

Abstract
Targeted drug delivery in response to external stimuli is therapeutically desirable, but long-term drug retention at the target site after stimulation is turned off remains a challenge. Herein, we present a targeted-delivery strategy via irreversible aggregation of drug carriers in response to mild external heating. We constructed two types of polymeric micelles, DBCO-TRM and Az-TRM, having a thermo-responsive polymer shell based on N-isopropylacrylamide (NIPAAm) and incorporating alkyne and azide moieties, respectively. Upon heating at 42 °C, the micelles aggregated through hydrophobic interaction between their dehydrated shells. Further, the azide moieties of Az-TRM become exposed on the surface due to the thermally shrinkage of the shells, thereby enabling crosslinking between the two types of micelles via azide-alkyne click chemistry to form irreversible aggregates. These aggregates were efficiently accumulated at tumor sites in mice by local heating after intravenous administration of a mixture of the micelles, and were well retained after cessation of heating due to their increased size. As proof of concept, we show that delivery of doxorubicin in this heat-guided drug delivery system dramatically improved the anti-tumor effect in a mouse model after a single treatment. Our results suggest that this platform could be an efficient tool for on-demand drug delivery.
Subject terms: Drug delivery, Nanocomposites, Nanoparticles, Drug delivery
Targeted drug delivery in response to external stimuli is therapeutically desirable, but long-term drug retention at the target site after stimulation is turned off remains a challenge. Here, the authors present a targeted delivery strategy via irreversible aggregation of drug carriers in response to mild external heating by constructing two types of polymeric micelles with a thermo-responsive polymer shell based on N-isopropylacrylamide and incorporating alkyne and azide moieties, respectively.
Introduction
Targeted drug delivery is an effective approach for increasing therapeutic efficiency and reducing off-target adverse effects. The two major strategies are passive targeting by modulating the physicochemical properties of the drug carrier, and active targeting by modifying the drug carrier with affinity ligands1,2. Both strategies can be employed for theranostics, which integrates therapy and diagnosis3,4. However, the delivery efficacy is dependent on factors such as heterogeneity in target tissues or cells, disease progression, and inter-individual differences. Therefore, to achieve on-demand targeted delivery, drug carriers responding to external stimuli (magnetic field, light, ultrasound, heat) have garnered attention5. The functionality of external stimulus-responsive carriers can be changed in situ, enabling the external control of drug pharmacokinetics. For example, magnetic drug targeting, in which magnetic drug carriers are guided to the target site in the body by external magnetic fields, is well established6,7. However, the attractive force disappears when the magnetic field is turned off8,9, leading to random diffusion of the carriers and off-target delivery via the systemic circulation10. Thus, we considered that a persistent change in drug carrier properties in response to external stimulation would improve the efficiency of stimulus-responsive targeting.
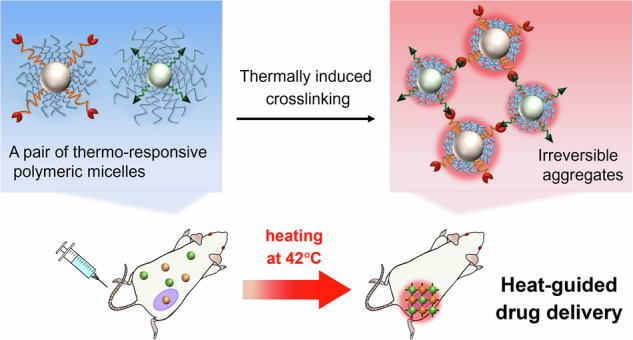
Herein, we aimed to develop a targeted delivery strategy based on irreversible aggregation of drug carriers in response to external heating. Nanoparticle aggregation triggered by tumor microenvironments (acidic pH11–14 or overexpression of certain enzymes15–18) has proven effective for long-term retention of the nanoparticles in tumor tissues, due to the increase in size19,20. We hypothesized that if drug carriers were irreversibly aggregated by crosslinking induced by external heating, the drug would be well retained at the heated site even after discontinuation of heating. To induce in situ crosslinking by heating, we designed a pair of thermo-responsive micelles, DBCO-TRM and Az-TRM, possessing bioorthogonal reaction handles, dibenzocyclooctyne (DBCO) and azide, respectively21, which could form a covalent bond between the micellar surfaces. The shells of both micelles mainly consist of a thermo-responsive polymer based on N-isopropylacrylamide (NIPAAm), which exhibits a phase transition at the lower critical solution temperature (LCST); namely, the polymer hydrates and extends below the LCST, while it dehydrates and shrinks above the LCST22,23. At temperatures below the LCST, both DBCO-TRM and Az-TRM are expected to circulate well in the blood due to their hydrophilic shells. Upon heating at a temperature above the LCST, the micelles aggregate at the heated site through hydrophobic interaction between their dehydrated thermo-responsive shells, and are trapped at the heated site due to their increased size. Further, the thermally shrinkage of the shell exposes the azide moieties of Az-TRM on the micelle surface, resulting in crosslinking between DBCO-TRM and Az-TRM via azide-alkyne click chemistry. This irreversible reaction enables retention of aggregates containing an encapsulated drug within the heated site after cessation of heating (Fig. 1). To validate this strategy, we employed it to deliver an anticancer drug in a tumor-bearing mouse model.
