

Abstract
Background:
Posterior cortical atrophy (PCA) is a rare syndrome characterized by early, prominent, and progressive impairment in visuoperceptual and visuospatial processing. PCA has been associated with underlying Alzheimer’s disease (AD) neuropathology, but large-scale biomarker and neuropathologic studies are lacking. We aimed to describe demographic, clinical, biomarker and neuropathologic correlates of PCA in a large international cohort.
Methods:
We contacted 55 research centers conducting PCA research identified in a literature review (n=1,353 papers) and an additional 7 sites recruited by advertising via the Alzheimer’s Association International Society to Advance Alzheimer’s Research and Treatment Atypical AD Professional Interest Area requesting de-identified, single-subject data from patients diagnosed with PCA. Inclusion criteria were: clinical diagnosis of PCA, and availability of AD biomarkers (PET or CSF) and/or autopsy diagnosis. Demographic, clinical, biofluid, neuroimaging, and neuropathologic data were collected.
Findings:
This exploratory meta-analysis included data from 1,092 patients evaluated at 36 sites in 16 countries. Mean age at symptom onset was 59·4 years [95%CI 58·9–59·8], 60% [95%CI 56–64] were women, and 80% [95%CI 72–89] presented with a “PCA pure” syndrome by Crutch 2017 consensus criteria. CSF amyloid-β (536 patients, 28 sites) was positive in 81% [95%CI 75–87] of patients, while CSF phosphorylated tau (503 patients, 29 sites) was positive in 65% [95%CI 56–75]. Amyloid-PET (299 patients, 24 sites) was positive in 94% [95%CI 90–97] while tau-PET (170 patients, 13 sites) was positive in 97% [95%CI 93–100]. At autopsy (145 patients, 13 sites), the most frequent neuropathologic diagnosis was AD (94% [95%CI 90–97]), with common co-pathologies: cerebral amyloid angiopathy (71%, [95%CI 54–88]), Lewy body disease (44% [95%CI 25–62]), and cerebrovascular injury (42% [95%CI 24,60]).
Interpretation:
PCA typically presents as a “pure,” young-onset dementia syndrome that is highly specific for underlying AD pathology.
Funding:
This primary meta-analysis was unfunded. Funding for all sites is available in Supplementary materials.
Introduction
Posterior cortical atrophy (PCA) is a clinically defined syndrome characterized by early, prominent, and progressive impairment of visuoperceptual/visuospatial processing due to progressive atrophy of parietal, posterior temporal, and occipital regions. The most recent consensus criteria 1 define the syndrome’s core clinical, cognitive and neuroimaging features. Although heterogeneity is found in both clinical and radiological presentations, most patients present with visual difficulties, such as space perception deficits, simultanagnosia, object perception deficit, constructional dyspraxia, and environmental agnosia. The disease is often associated with an early age of onset (< 65 years). Current diagnostic criteria define a “PCA pure” syndrome, which captures the syndrome’s core clinical and neuroimaging features. They also define a “PCA plus” syndrome, which additionally includes features suggestive of other neurodegenerative diseases, such as corticobasal degeneration (CBD) or Lewy body disease (LBD). At autopsy, the vast majority of PCA cases in the literature are attributed to Alzheimer’s disease (AD), though individual cases due to primary diffuse LBD, CBD and prion disease have been reported 2–4. PCA is often sporadic and is rarely present in autosomal dominant cases of AD. The apolipoprotein E ε4 allele (APOE4) is associated with increased risk of PCA, though the strength of the association is less than that observed in amnestic AD 5. In vivo bi omarkers (PET, CSF and plasma) of amyloid-beta (Aβ) or tau can provide evidence for or against the presence of AD neuropathology in patients presenting with clinical PCA 6, while brain imaging with MRI or 18F-fluorodeoxyglucose (18F-FDG) PET can support the diagnosis by demonstrating a characteristic pattern of atrophy or hypometabolism in parieto-occipital and parieto-temporal regions 7.
As a relatively rare syndrome, most reports of PCA have come from single sites, included modest sample sizes, and usually focused on specific clinical, genetic, neuroimaging or fluid biomarker features. A comprehensive clinical overview of the features of PCA in a large and representative sample is lacking. The goal of the present study was to describe demographic, clinical, biomarker and neuropathologic correlates in a large-scale international cohort of PCA patients by pooling together individual participant data.
Materials and methods
Search for centers and data collection
We contacted 55 research centers conducting PCA research identified in a literature review following the PRISMA (http://www.prisma-statement.org/) guidelines (n=1353 papers), and additional sites were recruited by advertising via the Alzheimer’s Association International Society to Advance Alzheimer’s Research and Treatment (ISTAART) Atypical AD Professional Interest Area (n=7 sites). The search was conducted on PubMed and the terms included: posterior cortical atrophy; PCA; Benson syndrome; visual variant of AD; progressive posterior cortical dysfunction; combined with: biomarkers; neuropathology; autopsy; cerebrospinal fluid; CSF; positron emission tomography; PET. Papers written in English were considered, and there was no restriction on the time period for the search. We requested de-identified, single-subject data from PCA patients (published and unpublished) at the first diagnostic visit. Inclusion criteria were: clinical diagnosis of PCA, and availability of AD biomarkers (PET or CSF) and/or autopsy diagnosis. Not all PCA patients fulfilled recent consensus criteria, being diagnosed using center-specific procedures and/or before development of consensus criteria. After contacting all potential sites, we surveyed potential collaborators to gather data and used the results to create the main database. All variables included in the database are presented in Supplementary Table 1. Demographic variables were: age at diagnostic visit, age at death, age at symptom onset, sex, education, handedness, and APOE4 carrier status. Clinical variables included: MMSE total score, CDR global score, any other severity/staging information, diagnosis (PCA pure or PCA plus by Crutch criteria), diagnosis details (other features if PCA plus), Mendez 8 and/or Tang-Wai criteria 2 (fulfilled or not), and other clinical and cognitive information based on the most recent consensus criteria 1. Sites were also asked which of the 4 non-visuospatial cognitive or neuropsychiatric domains were relatively spared at the time of diagnosis: anterograde memory, speech and nonvisual language, executive functions, and behavior. For the sake of the analyses, these variables were flipped to estimate the frequency of the domains being relatively impaired in patients with PCA.
Biomarker variables included CSF amyloid and p-tau, amyloid and tau PET, MRI scan showing predominant posterior atrophy, 18F-FDG-PET showing predominant posterior hypometabolism, and DaT-SPECT showing dopamine transporter nigrostriatal loss. All centers used their own thresholds and criteria for defining a biomarker as positive or negative.
Neuropathologic variables were collected according to the most recent diagnostic criteria for each neuropathology (see Supplementary Table 1) and included: main neuropathologic and contributing diagnoses, neurofibrillary tangle Braak stage, Aβ plaque Thal phase ), neuritic plaque CERAD score, LBD Braak staging, amygdala-predominant LBD, limbic-predominant age-related TDP-43 encephalopathy neuropathological change staging, argyrophilic grain disease staging, hippocampal sclerosis, vascular injury, cerebral amyloid angiopathy (, aging-related tau astrogliopathy, chronic traumatic encephalopathy, CBD and prion disease. All neuropathological variables were considered as binary variables (present/absent) due to the small sample sizes and differences in procedures across sites.
Institutional Review Board (IRB) review and data agreements
The University of California in San Francisco (UCSF) was the leading institution of this project. A waver or alteration of informed consent that applies to all subjects involved in the current study was acquired through the IRB of the Research Protection Program, which reviews and monitors research involving human subjects at UCSF and affiliated institutions to ensure the ethical treatment of the research subjects.
