

Abstract
Background
Percocypris pingi (Tchang) was classified as Endangered on the Red List of China′s Vertebrates in 2015 and is widely distributed in the Upper Yangtze River. Although breeding and release into wild habitats have been performed for this commercially important fish in recent years, low genetic diversity has been found in wild populations. Genomic resources are strongly recommended before formulating and carrying out conservation strategies for P. pingi. Thus, there is an urgent need to conserve germplasm resources and improve the population diversity of P. pingi. To date, the whole genome of P. pingi has not been reported.
Results
In our study, we constructed the first chromosome-level genome of P. pingi by high-throughput chromosome conformation capture (Hi-C) technology and PacBio long-read sequencing. The assembled genome was 1.7 Gb in size, with an N50 of 17,692 bp and a GC content from circular consensus sequencing of 37.67%. The Hi-C results again demonstrated that P. pingi was tetraploid (n = 98), with the genome consisting of 24-type and 25-type chromosomes. Chr.19 of the 24-type chromosomes in P. pingi resulted from the fusion of chr.19 and chr.22 in zebrafish. The divergence times between 24-type and 25-type chromosomes was around 6.1 million years ago. A total of 25,198 and 25,291 protein-coding genes were obtained from the 24-type and 25-type chromosomes, respectively. The ploidy of P. pingi is an allotetraploid. A total of 8,741 genes of P. pingi were clustered into 4,378 gene families that were shared with 14 other species, and the P. pingi genome had 68 unique gene families. Phylogenetic analyses indicated that P. pingi was most closely related to Schizothorax oconnori, and the genes were clustered on one branch. We identified 166 significantly expanded gene families and 173 significantly contracted gene families in P. pingi. The most enriched positive protein-coding genes, such as Bmp-4, Etfdh, homeobox protein HB9, and ATG3, were screened.
Conclusion
Our study provides a high-quality chromosome-anchored reference genome for P. pingi and provides sufficient information on the chromosomes, which will lead to valuable resources for genetic, genomic, and biological studies of P. pingi and for improving the genetic diversity, population size, and scientific conservation of endangered fish and other key cyprinid species in aquaculture.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-024-11100-9.
Keywords: Genome, Karyotype of chromosomes, Gene family, Endangered fish, Percocypris pingi
Background
Percocypris (Cyprinidae; Cypriniformes) is a genus of large, ferocious freshwater fish with a unique geographical pattern. Only one species or subspecies of Percocypris is distributed in each major water system in the southern part of the Qinghai‒Tibet Plateau (QTP), which provides an ideal model for verifying the hypothesis of ancient water system morphology and its impact on biological species on the southeastern margin of the QTP. The analysis of Rag2 (recombinant activation gene 2) [1, 2], S6K1 (ribosomal protein S6 kinase 1) [3], 16 S (16 S ribosomal small subunit genes) [1, 4], COI (cytochrome oxidase I) [1], Cyt b (cytochrome b) [1], and the complete mitogenome sequence [5] indicated that Percocypris is the sister group of Schizothorax. Based on three mitochondrial genes (16 S; COI; Cyt b) and one nuclear marker (Rag2), the species of Percocypris inhabiting five river drainages and one lake basin were reassigned to six species [1]. Three subspecies belonging to three species of Percocypris with the scientific names P. pingi, P. regani and P. tchangi are now widely accepted [6]. The genomes of aquatic organisms contain much genetic information, such as genome size, evolutionary information, and important functional genes. The genes or markers associated with economic traits can be screened from the genome and can be used to breed new varieties with excellent traits, ultimately improving the economic and ecological benefits of the aquaculture industry [7, 8]. There are several research hotspots for the fish genome, for example, the mechanisms of sex determination [9], phylogenetics and metamorphosis [10, 11], the immune system [12, 13], and adaptive evolution [14, 15]. Thus, whole-genome determination is helpful for studying the genetic evolution of Percocypris.
P. pingi (Tchang) is located in the Upper Yangtze River [1] and was classified as Endangered (EN) on the Red List of China’s Vertebrates in May 2015 [16]. P. pingi usually releases several thousands eggs in its lotic habitat once each spring [17]. Because of its large size and high nutritional value, it is a commercially important fish. Previous studies of P. pingi focused more on artificial propagation, anatomy, morphology, nutrition, and molecular genetic issues [18–22]. In 2011, on the basis of D-Loop sequences, the population genetic resources of P. pingi were deemed limited, and the population genetic structure was found to be simple in the Mianning section of the Yalong River [23]. In 2015, moderate to high levels of differentiations were detected between hatchery and wild populations and between hatchery populations using microsatellites and mtDNA markers [22]. P. pingi, a near-threatened cyprinid species, has suffered a dramatic decline due to anthropogenic factors [22]. Multiple factors, such as habitat deterioration, water pollution, hydropower station operation, overexploitation, and deterioration of conservation status, have dramatically decreased the population sizes and distribution ranges of P. pingi in recent years [24–26]. To date, the method of breeding and releasing has been used to increase the genetic diversity and population size of P. pingi, but these methods are not effective in wild populations [22]. Genome-wide analysis of genetic diversity, population structure, and functional genes is strongly recommended before formulating and carrying out conservation strategies for P. pingi.
In our study, we used high-resolution chromosome conformation capture (Hi-C) and PacBio long-read sequencing technologies to construct the first chromosome-level genome of P. pingi. We subsequently conducted comparative analyses with 14 published bony fish genomes, with a focus on gene families, hierarchical clustering of these families, and positive selection analyses of different groups. These results could be helpful for elucidating the molecular mechanisms underlying important economic traits in P. pingi, such as growth, high nutrition, disease resistance, and stress resistance, as well as for developing measures for its improvement and protection.
Results
Genome assembly
In this study, the whole genome of P. pingi was successfully obtained via Hi-C technology and PacBio long-read sequencing. Before the official launch of whole-genome sequencing, we conducted a genome survey and obtained a total of 203.20 Gb of raw data. The chromosomes of P. pingi showed tetraploidy, and the heterozygosity was 0.53%. In 3,836,199 of the circular consensus sequencing (CCS) reads, the N50 was 17,692 bp, and the GC content was 37.67%. The whole-genome sequences were divided into 24-type and 25-type chromosomes, for which the accuracy was 98.73% and 98.74%, respectively. After genome assembly by Hifiasm [27], the total length was 3,465,547,224 bp, the biggest sequence length was 36,547,708 bp, the N50 was 1,809,540 bp, and the GC content was 37.81% (Table 1). The 24- and 25-type chromosomes of P. pingi were determined by Hi-C (Fig. 1A and B), and the size of the genome of P. pingi was 1.7 Gb (Fig. 1C). The CCS reads were 1000–3000 bp (Fig. 1D).
Table 1.
Summary of the genome assembly of Percocypris pingi
| Genome assembly | N50 (size /number) | N90 (size /number) | Total length | |||
|---|---|---|---|---|---|---|
| Contigs | 180.9 kb /458 | 18.0 kb /2,569 | 3.5 Gb | |||
| Chromosomes | 49 chromosomes | 1.7 Gb | ||||
| TEs | Total length | Percent of genome (%) | ||||
| 24-type | 25-type | 24-type | 25-type | |||
| Total | 531.4 Mb | 520.6 Mb | 52.58 | 51.13 | ||
| DNA transposons | 300.3 Mb | 285.7 Mb | 29.71 | 28.06 | ||
| Retroelements | 204.1 Mb | 177.5 Mb | 20.2 | 17.45 | ||
| Satellites | 23.9 Mb | 22.0 Mb | 2.36 | 2.16 | ||
| Noncoding RNAs | Copies | Length | ||||
| 24-type | 25-type | 24-type | 25-type | |||
| rRNAs | 5,293 | 5,303 | 964.7 kb | 965.8 kb | ||
| miRNAs | 2,586 | 2,585 | 231.1 kb | 231.1 kb | ||
| tRNAs | 8,265 | 8,659 | 625.9 kb | 654.9 kb | ||
| Protein-coding genes | Total number | Annotated | Unannotated | |||
| 24-type 25-type | 25-type | 25-type | 24-type | 25-type | ||
| 25,198 25,291 | 24,913 | 24,999 | 285 | 292 | ||
Fig. 1.

