

ABSTRACT
Aims/Introduction
We previously showed that glucokinase haploinsufficiency improves the glucose tolerance of db/db mice by preserving pancreatic β‐cell mass and function. In the present study, we aimed to determine the effects of glucokinase haploinsufficiency on the β‐cell mass and function of long‐term high‐fat, high‐sucrose (HFHS) diet‐fed mice.
Materials and Methods
Four‐week‐old male glucokinase haploinsufficient (Gck +/− ) mice and 4‐week‐old male wild‐type (Gck +/+ ) mice (controls) were each divided into two groups: an HFHS diet‐fed group and a normal chow‐fed group, and the four groups were followed until 16, 40 or 60 weeks‐of‐age. Their glucose tolerance, glucose‐stimulated insulin secretion and β‐cell mass were evaluated. In addition, islets were isolated from 40‐week‐old mice, and the expression of key genes was compared.
Results
Gck +/− HFHS mice had smaller compensatory increases in β‐cell mass and glucose‐stimulated insulin secretion than Gck +/+ HFHS mice, and their glucose tolerance deteriorated from 16 to 40 weeks‐of‐age. However, their β‐cell mass and glucose‐stimulated insulin secretion did not decrease between 40 and 60 weeks‐of‐age, but rather, tended to increase, and there was no progressive deterioration in glucose tolerance. The expression of Aldh1a3 in pancreatic islets, which is high in several models of diabetes and is associated with an impairment in β‐cell function, was high in Gck +/+ HFHS mice, but not in Gck +/− HFHS mice.
Conclusions
Glucokinase haploinsufficiency prevents the progressive deterioration of pancreatic β‐cell mass/function and glucose tolerance in long‐term HFHS diet‐fed mice.
Keywords: β‐cell mass, Glucokinase, Glucose‐stimulated insulin secretion
We investigated the effects of glucokinase haploinsufficiency on the β‐cell mass and function in a mouse model of obesity and diabetes induced by a long‐term high‐fat, high‐sucrose diet. Our results showed that glucokinase haploinsufficiency prevents the progressive deterioration of pancreatic β‐cell mass/function and glucose tolerance in long‐term high‐fat, high‐sucrose diet‐fed mice.
INTRODUCTION
More than 500 million people around the world are currently living with diabetes, the majority of whom have type 2 diabetes 1 . Because diabetes remains a substantial public health issue, there is an urgent need to further elucidate the pathogenesis of type 2 diabetes and to identify effective treatments for the disease. Although the pathophysiology of type 2 diabetes is complex and not yet fully understood, the progressive loss of β‐cell mass and function are central to its pathophysiology 2 , 3 , 4 , 5 , 6 .
Findings obtained through studies on many animal and human genetic models have shown that the activation of glucokinase improves pancreatic β‐cell function and increases the mass 7 , 8 , 9 , 10 . Glucokinase is an enzyme responsible for the conversion of glucose to glucose 6‐phosphate, and is expressed in the pancreatic endocrine cells, liver, brain and gastrointestinal tract 11 , 12 . In particular, glucokinase in the pancreatic endocrine cells and liver cells plays an important role in the regulation of systemic glucose metabolism. Since 2003, a number of glucokinase activators have been developed, some of which have been shown to be effective and safe in phase III clinical trials 13 , 14 , 15 , 16 , 17 , 18 . In addition, glucokinase activators have been shown to enhance the function and proliferation of β‐cells in clinical trials and in our studies of mice 19 , 20 , 21 . However, it has been reported that the efficacy of glucokinase activators is not maintained over the long term 22 , 23 , 24 , 25 , 26 , suggesting that chronic glucokinase activation may in fact cause pancreatic β‐cell failure 27 , 28 .
Based on the above, we hypothesized that in the presence of conditions, such as type 2 diabetes, in which people are exposed to chronic hyperglycemia, the inactivation of glucokinase may preserve pancreatic β‐cell over the long term 29 . To test this hypothesis, we previously investigated the effects of glucokinase haploinsufficiency on the glucose tolerance and β‐cell mass/function of db/db mice, a genetically engineered model of obesity and diabetes 30 . The glucokinase‐haploinsufficient db/db mice showed less glucose intolerance and had superior β‐cell mass and insulin secretion than the db/db mice. Furthermore, deoxyribonucleic acid (DNA) microarray, real‐time quantitative polymerase chain reaction (PCR), immunohistochemical and metabolomic analyses have shown that glucokinase haploinsufficiency in the islets of db/db mice reduces the expression of stress‐related genes, increases the expression of transcription factors involved in β‐cell function, such as MafA and Nkx6.1, reduces mitochondrial damage and improves intracellular metabolic patterns. However, the protective effect of glucokinase inhibition on β‐cells has not been studied in a model of obesity and diabetes induced by an environmental factor, such as diet, which would be more consistent with the pathogenesis of type 2 diabetes in humans. Therefore, in the present study, we characterized the effects of glucokinase haploinsufficiency on the β‐cell mass and function of long‐term high‐fat, high‐sucrose (HFHS) diet‐fed mice.
MATERIALS AND METHODS
Study design
Four‐week‐old male glucokinase‐haploinsufficient (Gck +/− ) mice and 4‐week‐old male wild‐type (Gck +/+ ) mice (controls) were each divided into two groups: an HFHS diet‐fed group and a normal chow (NC)‐fed group, and these total four groups were followed until 16, 40 or 60 weeks‐of‐age. We measured the bodyweights and fed blood glucose concentrations of mice in each group over these periods, then carried out oral glucose tolerance testing (OGTT), followed by tissue collection, at 16, 40 or 60 weeks of age. Both glucose tolerance and glucose‐stimulated insulin secretion (GSIS) were assessed by OGTT, and pancreatic β‐cell mass was evaluated using pancreatic tissue sections. Subsequently, to identify differences in islet gene expression, we collected islets from 40‐week‐old Gck +/+ NC, Gck +/+ HFHS, Gck +/− NC and Gck +/− HFHS mice, and studied their gene expression using DNA microarray analysis (Gck +/+ HFHS vs Gck +/− HFHS) and real‐time quantitative PCR.