Statistical analysis
The primary goal of the study was to describe key demographic, clinical, biomarker, and neuropatholog-ical data from all patients by aggregating data across sites; we did so by using a meta-analysis framework. Mean values for continuous variables (e.g., age, MMSE) were combined using the inverse variance meta-analysis method; only sites with more than one participant on a variable were included. Pooled propor-tions were calculated for binary variables (e.g., sex, APOE4 status) using a restricted maximum likeli-hood model. In all meta-analyses, heterogeneity was quantified using the I2 statistics. In secondary anal-yses, we compared two groups of patients (e.g., amyloid positive versus negative, men versus women, PCA pure versus PCA plus) using linear mixed effect models for continuous or mixed-effect logistic regression for categorical outcomes. In both cases, a random intercept was included for each site, and 95% confidence intervals (CI) were estimated using likelihood profile method. As this is an exploratory descriptive study, correction for multiple comparisons was not applied. All statistics were conducted using Jamovi 1·2·27·0 (https://www.jamovi.org/) and Stata.
Results
We collected individual patient data from 1,092 patients evaluated at 36 sites in 16 countries (see Supplementary Table 2 for list of sites and corresponding number of patients). Note that 45% of all patients (and 72% of patients with autopsy) were evaluated in the United States (Supplementary Table 3).
Group-level pooled estimates for main demographic and clinical variables are presented in Table 1; gran-ular (i.e., site-level) data are presented in Supplementary Figures 1–10. The sample consisted of 60% [95%CI 56–64] women, mean age at symptom onset was 59·4 [95%CI 58·9–59·8], mean age at first diagnostic visit was 63·3 [95%CI 62·8–63·6]. 80% [95%CI 72–89] presented with a “PCA pure” and 20% [95%CI 11–28] with a “PCA plus” clinical syndrome, as defined by Crutch 2017 diagnostic criteria 1. At their diagnostic visit, patients had a mean MMSE score of 20·7 [95%CI 20·4–21·1], and 61%[95%CI 53–71] had a global CDR score ≥ 1. Most patients were right-handed (92% [95%CI 90–95) and 43% [95%CI 35–50] carried at least one copy of the APOE-ε4 allele. In the subsample of 228 patients (21 sites) who were reported to be deceased, mean age at death was 70·5 [95%CI 69.5–71.4].
Table 1.
Demographical and clinical description of the full sample
| Pooled estimate (mean/frequency) | 95%CI | Npatients | Nsites | I2 | |
|---|---|---|---|---|---|
|
| |||||
| Demographics | |||||
| Sex (% women) | 60% | [56, 64] | 1092 | 36 | 35.0 |
| Handedness (% right) | 93% | [90, 95] | 872 | 32 | 42.8 |
| Years of education | 14.1 | [13.9, 14.2] | 949 | 35 | 93.1 |
| APOE (% e4 carriers) | 43% | [35, 50] | 451 | 22 | 55.5 |
| Clinical | |||||
| Age at symptom onset, years | 59.4 | [58.9, 59.8] | 1031 | 34 | 77.4 |
| Age at diagnosis, years | 63.2 | [62.8, 63.6] | 1067 | 36 | 76.8 |
| Age at death, years | 70.5 | [69.5, 71.4] | 227 | 20 | 72.7 |
| Diagnosis (% PCA pure) | 80% | [72, 89] | 968 | 34 | 97.9 |
| MMSE at diagnosis, points (range : 0–30) | 20.7 | [20.4, 21.1] | 904 | 31 | 69.9 |
| CDR at diagnosis (% CDR≥1) | 62% | [53, 71] | 558 | 27 | 81.1 |
| Mendez criteria (% fulfilled) | 75% | [60, 90] | 535 | 21 | 99.4 |
| Tang-Wai criteria (% fulfilled) | 80% | [68, 92] | 535 | 21 | 98.6 |
| Early disturbance of posterior function (% yes) | 91% | [85, 97] | 992 | 36 | 98.1 |
| Insidious onset and gradual progression (% yes) | 99% | [98, 99] | 809 | 34 | 0 |
Mean values or frequency, and their corresponding 95% confidence intervals are derived from the meta-analysis of all available data. Sites with only one available datapoint (two sites for “MMSE”, one site for “age at death”) were not included in the meta-analysis. Raw data, including breakdown of each site, is presented in Supplementary Figures 1–10. Abbreviations: MMSE: Mini-Mental State Examination, CDR: Clinical Dementia Rating scale.
The frequencies of core features and involvement of additional cognitive or neuropsychiatric domains at the first diagnostic visit are shown in Figure 1A and 1B, respectively. Constructional dyspraxia was the most frequently reported (61% [95%CI 51–73]) core PCA clinical feature, followed by space perception deficit (49% [95%CI 38–61]), simultanagnosia (48% [95%CI 36–61]), and acalculia (47% [95%CI 38–57]). The less frequently reported core clinical features were finger agnosia (20% [95%CI 13–28]), ocu-lomotor apraxia (18% [95%CI 11–26]), and apperceptive prosopagnosia (17% [95%CI 10–24]). The as-sociations between core features are presented in Supplementary Figure 11. Besides visuoperceptual functions, at the time of diagnosis, 47% [95%CI 36–58] of patients had relatively impaired anterograde memory, 40% [95%CI 29–52] had relatively impaired executive functions, 33% [95%CI 21–44] had rel-atively impaired non-visual language and speech, and 32% [95%CI 22–42] had relatively impaired be-havior. It should be noted that data on the frequency of clinical features was highly heterogeneous across studies, as indicated by I2 >89%.
Figure 1.