Genome assembly and haplotype analysis of Percocypris pingi. A, Hi-C results for 24-type chromosomes. B, Hi-C results for 25-type chromosomes. C, statistics of genome annotation of P. pingi. From outside to inside, the results correspond to a, information on the genome; b, GC content; c, depth of second-generation high-throughput genome sequencing; d, depth of third-generation high-throughput genome sequencing; e, homozygous SNP distribution (outer ring), and heterozygous SNP distribution (inner ring); f, homozygous InDel distribution (outer ring) and heterozygous InDel distribution (inner ring); and g, complete comparison of the distribution of benchmarking universal single-copy orthologs (BUSCO) genes in the genome, where blue indicates single-copy BUSCOs and red indicates duplicated BUSCOs. D, circular consensus sequencing (CCS) read length distribution. E, K-mer depth of P. pingi genome and K-mer species frequency distribution. F, smudgeplots K-mer statistical distribution. G, spectral karyotyping of P. pingi chromosomes. H, comparative analysis of P. pingi and Danio rerio. Hi-C, high-throughput chromosome conformation capture; GC, genomic content unit; InDel, insertion‒deletion; SNP, single nucleotide polymorphism
Based on K-mer depth and K-mer species frequency distribution (Fig. 1E), there were three peaks on the curve, suggesting that P. pingi genome is polyploid. According to the Smudgeplot analysis and K-mer = 17, the K-mer proportions of AAAB, AABB, and AAAABBB were 0.49, 0.39, and 0.12 (Fig. 1F), respectively. Thus, the heterozygous genotype ratio of AAAB > AABB, indicating that the ploidy of P. pingi is an allotetraploid. Then, spectral karyotyping verified that the chromosome number of P. pingi is n = 98 (Fig. 1G). Finally, specifically, based on the genome data of zebrafish, chromosomal collinearity analysis revealed that chr.19 of the 24-type chromosomes resulted from the fusion of chr.19 and chr.22 in zebrafish (Fig. 1H). The 25-type chromosomes were completely mapped to the genome of zebrafish. The Ks values showed that, except for those of chr.11 and chr.19_22, the Ks values of the chromosomes were close to zero, which suggested that the differences among those chromosomes were very small. In addition, the Ks value of chr.19_20 was greater than that of chr.11.
To assess the accuracy of the genomic data obtained, the genomic data of P. pingi were compared with those of other species in the NT database (Table S2). The genome data of Cyprinus, Ctenopharyngodon, Sinocyclocheilus, Danio, and Carassius were matched. In particular, 84.77% of the genome data of P. pingi were from the Cyprinus genome, whereas less than 5% of the data were from other genera. When the second- and third-generation high-throughput genome sequencing data were compared to the assembled whole-genome data (Table S3 and Table S4), the comparison rates were 99.77% and 100%, and the coverages were 99.15% and 99.84%, respectively. The homozygous and heterozygous SNP rates were 0.000 and 0.001%, respectively, and both the homozygous and heterozygous InDel rates were 0.002%. Furthermore, 3,640 proteins (100%) were identified in the OrthoDB database by BUSCO assessment of genome integrity (Table S5). The above results indicate that the genome assembly of P. pingi was successful.
Genome annotation
The 24- and 25-type chromosomes were annotated. The 24- and 25-type chromosomes contained 52.28% and 51.15% transposable elements (TEs), respectively, including 29.71% and 28.06% DNA transposons, 10.19% and 8.14% LTRs, 9.51% and 8.82% LINEs, 0.5% and 0.495% SINEs, and 2.36% and 2.16% satellites, respectively (Fig. 2A, Table S6). The genome of P. pingi was subsequently matched to those of other fish species in the NR database for homology analysis, and the two sets of chromosomes were compared with protein-coding genes from Onychostoma macrolepis, Cyprinus carpio, Labeo rohita, Sinocyclocheilus graham, Carassium auratus and Danio rerio. After removing redundancies, a total of 25,198 (Table S7) and 25,291 (Table S7) protein-coding genes were obtained from the 24-type and 25-type chromosomes, respectively. The average gene length and CDS length of the protein-coding genes were similar for 24-type (19,671 bp and 1,677 bp, respectively) and 25-type (19,649 bp and 1,676 bp, respectively) chromosomes (Fig. 2B). Furthermore, based on the InterPro, GO, KEGG, SwissProt, TrEMBL, TF, Pfam, NR and KOG databases, functional annotations were obtained for 24,913 (98.87% of total) and 24,999 (98.85% of total) of the 24-type and 25-type chromosomes, respectively. Annotations of noncoding RNAs (Table S8), such as miRNAs (2586 and 2585 copies), tRNAs (8265 and 8659 copies), rRNAs (5293 and 5303 copies), and 5 S (4898 and 4908 copies), were obtained from the 24-type and 25-type chromosomes, respectively.
Fig. 2.

Genome annotations of Percocypris pingi. A, percentage of DNA transposons, LTRs, LINEs, and SINEs in the genome; B, length of genes, CDS, exons and introns in protein-coding genes; C, distribution of DNA transposons on chromosomes; D, distribution of miRNAs on chromosomes; E, distribution of tRNA transposons on chromosomes. LTRs, long interspersed elements; LINEs, long interspersed elements; SINEs, short interspersed elements; CDS, coding sequence; chr., chromosome; bp, base pair
The repeat sequences and noncoding RNAs had different chromosomal distributions. DNA transposons (Fig. 2C) and long terminal repeats (LTRs) (Figure S1) were widely distributed on the chromosomes. Long interspersed elements (LINEs) were mainly located on chr.20 (1–5 Mb) and chr.25 (25–34 Mb) (Figure S2). miRNAs and rRNAs were identified mainly on chr.10 (Fig. 2D, 1-5 Mb) and chr.20 (Figure S3, 1–5 Mb), respectively. Both the snRNAs (Figure S4, 1–8 Mb) and tRNAs (Figs. 2E, 1–17 Mb) were distributed mainly on chr.20. Furthermore, tRNAs were also identified primarily on chr.11 (1–15 Mb).
Fig. 5.

Gene family expansion and contraction analysis of the Percocypris pingi genome. A, the number of gene families showing expansion and contraction; B, enrichment of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for the expanded gene families; C, enrichment of KEGG pathways for the contracted gene families
Gene family analysis and construction of a phylogenetic tree
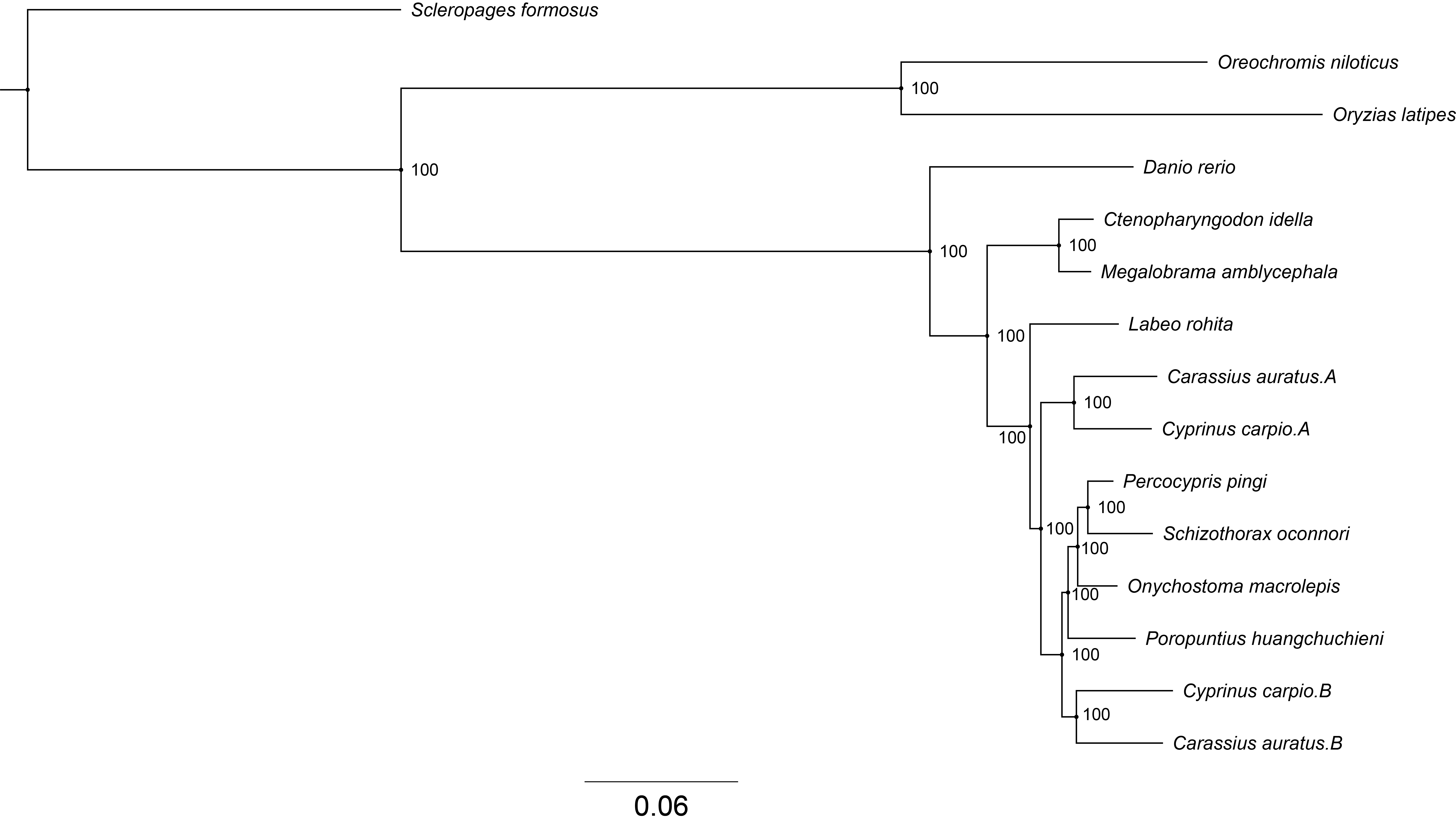
The 25,291 protein-coding genes of P. pingi were compared to the genes of the other 14 species via gene family analysis. In total, 8,741 genes from P. pingi were clustered into 4,378 gene families that were shared with 14 other species (Fig. 3A, Table S9). There were more unique families (68) in P. pingi than in C. auratus. A (12), C. auratus. B (31), C. idella (28), C. carpio A (7), C. carpio B (6), and L. rohita (54) (Fig. 3B). A total of 1,166 protein-coding genes were clustered into 68 unique families in P. pingi, and 214 of these genes were enriched in GO (Dataset S1) and KEGG (Dataset S2) functions. The common 8,795 gene families were identified among P. pingi, P. huangchuchieni, S. oconnori, and S. formosus (Fig. 3C). A total of 681 GO terms were enriched, including 401 related to biological processes, 62 related to cellular components, and 214 related to molecular functions. The most enriched GO terms included prolactin signaling pathway, prolactin receptor pathway, cellular response to cytokine stimulus, etc. Furthermore, 151 KEGG pathways were obtained, and the most enriched pathways were immune system (31 genes, 20 pathways; Fig. 3D) and signal transduction (38 genes, 23 pathways; Fig. 3E). In addition, 873 single-copy families were utilized to construct a maximum likelihood (ML) phylogenetic tree with a bootstrap support value of 100% for each clade (Fig. 3F). P. pingi and S. oconnori were clustered on one branch.