Animals
Gck +/+ mice and Gck +/− mice were generated as previously described 31 . The mutant Gck allele in these mice affects the expression of glucokinase in neuroendocrine cells, but not in hepatocytes 31 . In fact, the messenger ribonucleic acid (mRNA) expression of Gck in isolated islets from Gck +/− mice was only approximately 50% of that in the Gck +/+ mice (Figure S1). The mice were maintained at 25°C, under a 12‐h light/dark cycle, and had free access to drinking water and food.
Diet protocol
NC (MF; Oriental Yeast Co. Ltd., Tokyo, Japan) and the HFHS diet (58R3; TestDiet, Richmond, VA, USA) were used. Table S1 shows the energy composition of the diets.
Measurement of blood glucose concentration
Blood glucose concentration was measured using a Glutestmint (Sanwa Chemical, Nagoya, Japan).
Oral glucose tolerance testing
The OGTT was performed after 16 h of fasting. 1.5 mg of glucose per gram of bodyweight was administered orally, and blood samples were collected before, and 15, 30, 60 and 120 min after glucose loading. GSIS was assessed by plasma insulin concentration before and 15 min after glucose loading in the OGTT. Insulin concentration was determined with a Morinaga Ultra‐sensitive Mouse/Rat Insulin ELISA Kit (Moringa Institute of Biological Science, Yokohama, Japan).
Assessment of β‐cell mass with immunohistochemistry
Isolated pancreatic tissue was fixed in 4% paraformaldehyde at 4°C overnight. After paraffin embedding, 5‐μm sections were cut and mounted on glass slides. To inactivate endogenous peroxidase activity, the sections were immersed in phosphate‐buffered saline containing 3% (vol/vol) hydrogen peroxide and then in BLOXALL (Vector laboratories, Newark, NJ, USA) for 15 min each. The sections were then immunostained with goat anti‐rabbit insulin antibody (diluted 1:1,000; Proteintech, Rosemont, IL, USA) and counterstained with hematoxylin. Pancreatic area and β‐cell area were calculated using a BZ‐II analyzer (Keyence Corporation, Osaka, Japan), and β‐cell mass was assessed as follows: β‐cell mass (mg) = β‐cell area / pancreatic area × pancreatic weight (mg).
Islet isolation
Hanks' balanced salt solution (HBSS; Sigma‐Aldrich, St. Louis, MO, USA) containing 0.9 mg/mL of collagenase from Clostridium histolyticum (Sigma‐Aldrich) was prepared (collagenase‐supplemented HBSS). The liver and intestine of each mouse were exposed, the ampulla of Vater was clamped to block bile drainage into the duodenum, and 2.5 mL of collagenase‐supplemented HBSS was injected from the common bile duct toward the pancreatic duct. The distended pancreas was then removed and placed in a 50‐mL tube containing 7.5 mL of collagenase‐supplemented HBSS and incubated at 37°C for 24 min to digest the pancreas. After the digested pancreas had been dispersed by pipetting, it was washed with HBSS containing fetal bovine serum to stop the digestive process. The resulting solution, containing separated islets, was transferred to a Petri dish, and the islets were collected using a pipette under a microscope.
DNA microarray analysis
RNA was extracted from isolated islets using an miRNeasy Micro Kit (Qiagen, Hilden, Germany). The mRNA expression profile of each was characterized using a Mouse Clariom S Assay (Thermo Fisher Scientific, Waltham, MA, USA). Genes showing at least a 1.5‐fold difference in expression between groups were defined as differentially expressed genes. The gene expression profiles were established using Microarray Data Analysis Tool Ver. 3.2 (Filgen, Nagoya, Japan).
Real‐time quantitative PCR
RNA was isolated using an RNeasy mini kit (Qiagen), then complementary DNA was synthesized using SuperScript III (Invitrogen, Waltham, MA, USA) and Random primers (Promega, Madison, WI, USA). Real‐time quantitative PCR was carried out in duplicate using a 7500 Fast Real Time PCR system with FAST SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Target mRNA expression was quantified using the comparative cycle threshold method and normalized to that of Actb. Table S2 lists the primer sequences used.
Statistical analysis
The results are presented as the mean ± standard deviation. Comparisons between groups were carried out using one‐way anova, followed by the Bonferroni test. Statistical significance was accepted at P < 0.05.
RESULTS
Effects of glucokinase haploinsufficiency on the glucose tolerance, insulin secretion and β‐cell mass of mice consuming an HFHS diet
The bodyweights of the HFHS‐fed groups were significantly higher than those of the NC‐fed groups after 12 weeks‐of‐age, irrespective of their genotype (Figure 1a). The fed blood glucose concentrations of the Gck +/− NC mice were higher than those of the Gck +/+ NC mice, because of their genetic background, as previously described 10 , 30 , 31 , 32 . The Gck +/+ HFHS mice showed no deterioration in fed blood glucose concentration during the study up to 60 weeks‐of‐age. Gck +/− HFHS mice had high glucose concentrations, but showed no further increase over the long term (Figure 1b). OGTT showed that the glucose tolerance of the Gck +/+ HFHS mice had not deteriorated by 60 weeks‐of‐age (Figure 2). The glucose tolerance of the Gck +/− HFHS mice did not differ from that of the Gck +/− NC mice at 16 weeks‐of‐age (Figure 2a,d), but had deteriorated by 40 weeks‐of‐age (Figure 2b,e). Thereafter, however, the area under the glucose curve for the Gck +/− HFHS mice at 60 weeks‐of‐age was not larger than that at 40 weeks, implying that there was no deterioration in glucose tolerance between 40 and 60 weeks‐of‐age (Figure 2e,f). Insulin resistance, assessed using homeostasis model assessment for insulin resistance was higher in the HFHS‐fed than in the NC‐fed Gck +/+ and Gck +/− mice. However, there was no difference between the Gck +/+ HFHS mice and the Gck +/− HFHS mice (Figure S2).
Figure 1.
