death Differ. 2019;26(1):115–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Huang S, Zeng L, Li K, Yang L, Gao S, et al. Necroptosis in head and neck squamous cell carcinoma: characterization of clinicopathological relevance and in vitro cell model. Cell Death Dis. 2020;11(5):391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rioja-Blanco E, Arroyo-Solera I, Álamo P, Casanova I, Gallardo A, Unzueta U, et al. CXCR4-targeted nanotoxins induce GSDME-dependent pyroptosis in head and neck squamous cell carcinoma. J Exp Clin Cancer Res. 2022;41(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao YY, Lian JX, Lan Z, Zou KL, Wang WM, Yu GT. Ferroptosis promotes anti-tumor immune response by inducing immunogenic exposure in HNSCC. Oral Dis. 2021;29:14077. [DOI] [PubMed] [Google Scholar]
- 16.Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31(51): e1904197. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z, Zhu H, Zhao C, Liu D, Luo J, Ying Y, et al. DDIT4 promotes malignancy of head and neck squamous cell carcinoma. Mol Carcinog. 2023;62(3):332–47. [DOI] [PubMed] [Google Scholar]
- 18.Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12(8):599–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Nie L, Zhang Y, Yan Y, Wang C, Colic M, et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol. 2023. 10.1038/s41556-023-01091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Zhu H, Wang X, Lin S, Ruan C, Wang Q. A novel basement membrane-related gene signature for prognosis of lung adenocarcinomas. Comput Biol Med. 2023;154: 106597. [DOI] [PubMed] [Google Scholar]
- 21.Zhou J, Zhang B, Zhang X, Wang C, Xu Y. Identification of a 3-miRNA signature associated with the prediction of prognosis in nasopharyngeal carcinoma. Front Oncol. 2022;27(11): 823603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan S, Wang W, Zhu B, Pan X, Wu X, Tao W. Construction of nomograms for predicting pathological complete response and tumor shrinkage size in breast cancer. Cancer Manag Res. 2020;10(12):8313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran KA, Kondrashova O, Bradley A, Williams ED, Pearson JV, Waddell N. Deep learning in cancer diagnosis, prognosis and treatment selection. Genome Med. 2021;13(1):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X, Wang CY. From bulk, single-cell to spatial RNA sequencing. Int J Oral Sci. 2021;13(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Feng Y, Luo F, Peng G, Li Y. Positive regulators of T cell functions as predictors of prognosis and microenvironment characteristics of low-grade gliomas. Front Immunol. 2023;13:1089792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jia Q, Wu W, Wang Y, Alexander PB, Sun C, Gong Z, et al. Local mutational diversity drives intratumoral immune heterogeneity in non-small cell lung cancer. Nat Commun. 2018;9(1):5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28(11):1747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geeleher P, Cox N, Huang RS. pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE. 2014;9(9): e107468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao R, Bai S, Henderson YC, Lin Y, Schalck A, Yan Y, et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat Biotechnol. 2021;39(5):599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durante MA, Rodriguez DA, Kurtenbach S, Kuznetsov JN, Sanchez MI, Decatur CL, et al. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat Commun. 2020;11(1):496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergenstråhle J, Larsson L, Lundeberg J. Seamless integration of image and molecular analysis for spatial transcriptomics workflows. BMC Genomics. 2020;21(1):482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ru B, Huang J, Zhang Y, Aldape K, Jiang P. Estimation of cell lineages in tumors from spatial transcriptomics data. Nat Commun. 2023;2(14):568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ravi VM, Will P, Kueckelhaus J, Sun N, Joseph K, Salié H, et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell. 2022;40(6):639–55. [DOI] [PubMed] [Google Scholar]
- 36.Khan W, Haragannavar VC, Rao RS, Prasad K, Sowmya SV, Augustine D, et al. P-Cadherin and WNT5A expression in assessment of lymph node metastasis in oral squamous cell carcinoma. Clin Oral Investig. 2022;26(1):259–73. [DOI] [PubMed] [Google Scholar]