Fig. 3.

Gene family analysis of the Percocypris pingi genome and construction of a phylogenetic tree. A, orthology analysis of gene families in the P. pingi genome; B, common and unique gene families in 15 fish species; C, common and unique gene families in P. pingi, Poropuntius huangchuchieni, Schizothorax oconnori, and Scleropages formosus; D, Gene Ontology (GO) enrichment of 68 unique gene families in P. pingi. E, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment of 68 unique gene families in P. pingi. F, ML phylogenetic tree for 873 single-copy families in P. pingi. The genome information of 15 fish species were presented in Table S1
The estimation of divergence time
The divergence times of 12 fish species were calculated (Fig. 4). Three time-calibration points were established: (1) C. auratus.A and C. auratus.B (21.5–17.5 million years ago [Mya]), (2) D. rerio and C. idella (48.6–40.4 Mya), and (3) C. carpio. A and C. carpio.B (19.5-13.75 Mya). P. pingi was most closely related to S. oconnori, with a divergence time of approximately 7.9 million years ago (6.7–9.2 mya), and they were clustered on one branch. All the fish species in Cypriniformes were clustered in one large group and diverged approximately 31.6 million years ago (27.2–35.8 mya). Megalobrama amblycephala and Ctenopharyngodon idella were clustered in another group, and their divergence time was approximately 9.2 million years ago (4.8–15.6 mya). D. rerio and other species diverged approximately 44.3 million years ago (40.0–49.0 mya). In addition, the divergence times between 24-type and 25-type chromosomes was 6.1 million years ago (4.8–7.4 mya).
Fig. 4.

The divergence times among 12 fish species. The genome information of 12 fish species were presented in Table S1
Gene family expansion and contraction
Gene family expansion and contraction analysis of the P. pingi genome revealed that 376 gene families were expanded and 535 gene families were contracted, which were smaller than the corresponding numbers in S. oconnori (1,671 expanded gene families and 3,960 extracted gene families; Fig. 5A). In our study, 166 significantly expanded gene families (including 700 genes) and 173 significantly contracted gene families (including 109 genes) were identified (p<0.05). A total of 161 nonredundant genes from significantly expanded gene families in the P. pingi genome were highly enriched in 22 KEGG pathways (Fig. 5B, Dataset S3, p<0.05), 13 of which were related to immune disease and system (39 genes) and human diseases (88 genes). The AMPK signaling pathway (28 genes) was the most enriched among the significantly expanded gene families. Twenty-five nonredundant genes from significantly contracted gene families were enriched in 16 KEGG pathways (Fig. 5C, Dataset S3, p<0.05), 5 of which were involved in immune disease and system (12 genes) and 4 of which were enriched in lipid metabolism (5 genes). In addition, 10 genes in the significantly contracted gene families were enriched in apoptosis pathways.
The GO terms associated with the significantly expanded and contracted gene families of the P. pingi genome were analyzed. A total of 302 (in expanded gene families) and 39 (in contracted gene families) nonredundant genes were significantly enriched in 48 and 12 GO terms, respectively (p<0.05). Based on adjusted p values, ‘neurotransmitter: sodium symporter activity’, ‘neurotransmitter transport’, and ‘nucleosome’ were the most enriched GO terms for the significantly expanded gene families. Several immunity-related GO terms were also screened, such as ‘MHC class II protein complex’ (7 genes), ‘hemoglobin complex’ (6 genes), and ‘antigen processing and presentation’ (7 genes). Among the contracted gene families, the most enriched GO terms were ‘calcium-dependent cysteine-type endopeptidase activity’ (12 genes), ‘proteolysis’ (20 genes), and ‘intracellular anatomical structure’ (12 genes).
Positively selected genes
To study the positively selected protein-coding genes in the P. pingi genome, the sequences of genes belonging to single-copy gene families were utilized. In this study, 72 positively selected genes (p < 0.05) were identified. The number of positively selected sites was greatest for Ppi_9G0002070.1 (unknown protein, 231 sites), Ppi_18G0007580.1 [bone morphogenetic protein 4 (BMP-4), 39 sites], and Ppi_14G0004220.1 [electron-transferring-flavoprotein dehydrogenase (ETFDH), 103 sites]. Then, 24 positively selected genes (p < 0.05) were clustered into 40 KEGG pathways. Nine positively selected protein-coding genes were enriched in environmental information processing pathways (Fig. 6A), such as the Notch signaling pathway, MAPK signaling pathway, TGF-beta signaling pathway, and FoxO signaling pathway. Eight and seven positively selected protein-coding genes were enriched in human diseases (Fig. 6B) and cellular processes (Fig. 6C), respectively. In those enriched pathways, several important protein-coding genes were identified, such as Ppi_18G0007580.1 (BMP-4), Ppi_16G0004360.1 (SHC-transforming protein 1, SHC1), Ppi_1G0001150.1 (homeobox protein HB9), and Ppi_6G0001490.1 (ubiquitin-like-conjugating enzyme ATG3).
Fig. 6.

Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways associated with positively selected genes in Percocypris pingi genome. A, enrichment of environmental information processing; B, enrichment of human diseases; C, enrichment of cellular processes
Discussion
Most teleosts have undergone the teleost-specific genome duplication (TSGD) and harbor 24 to 25 chromosomes in their haploid genomes [28]. At present, it is generally believed that the basic chromosome number of diploid fish in the Cyprinidae family is 50 or 48, and 2n = 50 in diploid is the most primitive type of karyotype in Cyprinidae fish [29]. Percocypris lived in high-altitude regions, and their chromosomes evolved from 2n = 50 → 100 doubling [30]. Later, in the process of evolution or life, the chromosome functional mechanism tended to be complete or other genetic factors led to chromosome evolution from 2n = 100 → 98 due to Robertsonian translocation or centromere fusion [30]. In this study, we characterized a high-quality chromosome-anchored reference genome of P. pingi that consisted of 24- and 25-type chromosomes (n = 98). This finding is consistent with Yang et al. (2017) [31] and Zan et al. (1984) [32] that the number of chromosomes in the P. pingi genome was 98. The karyotypes of Teleostei species are different, and some of them are still controversial. Previous studies showed that the karyotype of the half-smooth tongue sole (C. semilaevis) genome [9] is n = 22, that of the Atlantic salmon genome [33] is n = 29, that of the C. carpio genome [28] is 2n = 100, that of the zebrafish genome [34] is 2n = 50, and that of the grass carp genome [35] is 2n = 48. In our study, P. pingi and S. oconnori were clustered in a lineage and closely related. The karyotype of S. oconnori has been reported as 2n = 92 or 2n = 106 [36]. Specifically, similar chromosome karyotypes were observed in grass carp, in which chromosome fusion occurred relative to zebrafish, and frequent crossovers were reported between the grass carp X and Y chromosomes [35]. Thus, the genome of P. pingi may have undergone fusion during the process of chromosome replication over the course of its long history of evolution, and as a result, this species may have strong adaptability to region-specific habitats.