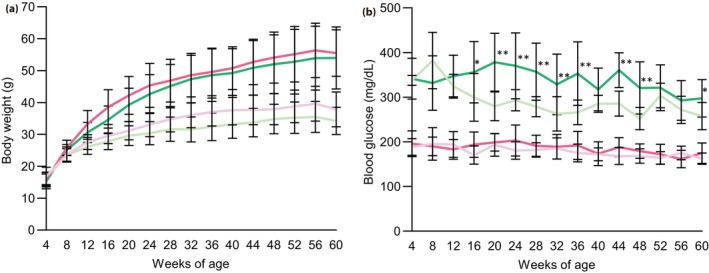
Bodyweights and blood glucose concentrations of mice in each group up to 60 weeks‐of‐age. (a) Bodyweights and (b) blood glucose concentrations of Gck +/+ normal chow (pale pink line), Gck +/+ high‐fat, high‐sucrose (pink line), Gck +/− normal chow (pale green line) and Gck +/− high‐fat, high‐sucrose (green line) mice (n = 11–20). Data are presented as mean ± standard deviation. **P < 0.01, *P < 0.05 versus Gck +/− normal chow.
Figure 2.
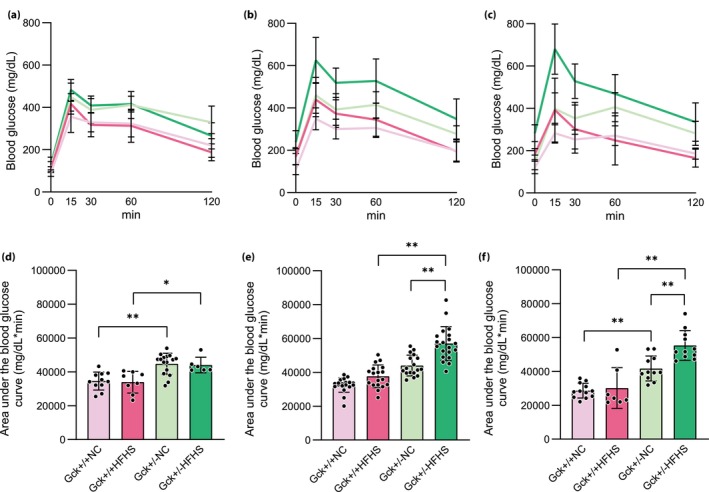
Glucose tolerance of the mice in each group over time. (a–c) Blood glucose concentrations during oral glucose tolerance testing of the Gck +/+ normal chow (NC; pale pink line), Gck +/+ high‐fat, high‐sucrose (HFHS; pink line), Gck +/− NC (pale green line), and Gck +/− HFHS (green line) groups at (a) 16 (n = 6–16), (b) 40 (n = 17–27) and (c) 60 (n = 7–12) weeks‐of‐age. (d–f) Areas under the blood glucose curves during oral glucose tolerance testing of the Gck +/+ NC (pale pink bar), Gck +/+HFHS (pink bar), Gck +/− NC (pale green bar) and Gck +/− HFHS (green bar) mice at (d) 16, (e) 40 and (f) 60 weeks‐of‐age. Data are presented as mean ± standard deviation. **P < 0.01, *P < 0.05.
The GSIS of the Gck +/+ HFHS mice was higher than that of Gck +/+ NC mice at 16 weeks‐of‐age, whereas that of the Gck +/− HFHS mice was not larger than that of the Gck +/− NC mice (Figure 3a). At 40 weeks‐of‐age, the GSIS of the Gck +/+ HFHS mice had further increased, and that of the Gck +/− HFHS mice was higher than that of the Gck +/− NC mice (Figure 3b). At 60 weeks‐of‐age, both the Gck +/+ HFHS and Gck +/− HFHS mice showed further increases in GSIS, and this was more marked in the Gck +/+ HFHS mice (Figure 3c). Thus, the GSIS of both the Gck +/+ HFHS and Gck +/− HFHS mice increases with time, but the increase in the Gck +/+ HFHS mice occurs earlier and is more marked.
Figure 3.
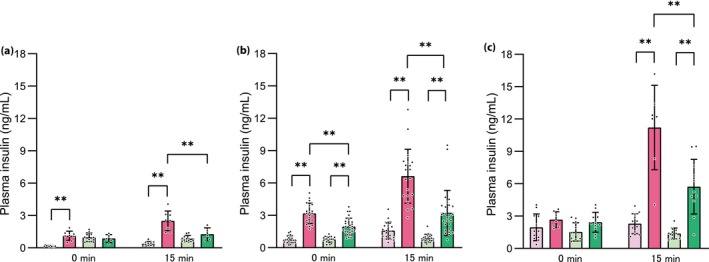
Glucose‐stimulated insulin secretion of the mice in each group over time. Plasma insulin concentrations before and 15 min after glucose loading in the Gck +/+ normal chow (pale pink bar), Gck +/+ high‐fat, high‐sucrose (pink bar), Gck +/− normal chow (pale green bar) and Gck +/− high‐fat, high‐sucrose (green bar) mice at (a) 16 (n = 5–11), (b) 40 (n = 17–26) and (c) 60 (n = 7–12) weeks‐of‐age. Data are presented as mean ± standard deviation. **P < 0.01.
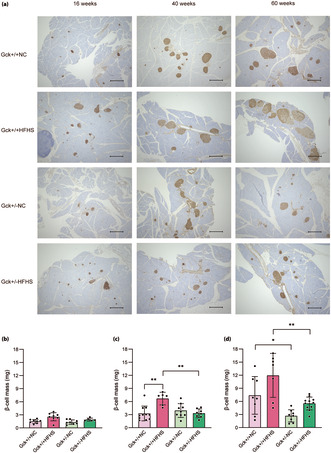
Figure 4a shows images of pancreatic sections stained for insulin (brown) as a marker of β‐cells. There were no significant differences in the β‐cell masses of the four groups of mice at 16 weeks‐of‐age (Figure 4b). At 40 weeks‐of‐age, the β‐cell mass of the Gck +/+ HFHS mice was larger than that of the Gck +/+ NC mice, but there was no difference in the masses of the Gck +/− HFHS and Gck +/− NC mice (Figure 4c). Subsequently, at 60 weeks‐of‐age, β‐cell mass was further increased in Gck +/+ HFHS mice, and an increasing trend was also observed in Gck +/− HFHS mice (Figure 4d). Briefly, β‐cell mass was significantly increased in Gck +/+ HFHS mice, but also showed an increasing trend in Gck +/− HFHS mice.
Figure 4.