Fig. 1. Schematic illustration of our heat-guided drug delivery system (DDS).

Upon external heating above the LCST, the pair of thermo-responsive micelles, DBCO-TRM and Az-TRM, are trapped at the heated site due to their increased size via hydrophobic aggregation between their dehydrated thermo-responsive shells. Further, irreversible crosslinking between DBCO-TRM and Az-TRM is induced by azide-alkyne click chemistry in response to the thermally exposure of the azide moieties on Az-TRM surface, leading to efficient delivery of the micelles after cessation of heating.
Results and Discussion
To construct the required micelles, amphiphilic diblock polymers were synthesized (Supplementary Scheme S1, S2 and Supplementary Table S1). As shown in Fig. 2a, DBCO-TRM consists of two polymers, P(NIPAAm-co-AAm)-b-PBMA, whose hydrophilic domain is comprised of a thermo-responsive polymer, and DBCO-P(NIPAAm-co-AAm)-b-PBMA, in which DBCO is introduced at the hydrophilic end of the polymer. By using DBCO-containing polymer with a longer chain length than the former polymer, DBCO is expected to be exposed on the surface of DBCO-TRM regardless of temperature. On the other hand, Az-TRM consists of P(NIPAAm-co-AAm)-b-PBMA and another polymer, P(Az-co-AAm)-b-PBMA, in which multiple azide moieties are introduced in the hydrophilic domain. To control the masking/exposure of azide by thermo-responsive extension/shrinkage of Az-TRM, the chain length of the azide-containing polymer was made moderately shorter than that of the thermo-responsive polymer.
Fig. 2. Thermo-responsiveness of the polymeric micelles.

a Molecular design of DBCO-TRM and Az-TRM. b Temperature-dependent change of optical transmittance of the micelles. c Reversible response of the micelles to heating. The size of the micelles was measured after holding the indicated temperature for 5 min.
DBCO-TRM and Az-TRM each exhibited phase transition upon heating (Fig. 2b), similar to previously reported polymeric micelles with NIPAAm-based shells24–26. To enable control of the pharmacokinetics of the micelles by external heating, the micellar LCSTs were tuned to a slightly higher than physiological temperature (Supplementary Table S2) by adjusting the copolymerization ratio of acrylamide (AAm) and NIPAAm22. The copolymerization ratio of the polymers and the mixing ratio of polymers in the micelles are shown in Supplementary Tables S1 and S2. As expected, when examined individually, DBCO-TRM (119 nm in mean diameter) and Az-TRM (114 nm) formed micellar aggregates with apparent sizes of 400 nm at 42 °C and more than 1 µm at 47 °C (Fig. 2c), likely due to increased hydrophobic interaction between the thermo-responsive shells. Indeed, the hydrodynamic diameter of the micelles was greatly increased at 42/47 °C (above the LCST) compared to that at 37 °C (below the LCST). Furthermore, the aggregation observed at 42/47°C was completely reversed after cooling to 32 °C (Fig. 2c, Supplementary Fig. S1).
Next, we examined the response of a mixture of DBCO-TRM and Az-TRM to heating. Micelles in the mixture increased in size upon heating above the LCST (42/47°C), showed little change upon cooling below the LCST (32 °C) and further increased in size upon repeated heating (Fig. 3a). In accordance with this, the optical transmittance of the micelle mixture decreased after heating above the LCST (Supplementary Fig. S2). In contrast to the behavior of the individual micelle types, micelle aggregation of the mixture was irreversible. To confirm that these aggregates contained both micelle species, DBCO-TRM and Az-TRM were fluorescently labeled by encapsulating different hydrophobic indocarbocyanine dyes, DiI and DiD, in the core. When the red solution of DiI-loaded DBCO-TRM and the blue solution of DiD-loaded Az-TRM were mixed, heated at 42 °C, and centrifuged at 25 °C, purple precipitates appeared (Fig. 3b). The same result was observed when DBCO-TRM was loaded with DiD instead of Dil and Az-TRM was loaded with Dil instead of DiD (Supplementary Fig. S3). The purple precipitates were dissolved in dimethyl sulfoxide (DMSO) and the fluorescence was measured. The results showed that DBCO-TRM and Az-TRM were aggregated at a ratio of 1:1 (Supplementary Table S3), and the aggregation ratio corresponded well to the mixing ratio of micelles (Supplementary Table S4), suggesting that the two types of micelles are crosslinked to each other in the aggregates, as expected.
Fig. 3. Thermally induced crosslinking between DBCO-TRM and Az-TRM.