TEs constitute a substantial proportion of the total genomic content and are biochemically active noncoding elements, such as noncoding RNAs and transcription factor-binding sites [37]. TEs have become a nearly ubiquitous feature of eukaryotic genomes and play important roles in host biology [38]. A previous study reported that small genomes were poor in TEs [9] and that the number of TEs in fish species was relatively large. In our study, the number of TEs in the P. pingi genome (genome size 1.7 Gb), observed 52.28% TEs in 24-type chromosomes and 51.15% TEs in 25-type chromosomes, was similar to that in the zebrafish (genome size 1.412 Gb, TEs 52.2%) [34], S. oconnori (1.94 Gb, TEs 56.65%) [39], and Atlantic salmon (2.97 Gb, TEs 58-60%) [33] genomes but greater than that in the C. carpio (1.69 Gb, 31.3% TEs) [28] and Oryzias latipes (700 Mb, TEs 30.68%) genomes [40]. In contrast, species presenting fewer TEs in their genomes include Gasterosteus aculeatus (genome size 463.3 Mb, TEs 13.48%) [14], Paralichthys olivaceus (535 Mb, TEs 10.37%) [11], Cynoglossus semilaevis (477 Mb, TEs 5.85%) [9], Fugu rubripes (365 Mb, TEs 2.7%) [41], Takifugu rubripes (322.5 Mb, TEs 7.1%) [41], and Tetraodon nigroviridis (340 Mb, 5.7%) [42]. The proportion difference of TEs between the grass carp and zebrafish genomes might be due to the exclusion of repetitive sequences located within unclosed gaps or within small fragments (< 200 bp) [35]. In addition, TEs frequently have tissue-specific functions [43]. Therefore, the above results suggest that more than 50% of the TEs in the P. pingi genome contribute raw sequences and can give rise to structures that shape chromatin organization [44].
Modern fishes (Teleostei) underwent an additional round of whole-genome duplication (WGD) [45], which is proposed to have facilitated teleost diversification and species radiation [9]. The ancestral genome of all teleost fish underwent a teleost-specific 3rd WGD [46, 47] (Ts3R, dated 225 to 333 Mya [48–50]). Previous studies have shown that this Ts3R is still present in modern teleost genomes [34, 40, 46]. As the sister species of P. pingi [1–4], S. oconnori is a QTP-endemic fish and a young tetraploid with a recent WGD event more recent than 1.23 Mya, and its 4R WGD occurred even later [39]. In addition, with the uplift of the QTP, the primitive suborder of barbfish gradually evolved into groups adapted to harsh environments such as those with cold, high altitudes and rapids [51]. According to its current evolutionary history and classification, P. pingi is a Barbinae fish. In this study, P. pingi and S. oconnori were found to have separated approximately 6.7–9.2 Mya, which indicates that before the last WSD event, they diverged into separate species. Then, the two haploid chromosome sets have a specific divergence time in fish, such as C. carpio [52]. The two diploid progenitors of C. carpio diverged around 23 Mya and merged around 12.4 Mya, and may have derived from a diploid Barbinae species [52]. In this study, 24-type and 25-type chromosomes of P. pingi diverged about 6.1 mya. Specially, the divergence time of two haploid chromosome sets in P. pingi was later than the separate time from S. oconnori. Furthermore, C. carpio [28] and C. auratus [53] have been determined to be typical allotetraploids. The polyploidization of S. oconnori was probably achieved by autopolyploidization [39]. Here, we have demonstrated that the polyploidization of P. pingi was achieved via AAAB type based on k-mers = 17. Furthermore, the Ks values for one-to-one chromosome comparisons indicate that nearly no differences exist between most of the 24- and 25-type chromosomes of P. pingi. These results suggest that P. pingi is autotetraploid .
Preferential retention and amplification of several gene families occurred over successive WGD events [34, 54, 55]. In this study, 376 expanded gene families and 535 contracted gene families were detected in the P. pingi genome, which were smaller than the numbers in the S. oconnori genome (1671 expanded gene families and 3,960 extracted gene families) [39]. Compared with a control set of fishes from low altitudes, the Tibetan fishes presented genome-wide accelerated evolution [56]. The S. oconnori genome experienced the latest, fourth round of whole-genome replication events [39, 51] and exhibited a high degree of chromosome synteny with D. rerio without obvious rearrangement. Thus, the genome of S. oconnori might have experienced accelerated evolution and accumulated more expanded and extracted gene families than that of P. pingi. In the functional enrichment analysis of the expanded gene families, several KEGG pathways (for example, Inflammatory bowel disease, Graft-versus-host disease, Asthma, and Antigen processing and presentation) and GO terms (such as MHC class II protein complex, hemoglobin complex and antigen processing and presentation) were related to immune disease and system and human diseases. In general, the number of expanded genes in the enriched immunity-related pathway was much greater than the number of contracted genes in this study. Multiple important immune-related genes, including histone H4, H2B, H3, major histocompatibility complex (class II), RAR-related orphan receptor alpha (ROR), and heat shock 70 kDa protein 1/8, were identified in this study. The expansion of core histone genes was observed in the genome of S. oconnori [39]. Gene families involved in the innate immune system have also been identified as expanded in other fish species, such as mudskippers [12], large yellow croaker [57], and Miichthys miiuy [13]. P. pingi may be a good model for studying the mechanism underlying immunity in fish. P. pingi can limit the trophont settlement and/or development of Ichthyophthirius multiffliis, which has almost no mortality and higher survival rates than southern catfish (Silurus meridionalis), grass carp, S. prenanti, and Culter alburnus [58]. Furthermore, hybridization with male P. pingi improved the cultivation characteristics of S. wangchiachii, such as a high crude protein content, and higher temperature tolerance and resistance to white spot disease (I. multiffliis), providing a new perspective and foundation for snow trout breeding [59]. The above results showed that the expanded gene families involved in immune pathways played a very important role in the evolution or life processes of P. pingi.
In this study, most of the positively selected genes were enriched in environmental information processing, human diseases, and cellular processes. Several important genes, such as Bmp-4, Etfdh, homeobox protein HB9, and ATG3, have been identified. BMP4 is a member of the BMP family that belongs to the transforming growth factor β (TGF-β) superfamily. BMPs are involved in the inhibition of cell growth, proliferation and the promotion of cell differentiation and apoptosis [60–63]. In zebrafish, Bmp4 has been reported to function in the regulation of immune responses, as Bmp4 can induce the production of type I IFNs in response to viral infection by promoting the phosphorylation of Tbk1 and Irf3 through the p38 MAPK pathway [61]. However, there have been few reports on the function of the Etfdh gene. A previous study revealed compound heterozygous mutations in the Etfdh gene in a case of late-onset riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency [64]. Thus, the function of the Etfdh gene may also be related to the immunity of P. pingi. Furthermore, the homeobox protein HB9 is essential for pancreatic and motor neuronal development [65]. In addition, ATG3-dependent autophagy has been implicated in mitochondrial homeostasis regulation in both pluripotency acquisition and maintenance and plays important roles in embryonic stem cell differentiation [66]. As a conserved degradation pathway, autophagy acts as a protective strategy for disposing of toxic cytoplasmic contents, prevents cellular damage in response to stress, and acts as a dynamic recycling system during normal development and differentiation by providing fuel for cellular renovation and homeostasis [66]. Therefore, these genes may play vital roles in the adaptation and survival of P. pingi.
Conclusions
To obtain the comprehensive, detailed, and systematic genetic research and germplasm resource evaluation, the detailed information in whole genome for P. pingi will be deeply explored in our future study. Our study provides a high-quality chromosome-anchored reference genome for P. pingi and provides sufficient information on the chromosomes, which will lead to valuable resources for genetic, genomic, and biological studies of P. pingi and for improving the genetic diversity, population size, and scientific conservation of endangered fish and other key cyprinid species in aquaculture.
Materials and methods
Sample collection
P. pingi (n = 10, body weight = 52.6 ± 0.28 g and body length = 15.35 ± 0.21 cm) were purchased from Zhou Gong He Ya Fish Co., LTD. P. pingi were cultured under natural conditions of 18.85 ± 0.02 ℃, and the water environment of the breeding system included dissolved oxygen (greater than 6 mg /L) and pH (6.8–7.5). One male and one female fish (1 year old) were selected and then anaesthetized with MS-222 (100 µg/mL, Syndel, WA, USA) for 10 min. All procedures and investigations were approved and reviewed by The Animal Research and Ethics Committees of Sichuan Agricultural University (approval no: 20220631). The muscles of 1 year old female P. pingi (the sex-determination type is XX/XY) were collected and cut into soybean-sized pieces (length, width, and height ≤ 0.5 cm) for DNA and Hi-C sequencing. Then, the muscles, skin, eyes, intestine, swim bladder, pituitary gland, brain, gonad (female and male), liver, spleen, kidney, and heart were collected from the same adult female of P. pingi and mixed to create one sample for transcriptome sequencing. All the samples were stored in liquid nitrogen until DNA and RNA extraction.