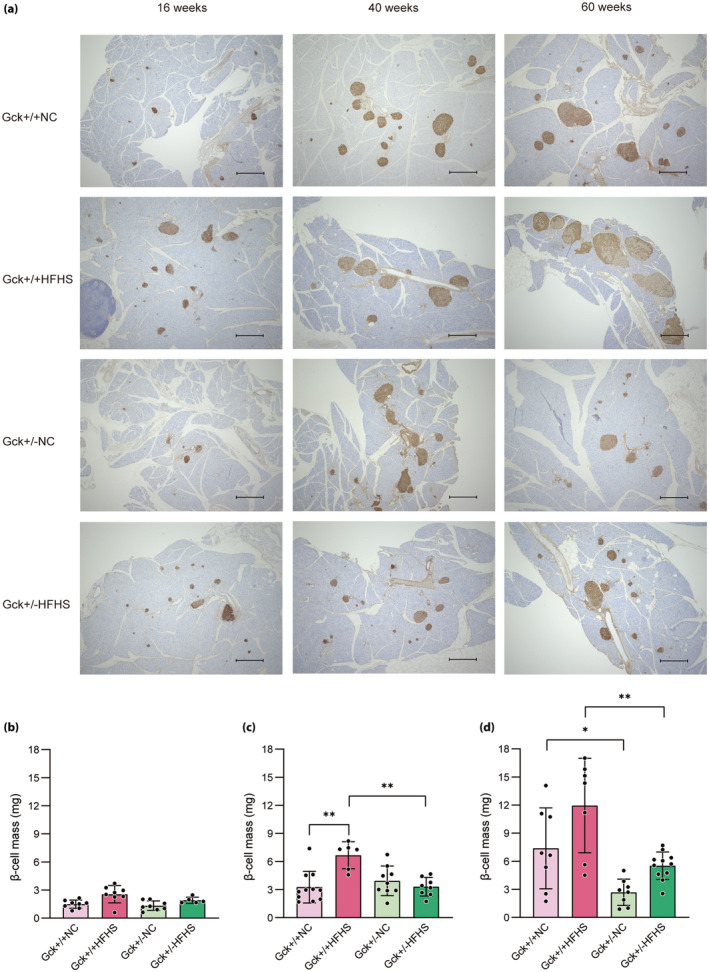
β‐Cell mass of mice in each group over time. (a) Immunohistological analysis of the pancreatic islets of mice in the four groups (Gck +/+ normal chow [NC], Gck +/+ high‐fat, high‐sucrose [HFHS], Gck +/− NC and Gck +/− HFHS mice) at 16, 40 and 60 weeks‐of‐age. β‐Cells are stained brown. Black scale bars represent 500 μm. (b–d) β‐Cell masses of the Gck +/+ NC (pale pink bar), Gck +/+HFHS (pink bar), Gck +/− NC (pale green bar) and Gck +/− HFHS (green bar) groups at (b) 16 (n = 5–8), (c) 40 (n = 6–11) and (d) 60 (n = 7–11) weeks‐of‐age. Data are presented as mean ± standard deviation. **P < 0.01, *P < 0.05.
Effects of glucokinase haploinsufficiency on the islet gene expression of mice consuming an HFHS diet
When we compared the pancreatic islet gene expression profiles of the Gck +/− HFHS and Gck +/+ HFHS mice by DNA microarray analysis, we identified statistically significant, 1.5‐fold differences in the expression of 356 of 22,206 genes. Of these, the expression of 128 genes was upregulated and that of 228 genes was downregulated in Gck +/− HFHS mice versus Gck +/+ HFHS mice. The top 20 up‐ and downregulated genes are listed in Tables 1 and 2. The most downregulated genes included Spp1, Fos, Aldh1a3 and Nupr1, which have been reported to be upregulated by stress in pancreatic β‐cells 33 , 34 , 35 , 36 , 37 , 38 . Pathway analysis was carried out on the up‐ and downregulated genes, and significant differences were identified in six pathways, including the Jun‐Dmp1 and transforming growth factor‐β pathways (Table 3).
Table 1.
Top 20 upregulated genes in the microarray analysis
| Gene symbol | Gene accession | P‐value | Ratio |
|---|---|---|---|
| Nrcam | NM_001146031 | 0.02950 | 4.529 |
| Gria3 | NM_001281929 | 0.02421 | 2.853 |
| Pdk4 | NM_013743 | 0.00748 | 2.468 |
| Rbp4 | NM_001159487 | 0.02038 | 2.303 |
| Slc16a7 | NM_011391 | 0.04280 | 2.233 |
| Hrh3 | NM_133849 | 0.00395 | 2.204 |
| Rab3c | NM_023852 | 0.02065 | 2.198 |
| 1700086L19Rik | NR_030733 | 0.04217 | 2.191 |
| Pcdh15 | NM_001142735 | 0.03069 | 2.181 |
| Myo3a | NM_148413 | 0.00016 | 2.175 |
| Dpp10 | NM_199021 | 0.04415 | 2.154 |
| Fbxo47 | NM_001081435 | 0.03417 | 2.098 |
| Galnt13 | NM_173030 | 0.02984 | 2.078 |
| Scn3b | NM_001083917 | 0.04781 | 2.075 |
| Ptprz1 | NM_001081306 | 0.03182 | 2.049 |
| Asic2 | NM_001034013 | 0.01799 | 2.027 |
| Epb41l4a | NM_013512 | 0.00189 | 2.023 |
| Hcar1 | NM_175520 | 0.02590 | 2.022 |
| Ankrd34c | NM_207260 | 0.00601 | 2.006 |
| Dpp6 | NM_001136060 | 0.03635 | 2.003 |
Table 2.
Top 20 downregulated genes in the microarray analysis
| Gene symbol | Gene accession | P‐value | Ratio |
|---|---|---|---|
| Spp1 | NM_001204201 | 0.00355 | 0.071 |
| Gucy2c | NM_001127318 | 0.01929 | 0.208 |
| Fos | NM_010234 | 0.00940 | 0.283 |
| Necab2 | NM_054095 | 0.00305 | 0.290 |
| Dapl1 | NM_029723 | 0.00745 | 0.306 |
| Rasd2 | NM_029182 | 0.00259 | 0.312 |
| Gm3298 | ENSMUST00000165289 | 0.02709 | 0.342 |
| Aldh1a3 | NM_053080 | 0.01939 | 0.350 |
| Fabp4 | NM_024406 | 0.01750 | 0.360 |
| Serpina7 | NM_177920 | 0.00559 | 0.365 |
| Nupr1 | NM_019738 | 0.02311 | 0.374 |
| Cpne4 | NM_028719 | 0.00799 | 0.374 |
| Creld2 | NM_029720 | 0.04892 | 0.378 |
| Rasl10b | NM_001013386 | 0.01203 | 0.387 |
| Jchain | NM_152839 | 0.03197 | 0.393 |
| Apof | NM_133997 | 0.00135 | 0.397 |
| Gm8281 | ENSMUST00000100893 | 0.03760 | 0.405 |
| Gabra4 | NM_010251 | 0.00058 | 0.411 |
| Pcp4 | NM_008791 | 0.04728 | 0.413 |
| Thbs1 | NM_011580 | 0.01510 | 0.419 |
Table 3.