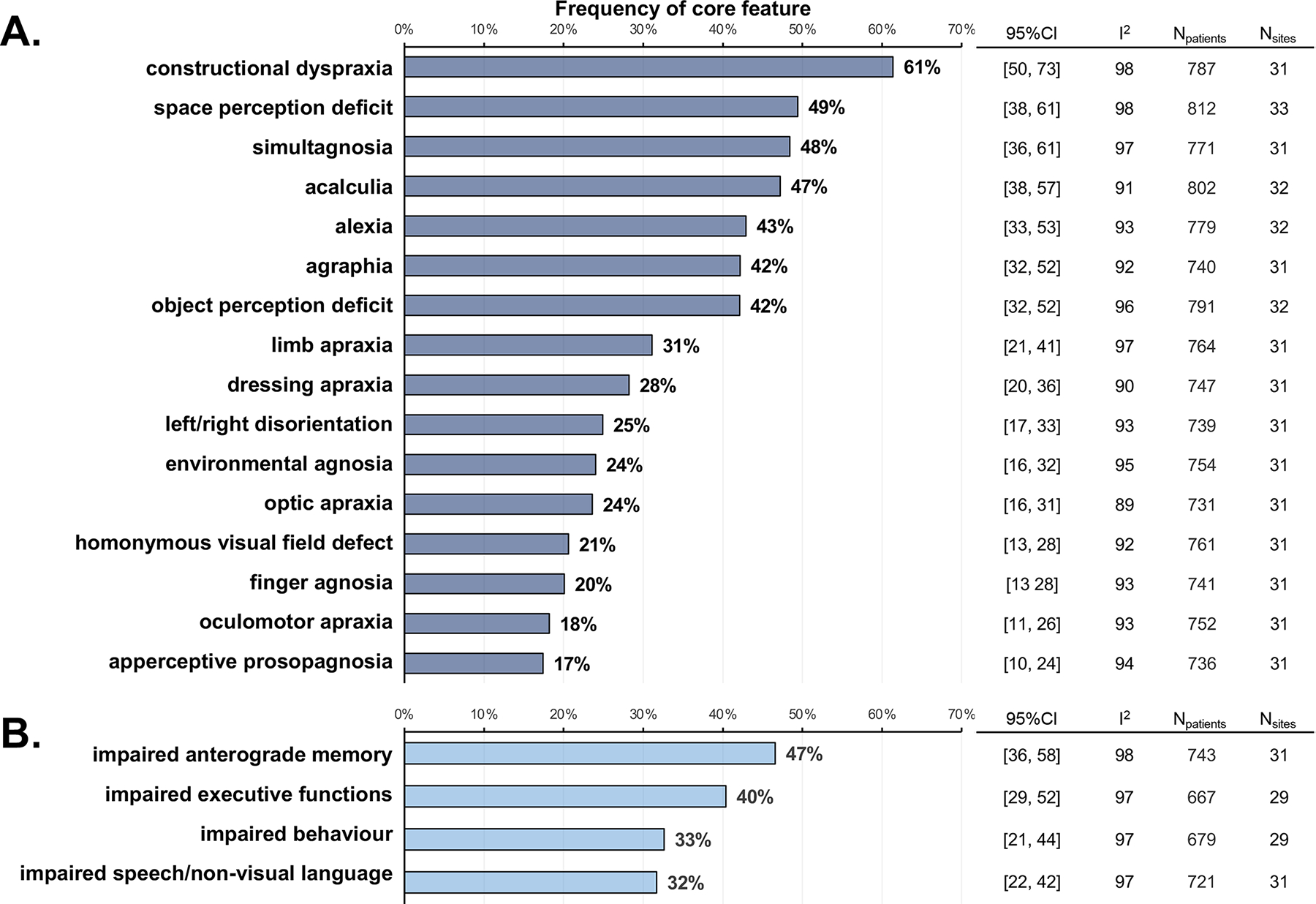
Frequencies of core features and involvement of additional cognitive or neuropsychiatric domains at the first diagnostic visit. Biomarker findings are reported in figure 2 (site-level data are in the appendix pp 20–27). When reported, CSF amyloid β was positive (ie, in the range consistent with underlying brain amyloid or tau deposition) in 81% (95% CI 75–87) of participants and CSF p-tau was positive in 65% (56–75), but findings were heterogeneous across research centres (I2 >75%). Amyloid-PET and tau-PET were positive for most participants (amyloid-PET, 94% [95% CI 90–97]; tau-PET 97% [93–100]), and heterogeneity statistics were low (I2 ≤15%). Predominant posterior cortical atrophy on MRI was found in 85% (79–91) of participants, and predominant posterior [18F]FDG-PET hypometabolism was reported for 97% (95–98). DaT-SPECT scan results were reported in a small subsample (72 participants from 15 research centres) and showed evidence of nigrostriatal loss in 51% (95% CI 33–69) of participants.
Biomarker findings are reported in Figure 2 (see also Supplementary Figures 12–19 for site-level data and forest plots). When reported, CSF Aβ was positive (i.e., in the AD range) in 81% [95%CI 75–87] and CSF p-tau was positive in 65% [95%CI 56–75], with marked heterogeneity across sites (I2 >75%). PET biomarkers for amyloid and tau were highly positive (amyloid: 94% [95%CI 91–97], tau: 97% [95%CI 93–100]) and the heterogeneity statistics were low (I2 <15%). Predominant posterior cortical atrophy was found in 85% [95%CI 79–91] patients while predominant posterior 18F-FDG-PET hypome-tabolism was reported in 97% [95%CI 95–98] of cases. DaT-SPECT scan results were reported in a small subsample (72 patients from 15 sites) and showed evidence of nigrostriatal loss in 51% [95%CI 33–69] of patients.
Figure 2.
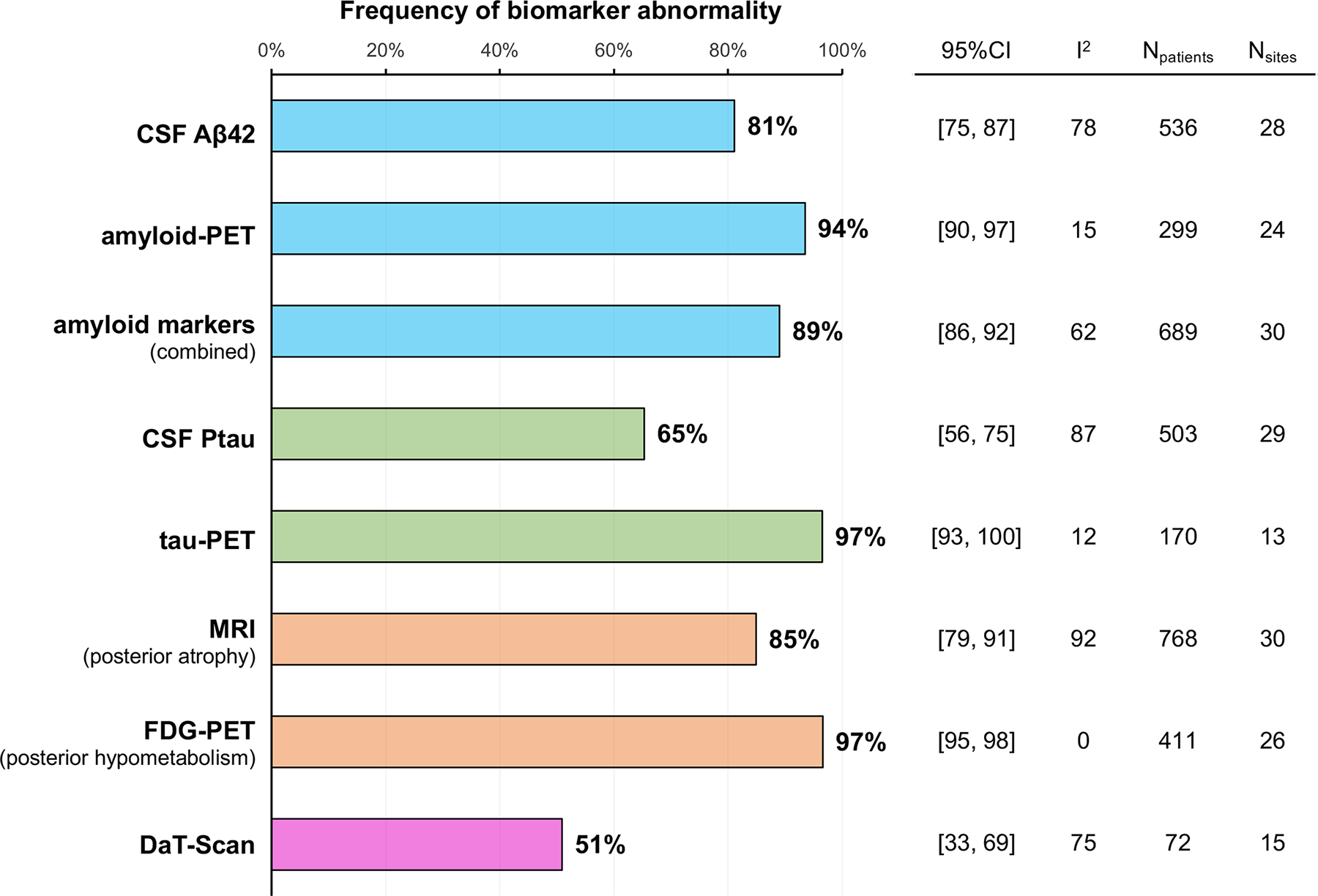
Frequency of biomarker abnormality. To assess associations between Alzheimer’s disease biomarker results and clinical variables, 689 participants from 30 research centres who had either amyloid-PET or CSF amyloid biomarker data (or both) were classified as amyloidpositive when at least one of the biomarkers was positive, and as amyloid-negative when both markers (or the only available marker) were negative. Group comparisons are shown in table 2. Patients who were amyloid-positive were more likely to have posterior cortical atrophy pure than were amyloid-negative patients (95% vs 81%, p<0·0001). Age at symptom onset and age at diagnosis, sex, and other clinical features did not differ by amyloid status.