a Irreversible response to heating of a mixture of DBCO-TRM and Az-TRM. b Photographs of the micellar aggregates. c Effect of inhibition of azide-alkyne click chemistry. d Comparison of micelle aggregation using DBCO-TRM with a higher LCST. e Effect of azide-containing polymer length on the micellar aggregation. f Control of micellar aggregation by temperature in the presence of serum. These data were obtained by measuring micelle mixture at 1 mg/mL (mean ± SD, n = 3, Student’s t test).
We also confirmed the involvement of azide-alkyne click chemistry in the thermally induced crosslinking. As a control without azide, TRM was prepared from only P(NIPAAm-co-AAm)-b-PBMA (Supplementary Table S2, Supplementary Fig. S4) and reacted with DBCO-TRM. Alternatively, DBCO-TRM was treated with excess NaN3 (DBCO-TRM(N3)) (Supplementary Fig. S5) before reaction with Az-TRM to inhibit the reactivity of DBCO. In both cases, the increases of micellar size upon heating above the LCST were transient (Supplementary Figs. S6, S7) and reversed after cooling (Fig. 3c). To further investigate the role of the phase transition, DBCO-TRM(high) with a higher LCST (52 °C) than 47 °C was prepared by increasing the ratio of AAm in the polymer (Supplementary Fig. S8). Upon heating at 47 °C, the efficiency of crosslinking of DBCO-TRM(high) with Az-TRM was much lower than that of DBCO-TRM (Fig. 3d). This result indicates that hydrophobic aggregation above the LCST drives the micellar crosslinking. In addition, two control micelles, Az-TRM(long) and Az-TRM(short), were prepared using azide-containing polymers with longer and shorter chain lengths than that used in Az-TRM, respectively. The LCSTs of these micelles were between 39 and 40 °C (Supplementary Fig. S9). At 37 °C, Az-TRM(long) was partly aggregated with DBCO-TRM compared with Az-TRM (Fig. 3e). This result also supports the idea that hydrophobic interactions above the LCST promote the micellar crosslinking. In contrast, the efficiency of crosslinking with DBCO-TRM at 47 °C was decreased in Az-TRM(short) (Fig. 3e). These results support the idea that the change of exposure of azide upon transition across the LCST is deeply related to the thermal control of micellar crosslinking.
To utilize thermally induced crosslinking of drug carriers for targeted delivery, precise control of aggregation in vivo is indispensable. Thus, we evaluated micellar aggregation in serum-containing buffer. Although the LCSTs were slightly increased in the presence of serum (Supplementary Figs. S10, S11), the size of the aggregates of DBCO-TRM and Az-TRM after heating at 42 °C increased with increasing serum concentration (Supplementary Fig. S12). To examine the possibility of trapping serum component into the aggregates, FITC-labeled dextrans were mixed and heated with DBCO-TRM and Az-TRM, where the ratio of fluorescence intensity suggested that neglectable amounts of dextrans were incorporated (Supplementary Fig. S13). Furthermore, serum similarly increased the size of aggregates during heating when TRM (non-azide control) was used instead of Az-TRM (Supplementary Fig. S14). These results suggest that micellar aggregation above the LCST may be enhanced by hydrophobic interaction between the aggregates and serum proteins. In PBS containing 70% FBS, only small aggregates were formed after heating at 37 °C, and nonspecific aggregation with serum components did not occur (Fig. 3f). In contrast, the micelle mixture (129 nm in mean diameter) formed aggregates with an apparent size of 2 µm after heating at 42 °C. Further, no aggregation was observed in a mixture of DBCO-TRM and TRM (non-azide control) after heating at 42 °C (Supplementary Fig. S15). Thus, crosslinking of DBCO-TRM and Az-TRM can be controlled by heating at 42 °C.
As proof-of-concept, we evaluated a mixture of DBCO-TRM and Az-TRM for tumor-targeted delivery in vivo. A hydrophobic anticancer drug, doxorubicin (Dox), was encapsulated in DBCO-TRM (Supplementary Table S2), and we confirmed the resulting DBCO-TRM@Dox was crosslinked with Az-TRM by heating (Supplementary Fig. S17). Further, although the release rate of Dox showed some decrease by pre-heating in the in vitro assay, sufficient release of Dox from the micelles was observed even after the crosslinking (Supplementary Fig. S18). Then, as illustrated in Fig. 4a, a mixture of DBCO-TRM@Dox and DiD-loaded Az-TRM was intravenously administered to tumor-bearing model mice, and the subcutaneous tumor was heated for 30 min at 42 °C from 1 min after the injection (Supplementary Fig. S19). This procedure is feasible for clinical hyperthermia treatment27–29. At 1.5 h after the coadministration of DBCO-TRM@Dox and Az-TRM followed by heating, micelle accumulation in the tumor was increased more than 10-fold (Fig. 4b), and this enhancement was retained at 48 h postinjection. A similar result was obtained for Dox accumulation (Supplementary Fig. S20). When TRM (non-azide control) instead of Az-TRM was coadministered with DBCO-TRM@Dox, micelle accumulation was not significantly increased upon heating. This result confirms that thermally induced crosslinking between DBCO-TRM and Az-TRM is effective for targeted delivery to heated tissue. Interestingly, infarction of surface blood vessels was seen on the heated tumor (body surface side) in the mice at 48 h after coadministration of DBCO-TRM@Dox and Az-TRM (Fig. 4c). This was clearly different from the appearance in mice treated with DBCO-TRM@Dox and Az-TRM but without heating, or in heated mice treated with DBCO-TRM and TRM, which do not undergo crosslinking.