Genomic DNA was extracted from the samples using the standard phenol–chloroform extraction method for preparing the DNA sequencing library. The quality and concentration of the genomic DNA were checked via 1% agarose gel electrophoresis and a Pultton DNA/Protein Analyzer [67]. Total RNA from mixed tissues was purified using TRIzol reagent (Invitrogen). Then, RNA quality was evaluated by a 2100 Bioanalyzer (Agilent Technologies) and NanoDrop ND-1000 spectrophotometer (Labtech). The cDNA library was constructed by Thermo Scientific RevertAid First Strand cDNA Synthesis Kit (ThermoFisher, USA), and cDNA fragments were sequenced by Illumina HiSeqTM platform. Raw data was obtained after removing the adaptor and low-quality reads.
Hi-C-assisted genome assembly
Samples were treated with paraformaldehyde to fix the conformation of the DNA. After lysis of the tissues, the cross-linked DNA was treated with restriction endonuclease to produce sticky ends, which were repaired. Biotin was introduced at the same time to mark the ends of the oligonucleotides. DNA ligase linked the DNA fragments, and then the cross-linked DNA was removed by protease digestion. The DNA was purified and randomly sheared into 300–500 bp fragments. Avidin magnetic beads captured the labeled DNA for second-generation library sequencing. The DNA containing biotin was collected, and the DNA fragments were end-repaired. A was added to DNA fragment, and splicing was performed. The number of PCR amplification cycles was evaluated, and the entire library was purified and constructed. A Qubit 2.0 instrument was used for preliminary quantification, and the library was diluted to a concentration of 1 ng/µL. The insert size of the library was determined with an Agilent 2100 instrument. Once the insert size matched the expected size, q-PCR was used to quantify the effective concentration of the library accurately (effective concentration of the library > 2 nM) to ensure the quality of the library. The different libraries were subsequently pooled according to the effective concentration and volume requirements for second-generation sequencing. The original image data files obtained by the second-generation sequencing platform were analyzed by base calling and converted into sequenced reads. The results were stored in FASTQ (fq) file format. For data quality control, reads containing junction sequences were filtered out, and bases with a continuous mass less than 20 at both ends of the sequencing read were removed. When the final length of the sequencing read was less than 50 bp, the read was removed, and only paired reads were retained.
The contigs or scaffolds were segmented, anchored, sequenced, directed, and merged to obtain the chromosome-level genome. First, small scaffolds were filtered out, and the remaining scaffolds were analyzed for locus interaction frequency consistency. Scaffolds with consistent remote interaction patterns were retained, and those with inconsistent patterns were removed. Scaffolds of chromosome length were anchored, sequenced, and oriented according to the strength of interaction between a pair of interacting sequences. Then, the preliminary and credible scaffolds of chromosome length were established. Overlapping genomic regions were identified based on sequence homology and high similarity of long-distance interaction patterns; and scaffolds and contigs were combined according to overlapping regions to obtain the final chromosome length. After the assisted assembly of the genome, Juicer (version 1.5) [68] constructed an interaction map, and JuiceBox (version 1.9.8) [69] was utilized to visually correct the map. The distribution of chromosomes and whether there are interactions between chromosomes were analyzed.
Genome assembly
For the detection of qualified P. pingi DNA samples, fragments of 300–350 bp were randomly interrupted by ultrasonic fragmentation. The whole library was prepared by end repair, A-tail addition, sequencing joint addition, purification, and PCR amplification. After the library was constructed, the PacBio Sequel II platform and Illumina NovaSeq-6000 platform were used for genomic sequencing to generate long and short reads, respectively. Blastn (version 2.11.0+) [70] was used to determine whether the library was contaminated. Based on the sequencing data produced and quality control, invalid data were filtered and excluded. A total of 10,000 pairs of reads were randomly selected from the filtered high-quality data. Blast software (version 2.2.26) [71] was used to map the reads against the NCBI nucleotide database (NT library), and the top 80% of the species were displayed. The sequencing data were filtered and corrected for redundancy by Fastp (version 0.23.2) [72] for a total of 203.20 Gb. K-mer-based analysis was performed using GCE (version 1.0.0) [67] software, after which the genome size, heterozygosity rate and repeat sequence abundance were estimated before genome assembly. Smudgeplot (version 0.2.3dev) [73] was used to estimate the ploidy of P. pingi, and the number of hybrid k-mer pairs was counted by comparing the total coverage of k-mer pairs (CovA + CovB) and the relative coverage (CovB/(CovA + CovB)). By setting the haploid genome coverage as λ and assuming λ = 100, the sum of coverage and coverage ratio of the haploid genome were obtained, and the ploidy type of P. pingi was preliminarily estimated. The purity (OD260/280 and OD260/230), concentration and nucleic acid absorption peak of the genomic DNA were detected with a NanoDrop spectrophotometer; the concentration of the genomic DNA was accurately determined via a Qubit spectrophotometer. The concentration determined via Qubit was compared to the concentration determined via Nanodrop to determine the purity of the sample. The integrity of the genomic DNA was tested by electrophoresis.
For samples that passed testing, gTUBE was used to interrupt the genomic DNA, exonuclease VII was utilized for digestion, and the 3’ single-strand ends were removed. A damage repair kit was used to repair single-strand breaks or base deletions via oxidation; DNA was repaired to flat ends by terminal repair. SMRT dumbbell connectors were connected, after which exonuclease digestion was performed. The fragments were not connected to the SMRT dumbbell connectors that were removed. Furthermore, 0.45X PB magnetic beads were used for secondary screening and purification, and sequencing libraries were obtained. After library construction was completed, Qubit 3.0 and Agilent 2100 instruments were used for accurate quantification and size detection of the library, respectively. Only after the size of the library matched the expected size was computer sequencing carried out. The qualified library was sequenced using the PacBio Sequel II system according to the target data volume. Hifiasm (version 0.16.1-r375) [27] software was used to assemble the genome of P. pingi. The genome sequence was compared with the NT database, second- and third-generation data and transcriptome data, and the overall genome assembly quality was evaluated by using BUSCO (version 5.3.2) [74] and other methods and metrics, including basic information statistics, data correctness evaluation, sequence consistency evaluation, assembly integrity evaluation, accuracy evaluation and heterozygosity evaluation.
Karyotype analysis of chromosomes
The pectoral fin of P. pingi was injected with phytohemagglutinin (PHA, 10 µg/µL), followed by 2 to 2.5 µL of PHA/1 g of fish. One-half of the needle was inserted backwards into the thinnest part at a 45-degree angle, and the procedure was repeated 12 h later; then, colchicine (10 µg/µL, 100 µL) was injected 12 h later. After 4 h, the branchial arch was cut off, and the fish was sacrificed. In the meantime, the head kidney (a small amount) was collected into a Petri dish containing 2 mL of NaCl solution (0.9%) and cut with scissors; then, the broken tissues were blown with a 1 mL gun, precipitated naturally for 5 min, and centrifuged at 1500 rpm/min for 8 min at 20 °C. After removal of the supernatants, the tissues were gently resuspended in 8 mL of 0.075 M KCl and stored at 37 °C for 1 h. During this process, 10 mL/sample fixative solution (methanol: glacial acetic acid = 3:1) was prepared. Two milliliters of fresh fixing solution were used to fix the tissues at room temperature for 5 min, after which the tissues were centrifuged at 4 °C and 1500 rpm for 8 min. The supernatants were subsequently removed, and the remaining samples were gently precipitated, resuspended, fixed with 2 mL of fixing solution at 4 °C for 15 min, and centrifuged at 1500 rpm for 8 min at 4 °C; these steps were repeated 2–3 times. Finally, an appropriate amount of fresh fixing liquid (approximately 1–2 mL of resuspended tissue) was taken from the − 20 °C freezer to remove frozen clean slide drops (the slide was first washed with pure ethanol, burned on an alcohol lamp, and subsequently stored at -20 °C one day in advance). The slides with sample droplets were dried at 42 °C and stored at -20 °C for later use [75]. Observations were performed by a microscope (Zeiss Axio Imager 2).
Genome annotations
The genome annotation process included four main steps: repeat sequence recognition, noncoding RNA prediction, gene structure prediction and functional annotation. Repetitive sequence annotation was combined with the homology prediction method by using the Repbase library [http://www.girinst.org/repbase, RepeatMasker (version 4.0.7) [76]; http://www.repeatmasker.org, RepeatProteinMask (version 4.1.0)] and de novo ab initio prediction methods based on self-sequence alignment [RepeatModeler (version 2.0) [77], Piler, RepeatScount] and repeated sequence features (software: Trf (version 4.09) [78] and LTR-FINDER (version 1.0.2) [79]). Coding gene structure was predicted via homologous prediction (at least 2 or 3 near-source species were selected) and de novo ab initio prediction [software: Augustus (version 2.7) [80], Genscan (version 1.0) [81], GlimmerHMM (version 3.0.4) [82], and cDNA/EST prediction]. Moreover, RNA-seq data were obtained via TopHat [83] comparison and Cufflinks [84] assembly of transcripts. With the assistance of MAKER (version 2.31.10) [85] software, the predicted gene sets were subsequently integrated into a nonredundant and more complete gene set. The final reliable gene set was obtained through the integration of CEGMA results and the HiCESAP process. Finally, the proteins in the gene set were annotated by several protein databases (SwissProt, TrEMBL, KEGG, InterPro and GO). In the annotation process for noncoding RNA, tRNAscan-SE (version 1.3.1) [86] was used to identify tRNA sequences in the genome according to structural characteristics. Because the highly conserved rRNA sequences of related species were selected as reference sequences, the rRNA in the genome was searched by BLASTN comparison.