To assess the associations between AD biomarker results and clinical variables, we classified the 689 patients (30 sites) with available (PET or CSF) amyloid biomarker as ‘amyloid-positive’ when at least one of the Aβ biomarkers was positive and ‘amyloid-negative’ when both markers, or the only available marker was negative. Group comparisons are shown in Table 2. Compared to amyloid-negative patients, amyloid-positive patients were more likely to have a PCA-pure (86% vs 62%, p<0·0001) than a PCA-plus syndrome; were slightly younger at symptom onset and diagnosis; and tended to have a higher pro-portion of women (63%, vs, 53%, p=0.085). Other features were similar in Aβ-positive and negative groups.
Table 2.
Comparison of the demographic and clinical characteristics of amyloid-positive and amyloid-negative patients
| Npatients | Nsites | Estimate [95%CI] | p | Marginal means [95%CI] | ||
|---|---|---|---|---|---|---|
| Aβ-negative | Aβ-positive | |||||
|
| ||||||
| Age at symptom onset | 644 | 28 | −1.322 [−3.130, 0.477] | 0.15 | 60.4 [58.4, 62.4] | 59.1 [57.8, 60.4] |
| Age at diagnosis | 671 | 30 | −1.415 [−3.18, 0.348] | 0.12 | 63.9 [62.0, 65.8] | 62.5 [61.4, 63.6] |
| MMSE at diagnosis | 596 | 28 | −0.059 [−1.350, 1.290] | 0.93 | 20.8 [19.5, 22.2] | 20.7 [19.9, 21.6] |
| CDR at diagnosis (%CDR≥1) | 413 | 21 | 0.118 [−0.573, 0.818] | 0.74 | 61% [42, 77] | 58% [46, 70] |
| Sex (% Women) | 689 | 30 | 0.406 [−0.058, 0.867] | 0.085 | 53% [42, 63] | 63% [59, 67] |
| APOE (% e4 carriers) | 342 | 22 | 0.089 [−0.639, 0.818] | 0.81 | 45% [29, 62] | 47.5% [40, 55] |
| PCA diagnosis (% PCA pure) | 625 | 29 | 1.506 [0.769, 2.26] | <0.0001 | 81% [56, 94] | 95% [86, 98] |
| MRI (% posterior atrophy) | 543 | 27 | 0.226 [−0.605, 1.060] | 0.59 | 92% [78, 97] | 94% [85, 97] |
| FDG-PET (% post. hypometabolism) | 324 | 22 | 0.578 [−0.896, 2.05] | 0.44 | 97% [84, 99] | 98% [94, 99] |
For each variable, a separate linear mixed model (continuous outcomes) or generalized mixed effect model (binary outcomes) was run, using amyloid status as a fixed effect and site as a random effect. Estimate 95%CI were obtained using the likelihood profile method. Number of participants and sites with available data varies from one variable to the other. Aβ-positive patients included those who received a positive result on CSFAβ42 and/or amyloid-PET, and Aβ-negative patients received negative results from CSF or PET. This sample included 390 with CSF only, 153 with PET only, and 146 with both; in this subgroup 27 patients had discrepant results (3 CSF+/PET-, 24 CSF-/PET+) and were included in the amyloid-positive group.
Thirteen sites shared autopsy results from a total of 145 participants. This subsample included 50% women [95%CI 38–61], with a mean age at symptom onset of 58·6 [95%CI 57·4–59·8], and a mean age at death of 69·4 [95%CI 68·2–70·6]. Across sites, the primary neuropathological diagnosis was AD with a pooled estimate of 94% [95%CI 90–97] and minimal heterogeneity (I2=0); see Figure 3a. Most patients with primary AD were found to have one or more co-pathologies, the most common being cerebral am-yloid angiopathy (71%), Lewy body disease (44%), and cerebrovascular injury (42%); see Figure 3B and Supplementary Figure 20.
Figure 3.
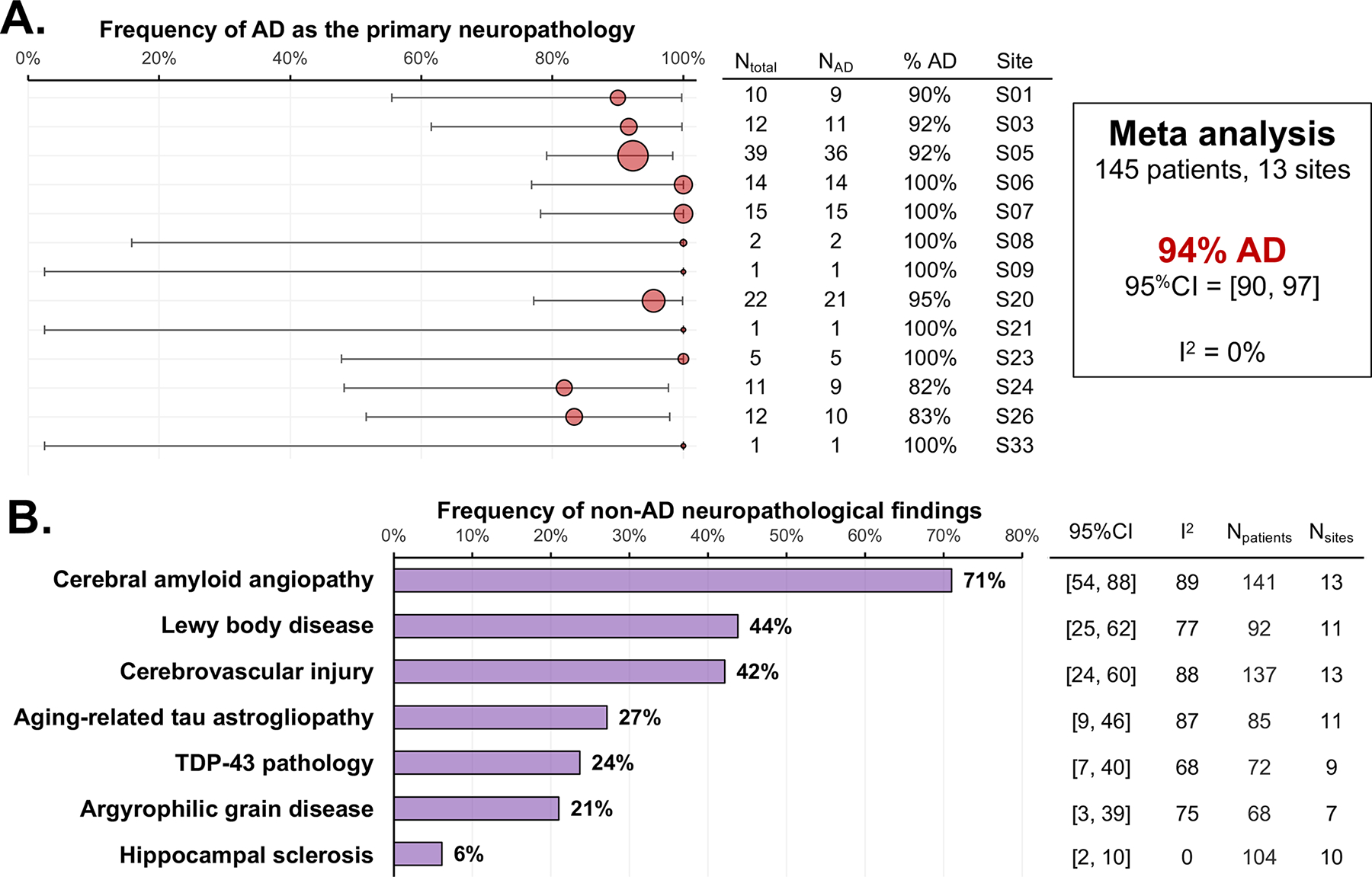
Frequency of neuropathological findings in the sample. Only ten participants from six research centres had a primary neuropathological diagnosis that was not Alzheimer’s disease (table 3). Four had a primary neuropathological diagnosis of Lewy body disease, all of whom also had significant levels of Alzheimer’s disease neuropathological changes. Other primary diagnoses were frontotemporal lobar degeneration with non-Alzheimer’s disease tauopathy (n=3, two with corticobasal degeneration and one with Pick’s disease) or TDP-43 type A (n=1, due to a pathogenic granulin [GRN] mutation), and brain infarct with minimal comorbid Alzheimer’s disease neuropathology (n=2; both cases had late age at onset [ages 88 years and 90 years] and were from the same research centre).