Fig. 4. Heat-guided delivery in vivo.

a Schematic illustration of the treatment procedure. b Accumulation of micelles at the tumor site (mean ± SD, n = 5, Student’s t test). c Photographs of the tumor and subcutaneous tissue at 48 h postinjection. d Tumor growth curves (mean + SD, n = 8−10) and e survival curves of the tumor-bearing mice after different treatments at day 0 (mean ± SD, n = 8−10, log-rank test).
We then examined the anticancer effect of heat-guided DDS using DBCO-TRM and Az-TRM. Tumor-bearing mice were treated with micelles and local heating at day 0. Compared with PBS-treated mice, tumor growth was partially inhibited by coadministration of DBCO-TRM@Dox and Az-TRM (Fig. 4d). Furthermore, the combination of local heating with coadministration of these micelles almost completely suppressed tumor growth for 22 days after the treatment. The therapeutic effects of anticancer drugs are enhanced by hyperthermia30–32, but the enhancing effect of heating was smaller in the group treated with Dox (without micelles) than in the group treated with DBCO-TRM@Dox and Az-TRM (Supplementary Fig. S21). These results indicate that the efficient delivery of Dox by thermally induced micellar crosslinking contributes to the therapeutic effect. The survival of tumor-bearing mice was also significantly prolonged by the combination of DBCO-TRM@Dox and Az-TRM with heating (Fig. 4e). Indeed, the tumors of 7 of the 10 mice became unmeasurably small (less than 1 mm in diameter) by day 24 after a single treatment, and no regrowth was seen at day 180. Since embolization with microspheres is used for cancer therapy based on the selective occlusion of blood vessels feeding tumors33–35, infarction in the locally heated tumor might also play a role in this dramatic therapeutic effect. However, application of DBCO-TRM (without Dox) and Az-TRM with heating had no marked therapeutic effect (Fig. 4e) and the blood was also unaffected (Supplementary Fig. S22), suggesting that not only micellar crosslinking but also Dox was required for this embolization. We consider that the potent therapeutic effect of our system using DBCO-TRM@Dox and Az-TRM with heating may be due to a synergic effect of three therapeutic modalities, i.e., chemotherapy, hyperthermia, and embolization. Importantly, treated groups showed no body weight loss (Supplementary Fig. S23) or change in serum biochemical parameters containing D-dimer (a biomarker of thrombus)36 (Supplementary Table S5), suggesting our system did not cause acute systemic toxicity.
Conclusions
We have achieved targeted drug delivery guided by local heating to induce irreversible crosslinking between drug carriers. We term this system heat-guided DDS. Our constructed micelles, DBCO-TRM and Az-TRM, each have diameters of about 100 nm, within the favorable size range for blood circulation37,38. Upon heating at 42 °C, the two types of micelles rapidly aggregate with each other, affording an apparent particle size of 2 µm, which is larger than the cutoff size of tumor vessels (200 nm to 1.2 µm)39,40, thereby facilitating retention in the tumor. The backbone of the polymers (C-C bonds) is not biodegradable, so we consider that the disappearance of the micelle aggregates may be mainly caused by the gradual release of polymer from the micelles. We confirmed efficient delivery of the micelles to tumor tissues by local heating, and showed that delivery of Dox by thermally induced crosslinking dramatically improved the anti-tumor effect in a mouse model. Further studies are needed to understand the precise mechanism of the therapeutic effect, for example, whether Dox delivered to the tumor directly damages the tumor cells, or whether it causes the occlusion of the blood vessels feeding the tumors. We believe this design platform may prove widely applicable for on-demand drug delivery.
Method
Materials
N-Isopropylacrylamide (NIPAAm), glycidyl acrylate (GA) and DMT-MM were purchased from Tokyo Chemical Industries (Tokyo, Japan). NIPAAm was purified by recrystallization from n-hexane. Acrylamide (AAm) was purchased from Nacalai tesque (Kyoto, Japan) and used without further purification. 4-Cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl] pentanoic acid (CDTPA) was purchased from Sigma-Aldrich (St. Louis, MO, USA). V-501 and butyl methacrylate (BMA) were purchased from Fujifilm Wako Pure Chemical Corporation (Osaka, Japan). GA and BMA were passed through an inhibitor removers packed column (Sigma-Aldrich) before use. Sulfo DBCO-amine was purchased from Vector Laboratories (Burlingame, CA, USA). 1,1’-Dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI) was obtained from AAT Bioquest (Pleasanton, CA, USA) and 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindodicarbocyanine perchlorate (DiD) was obtained from Setareh Biotech (Eugene, OR, USA). Doxorubicin hydrochloride was purchased from BLD Pharmatech (Shanghai, China). Dialysis membranes (Spectra/Por 7, molecular weight cut-off (MWCO) 1 kDa and 15 kDa) were purchased from Spectrum Laboratories (Rancho Dominguez, CA, USA). Hydrophilic syringe filter (Minisart RC25) was obtained from Sartorius (Goettingen, Germany). Other organic solvents and chemical reagents were purchased from Fujifilm Wako Pure Chemical Corporation. RPMI-1640 medium and Dulbecco’s phosphate-buffered saline without calcium and magnesium (PBS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Fetal bovine serum (FBS) was obtained from Biosera (Nuaille, France).