Results of the pathway analysis carried out using the microarray data
| Pathway | Total | Up | Down | P‐value | Up list | Down list |
|---|---|---|---|---|---|---|
| Mm_Triacylglyceride_synthesis_WP386_117951 | 23 | 2 | 1 | 0.009607 | Lpl, Gk | Plpp1 |
| Mm_Novel_Jun‐Dmp1_pathway_WP3654_116721 | 26 | 0 | 3 | 0.013013 | Jun, Fos, Myc | |
| Mm_TGF‐beta_signaling_pathway_WP113_116497 | 52 | 0 | 4 | 0.015194 | Thbs1, Jun, Spp1, Fos | |
| Mm_Electron_transport_chain_WP295_117879 | 94 | 0 | 5 | 0.027087 | Cox6a1, Ndufs7, Atp5o, Ndufs8, Cox8a | |
| Mm_Retinol_metabolism_WP1259_106842 | 39 | 2 | 1 | 0.034876 | Lpl, Rbp4 | Aldh1a3 |
| Mm_GPCRs,_peptide_WP234_117936 | 69 | 3 | 1 | 0.036202 | Avpr1b, Cxcr6, Sstr2 | Ednrb |
We next used real‐time quantitative PCR analysis to measure the expression of Ki67, which is involved in cell proliferation; Mafa, Nkx6.1 and Pdx1, which are transcription factors involved in β‐cell function; Slc2a2, which is responsible for the uptake of glucose by β‐cells; and Spp1, Fos, Aldh1a3 and Nupr1, which were substantially downregulated according to the results of the microarray analysis (Figure 5). Although we showed that Gck +/+ HFHS mice had high β‐cell mass, Ki67 gene expression, evaluated using real‐time quantitative PCR, was higher in Gck +/− HFHS mice than in Gck +/− NC mice. However, there was no significant difference in the expression of this gene between Gck +/+ HFHS mice and Gck +/+ NC mice (Figure 5a). The expression of Mafa, Nkx6.1 and Pdx1 was not decreased by HFHS diet‐feeding in either the Gck +/+ or Gck +/− mice (Figure 5b–d). Rather, the expression of Pdx1 was higher in Gck +/− HFHS mice than in Gck +/− NC mice, and a similar trend was obtained with respect to the expression of Mafa (Figure 5b,d). There were no differences in the expression of Slc2a2 between the four groups of mice (Figure 5e). Of note, the real‐time quantitative PCR analysis showed that Aldh1a3 was upregulated in Gck +/+ HFHS mice vs Gck +/+ NC mice, but not in Gck +/− HFHS versus Gck +/− NC mice. Its expression was also significantly higher in Gck +/+ HFHS mice than in Gck +/− HFHS mice, consistent with the results of the microarray analysis (Figure 5f). With respect to the other downregulated genes that have previously been reported to be upregulated in pancreatic β‐cells in response to stress, the expression of Nupr1, but not that of Spp1 or Fos, was significantly lower in Gck +/− HFHS mice than in Gck +/+ HFHS mice (Figure 5g–i).
Figure 5.
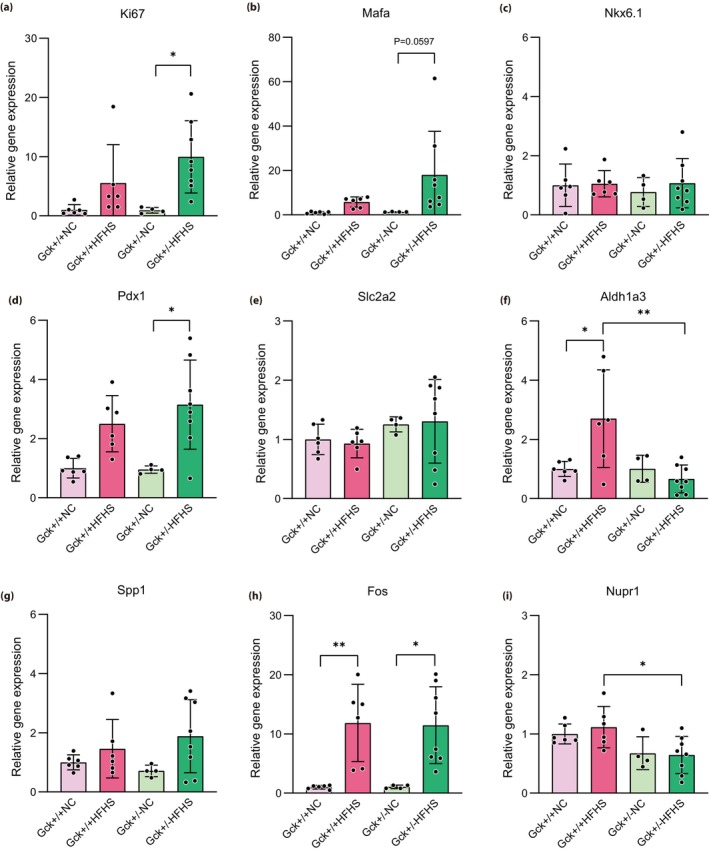
Expression of key genes in the pancreatic islets of mice in each group at 40 weeks‐of‐age. Expression of the Ki67, Mafa, Nkx6.1, Pdx1, Slc2a2, Aldh1a3, Spp1, Fos and Nupr1 genes in the Gck +/+ normal chow (NC; pale pink bar), Gck +/+ high‐fat, high‐sucrose [HFHS; pink bar), Gck +/− NC (pale green bar) and Gck +/− HFHS (green bar) groups at 40 weeks‐of‐age (n = 4–8). Data are normalized to the expression of Actb and are presented as mean ± standard deviation. **P < 0.01, *P < 0.05.