In addition, the covariance model of the Rfam 12.0 [87] family was used to predict the sequence information of miRNAs and snRNAs in the genome using INFERNAL (version 1.1) [88]. The gene annotations included the structures and functions. First, the position and structure of genes were predicted by various methods (homology-based prediction, de novo prediction and transcriptome-based prediction), and functional annotations were subsequently performed to determine the biological function of the products and metabolic pathways involved. BUSCO (version 5.3.2) [89] software was applied to construct single-copy gene sets for several large evolutionary branches from the OrthoDB database. The gene set splicing results were compared with the gene set, the accuracy and completeness of the splicing data were evaluated according to the proportion and completeness of the comparison, and assembly annotation was performed.
Phylogenetic analysis and divergence time evaluation
First, gene family identification was performed, and gene sets of the species involved in the analysis were screened. If there were multiple transcripts of a gene (variable splicing), only the transcript with the longest coding region was retained. Moreover, coding genes for proteins with fewer than 30 amino acids or genes with internal stop codons were filtered out. All-vs.-all BLASTP (version 2.11.0+) (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used for all species (Table S1) to assess protein sequence similarity with an e value of 1e-5. OrthoMCL (version 2.0.9) [90] was utilized to cluster the above results, and the expansion coefficient was 1.5. The genes in single-copy gene families were selected. The protein sequences for genes in each single-copy gene family were compared via MAFFT (version 7.487) [91] software and subsequently reversed for CDS multiple sequence comparison. By combining the results for all the single-copy genes, a superalignment matrix was constructed in three ways, including directly connecting the comparison results of all the single-copy genes, extracting conserved sequences by connecting the comparison results of all the single-copy genes using Gblocks (version 0.91b) [92], and identifying conserved sequences in the comparison results of each single-copy gene by Gblocks (version 0.91b) [92]. These conserved sequences were subsequently combined. As a result, based on the above three methods, three sets of data were obtained for all sites not processed by the superalignment matrix, and the phase 1 sites and 4D sites (quadruple degeneracy sites) were obtained. For the above 9 sets of data, RAxML (version 8.2.12) [93] was used to construct phylogenetic trees of species using the maximum likelihood (ML) method. The comparative genomic analysis of the species is detailed in Table S1.
Based on the gene sequences of the single-copy gene families, the differentiation time was estimated and phylogenetic tree was constructed with MCMCTree (version 4.9) [94] in PAML (version 4.9) (--burnin 1,000,000, --sampfreq 10, --nsample 500,000) [95], and the time marker (correction point) was obtained from the TimeTree website. Time correction points are often used on the basis of species divergence times clearly identified by accurate fossil evidence or molecular biological evidence to correct the species divergence times in the phylogenetic tree. 4D sites in single-copy genes were extracted to determine the differentiation time of related species on the TimeTree website. MCMCTree was used to estimate the parameter gradient and Hessian matrix, select the correlated molecular clock and JC69 model, and then calculate the divergence time.
Expansion and contraction of gene families
CAFE (version 5) [96] was used to calculate a p value related to the size of each gene family in living species based on a model of gene family evolution. The presence of large numerical differences, especially among closely related species, may result in low p values. Gene families with low p values exhibit significant contraction or expansion, which may be associated with natural selection or with large copies or deletions of chromosomes containing multiple related genes.
The gene families exhibiting significant expansion and contraction were selected (p < 0.05), and GO and KEGG enrichment analyses of the genes in these gene families were performed to analyze the important biological processes of the species. The enrichment value was determined by the Rich factor, the q value and the number of genes enriched in the pathway/GO term. The Rich factor refers to the ratio of the number of genes in the expanded/contracted gene family enriched for the pathway/GO term to the number of annotated genes. According to the GO and KEGG annotations and official classification, the genes were classified into functional and biological pathways. Moreover, the clusterProfiler function in R software was used for enrichment analysis; the p value was calculated, and then FDR correction was performed on the p value. The significant terms were screened according to p < 0.05.
Positive selection analysis
Positive selection on protein-coding sequences was detected by the CodeML module in PAML (version 4.9). Multiple sequence alignments of protein sequences for each single-copy gene family were performed with MAFFT (version 7.471) [97] software, and the results were subsequently reverted to the results of multiple sequence alignments of CDS. The target species was identified as the foreground branch, and the other species were identified as background branches. Based on the two models, Model A (assuming that the foreground branch ω was under positive selection, ω > 1) and the null model (no ω value at any site was allowed to be greater than 1), the likelihood values were calculated. Then, likelihood ratio tests (LRTs) were carried out for the above likelihood values through the chi2 program in PAML (version 4.9). The p value was corrected to detect significant differences (FDR < 0.05), and the Bayes empirical Bayes (BEB) method was used to obtain the posterior probability of a site being positively selected (values greater than 0.95 generally indicated a site that was significantly positively selected). The positively selected genes (FDR < 0.05) were enriched according to KEGG and GO analyses to reveal possible biological mechanisms.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We would like to thank Wuhan Wanmo Technology Company for their assistance for bioinformatic analyses.
Author contributions
D.Y.Y and T.M.Y. conceived and designed the study. K.G., X.B. Z., and Q.Q.C. collected the samples. D.Y.Y., H.L. W., X.Y.W. and T.Z.T. analyzed the data. Z.H. and C.X.L. wrote the paper. M.W.Z., D.Y.Y. and C.X.L. critical revised the article. All authors discussed the data and read and approved the final version of the manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (grant numbers 31972777, and 31402286).
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive [98] at the National Genomics Data Center [99], China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA014865), and are publicly accessible at https://ngdc.cncb.ac.cn/gsa.
Declarations
Ethics approval and consent to participate
Animal experiments were approved by the Animal Research and Ethics Committees of Sichuan Agricultural University (Sichuan, China), and all experimental procedures strictly followed the related laboratory regulations and the relevant guidelines. The study adheres to the ARRIVE 2.0 guidelines for reporting animal research.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Zhi He and Chunxia Li contributed equally to this work.
Contributor Information
Deying Yang, Email: deyingyang@sicau.edu.cn.
Taiming Yan, Email: yantaiming@sicau.edu.cn.