Synthesis of DBCO-P(NIPAAm-co-AAm)-b-PBMA
Carboxyl-terminated amphiphilic diblock polymer was prepared by two-step reversible addition−fragmentation chain transfer (RAFT) polymerization. NIPAAm, AAm, and CDTPA as a chain transfer agent (CTA; 1.0 eq.) were dissolved in 1,4-dioxane in a two-necked flask (initial total monomer: 3000 mM). Then, V-501 (0.1 eq.) as a radical initiator was added to the solution. The reaction mixture was degassed by bubbling with argon for 30 min, and stirred for 16 h at 74 °C. Then, the reaction solution was diluted with acetone and poured into diethyl ether to precipitate the polymer, which was collected and dried under vacuum to afford P(NIPAAm-co-AAm) as a pale yellow solid. In the second block preparation, P(NIPAAm-co-AAm) as the macro-CTA, BMA (50 eq.), and V-501 (0.2 eq.) were reacted in 1,4-dioxane for 24 h at 74 °C, and then the reaction solution was dialyzed against methanol using dialysis membranes (MWCO 1 kDa) at room temperature for 3 days. The resulting solution was evaporated to dryness. Next, DBCO was introduced via amide condensation at the carboxyl terminal of the diblock polymer. To a solution of P(NIPAAm-co-AAm)-b-PBMA (67 mg/mL) in tetrahydrofuran (THF) was added sulfo DBCO-amine (1.2 eq.) and triethylamine (10 eq.). To this mixture was added DMT-MM (10 eq.) and the whole was stirred for 16 h at 4 °C. After further reaction for 16 h at room temperature, the polymer was purified using the same method as described for the second block preparation.
Synthesis of P(Az-co-AAm)-b-PBMA
P(GA-co-AAm)-b-PBMA was prepared by two-step RAFT polymerization as described above. GA was copolymerized with AAm in the first block (initial total monomer: 1000 mM). The molar equivalent ratios of total monomer and initiator to CTA were 200 and 0.2, respectively. In the second block preparation, BMA (50 eq.) was reacted with P(GA-co-AAm). Next, azide moieties were introduced by ring-opening reaction of epoxides in GA. To a solution of P(GA-co-AAm)-b-PBMA (2 mM) in dimethyl sulfoxide (DMSO) was added NaN3 (5 eq. to epoxide in polymer) and NH4Cl (5 eq.) and the mixture was stirred for 24 h at 50 °C. The polymer was dialyzed against water and lyophilized to afford P(Az-co-AAm)-b-PBMA.
Characterization of the polymers
The molecular weight of polymers was determined by gel permeation chromatography (GPC) (GPC-8020 system, TOSOH, Tokyo, Japan) using a TSKgel guard column α and TSKgel column α-M with N,N-dimethylformamide (DMF) containing 10 mM LiCl as a mobile phase. Calibration was done with polyethylene oxide standards. 1H NMR spectra were recorded at 500 MHz using a AVANCE500 spectrometer (Bruker, Billerica, MA, USA).
Preparation of the polymeric micelles
15 mg of diblock copolymer was dissolved in 2.0 mL N,N-dimethylacetamide (DMAc) containing a fluorescent dye (30 µg of DiI or DiD) and the solution was stirred overnight at room temperature. The polymer solution was slowly added to 6.0 mL of deionized water stirred at 750 rpm using a syringe pump at 0.1 mL/min. The resulting solution was dialyzed against deionized water using dialysis membranes (MWCO 15 kDa) at 20 °C for 1 day. After removal of un-loaded dye through a 0.45 µm pore syringe filter, the micelle solutions were stored at 4 °C until use. For preparation of Dox-loaded DBCO-TRM (DBCO-TRM@Dox), doxorubicin hydrochloride was neutralized with triethylamine (5 eq.) in DMSO overnight at room temperature, and the Dox solution (5 mg/mL) was mixed with the diblock copolymer (10 wt%) instead of the dye. The drug-loading content in the micelle was calculated to approximately 2.5 wt% from the fluorescence intensity of Dox measured using a fluorometer (FP-6300; JASCO, Tokyo, Japan). To check the homogeneity and reproducibility of the prepared micelles, the hydrodynamic diameters and their distribution were measured, and the efficiency of crosslinking of DBCO-TRM and Az-TRM was evaluated prior to use in the following experiments.