Overall, only 10 cases (from 6 sites) had a non-AD primary neuropathological diagnosis; see Table 3 for details. Briefly, 4 patients had a primary neuropathological diagnosis of Lewy body disease, all of whom also had significant levels of AD neuropathological changes. Other primary etiologies included fronto-temporal lobar degeneration with non-AD tauopathy (n=2 with corticobasal degeneration, n=1 with Pick’s disease) or with TDP-43 pathology type A (n=1, who was due to a pathogenic granulin (GRN) mutation). Lastly, two patients had a primary diagnosis of brain infarct with minimal comorbid AD neu-ropathology; note that both cases were older (onset at 88 and 90 years old) and were enrolled at the same site.
Table 3.
Clinical characteristics of the 10 patients with non-AD primary neuropathological diagnosis
| Primary neuropathological diagnosis | Age at symptom onset | Age at death | Diagnosis | sex | APOE4 status | Thal phase | NFT Braak stage | CERAD score | ADNC level | Site |
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| LBD | 67 | 71 | PCA plus | M | e4 carrier | 5 | IV | moderate | intermediate | S01 |
| LBD | 61 | 70 | PCA pure | F | - | - | IV | frequent | intermediate | S03 |
| LBD | 58 | 68 | - | M | e4 carrier | 5 | VI | frequent | high | S05 |
| LBD | 79 | 87 | - | M | non-carrier | 4 | V | frequent | high | S05 |
| FTLD-tau (CBD) | 58 | 64 | PCA plus | M | e4 carrier | 2 | II | sparse | low | S20 |
| FTLD-tau (CBD) | 51 | 57 | PCA pure | M | non-carrier | 0 | 0 | none | none | S24 |
| Brain infarct | 90 | 91 | - | F | - | 1 | 0 | sparse | low | S26 |
| Brain infarct | 88 | 94 | - | F | - | 0 | 0 | none | none | S26 |
| FTLD-TDP43 Type A | 59 | 68 | PCA pure | M | - | 0 | 0 | none | none | S24 |
| FTLD-tau (Pick’s) | 58 | 68 | PCA pure | F | e4 carrier | 2 | I | moderate | low | S05 |
Abbreviations: LBD: Lewy body disease, FTLD: frontotemporal lobar degeneration, CBD: corticobasal degeneration, TDP-43: TAR DNA-binding protein 43, APOE4: allele ε4 of the apolipoprotein E gene, NFT: Neurofibrillary Tangles, CERAD: Consortium to Establish a Registry for Alzheimer’s Disease, ADNC: Alzheimer’s Disease Neuropathological changes, MMSE: Mini-Mental State Examination; CDR: Clinical Dementia Rating scale. Corresponding site descriptions can be found in Supplementary Table 2.
We investigated the associations between sex and key clinical, biomarker, and neuropathological variables. On average, women had lower MMSE scores at their first diagnostic visit (difference = 1.08 [95% CI 0.3–1.8], p=0·0046), and were more likely to have a PCA pure syndrome compared to men (95% [95%CI 97–98] versus 91% [95%CI 78–97], p=0·0048). As mentioned above, women tended to be more amyloid-positive compared to men: 91% [95%CI 96–94] versus 87% [95%CI 80–92], p=0.089. No other sex dif-ferences were observed (see Supplementary Table 4).
Lastly, we assessed the relationships between the diagnosis of PCA pure versus PCA plus on key clinical, biomarker, and neuropathological variables. Overall, patients with PCA pure had a younger age of symptom onset (difference = 2.4 years [95%CI 0.9–3.9], p = 0.0018) and age at diagnostic visit (difference =3.6 years [95CI 2.1–5.0], p<0.0001) compared to patients with PCA plus. The PCA plus group had a lower frequency of women (non-significant trend) and a significantly lower rate of positive amyloid biomarkers compared to the PCA pure group (see Supplementary Table 5 for details).
Discussion
In this multi-site, international study involving 1,092 participants diagnosed with PCA who had available AD biomarkers and/or autopsy data, we found that the syndrome generally has an early age-of-onset, disproportionately affects women, and often presents in its pure form (i.e., without clinical features of other neurodegenerative diseases) per Crutch 2017 diagnostic criteria.1 By the time of diagnosis, patients are usually at the dementia stage, and additional cognitive domains are often impaired. While APOE4 genotype prevalence is enriched compared to a normal population, it is lower than in amnestic AD,9 suggesting a weaker link between APOE genotype and PCA. Importantly, amyloid biomarkers are positive in over 89% of individuals, and AD was the primary diagnosis in 94% of patients with autopsy data, underscoring that the PCA clinical syndrome is caused by underlying AD neuropathology in most cases.
Our cohort consisted of 60% women versus 40% men. This ratio is consistent with previous findings from smaller studies, which showed that PCA affects women to a larger extent than men 5. Reports on sex predilection have varied across AD variants, with a higher proportion of women to men in amnestic (just over 50% women in most studies10 and dysexecutive AD (62% women)11 and a higher proportion of men in logopenic-variant primary progressive aphasia (lvPPA; 52% men)12 and behavioral-variant AD (62% men).13 A previous study reported that the prevalence of mathematical and visuospatial learning disabilities is higher in PCA patients than in other clinical presentations of AD 14. Given that the prevalence of mathematical learning disabilities is higher in girls versus boys during schooling 15, women could have a greater cognitive vulnerability to the PCA syndrome. We found that the frequency of acalculia was significantly higher in women versus men, which is consistent with this hypothesis.
PCA is often associated with an early age of onset 16,17. In our study, average age of onset was 59.4 years old and 75% of patients had an age of onset less than 65 (the 3rd quartile of the distribution, see Supplementary Figure 5), the common threshold for early-onset dementia. At the time of diagnosis, mean MMSE was 21 and mean global CDR was 1 (mild dementia), suggesting that symptoms are relatively advanced at the time of first diagnosis. 18 Consistent with this, some clinicians reported involvement of additional cognitive or behavioral domains at first diagnostic visit, likely reflecting progression from an initial pure visuoperceptual/visuospatial syndrome to a multi-domain dementia. Patients with PCA often face a delay in diagnosis due to their young age and visual-predominant symptoms 19. Better awareness of the syndrome of PCA amongst primary care providers, optometrists/ophthalmologists and neurologists is needed for earlier detection and treatment.