References
- 1.Wang M, Yang JX, Chen XY. Molecular phylogeny and biogeography of percocypris (Cyprinidae, Teleostei). PLoS ONE. 2013;8(6):e61827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Li J, He S. Molecular evidence for the monophyly of east Asian groups of Cyprinidae (Teleostei: Cypriniformes) derived from the nuclear recombination activating gene 2 sequences. Mol Phylogenet Evol. 2007;42(1):157–70. [DOI] [PubMed] [Google Scholar]
- 3.Kong X, Wang X, Gan X, Li J, He S. Phylogenetic relationships of Cyprinidae (Teleostei: Cypriniformes) inferred from the partial S6K1 gene sequences and implication of indel sites in intron 1. Sci China C Life Sci. 2007;50(6):780–8. [DOI] [PubMed] [Google Scholar]
- 4.Li J, Wang X, Kong X, Zhao K, He S, Mayden RL. Variation patterns of the mitochondrial 16S rRNA gene with secondary structure constraints and their application to phylogeny of cyprinine fishes (Teleostei: Cypriniformes). Mol Phylogenet Evol. 2008;47(2):472–87. [DOI] [PubMed] [Google Scholar]
- 5.Li Y, Wang J, Peng Z. The complete mitochondrial genome of Percocypris pingi (Teleostei, Cypriniformes). Mitochondrial DNA. 2013;24(1):40–2. [DOI] [PubMed] [Google Scholar]
- 6.Kottelat M. Freshwater fishes of Northern Vietnam: a preliminary check-list of the fishes known or expected to Occur in Northern Vietnam: with comments on Systematics and nomenclature. Washington, DC: World Bank, Environment and Social Development Unit, East Asia and Pacific Region; 2001. [Google Scholar]
- 7.Bogen C, Al-Dilaimi A, Albersmeier A, Wichmann J, Grundmann M, Rupp O, Lauersen KJ, Blifernez-Klassen O, Kalinowski J, Goesmann A, et al. Reconstruction of the lipid metabolism for the microalga Monoraphidium neglectum from its genome sequence reveals characteristics suitable for biofuel production. BMC Genomics. 2013;14:926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collén J, Porcel B, Carré W, Ball SG, Chaparro C, Tonon T, Barbeyron T, Michel G, Noel B, Valentin K, et al. Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the Archaeplastida. Proc Natl Acad Sci U S A. 2013;110(13):5247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen S, Zhang G, Shao C, Huang Q, Liu G, Zhang P, Song W, An N, Chalopin D, Volff JN, et al. Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat Genet. 2014;46(3):253–60. [DOI] [PubMed] [Google Scholar]
- 10.Reichwald K, Petzold A, Koch P, Downie BR, Hartmann N, Pietsch S, Baumgart M, Chalopin D, Felder M, Bens M, et al. Insights into sex chromosome evolution and aging from the genome of a short-lived fish. Cell. 2015;163(6):1527–38. [DOI] [PubMed] [Google Scholar]
- 11.Shao C, Bao B, Xie Z, Chen X, Li B, Jia X, Yao Q, Orti G, Li W, Li X, et al. The genome and transcriptome of Japanese flounder provide insights into flatfish asymmetry. Nat Genet. 2017;49(1):119–24. [DOI] [PubMed] [Google Scholar]
- 12.You X, Bian C, Zan Q, Xu X, Liu X, Chen J, Wang J, Qiu Y, Li W, Zhang X, et al. Mudskipper genomes provide insights into the terrestrial adaptation of amphibious fishes. Nat Commun. 2014;5:5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu T, Xu G, Che R, Wang R, Wang Y, Li J, Wang S, Shu C, Sun Y, Liu T, et al. The genome of the miiuy croaker reveals well-developed innate immune and sensory systems. Sci Rep. 2016;6:21902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, Swofford R, Pirun M, Zody MC, White S, et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature. 2012;484(7392):55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tine M, Kuhl H, Gagnaire PA, Louro B, Desmarais E, Martins RS, Hecht J, Knaust F, Belkhir K, Klages S, et al. European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nat Commun. 2014;5:5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang ZG, Jiang JP, Wang YZ, Zhang E, Zhang YY, Li LL, Xie F, Cai B, Cao L, Zheng GM. Red List of China’s vertebrates. Biodivers Sci. 2016;24(5):500–51. [Google Scholar]
- 17.Ding RH. The fishes of Sichuan. Chengdu: Sichuan Publishing House of Science and Technology; 1994. [Google Scholar]
- 18.Deng YP, Yang K, Gan WX, Zeng RK, Yan PF, Song ZB. Development of 12 tetranucleotide microsatellite markers for the tetraploidy fish Percocypris pingi (Tchang). Conserv Genet Resour. 2015;7:99–101. [Google Scholar]
- 19.Lai JS, Du J, Zhao G, Deng XC, Li H, He XH, Zhao HP. Embryonic and Postembryonic Development of Percocypris pingi pingi(Tchang). Southwest China J Agricultural Sci. 2014;27(3):1326–31. [Google Scholar]
- 20.Lorenzen K, Beveridge MC, Mangel M. Cultured fish: integrative biology and management of domestication and interactions with wild fish. Biol Rev Camb Philos Soc. 2012;87(3):639–60. [DOI] [PubMed] [Google Scholar]
- 21.Li S. The early development of digestive system and the activity of digestive enzymes and gene expression in Percocypris pingi. Wenjiang: Sichuan Agricultural Unversity; 2018. [Google Scholar]
- 22.Li X, Deng Y, Yang K, Gan W, Zeng R, Deng L, Song Z. Genetic Diversity and structure analysis of Percocypris pingi (Cypriniformes: Cyprinidae): implications for Conservation and Hatchery Release in the Yalong River. PLoS ONE. 2016;11(12):e0166769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yue XJ, Shi JR, Wang YM, Li B, Zou YC, Qing CJ, Xie BW, Wang Y, Qi ZM. Population genetic diversity of Percocypris pingi in Yalong River, the tributary of the Yangtze River. Freshw Fisheries. 2015;45(3):14–8. [Google Scholar]
- 24.Huang F, Xia ZQ, Zhang N, Lu ZH. Does hydrologic regime affect fish diversity?-A case study of the Yangtze Basin (China). Environ Biol Fish. 2011;92(4):569–84. [Google Scholar]
- 25.Jiang H, Xie SG, Zhao WQ, Chang JB. Changes of fish assemblages after construction of Ertan Reservoir in Yalongjiang River. Acta Hydrobiol Sin. 2007;31(4):532–9. [Google Scholar]
- 26.Zhang X, Gao X, Wang JW, Cao WX. Extinction risk and conservation priority analyses for 64 endemic fishes in the Upper Yangtze River, China. Environ Bio Fish. 2015;98(1):261–72. [Google Scholar]
- 27.Cheng H, Concepcion GT, Feng X, Zhang H, Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 2021;18(2):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu P, Zhang X, Wang X, Li J, Liu G, Kuang Y, Xu J, Zheng X, Ren L, Wang G, et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat Genet. 2014;46(11):1212–9. [DOI] [PubMed] [Google Scholar]
- 29.Arai R. A chromosome study on two Cyprinid fishes, Acrossocheilus labiatus and Pseudorasbora pumila pumila, with notes on eurasian cyprinids and their karyotypes. Bull Natl Sci Museum. 1982;8(3):131–52. [Google Scholar]
- 30.Chen ZM, Chen YF. Genetic relationships of the specialized Schizothoracine fishes inferred from random amplified polymorphic and analysis. Zoological Res. 2000;21(4):262–8. [Google Scholar]
- 31.Li S, Tie HM, Duan J, Zhao ZM, Yang S, Guo XL, Yang SY. The Karyotype and C-Banding of Percocypris pingi. Progress Fish Sci. 2017;38(5):19–24. [Google Scholar]
- 32.Zan RG, Song Z, Liu WG. Studies of karyotypes of seven species of fish in Barbinae, with a discussion on identification of fish polyploidy. Zoological Res. 1984;5(1):82–90. [Google Scholar]
- 33.Lien S, Koop BF, Sandve SR, Miller JR, Kent MP, Nome T, Hvidsten TR, Leong JS, Minkley DR, Zimin A, et al. The Atlantic salmon genome provides insights into rediploidization. Nature. 2016;533(7602):200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, McLaren K, Matthews L, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496(7446):498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Lu Y, Zhang Y, Ning Z, Li Y, Zhao Q, Lu H, Huang R, Xia X, Feng Q, et al. The draft genome of the grass carp (Ctenopharyngodon Idellus) provides insights into its evolution and vegetarian adaptation. Nat Genet. 2015;47(6):625–31. [DOI] [PubMed] [Google Scholar]
- 36.Yunfei W, Bin K, Qiang M, Cuizhen W. Chromosome diversity of tibetan fishes. Zoological Res. 1999;20(4):258–64. [Google Scholar]
- 37.Chuong EB, Elde NC, Feschotte C. Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet. 2017;18(2):71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayward A, Gilbert C. Transposable elements. Curr Biol. 2022;32(17):R904–9. [DOI] [PubMed] [Google Scholar]
- 39.Xiao S, Mou Z, Fan D, Zhou H, Zou M, Zou Y, Zhou C, Yang R, Liu J, Zhu S et al. Genome of Tetraploid Fish Schizothorax o’connori Provides Insights into Early Re-diploidization and High-Altitude Adaptation. iScience 2020, 23(9):101497. [DOI] [PMC free article] [PubMed]
- 40.Kasahara M, Naruse K, Sasaki S, Nakatani Y, Qu W, Ahsan B, Yamada T, Nagayasu Y, Doi K, Kasai Y, et al. The medaka draft genome and insights into vertebrate genome evolution. Nature. 2007;447(7145):714–9. [DOI] [PubMed] [Google Scholar]
- 41.Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu Rubripes. Science. 2002;297(5585):1301–10. [DOI] [PubMed] [Google Scholar]
- 42.Van de Peer Y. Tetraodon genome confirms Takifugu findings: most fish are ancient polyploids. Genome Biol. 2004;5(12):250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gebrie A. Transposable elements as essential elements in the control of gene expression. Mob DNA. 2023;14(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawson HA, Liang Y, Wang T. Transposable elements in mammalian chromatin organization. Nat Rev Genet. 2023;24(10):712–23. [DOI] [PubMed] [Google Scholar]
- 45.Meyer A, Schartl M. Gene and genome duplications in vertebrates: the one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr Opin Cell Biol. 1999;11(6):699–704. [DOI] [PubMed] [Google Scholar]