Characterization of the polymeric micelles
The hydrodynamic diameters and their distribution of DiI-loaded micelles (1 mg/mL in PBS) were measured at 25 °C by dynamic light scattering (DLS) using a Zetasizer Nano-ZS (Malvern Instrument, Worcestershire, UK) equipped with a 633 nm laser at a scattering angle of 173°. The optical transmittance of the DiI-loaded micelles (1 mg/mL in PBS) was measured at 750 nm using a UV-Vis spectrophotometer V-630 (JASCO) and a water circulation bath CTU-100 (JASCO) at a heating rate of 1.0 °C/min. The LCST of the micelle solution was defined as the temperature showing a 50% decrease in optical transmittance. The critical micelle concentration (CMC) of the micelles was determined using pyrene as a fluorescence probe. Pyrene solution in acetone (120 µM, 7.5 µL) was added to micelles without fluorescent dye in PBS (1500 µL) at various concentrations (1−1000 µg/mL) and the mixtures were incubated at 20°C for 16 h. The fluorescence spectra were recorded using a fluorescence spectrophotometer F-7000 (Hitachi High-Tech, Tokyo, Japan) (excitation: 338 nm, emission: 350−450 nm) and the CMC value was estimated by extrapolating the crossing-point of the intensity ratio I375/ I386.
Aggregation of the polymeric micelles
DBCO-TRM and Az-TRM were mixed equally (1.0 mg/mL in PBS), and the temperature was repeatedly cycled between 32 °C (cooling) and the indicated temperature (heating). The size of the micelles in the mixture (100 µL) was measured 5 min after reaching the target temperature by DLS. After a total of 10 min at the indicated value, the temperature was changed to the next level. For evaluation of the irreversibility of aggregation by heating, the size of micelles in the mixture was measured at 32 °C after two heating/cooling cycles (10 min at each step).
In vitro release of Dox from the polymeric micelles
DBCO-TRM@Dox and Az-TRM were mixed in equal amounts (1.0 mg/mL in PBS, 0.75 mL each), heated twice at 42 °C for 10 min, and transferred into a dialysis tube (MWCO 12−14 kDa). The dialysis tube was immersed in 30 mL of PBS (pH 7.4) containing 1% Tween-20 and continuously shaken at 100 rpm and 37 °C. At 0.5, 1, 3, 8, 24, 48, 72 and 96 h after the immersion, 1.5 mL of the outside solution was taken out and replaced with an equal volume of fresh buffer. The release solution was lyophilized and the residue was redissolved in 90% DMSO, then the fluorescence intensity was measured using a fluorescence spectroscopy F-7000 (excitation: 480 nm, emission: 550 nm).
Cell culture
Murine colon carcinoma (Colon-26; RCB2657) cells were obtained from RIKEN Cell Bank (Ibaraki, Japan). Colon-26 cells were cultured on non-coated dishes in RPMI-1640 supplemented with 10% FBS, 100 units/mL penicillin G and 100 mg/L streptomycin. Cells were grown in a CO2 incubator (humidified atmosphere of 5% CO2 in air at 37 °C) and subcultured twice a week after washing with PBS and detaching with 0.25% trypsin-EDTA.
Animal model
All animal experiments were carried out in accordance with the Guidelines on Animal Experimentations of Keio University. BALB/cCrSlc mice (female, 6-week-old, 16−18 g) were purchased from Sankyo Labo Service (Tokyo, Japan). The mice were housed in the animal care facility with food and water ad libitum and were used for experiments at 7–8 weeks old. To establish the tumor-bearing model, 2.5 × 105 Colon-26 cells in 50 μL of PBS were subcutaneously transplanted into the right hind limb of each mouse using a 29 G needle (Terumo, Tokyo, Japan). The length (L) and width (W) of the tumor were measured using an electronic caliper, and the tumor volume (V) was calculated as 0.5 × L × W2. When the tumor volume reached 50−100 mm3, the mice were assigned to in vivo experiments.
In vivo tumor accumulation
A PBS solution containing equal amounts of DBCO-TRM@Dox and DiD-loaded Az-TRM was injected via the tail vein (200 µL/mouse, 240 mg polymer/kg body weight, 5 animals/group) using a 1.0 mL insulin syringe with a 29 G needle (Terumo). At 1 min after the injection, an electric hand warmer (XY-101; ZeRay) was placed over the tumor site for 30 min for local heating at 42 °C (the setting was the first gear). During the procedure, mice were anesthetized with isoflurane and kept on a heating pad (Heater Mat type III; Natsume Seisakusho, Tokyo, Japan). At 48 h after the injection, tumor tissue was collected and homogenized, and the fluorescence of DiD and Dox in the tissue lysate was measured using a fluorescence spectroscopy (FP-6300; JASCO). The accumulation of micelles in the tumor is shown as % ID/g (percent of injected dose per gram of tissue). Standard samples were prepared for each type of micelle to estimate the value corresponding to 1% of injected dose: tissue lysate from untreated tumor-bearing mice was mixed with the micelle solution.