- 37.Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368(6487):5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y, Tao L, Xin J, Dai Y, Chen X, Zou J, et al. Development and experimental verification of a prognosis model for disulfidptosis-associated genes in HNSCC. Medicine (Baltimore). 2024;103(12): e37308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xue H, Sun Q, Zhang H, Huang H, Xue H. Disulfidptosis features and prognosis in head and neck squamous cell carcinoma patients: unveiling and validating the prognostic signature across cohorts. J Cancer Res Clin Oncol. 2024;150(3):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanzaki R, Pietras K. Heterogeneity of cancer-associated fibroblasts: opportunities for precision medicine. Cancer Sci. 2020;111(8):2708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Z, Sun C, Qin Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics. 2021;11(17):8322–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Becker LM, O’Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep. 2020;31(9): 107701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desbois M, Wang Y. Cancer-associated fibroblasts: key players in shaping the tumor immune microenvironment. Immunol Rev. 2021;302(1):241–58. [DOI] [PubMed] [Google Scholar]
- 45.Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, et al. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors. Cell Cycle. 2011;10(15):2504–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pereira BA, Vennin C, Papanicolaou M, Chambers CR, Herrmann D, Morton JP, et al. CAF subpopulations: a new reservoir of stromal targets in pancreatic cancer. Trends Cancer. 2019;5(11):724–41. [DOI] [PubMed] [Google Scholar]
- 47.Meisel R, Zibert A, Laryea M, Göbel U, Däubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103(12):4619–21. [DOI] [PubMed] [Google Scholar]
- 48.Niu D, Luo T, Wang H, Xia Y, Xie Z. Lactic acid in tumor invasion. Clin Chim Acta. 2021;522:61–9. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Z, Hu Y, Chen Y, Chen Z, Zhu Y, Chen M, et al. Immunometabolism in the tumor microenvironment and its related research progress. Front Oncol. 2022;12:1024789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martínez-Reyes I, Chandel NS. Cancer metabolism: looking forward. Nat Rev Cancer. 2021;21(10):669–80. [DOI] [PubMed] [Google Scholar]
- 51.Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov. 2022;21(2):141–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between metabolism and cancer biology. Cell. 2017;168(4):657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song Z, Xiaoli AM, Li Y, Siqin G, Wu T, Strich R, et al. The conserved Mediator subunit cyclin C (CCNC) is required for brown adipocyte development and lipid accumulation. Mol Metab. 2022;64: 101548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Broude EV, Győrffy B, Chumanevich AA, Chen M, McDermott MS, Shtutman M, et al. Expression of CDK8 and CDK8-interacting genes as potential biomarkers in breast cancer. Curr Cancer Drug Targets. 2015;15(8):739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fang S, Jin X, Zhou C, Gong Z. Cyclin C: anew responser for chemosensitivity in cancer. Clin Transl Med. 2022;12(4): e833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crespo-Yàñez X, Aguilar-Gurrieri C, Jacomin AC, Journet A, Mortier M, Taillebourg E, et al. CHMP1B is a target of USP8/UBPY regulated by ubiquitin during endocytosis. PLoS Genet. 2018;14(6): e1007456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang M, An S, Wang D, Ji H, Geng M, Guo X, et al. Quantitative proteomics identify the possible tumor suppressive role of protease-activated receptor-4 in esophageal squamous cell carcinoma cells. Pathol Oncol Res. 2019;25(3):937–43. [DOI] [PubMed] [Google Scholar]
- 58.Ma L, Yu H, Zhu Y, Xu K, Zhao A, Ding L, et al. Isolation and proteomic profiling of urinary exosomes from patients with colorectal cancer. Proteome Sci. 2023;21(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cadamuro M, Brivio S, Mertens J, Vismara M, Moncsek A, Milani C, et al. Platelet-derived growth factor-D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J Hepatol. 2019;70(4):700–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material1 Table S1. Disulfidptosis-related genes. (XLS 22 KB)
Supplementary material2 Table S2. Five risk genes. (DOC 15 KB)
Supplementary material3 Table S3. The Primers. (DOC 17 KB)
Supplementary material4 Fig. S1 NMF method. (A) Parameter changes. (B) Classification of patients with different ranks. (JPG 1465 KB)
Supplementary material5 Fig. S2 Characteristic of disulfidptosis-related genes. (A) The results of PCA. (B) ESTIMATE results. (C) CIRBERSORT results. (D) ssGSEA results. (JPG 1264 KB)
Supplementary material6 Fig. S3 Mutation of disulfidptosis-related risk genes. (A) The circle chart showed the location and CNV of some disulfidptosis-related genes. Red represents the increase in copy number, and blue represents the decrease. (B) PPI network. (JPG 3402 KB)
Supplementary material7 Fig. S4 Comparison responses to chemotherapy between two groups. PRRophitic results showed sensitivity to drug treatment. (A) The drugs to which the high-risk group was more sensitive, (B) those to which the low-risk group was more sensitive. (JPG 763 KB)
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
R software was used in this study. The relevant R package was explained in the Materials and Methods.