- 46.Jaillon O, Aury JM, Brunet F, Petit JL, Stange-Thomann N, Mauceli E, Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A, et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004;431(7011):946–57. [DOI] [PubMed] [Google Scholar]
- 47.Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang YL, et al. Zebrafish hox clusters and vertebrate genome evolution. Science. 1998;282(5394):1711–4. [DOI] [PubMed] [Google Scholar]
- 48.Near TJ, Eytan RI, Dornburg A, Kuhn KL, Moore JA, Davis MP, Wainwright PC, Friedman M, Smith WL. Resolution of ray-finned fish phylogeny and timing of diversification. Proc Natl Acad Sci U S A. 2012;109(34):13698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santini F, Harmon LJ, Carnevale G, Alfaro ME. Did genome duplication drive the origin of teleosts? A comparative study of diversification in ray-finned fishes. BMC Evol Biol. 2009;9:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hurley IA, Mueller RL, Dunn KA, Schmidt EJ, Friedman M, Ho RK, Prince VE, Yang Z, Thomas MG, Coates MI. A new time-scale for ray-finned fish evolution. Proc Biol Sci. 2007;274(1609):489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma BS, Wei KJ, Zhao TY, Pei FC, Bin H. Research progress on the systematic evolution and plateau adaptation of schizothoracine fishes. J Lake Sci. 2023;35(3):1–18. [Google Scholar]
- 52.Xu P, Xu J, Liu GJ, Chen L, Zhou ZX, Peng WZ, Jiang YL, Zhao ZX, Jia ZY, Sun YH, et al. The allotetraploid origin and asymmetrical genome evolution of the common carp Cyprinus carpio. Nat Commun. 2019;10(1):4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang J, Chen X, Bai J, Fang D, Qiu Y, Jiang W, Yuan H, Bian C, Lu J, He S, et al. The Sinocyclocheilus cavefish genome provides insights into cave adaptation. BMC Biol. 2016;14:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aury JM, Jaillon O, Duret L, Noel B, Jubin C, Porcel BM, Segurens B, Daubin V, Anthouard V, Aiach N, et al. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature. 2006;444(7116):171–8. [DOI] [PubMed] [Google Scholar]
- 55.Maere S, De Bodt S, Raes J, Casneuf T, Van Montagu M, Kuiper M, Van de Peer Y. Modeling gene and genome duplications in eukaryotes. Proc Natl Acad Sci U S A. 2005;102(15):5454–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang L, Wang Y, Sun N, Chen J, He S. Genomic and functional evidence reveals convergent evolution in fishes on the Tibetan Plateau. Mol Ecol. 2021;30(22):5752–64. [DOI] [PubMed] [Google Scholar]
- 57.Wu C, Zhang D, Kan M, Lv Z, Zhu A, Su Y, Zhou D, Zhang J, Zhang Z, Xu M, et al. The draft genome of the large yellow croaker reveals well-developed innate immunity. Nat Commun. 2014;5:5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li LZ, Li MH, Zeng RK, Deng LJ, Yang K, Song ZB. High resistance to Ichthyophthirius multifiliis in Percocypris pingi, an endemic fish in upper Yangtze River. Aquaculture. 2023;562:738767. [Google Scholar]
- 59.Gu HR, Wang HY, Deng SH, Dai XY, He XF, Wang ZJ. Hybridization with Percocypris pingi male can improve the cultivation characteristics of snow trout (Schizothorax wangchiachii), especially the resistance to white spot disease (Ichthyophthirius multifiliis). Aquaculture. 2023;562:738805. [Google Scholar]
- 60.Bragdon B, Moseychuk O, Saldanha S, King D, Julian J, Nohe A. Bone morphogenetic proteins: a critical review. Cell Signal. 2011;23(4):609–20. [DOI] [PubMed] [Google Scholar]
- 61.Crisan M, Kartalaei PS, Vink CS, Yamada-Inagawa T, Bollerot K, van Linden IW, de Sousa Lopes R, Monteiro SM, Mummery R. BMP signalling differentially regulates distinct haematopoietic stem cell types. Nat Commun. 2015;6:8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu M, Chen G, Li YP. TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016;4:16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu Y, Mutlu AS, Liu H, Wang MC. High-throughput screens using photo-highlighting discover BMP signaling in mitochondrial lipid oxidation. Nat Commun. 2017;8(1):865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wen B, Li D, Li W, Zhao Y, Yan C. Multiple acyl-CoA dehydrogenation deficiency as decreased acyl-carnitine profile in serum. Neurol Sci. 2015;36(6):853–9. [DOI] [PubMed] [Google Scholar]
- 65.Ingenhag D, Reister S, Auer F, Bhatia S, Wildenhain S, Picard D, Remke M, Hoell JI, Kloetgen A, Sohn D, et al. The homeobox transcription factor HB9 induces senescence and blocks differentiation in hematopoietic stem and progenitor cells. Haematologica. 2019;104(1):35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu K, Zhao Q, Liu P, Cao J, Gong J, Wang C, Wang W, Li X, Sun H, Zhang C, et al. ATG3-dependent autophagy mediates mitochondrial homeostasis in pluripotency acquirement and maintenance. Autophagy. 2016;12(11):2000–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu B, Shi Y, Yuan J, Hu X, Zhang H, Li N, Li Z, Chen Y, Mu D, Fan W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quant Biology. 2013;35(s 1–3):62–7. [Google Scholar]
- 68.Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, Aiden EL. Juicer provides a one-click system for analyzing Loop-Resolution Hi-C experiments. Cell Syst. 2016;3(1):95–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016;3(1):99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. 1990. [DOI] [PubMed]
- 71.Mount DW. Using the Basic Local Alignment Search Tool (BLAST). CSH Protoc 2007, 2007:pdb.top17. [DOI] [PubMed]
- 72.Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Cold Spring Harbor Lab. 2018;17:i884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and smudgeplots: reference-free profiling of polyploid genomes. Cold Spring Harbor Lab 2019(1):1432. [DOI] [PMC free article] [PubMed]
- 74.Simo FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinf (Oxford England). 2015;31(19):3210–2. [DOI] [PubMed] [Google Scholar]
- 75.Gui ZJF. Identification of genome organization in the unusual allotetraploid form of Carassius auratus Gibelio. Aquaculture 2007:109–17.
- 76.Tarailo-Graovac M, Chen N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Current Protocols in Bioinformatics 2009, 25(1):4.10.11–14.10.14. [DOI] [PubMed]
- 77.Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci. 2020;117(17):9451–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gary B. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 1999(2):573–80. [DOI] [PMC free article] [PubMed]
- 79.Zhao X. W Hao 2007 LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35 Web Server issue W265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mario S, Oliver K, Irfan G, Alec H, Stephan W, Burkhard M. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 2006, 34(Web Server issue):W435–439. [DOI] [PMC free article] [PubMed]
- 81.Burge C. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268(1):78–94. [DOI] [PubMed] [Google Scholar]
- 82.Majoros W, Pertea M, Salzberg S. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20(16):2878–9. [DOI] [PubMed] [Google Scholar]
- 83.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Trapnell CRA, Goff L, Pertea G, Kim D, Kelley DR. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cantarel BL, Korf I, Robb SMC, Parra G, Ross E. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18(1):188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lowe TM, Chan PP. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res 2016(W1):W54–7. [DOI] [PMC free article] [PubMed]
- 87.Nawrocki EP, Burge SW, Alex B, Jennifer D, Eberhardt RY, Eddy SR, Floden EW, Gardner PP, Jones TA, John T. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res 2015(D1):D130. [DOI] [PMC free article] [PubMed]
- 88.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25(10):1335–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Waterhouse ASF, Panagiotis RM, Kriventseva I, Zdobnov EV. EM: BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015(19):3210–2. [DOI] [PubMed]
- 90.Li L. OrthoMCL: identification of Ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nakamura T, Yamada, Kazunori D. Tomii, Kentaro, Katoh, Kazutaka: parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics. 2018;14(34):2490–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gerard T, Jose C. Improvement of Phylogenies after removing Divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56(4):564–77. [DOI] [PubMed] [Google Scholar]
- 93.Alexandros S. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ziheng Y. PAML 4: phylogenetic analysis by Maximum Likelihood. Mol Biol Evol. 2007;24(8):1586–91. [DOI] [PubMed] [Google Scholar]
- 95.Cummings MP. PAML (phylogenetic analysis by Maximum Likelihood): Dictionary of Bioinformatics and Computational Biology. Mol Biol Evol. 2016;24:1586–91. [DOI] [PubMed] [Google Scholar]
- 96.Mendes FK, Vanderpool D, Fulton B, Hahn MW. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics. 2020;36(22–23):5516–8. [DOI] [PubMed] [Google Scholar]
- 97.Katoh K, Asimenos G, Toh H. Multiple alignment of DNA sequences with MAFFT. Methods Mol Biol. 2009;537(8):39–64. [DOI] [PubMed] [Google Scholar]
- 98.Chen T, Chen X, Zhang S, Zhu J, Tang B, Wang A, Dong L, Zhang Z, Yu C, Sun Y, et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics Proteom Bioinf. 2021;19(4):578–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Partners C-NM. Database resources of the National Genomics Data Center, China National Center for Bioinformation in 2024. Nucleic Acids Res. 2024;52(D1):D18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive [98] at the National Genomics Data Center [99], China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA014865), and are publicly accessible at https://ngdc.cncb.ac.cn/gsa.