Anti-tumor effect in vivo
The subcutaneous tumor-bearing model mice (8−10 animals/group) were injected with the mixed solution of micelles and locally heated as described above. The dose of Dox was 3 mg/kg. The day of this single treatment was set as day 0, and the tumor volume and the body weight were measured every other day. Mice with tumor volume exceeding 2000 mm3 were euthanized as an experimental end point. Survival rates were analyzed by the Kaplan−Meier method and median survival time in days was calculated for each group. To evaluate the safety of the micelles, blood samples collected at 48 h after the injection, and serum alanine aminotransferase (ALT), aspartate transaminase (AST), blood urea nitrogen (BUN) and creatinine (Cre) were measured at Oriental Yeast Co. (Tokyo, Japan). Mouse D-dimer was measured using an ELISA kit (Cloud-Clone Corp, Houston, TX, USA).
Statistical analysis
All data are shown as mean ± standard deviation (SD) obtained from three samples. The differences between two groups were analyzed by Student’s t test, and the survival rate was analyzed by the log-rank test with the Bonferroni correction using EZR software [SR1]. Values of p > 0.05 were considered statistically not significant (n.s.).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work was supported in part by JSPS KAKENHI Grant Numbers JP23H02613, JP21H05262, JP23K17389 and JP23K20040 to K.H, a grant from the Japan Agency for Medical Research and Development (AMED) (JP23ak0101182h0003, JP23wm0325046s0103 and JP23gm1510012s0201) to K.H., JST CREST to K.H., Nakatani Foundation for Advancement of Measuring Technologies in Biomedical Engineering, The Uehara Memorial Foundation and Shiono Wellness Foundation to K.H., Program for the Advancement of Next Generation Research Projects (Keio University) and Academic Development Fund (Keio University Academic Development Funds) to K.H.
Author contributions
Sota Yamada: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing –original draft. Eita Sasaki: Visualization, Writing – review & editing. Hisashi Ohno: Writing – review & editing. Kenjiro Hanaoka: Funding acquisition, Project administration, Supervision, Writing – review & editing.
Peer review
Peer review information
Communications Chemistry thanks Kanjiro Miyata and the other, anonymous, reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
The data that support the findings of this study are available from the corresponding author, K.H., upon reasonable request.
Competing interests
The authors declare no competing interests
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-024-01383-0.
References
- 1.Rosenblum, D., Joshi, N., Tao, W., Karp, J. M. & Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun.9, 1410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Attia, M. F., Anton, N., Wallyn, J., Omran, Z. & Vandamme, T. F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol.71, 1185–1198 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Hapuarachchige, S. & Artemov, D. Theranostic Pretargeting Drug Delivery and Imaging Platforms in Cancer Precision Medicine. Front. Oncol.10, 1131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burkett, B. J. et al. A Review of Theranostics: Perspectives on Emerging Approaches and Clinical Advancements. Radio. Imaging Cancer5, e220157 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armenia, I. et al. Photonic and magnetic materials for on-demand local drug delivery. Adv. Drug Deliv. Rev.191, 114584 (2022). [DOI] [PubMed] [Google Scholar]
- 6.Price, P. M., Mahmoud, W. E., Al-Ghamdi, A. A. & Bronstein, L. M. Magnetic Drug Delivery: Where the Field Is Going. Front. Chem.6, 619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tong, S., Zhu, H. & Bao, G. Magnetic iron oxide nanoparticles for disease detection and therapy. Mater. Today31, 86–99 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mody, V. V. et al. Magnetic nanoparticle drug delivery systems for targeting tumor. Appl. Nanosci.4, 385–392 (2014). [Google Scholar]
- 9.Kianfar, E. Magnetic Nanoparticles in Targeted Drug Delivery: a Review. J. Supercond. Nov. Magn.34, 1709–1735 (2021). [Google Scholar]
- 10.Schneider-Futschik, E. K. & Reyes-Ortega, F. Advantages and Disadvantages of Using Magnetic Nanoparticles for the Treatment of Complicated Ocular Disorders. Pharmaceutics13, 1157 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao, X. et al. Guiding Brain‐Tumor Surgery via Blood–Brain‐Barrier‐Permeable Gold Nanoprobes with Acid‐Triggered MRI/SERRS Signals. Adv. Mater.29, 1603917 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Li, H. et al. Hemoglobin as a Smart pH-Sensitive Nanocarrier to Achieve Aggregation Enhanced Tumor Retention. Biomacromolecules19, 2007–2013 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Zhang, Y. et al. Enhanced Radiosensitization by Gold Nanoparticles with Acid‐Triggered Aggregation in Cancer Radiotherapy. Adv. Sci.6, 1801806 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai, Y. et al. Twins‐like nanodrugs synchronously transport in blood and coalesce inside tumors for sensitive ultrasound imaging and triggerable penetrative drug delivery. Aggregate5, e476 (2024). [Google Scholar]