DISCUSSION
In the present study, HFHS diet‐feeding resulted in a deterioration of glucose tolerance in Gck +/− mice because of smaller compensatory increases in β‐cell mass and insulin secretion. However, over the long term, there were no further decreases in β‐cell mass or insulin secretion, but rather, tendencies for these to increase, and no progressive deterioration of glucose tolerance was identified. Terauchi et al. 10 showed that glucokinase is important for the compensatory increase in β‐cell mass associated with the feeding of Gck +/+ and Gck +/− mice with a high‐fat diet for 20 weeks. This finding would suggest that high‐fat diet‐feeding would lead to a progressive deterioration in the glucose tolerance of Gck +/− mice, and indeed, between 16 and 40 weeks‐of‐age, the Gck +/− HFHS mice in the present study showed a deterioration in glucose tolerance, which was associated with a less substantial increase in β‐cell mass than that of Gck +/+ HFHS mice. Because there was no difference in the insulin resistance of the Gck +/+ HFHS mice and the Gck +/− HFHS mice (Figure S2), we can conclude that glucokinase plays an important role in the compensatory increase in β‐cell mass and function that is associated with HFHS diet‐feeding, as well as with high‐fat diet‐feeding. However, over the long term, the Gck +/− HFHS mice also showed upward trends in β‐cell mass and insulin secretion, suggesting that the pathogenesis of the deterioration in glucose tolerance differs from that of the non‐compensatory phase of pancreatic β‐cell function, and mass that characterizes in type 2 diabetes (Figure S3).
We fed the same mice used in the high‐fat diet‐feeding experiment described above a high‐starch diet for 15 weeks, and found that glucokinase plays an important role in the compensatory increase in β‐cell mass, even when fed this diet 39 . Thus, it has already been shown that the same mechanism underpins the compensatory increases in β‐cell mass that occur in response to high‐fat and high‐starch diet‐feeding. What makes the present study different from the previous two studies is the composition of the diet and the long study period. The reason we used an HFHS diet was to increase the level of stress placed on the β‐cells. The HFHS diet used in the present study contains a high proportion of saturated fatty acids. Palmitic acid, a saturated fatty acid, has been reported to induce the dysfunction of β‐cells 40 . However, the present study was carried out under ad libitum feeding conditions, and the increases in bodyweight and blood glucose concentration seemed to be smaller than those achieved by high‐fat diet‐feeding (data not shown). Therefore, it is not known whether the HFHS diet‐feeding increased the stress on the β cells. However, the length of the study period is a strength of the present study. Through longer‐term observations, it has become clear that glucokinase haploinsufficiency slows the development of the phenotype, but does not result in a loss of the potential for an increase in β‐cell mass over the long term.
Another important finding of the present study is that the Aldh1a3 expression in pancreatic islets was high in Gck +/+ HFHS mice, but not in Gck +/− HFHS mice. Aldehyde dehydrogenase activities typically increase in response to stress, and, therefore, the metabolism of endogenous and exogenous aldehydes increases, thereby reducing oxidative stress and lipid peroxidation 35 . Although no previous studies have identified the mechanism by which Aldh1a3 expression is upregulated in pancreatic islets, Shimamura et al. 36 showed that Aldh1a3 expression in pancreatic islets is high in mice with high‐fat diet‐induced diabetes and db/db mice with poor glucose tolerance. Another study showed that Aldh1a3 expression is high in various models of diabetes, and that high Aldh1a3 expression is a common feature of diabetic β‐cells 41 . Indeed, Son et al. 42 showed that db/db mice with β‐cell‐specific knockout of Aldh1a3 show superior glucose tolerance and insulin secretion. These data imply that Aldh1a3 expression is associated with pancreatic β‐cell function.
In the present study, although the β‐cell mass and insulin secretion of the Gck +/+ HFHS mice were substantially higher than those of mice fed normal chow, the high expression of Aldh1a3 suggests that a higher level of metabolic stress and poorer pancreatic β‐cell function were also present. In contrast, Gck +/− HFHS mice did not show high Aldh1a3, suggesting that even when their β‐cells are challenged by a high glucose load, they have a low level of metabolic stress, owing to less post‐glucokinase metabolic signaling, which prevents the deterioration of pancreatic β‐cell mass and function. The associated upregulation of the Jun‐Dmp1 and transforming growth factor‐β pathways identified using pathway analysis of the DNA microarray analysis data (the appropriate genes were downregulated in Gck +/− HFHS mice vs Gck +/+ HFHS mice), and the low Nupr1 expression identified using DNA microarray analysis and real‐time quantitative PCR analysis are consistent with this contention. Aldh1a3 expression is also known to be upregulated in association with β‐cell dedifferentiation 43 . The higher Aldh1a3 mRNA expression in the Gck +/+ HFHS mice versus the Gck +/− HFHS mice might indicate that the HFHS diet induces such dedifferentiation in the Gck +/+ mice. However, there was no difference in the expression of Neurog3, a marker of islet progenitor cells, between the Gck +/+ HFHS mice and the Gck +/− HFHS mice, as shown by DNA microarray analysis (ratio 1.209, P = 0.40) and real‐time quantitative PCR (Figure S4). Therefore, to determine whether dedifferentiation is part of the phenotype, further research is needed.
As mentioned in the Introduction, we previously established and analyzed Gck +/− db/db mice, and reported that glucokinase haploinsufficiency improves intracellular metabolism, and preserves pancreatic β‐cell mass and function in db/db mice 29 , 30 . The results of the present study of HFHS‐fed mice suggest that glucokinase haploinsufficiency also prevents the progressive decline in pancreatic β‐cell mass and function in this model. However, we identified some differences between the findings obtained using this model and those of our previous study of db/db mice. We found that Gck +/+ db/db mice experience a marked deterioration in glucose tolerance, but the glucose tolerance of Gck +/+ HFHS mice was found not to deteriorate up to 60 weeks‐of‐age. Furthermore, the db/db mice showed significantly lower expression of the Ki67, Mafa, Nkx6.1 and Pdx1 genes in the pancreatic islets of Gck +/+ db/db mice than in those of Gck +/− db/db mice, although there were no significant differences between Gck +/+ HFHS and Gck +/− HFHS mice in the present study. This might be because the glucose load on the β‐cells induced by long‐term HFHS diet‐feeding was much lower than that of db/db mice. Therefore, another model of diet‐induced obesity and diabetes in which glucose tolerance is impaired should be established for future studies. This model would more clearly show the protective effect of glucokinase inhibition on pancreatic β‐cells.