Previous clinicopathologic studies have shown that AD is the most common neuropathologic cause of PCA, though some cases are due to LBD, CBD and prion disease 2–4. Although these studies emerged from various sites, the samples were generally small due to the rareness of the syndrome and the challenges associated with postmortem data collection. In this larger multi-site sample, we found a very strong correlation between PCA syndrome and AD neuropathology, which is stronger than the reported relationships between other clinical variants and underlying AD. 12,20 Indeed, previous studies reported that AD pathology is found in about 70% of patients with an amnestic presentation, and 76% of patients presenting with lvPPA. Here, we found that AD pathology is found in 94% of patients with PCA. Only ten patients received a primary neuropathologic diagnosis that was not AD, and half of them were also found to have intermediate-high ADNC. Therefore, PCA might be the most predictive clinical syndrome for underlying AD. As has been shown in smaller series, other neuropathologies that can be considered in the differential diagnosis of PCA include LBD, CBD, FTLD-TDP Type A and Pick’s disease. No cases of prion disease were reported in our sample, but these have been reported previously. Two cases of PCA due to brain infarcts were reported in this series, both occurring in patients in their 90s. It is unclear if these cases would have met current clinical criteria for PCA, which exclude acute or non-progressive presentations. These cases do raise the possibility of alternative etiologies for PCA in the very old.
Biomarkers are useful for supporting or excluding AD as the cause of the dementia syndrome during life, particularly in patients presenting with non-amnestic syndromes or early age-of-onset 28. Like other clinical variants of AD, CSF Aβ 1–42 concentrations or the ratio of Aβ1–42 / Aβ1–40 are lowered, and total and phosphorylated tau 181 levels concentrations are elevated in the early stages of PCA21–23. Similarly, previous studies have reported high proportions of amyloid and tau PET positivity in PCA cohorts. In our study, CSF Aβ, amyloid and tau PET all were very frequently in the AD range in patients with PCA, including those with autopsy-confirmed AD. In contrast, CSF p-tau showed limited sensitivity in our cohort. Previous studies have indicated limited sensitivity of CSF p-tau as a “stand-alone” biomarker for ADNC, and diagnostic accuracy is improved by calculating the ratio of CSF p-tau/Aβ1–42 23. We found atrophy and hypometabolism in posterior cortical regions in 89% and 97% of participants, making both MRI and 18F-FDG-PET techniques robust techniques to help with PCA diagnosis in vivo by establishing a neurodegenerative basis and posterior cortical localization. Importantly, the prevalence of “PCA-plus” features was significantly higher in amyloid biomarker-negative compared to amyloid-positive individuals, which suggests that non-AD pathologies could be mostly responsible for the “plus” clinical findings (ex: limb rigidity, parkinsonism, etc.).
Our results corroborate syndrome and disease level descriptions outlined in PCA consensus classification in an international multi-center study. The frequency of each of the core clinical features was very similar to what was found in the recent consensus classification paper, and predominantly included mixed ventral and dorsal visual stream features1. Constructional dyspraxia, space perception deficit, simultanagnosia, and acalculia were commonly reported (>50%). In the consensus classification paper, the most frequent clinical features were also constructional dyspraxia, space perception deficit and simultanagnosia, although acalculia was slightly less common. Less frequent features in both cohorts, include finger agnosia, oculomotor apraxia and apperceptive prosopagnosia.
One of the major strengths of this study is the size and the geographical diversity of our sample; we collected data from 1,092 patients in 16 different countries and 36 individual sites, which represents by far the largest and most representative study conducted on PCA to date. Another strength of this study is the number of cases with autopsy data. We obtained the main neuropathologic diagnosis for 145 participants and quantified the prevalence of other common neuropathologies in subsamples ranging from 50 to 145 participants.
Our study also has limitations. This is a retrospective study that aggregated data from multiple centers without a standard clinical protocol. All data, including most notably the diagnosis of PCA, were based on the standards applied at the local site. This enhances generalizability, but also can lead to high heterogeneity (as evidenced by high I2 values for some variables) as well as non-randomness of missing data. It is likely that some biases impacted whether certain clinical features were assessed or whether bi-omarkers were ordered (e.g., DaT-SPECT imaging was only available in 74 patients and may have been ordered only in patients with suspected LBD). We took a conservative approach by assuming clinical features were absent if missing, which may lead to under-estimation of their true prevalence. Data were aggregated at sites over years, during which clinical definitions of PCA evolved. The site data survey was designed to minimize site burden, resulting in less available detail. Future prospective studies of PCA should promote cross-center comparability through standardized protocols and include age- and severity-matched disease control groups (especially with other variants of AD) for comparison 27. Future work should also include data on race/ethnicity in PCA and provide more detailed information on bi-omarkers (ex: specific CSF assays used). Lastly, as this is an exploratory descriptive study, correction for multiple comparisons in the statistical models was not applied, and no adjustment for confounders was included in the analyses.
In conclusion, this multi-site study refines our understanding of the relationship between pathology, biomarkers, and clinical features in PCA, and provides up-to-date descriptive statistics related to this syndrome. Our results highlight the strong link between PCA and underlying AD and emphasize the im-portance of AD biomarker testing as part of the diagnostic evaluation of patients with PCA.
Supplementary Material
Research in context panel.
Evidence before this study:
We searched PubMed for articles on posterior cortical atrophy (PCA) with no language restrictions from database inception up to August 1st, 2022, using the following terms: “posterior cortical atrophy”, “posterior cortical atrophy pathology”, “posterior cortical atrophy Alzheimer’s disease”, “visual variant of Alzheimer’s disease”, “atypical Alzheimer’s disease”, “non-amnestic Alzheimer’s disease”, focusing on studies that reported neuropathologic findings in PCA. A few studies investigated the neuropathologic profiles of patients with PCA and have shown a high prevalence of cases due to Alzheimer’s disease (AD) pathology, although some case series have reported cases due to Lewy body pathology, corticobasal degeneration and prion disease. However, these studies had small sample sizes (ranging from 20 to 40 patients) and none of them systematically reported and analyzed clinical, biomarker and neuropathologic data in a large, multi-site sample of PCA patients.
Added value of this study:
We collected demographic, clinical, biomarker and neuropathologic data in 1,092 patients with PCA in 16 countries and 36 different sites. Variables included age at symptom onset, at diagnostic visit and at death, apolipoprotein E4 (APOE4) status, Mini-Mental State Examination (MMSE) and Clinical Dementia Rating (CDR) global scores, diagnostic details per the most recent research criteria (i.e., PCA pure versus PCA plus), individual symptoms, other cognitive domains relatively spared, imaging and fluid markers ((Magnetic Resonance Imaging (MRI), Dopamine Transporter (DaT)-Single-photon emission computed tomography (SPECT), 18F-fluorodeoxyglucose (18F-FDG) Positron Emission Tomography (PET), amyloid-PET, tau-PET, cerebrospinal fluid (CSF) amyloid and tau)) and neuropathologic findings (main neuropathologic diagnosis, presence/absence of common neuropathologies, staging of specific pathologies). This multi-site study refined our understanding of the relationship between pathology, biomarkers and clinical features in PCA. Our findings highlight the early age-of-onset and female predominance of this syndrome. We showed that AD is by far the most common underlying neuropathology and that PCA may be the most predictive syndrome for AD neuropathology. We also showed that co-pathologies are frequent.
Implications of all the available evidence:
Our study provides up-to-date demographic, clinical, biomarker and neuropathologic data in PCA. Our findings show a clear added value of in vivo biomarkers for AD, and other imaging tools to capture atrophy and hypometabolism patterns, which closely mirror the symptomatology in PCA. Our results are also consistent with the most recent consensus criteria, which stated the importance of distinguishing “PCA pure” (core PCA syndrome only) versus “PCA plus” (core PCA syndrome and core features of another neurodegenerative syndrome) presentations, as these two groups may reflect distinctive pathophysiological processes. Further work is needed to understand what drives cognitive vulnerability and progression rates by investigating the contribution of sex, genetics, premorbid cognitive strengths/weaknesses, and brain network integrity. The present study will provide clinicians, patients, and caregivers with a better understanding of the specific clinical features of PCA and their associations with the underlying disease(s).