- 15.Ruan, S. et al. Increased Gold Nanoparticle Retention in Brain Tumors by in Situ Enzyme-Induced Aggregation. ACS Nano10, 10086–10098 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Hu, Q. et al. Tumor Microenvironment-Mediated Construction and Deconstruction of Extracellular Drug-Delivery Depots. Nano Lett.16, 1118–1126 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Sun, Z. et al. Controlled Nano-Bio Interface of Functional Nanoprobes for in Vivo Monitoring Enzyme Activity in Tumors. ACS Nano13, 1153–1167 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Yang, K. et al. Enzyme-induced in vivo assembly of gold nanoparticles for imaging-guided synergistic chemo-photothermal therapy of tumor. Biomaterials223, 119460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu, W., Liu, R., Zhou, Y. & Gao, H. Size-Tunable Strategies for a Tumor Targeted Drug Delivery System. ACS Cent. Sci.6, 100–116 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng, G. et al. Stimuli-responsive size-changeable strategy for cancer theranostics. Nano Today38, 101208 (2021). [Google Scholar]
- 21.Mitry, M. M. A., Greco, F. & Osborn, H. M. I. In Vivo Applications of Bioorthogonal Reactions: Chemistry and Targeting Mechanisms. Chem. Eur. J.29, e202203942 (2023). [DOI] [PubMed] [Google Scholar]
- 22.Lanzalaco, S. & Armelin, E. Poly(N-isopropylacrylamide) and Copolymers: A Review on Recent Progresses in Biomedical Applications. Gels3, 36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sponchioni, M., Capasso Palmiero, U. & Moscatelli, D. Thermo-responsive polymers: Applications of smart materials in drug delivery and tissue engineering. Mater. Sci. Eng. C. Mater. Biol. Appl102, 589–605 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Chung, J. E. et al. Thermo-responsive drug delivery from polymeric micelles constructed using block copolymers of poly(N-isopropylacrylamide) and poly(butylmethacrylate). J. Control Rel.62, 115–127 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Akimoto, J., Nakayama, M., Sakai, K. & Okano, T. Temperature-induced intracellular uptake of thermoresponsive polymeric micelles. Biomacromolecules10, 1331–1336 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Hiruta, Y., Kanda, Y., Katsuyama, N. & Kanazawa, H. Dual temperature-and pH-responsive polymeric micelle for selective and efficient two-step doxorubicin delivery. RSC Adv.7, 29540–29549 (2017). [Google Scholar]
- 27.Hegyi, G., Szigeti, G. P. & Szász, A. Hyperthermia versus Oncothermia: Cellular Effects in Complementary Cancer Therapy. Evid. Based Complement Altern. Med.2013, 672873 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Behrouzkia, Z., Joveini, Z., Keshavarzi, B., Eyvazzadeh, N. & Aghdam, R. Z. Hyperthermia: How Can It Be Used? Oman Med. J.31, 89–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobšíček Trefná, H. et al. Quality assurance guidelines for interstitial hyperthermia. Int. J. Hyperth.36, 277–294 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Issels, R. D. Hyperthermia adds to chemotherapy. Eur. J. Cancer44, 2546–2554 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Datta, N. R. et al. Local hyperthermia combined with radiotherapy and-/or chemotherapy: recent advances and promises for the future. Cancer Treat. Rev.41, 742–753 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Issels, R. D. et al. Effect of neoadjuvant chemotherapy plus regional hyperthermia on long-term outcomes among patients with localized high-risk soft tissue sarcoma the EORTC 62961-ESHO 95 randomized clinical trial. JAMA Oncol.4, 483–492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caine, M. et al. Review of the Development of Methods for Characterization of Microspheres for Use in Embolotherapy: Translating Bench to Cathlab. Adv. Health. Mater.6, 1601291 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Nuzulia, N. A., Mart, T., Ahmed, I. & Sari, Y. W. The Use of Microspheres for Cancer Embolization Therapy: Recent Advancements and Prospective. ACS Biomater. Sci. Eng.10, 637–656 (2024). [DOI] [PubMed] [Google Scholar]
- 35.Welling, M. M. et al. Microspheres as a Carrier System for Therapeutic Embolization Procedures: Achievements and Advances. J. Clin. Med.12, 918 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olson, J. D. D-dimer: An Overview of Hemostasis and Fibrinolysis, Assays, and Clinical Applications. Adv. Clin. Chem.69, 1–46 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Hoshyar, N., Gray, S., Han, H. & Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine11, 673–692 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haripriyaa, M. & Suthindhiran, K. Pharmacokinetics of nanoparticles: current knowledge, future directions and its implications in drug delivery. Futur. J. Pharm. Sci.9, 113 (2023). [Google Scholar]
- 39.Yuan, F. et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res.55, 3752–3756 (1995). [PubMed] [Google Scholar]
- 40.Hobbs, S. K. et al. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc. Natl Acad. Sci. USA95, 4607–4612 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, K.H., upon reasonable request.