In conclusion, glucokinase haploinsufficiency prevents the progressive deterioration of pancreatic β‐cell mass/function and glucose tolerance in long‐term HFHS diet‐fed mice.
DISCLOSURE
The authors declare no conflict of interest.
Approval of the research protocol: N/A.
Informed Consent: N/A.
Approval date of Registry and the Registration No. of the study/trial: N/A.
Animal Studies: The study was approved by the Animal Use Committee of Hokkaido University Graduate School of Medicine, and was carried out in accordance with the Animal Use Guidelines of Hokkaido University (approval numbers: 18‐0033 [until 3/31/2023] and 23‐0078 [from 4/1/2023]).
Funding
This work was supported by Grants in Scientific Research (C), numbers 19K08992 and 23 K07959, from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan; Grants from the Japan Association for Diabetes Education and Care; and Grants‐in‐Aid from the Japan Diabetes Foundation, the Takeda Science Foundation and the Manda Memorial Foundation (to AN).
Supporting information
Figure S1. Expression of Gck in the pancreatic islets of mice in NC‐fed groups.
Figure S2. HOMA‐IR of the mice in each group at 40 weeks of age.
Figure S3. Changes of β‐cell mass and function over time in HFHS‐fed groups.
Figure S4. Expression of Neurog3 in the pancreatic islets of mice in the HFHS‐fed groups at 40 weeks of age.
Table S1. Energy composition of the diets.
Table S2. Sequences of the primers used for real‐time quantitative PCR.
Acknowledgment
We thank Marika Watanabe, Mari Azuma and Natsumi Fujimori for their technical support. We also thank Mark Cleasby, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
References
- 1. Collaborators GBDD . Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the Global Burden of Disease Study 2021. Lancet 2023; 402: 203–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weir GC, Gaglia J, Bonner‐Weir S. Inadequate beta‐cell mass is essential for the pathogenesis of type 2 diabetes. Lancet Diabetes Endocrinol 2020; 8: 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eizirik DL, Pasquali L, Cnop M. Pancreatic beta‐cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat Rev Endocrinol 2020; 16: 349–362. [DOI] [PubMed] [Google Scholar]
- 4. Yagihashi S, Inaba W, Mizukami H. Dynamic pathology of islet endocrine cells in type 2 diabetes: Beta‐cell growth, death, regeneration and their clinical implications. J Diabetes Investig 2016; 7: 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kendall DM, Cuddihy RM, Bergenstal RM. Clinical application of incretin‐based therapy: Therapeutic potential, patient selection and clinical use. Am J Med 2009; 122: S37–S50. [DOI] [PubMed] [Google Scholar]
- 6. Butler AE, Janson J, Bonner‐Weir S, et al. Beta‐cell deficit and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52: 102–110. [DOI] [PubMed] [Google Scholar]
- 7. Kassem S, Bhandari S, Rodriguez‐Bada P, et al. Large islets, beta‐cell proliferation, and a glucokinase mutation. N Engl J Med 2010; 362: 1348–1350. [DOI] [PubMed] [Google Scholar]
- 8. Nakamura A. Glucokinase as a therapeutic target based on findings from the analysis of mouse models. Endocr J 2022; 69: 479–485. [DOI] [PubMed] [Google Scholar]
- 9. Porat S, Weinberg‐Corem N, Tornovsky‐Babaey S, et al. Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab 2011; 13: 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Terauchi Y, Takamoto I, Kubota N, et al. Glucokinase and IRS‐2 are required for compensatory beta cell hyperplasia in response to high‐fat diet‐induced insulin resistance. J Clin Invest 2007; 117: 246–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matschinsky FM, Wilson DF. The central role of glucokinase in glucose homeostasis: A perspective 50 years after demonstrating the presence of the enzyme in islets of Langerhans. Front Physiol 2019; 10: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matschinsky FM, Magnuson MA, Zelent D, et al. The network of glucokinase‐expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes 2006; 55: 1–12. [PubMed] [Google Scholar]
- 13. Zhu D, Li X, Ma J, et al. Dorzagliatin in drug‐naive patients with type 2 diabetes: A randomized, double‐blind, placebo‐controlled phase 3 trial. Nat Med 2022; 28: 965–973. [DOI] [PubMed] [Google Scholar]
- 14. Yang W, Zhu D, Gan S, et al. Dorzagliatin add‐on therapy to metformin in patients with type 2 diabetes: A randomized, double‐blind, placebo‐controlled phase 3 trial. Nat Med 2022; 28: 974–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Toulis KA, Nirantharakumar K, Pourzitaki C, et al. Glucokinase activators for type 2 diabetes: Challenges and future developments. Drugs 2020; 80: 467–475. [DOI] [PubMed] [Google Scholar]
- 16. Vella A, Freeman JLR, Dunn I, et al. Targeting hepatic glucokinase to treat diabetes with TTP399, a hepatoselective glucokinase activator. Sci Transl Med 2019; 11: eaau3441. [DOI] [PubMed] [Google Scholar]
- 17. Zhu D, Gan S, Liu Y, et al. Dorzagliatin monotherapy in Chinese patients with type 2 diabetes: A dose‐ranging, randomised, double‐blind, placebo‐controlled, phase 2 study. Lancet Diabetes Endocrinol 2018; 6: 627–636. [DOI] [PubMed] [Google Scholar]
- 18. Grimsby J, Sarabu R, Corbett WL, et al. Allosteric activators of glucokinase: Potential role in diabetes therapy. Science 2003; 301: 370–373. [DOI] [PubMed] [Google Scholar]
- 19. Zhu XX, Zhu DL, Li XY, et al. Dorzagliatin (HMS5552), a novel dual‐acting glucokinase activator, improves glycaemic control and pancreatic beta‐cell function in patients with type 2 diabetes: A 28‐day treatment study using biomarker‐guided patient selection. Diabetes Obes Metab 2018; 20: 2113–2120. [DOI] [PubMed] [Google Scholar]