Funding source
This manuscript was facilitated by the Alzheimer’s Association International Society to Advance Alzheimer’s Research and Treatment (ISTAART), through the Atypical AD Professional Interest Area. The views and opinions expressed by authors in this publication represent those of the authors and do not necessarily reflect those of the PIA membership, ISTAART or the Alzheimer’s Association.
M.C. received research support from the Fonds de Recherche du Québec – Santé (FRQS). M.F., F.A., E.C., F.C. and G.M. receive research support from the Foundation Research on Alzheimer Disease and Italian Ministry of Healthy (#GR-2010–2303035). M.E.M. receives research support from the NIH (R01 AG054449, R01 AG075802, U01-AG057195 and P30 AG062677). Data from Mayo Clinic (Jacksonville) was supported by the State of Florida Alzheimer’s Disease Initiative and the Mayo Clinic Alzheimer’s Disease Research Center. B.D. receives research funding from NIA R21-AG051987, P50-AG005134, and R01-DC014296 and philanthropic funding to the MGH FTD Unit including the Mooney Family Fund. K.Y. is an Etherington PCA Senior Research Fellow and is funded by the Alzheimer’s Society, grant number 453 (AS-JF-18–003).The work was also supported by an Alzheimer’s Research UK Senior Research Fellowship and ESRC/NIHR (ES/L001810/1) grant to S.C.J.M.S acknowledges the support of the National Institute for Health Research University College London Hospitals Biomedical Research Centre, ARUK (ARUK-PG2017–1946), Weston Brain Institute (UB170045), Medical Research Council, and British Heart Foundation. T.L. is supported by an Alzheimer’s Research UK senior fellowship. The Queen Square Brain Bank is supported by the Reta Lila Weston Institute for Neurological Studies and the Medical Research Council.The Dementia Research Centre is supported by Alzheimer’s Research UK, Brain Research Trust, and The Wolfson Foundation. This work was also supported by the NIHR Queen Square Dementia Biomedical Research Unit, and the NIHR UCL/H Biomedical Research Centre.This work was supported by the MRC Dementia Platform UK and the UK Dementia Research Institute at UCL, which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. D.G., A.H. and G.L. receive research support from Shiley-Marcos ADRC P30 AG062429. K.J., J.W. and J.G.R. receive research funding from the NIH (R01-AG50603). B.D.C.B. receives funding from Alzheimer Nederland (#WE.15–2019-13, WE.03–2021-15, and #WE.06–2023-01). M.M., J.R. and K.A. would like to thank Dr Annelies Quaegebeur. Their research was supported by the NIHR Cambridge Biomedical Research Centre including the Cambridge Brain Bank (BRC-1215–20014. The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care) and the Race Against Dementia. M.F.M. receives research support from the NIA (1RF1AG050967). R.M. is supported by France Alzheimer, Fondation Recherche Alzheimer, Philippe Chatrier Foundation and by Rosita Gomez Association. O.H. has acquired research support (for the institution) from ADx, AVID Radiopharmaceuticals, Biogen, Eli Lilly, Eisai, Fujirebio, GE Healthcare, Pfizer, and Roche. In the past 2 years, he has received consultancy/speaker fees from AC Immune, Amylyx, Alzpath, BioArctic, Biogen, Cerveau, Fujirebio, Genentech, Novartis, Roche, and Siemens. Work at the Lund University was supported by the Swedish Research Council (2016–00906), the Knut and Alice Wallenberg foundation (2017–0383), the Marianne and Marcus Wallenberg foundation (2015·0125), the Strategic Research Area MultiPark (Multidisciplinary Research in Parkinson’s disease) at Lund University, the Swedish Alzheimer Foundation (AF-939932), the Swedish Brain Foundation (FO2021–0293), The Parkinson foundation of Sweden (1280/20), the Konung Gustaf V:s och Drottning Victorias Frimurarestiftelse, the Skåne University Hospital Foundation (2020-O000028), Regionalt Forskningsstöd (2020–0314) and the Swedish federal government under the ALF agreement (2018-Projekt0279). P.N. is supported by The Mater Foundation. D.P., S.P.C. and G.T. are supported by The Italian Ministry of Health (Ricerca Finalizzata Progetto Reti Nazionale AD NET-2011–02346784). O.P., N.C., J.B., J.H. and D.F. were supported by funding to ForeFront, a collaborative research group dedicated to the study of frontotemporal dementia and motor neuron disease, from the National Health and Medical Research Council (NHMRC) (GNT1037746) and the Australian Research Council (ARC) Centre of Excellence in Cognition and its Disorders Memory Program (CE11000102). OP is supported by an NHMRC Leadership Fellowship (GNT2008020). L.A. receives research support from Indiana Alzheimer’s Disease Research Center (P30 AG010133 and LEADS (U01AG6057195). E.R., S.W. and M.M. received research support from the NIH (P30AG13854 and P30AG072977). B.D.C.B. receives funding from Alzheimer Nederland (#WE.15–2019-13, WE.03–2021-15, and #WE.06–2023-01). Data from Geneva was supported by the following Alzheimer’s Disease Research Center (ADRC) grants: NIA P50 AG005138 and P30 AG066514. Data from Xuanwu Hospital was supported by the National Natural Science Foundation of China (81971011) and Beijing Municipal Science and Technology Committee (7202060). Research of Alzheimer center Amsterdam has been funded by ZonMW, NWO, EU-FP7, EU-JPND, Alzheimer Nederland, Hersenstichting CardioVascular Onderzoek Nederland, Health~Holland, Topsector Life Sciences & Health, stichting Dioraphte, Gieskes-Strijbis fonds, stichting Equilibrio, Edwin Bouw fonds, Pasman stichting, stichting Alzheimer & Neuropsychiatrie Foundation, Philips, Biogen MA Inc, Novartis-NL, Life-MI, AVID, Roche BV, Fujifilm, Combinostics. Data from Penn Frontotemporal Degeneration Center was funded by the following grants: R01-AG054519 and K01-AG061277. Data from M.A.S.S.’s group comes from The Sant Pau Initiative on Neurodegeneration (SPIN) cohort. L.T.G receives research support from the NIH (K24053435) and R01AG075802. G.D.R. receives research support from NIH/NIA R35 AG072362 and NIH/NIA P30 AG062422, U01-AG057195, and P01-AG019724. Other support includes NINDS, Alzheimer’s Association, American College of Radiology, Rainwater Charitable Foundation, Shanendoah Foundation, Avid Radiopharmaceuticals, GE Healthcare, Life Molecular Imaging, Genentech.
Footnotes
Declaration of interests
L.A. has received personal compensation for serving as a consultant for Biogen, Two Labs, FL Dept Health, Genentech, NIH Biobank, Eli Lilly, GE Healthcare, Eisai, Roche Diagnostics, and for serving on a Data Safety and Monitoring Board for IQVIA. L.A. receives research support from the National Institute on Aging, the Alzheimer’s Association, Roche Diagnostics, AVID radiopharmaceuticals, Life Molecular Imaging, and Eli Lilly. M.F.M. is the section editor for Behavioral Neurology for UpToDate. J.G.R. serves on the Drug Safety Monitoring Board (DSMB) for the National Institute of Neurological Disorders and Stroke (NINDS) StrokeNET. K.A.J. is an Associate Editor for Annals of Clinical and Translational Neurology and is on the editorial boards of Journal of Neurology, Acta Neuropathologica and Neuropathology and Applied Neurobiology. J.L.W is an Associate Editor for Brain Connectivity and Journal of Alzheimer’s Disease. R.O. has received research support from Avid Radiopharmaceuticals, has given lectures in symposia sponsored by GE Healthcare and is an editorial board member of Alzheimer’s Research & Therapy and the European Journal of Nuclear Medicine and Molecular Imaging. F. Agosta is Associate Editor of NeuroImage: Clinical, has received speaker honoraria from Biogen Idec, Italfarmaco, Roche and Zambon, and receives or has received research supports from the Italian Ministry of Health, the Italian Ministry of University and Research, AriSLA (Fondazione Italiana di Ricerca per la SLA), the European Research Council and Foundation Research on Alzheimer Disease. Prof. Filippi is Editor-in-Chief of the Journal of Neurology, Associate Editor of Human Brain Mapping, Neurological Sciences, and Radiology; received compensation for consulting services from Alexion, Almirall, Biogen, Merck, Novartis, Roche, Sanofi; speaking activities from Bayer, Biogen, Celgene, Chiesi Italia SpA, Eli Lilly, Genzyme, Janssen, Merck-Serono, Neopharmed Gentili, Novartis, Novo Nordisk, Roche, Sanofi, Takeda, and TEVA; participation in Advisory Boards for Alexion, Biogen, Bristol-Myers Squibb, Merck, Novartis, Roche, Sanofi, Sanofi-Aventis, Sanofi-Genzyme, Takeda; scientific direction of educational events for Biogen, Merck, Roche, Celgene, Bristol-Myers Squibb, Lilly, Novartis, Sanofi-Genzyme; he receives research support from Biogen Idec, Merck-Serono, Novartis, Roche, Italian Ministry of Health, and Fondazione Italiana Sclerosi Multipla. G.D.R. has served on Scientific Advisory Boards for Alector, Eli Lilly, Genentech, Merck, Roche. He serves on a Data Safety and Monitoring Board for Johnson & Johnson. He is an Associate Editor for JAMA Neurology. No other potential conflicts of interest relevant to this article exist.
Data sharing
Applications for data sharing by qualified investigators can be made to the UCSF Alzheimer’s Disease Research Center (https://memory.ucsf.edu/research-trials/professional/open-science). Data sharing requests will be subject to the limitations specified in data transfer agreements between UCSF and the individual sites providing data for this single-subject meta-analysis.
References
- 1.Crutch SJ, Schott JM, Rabinovici GD, et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement 2017; 13(8): 870–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang-Wai DF, Graff-Radford NR, Boeve BF, et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 2004; 63(7): 1168–74. [DOI] [PubMed] [Google Scholar]
- 3.Alladi S, Xuereb J, Bak T, et al. Focal cortical presentations of Alzheimer’s disease. Brain 2007; 130(Pt 10): 2636–45. [DOI] [PubMed] [Google Scholar]
- 4.Renner JA, Burns JM, Hou CE, McKeel DW, Jr., Storandt M, Morris JC. Progressive posterior cortical dysfunction: a clinicopathologic series. Neurology 2004; 63(7): 1175–80. [DOI] [PubMed] [Google Scholar]
- 5.Schott JM, Crutch SJ, Carrasquillo MM, et al. Genetic risk factors for the posterior cortical atrophy variant of Alzheimer’s disease. Alzheimers Dement 2016; 12(8): 862–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zou K, Abdullah M, Michikawa M. Current Biomarkers for Alzheimer’s Disease: From CSF to Blood. J Pers Med 2020; 10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitwell JL, Jack CR Jr., Kantarci K, et al. Imaging correlates of posterior cortical atrophy. Neurobiol Aging 2007; 28(7): 1051–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mendez MF, Ghajarania M, Perryman KM. Posterior cortical atrophy: clinical characteristics and differences compared to Alzheimer’s disease. Dement Geriatr Cogn Disord 2002; 14(1): 33–40. [DOI] [PubMed] [Google Scholar]
- 9.Crean S, Ward A, Mercaldi CJ, et al. Apolipoprotein E epsilon4 prevalence in Alzheimer’s disease patients varies across global populations: a systematic literature review and meta-analysis. Dement Geriatr Cogn Disord 2011; 31(1): 20–30. [DOI] [PubMed] [Google Scholar]
- 10.Petersen RC, Aisen PS, Beckett LA, et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 2010; 74(3): 201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Townley RA, Graff-Radford J, Mantyh WG, et al. Progressive dysexecutive syndrome due to Alzheimer’s disease: a description of 55 cases and comparison to other phenotypes. Brain Commun 2020; 2(1): fcaa068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergeron D, Gorno-Tempini ML, Rabinovici GD, et al. Prevalence of amyloid-beta pathology in distinct variants of primary progressive aphasia. Ann Neurol 2018; 84(5): 729–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ossenkoppele R, Singleton EH, Groot C, et al. Research Criteria for the Behavioral Variant of Alzheimer Disease: A Systematic Review and Meta-analysis. JAMA Neurol 2022; 79(1): 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller ZA, Rosenberg L, Santos-Santos MA, et al. Prevalence of Mathematical and Visuospatial Learning Disabilities in Patients With Posterior Cortical Atrophy. JAMA Neurol 2018; 75(6): 728–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dirks E, Spyer G, van Lieshout EC, de Sonneville L. Prevalence of combined reading and arithmetic disabilities. J Learn Disabil 2008; 41(5): 460–73. [DOI] [PubMed] [Google Scholar]
- 16.Suarez-Gonzalez A, Lehmann M, Shakespeare TJ, et al. Effect of age at onset on cortical thickness and cognition in posterior cortical atrophy. Neurobiol Aging 2016; 44: 108–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schott JM, Crutch SJ. Posterior Cortical Atrophy. Continuum (Minneap Minn) 2019; 25(1): 52–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Fox NC. Posterior cortical atrophy. Lancet Neurol 2012; 11(2): 170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graff-Radford J, Yong KXX, Apostolova LG, et al. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol 2021; 20(3): 222–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J Neuropathol Exp Neurol 2012; 71(4): 266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grothe MJ, Moscoso A, Ashton NJ, et al. Associations of Fully Automated CSF and Novel Plasma Biomarkers With Alzheimer Disease Neuropathology at Autopsy. Neurology 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seeburger JL, Holder DJ, Combrinck M, et al. Cerebrospinal fluid biomarkers distinguish postmortem-confirmed Alzheimer’s disease from other dementias and healthy controls in the OPTIMA cohort. J Alzheimers Dis 2015; 44(2): 525–39. [DOI] [PubMed] [Google Scholar]
- 23.Mattsson-Carlgren N, Grinberg LT, Boxer A, et al. Cerebrospinal Fluid Biomarkers in Autopsy-Confirmed Alzheimer Disease and Frontotemporal Lobar Degeneration. Neurology 2022; 98(11): e1137–e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karanth S, Nelson PT, Katsumata Y, et al. Prevalence and Clinical Phenotype of Quadruple Misfolded Proteins in Older Adults. JAMA Neurol 2020; 77(10): 1299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson JL, Lee EB, Xie SX, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018; 141(7): 2181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spina S, La Joie R, Petersen C, et al. Comorbid neuropathological diagnoses in early versus late-onset Alzheimer’s disease. Brain 2021; 144(7): 2186–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Apostolova LG, Aisen P, Eloyan A, et al. The Longitudinal Early-onset Alzheimer’s Disease Study (LEADS): Framework and methodology. Alzheimers Dement 2021; 17(12): 2043–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmqvist S, Zetterberg H, Mattsson N, et al. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology 2015; 85(14): 1240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Applications for data sharing by qualified investigators can be made to the UCSF Alzheimer’s Disease Research Center (https://memory.ucsf.edu/research-trials/professional/open-science). Data sharing requests will be subject to the limitations specified in data transfer agreements between UCSF and the individual sites providing data for this single-subject meta-analysis.