- 20. Nakamura A, Togashi Y, Orime K, et al. Control of beta cell function and proliferation in mice stimulated by small‐molecule glucokinase activator under various conditions. Diabetologia 2012; 55: 1745–1754. [DOI] [PubMed] [Google Scholar]
- 21. Nakamura A, Terauchi Y, Ohyama S, et al. Impact of small‐molecule glucokinase activator on glucose metabolism and beta‐cell mass. Endocrinology 2009; 150: 1147–1154. [DOI] [PubMed] [Google Scholar]
- 22. Kawata S, Nakamura A, Miyoshi H, et al. Glucokinase activation leads to an unsustained hypoglycaemic effect with hepatic triglyceride accumulation in db/db mice. Diabetes Obes Metab 2022; 24: 391–401. [DOI] [PubMed] [Google Scholar]
- 23. Nakamura A, Terauchi Y. Present status of clinical deployment of glucokinase activators. J Diabetes Investig 2015; 6: 124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilding JP, Leonsson‐Zachrisson M, Wessman C, et al. Dose‐ranging study with the glucokinase activator AZD1656 in patients with type 2 diabetes mellitus on metformin. Diabetes Obes Metab 2013; 15: 750–759. [DOI] [PubMed] [Google Scholar]
- 25. Kiyosue A, Hayashi N, Komori H, et al. Dose‐ranging study with the glucokinase activator AZD1656 as monotherapy in Japanese patients with type 2 diabetes mellitus. Diabetes Obes Metab 2013; 15: 923–930. [DOI] [PubMed] [Google Scholar]
- 26. Meininger GE, Scott R, Alba M, et al. Effects of MK‐0941, a novel glucokinase activator, on glycemic control in insulin‐treated patients with type 2 diabetes. Diabetes Care 2011; 34: 2560–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tornovsky‐Babeay S, Weinberg‐Corem N, Ben‐Haroush Schyr R, et al. Biphasic dynamics of beta cell mass in a mouse model of congenital hyperinsulinism: Implications for type 2 diabetes. Diabetologia 2021; 64: 1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tornovsky‐Babeay S, Dadon D, Ziv O, et al. Type 2 diabetes and congenital hyperinsulinism cause DNA double‐strand breaks and p53 activity in beta cells. Cell Metab 2014; 19: 109–121. [DOI] [PubMed] [Google Scholar]
- 29. Nakamura A, Omori K, Terauchi Y. Glucokinase activation or inactivation: Which will lead to the treatment of type 2 diabetes? Diabetes Obes Metab 2021; 23: 2199–2206. [DOI] [PubMed] [Google Scholar]
- 30. Omori K, Nakamura A, Miyoshi H, et al. Glucokinase inactivation paradoxically ameliorates glucose intolerance by increasing beta‐cell mass in db/db mice. Diabetes 2021; 70: 917–931. [DOI] [PubMed] [Google Scholar]
- 31. Terauchi Y, Sakura H, Yasuda K, et al. Pancreatic β‐cell‐specific targeted disruption of glucokinase gene. J Biol Chem 1995; 270: 30253–30256. [DOI] [PubMed] [Google Scholar]
- 32. Terauchi Y, Iwamoto K, Tamemoto H, et al. Development of non‐insulin‐dependent diabetes mellitus in the double knockout mice with disruption of insulin receptor substrate‐1 and beta cell glucokinase genes. Genetic reconstitution of diabetes as a polygenic disease. J Clin Invest 1997; 99: 861–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Icer MA, Gezmen‐Karadag M. The multiple functions and mechanisms of osteopontin. Clin Biochem 2018; 59: 17–24. [DOI] [PubMed] [Google Scholar]
- 34. Priya Dharshini LC, Vishnupriya S, Sakthivel KM, et al. Oxidative stress responsive transcription factors in cellular signalling transduction mechanisms. Cell Signal 2020; 72: 109670. [DOI] [PubMed] [Google Scholar]
- 35. Singh S, Brocker C, Koppaka V, et al. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic Biol Med 2013; 56: 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shimamura M, Karasawa H, Sakakibara S, et al. Raldh3 expression in diabetic islets reciprocally regulates secretion of insulin and glucagon from pancreatic islets. Biochem Biophys Res Commun 2010; 401: 79–84. [DOI] [PubMed] [Google Scholar]
- 37. Path G, Mehana AE, Pilz IH, et al. NUPR1 preserves insulin secretion of pancreatic beta‐cells during inflammatory stress by multiple low‐dose streptozotocin and high‐fat diet. Am J Physiol Endocrinol Metab 2020; 319: E338–E344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mallo GV, Fiedler F, Calvo EL, et al. Cloning and expression of the rat p8 cDNA, a new gene activated in pancreas during the acute phase of pancreatitis, pancreatic development, and regeneration, and which promotes cellular growth. J Biol Chem 1997; 272: 32360–32369. [DOI] [PubMed] [Google Scholar]
- 39. Tsuchida K, Nakamura A, Miyoshi H, et al. Glucokinase is required for high‐starch diet‐induced beta‐cell mass expansion in mice. J Diabetes Investig 2021; 12: 1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eguchi K, Manabe I, Oishi‐Tanaka Y, et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab 2012; 15: 518–533. [DOI] [PubMed] [Google Scholar]
- 41. Kim‐Muller JY, Fan J, Kim YJ, et al. Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic beta cells in diabetic mice. Nat Commun 2016; 7: 12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Son J, Du W, Esposito M, et al. Genetic and pharmacologic inhibition of ALDH1A3 as a treatment of beta‐cell failure. Nat Commun 2023; 14: 558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cinti F, Bouchi R, Kim‐Muller JY, et al. Evidence of beta‐cell dedifferentiation in human type 2 diabetes. J Clin Endocrinol Metab 2016; 101: 1044–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Expression of Gck in the pancreatic islets of mice in NC‐fed groups.
Figure S2. HOMA‐IR of the mice in each group at 40 weeks of age.
Figure S3. Changes of β‐cell mass and function over time in HFHS‐fed groups.
Figure S4. Expression of Neurog3 in the pancreatic islets of mice in the HFHS‐fed groups at 40 weeks of age.
Table S1. Energy composition of the diets.
Table S2. Sequences of the primers used for real‐time quantitative PCR.