

Summary
The availability of multi-omics data applied to profile cancer cohorts is rapidly increasing. Here, we present a protocol for Multiomics2Targets, a computational pipeline that can identify driver cell signaling pathways, protein kinases, and cell-surface targets for immunotherapy. We describe steps for preparing the data, uploading files, and tuning parameters. We then detail procedures for running the workflow, visualizing the results, and exporting and sharing reports containing the analysis.
For complete details on the use and execution of this protocol, please refer to Deng et al.1
Subject areas: bioinformatics, cancer, systems biology, computer sciences
Graphical abstract
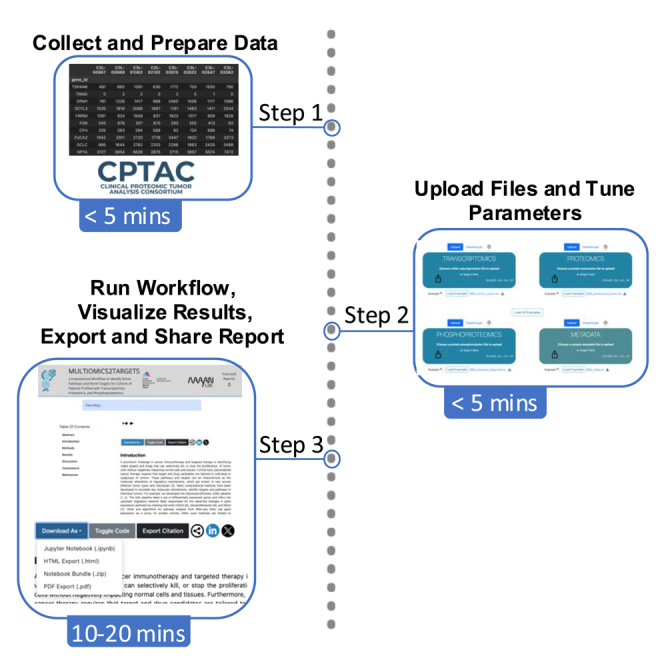
Highlights
-
•
A comprehensive workflow to analyze data from cancer patient cohorts
-
•
Workflow to analyze transcriptomics, proteomics, and phosphoproteomics
-
•
Results are produced in a research-paper-like report that can be shared
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
The availability of multi-omics data applied to profile cancer cohorts is rapidly increasing. Here, we present a protocol for Multiomics2Targets, a computational pipeline that can identify driver cell signaling pathways, protein kinases, and cell-surface targets for immunotherapy. We describe steps for preparing the data, uploading files, and tuning parameters. We then detail procedures for running the workflow, visualizing the results, and exporting and sharing reports containing the analysis.
Before you begin
With the growing availability of tumors profiled with multiple-omics technologies, there is a need for tools to analyze and integrate this data to produce actionable insights towards the development of personalized therapeutics. Multiomics2Targets1 is a platform that enables users with little or no programming background to perform integrative analysis to identify cell surface proteins aberrantly expressed on tumor cells, and driver cell signaling pathways, including protein kinases, for cancer subtypes. The pipeline produces a paper-like report automatically as the output of the platform along with numerous figures and tables, all of which are available for download, additional analysis, and reuse. The following protocol describes the specific steps involved in analyzing the data collected by the Clinical Proteomic Tumor Analysis Consortium (CPTAC).2 However, the workflow can be applied to any collections of transcriptomics, proteomics, and phosphoproteomics datasets individually or in combination. In other words, although uploading matched transcriptomics, proteomics, and phosphoproteomics produces the most robust and complete analysis, only one data type is needed to perform a partial run of the workflow.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| CPTAC3 cohort dataset (open-access data; no approval is required) | Li et al.3 |
https://pdc.cancer.gov/pdc/cptac-pancancer, https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001287.v16.p6 |
| Software and algorithms | ||
| The source code for Multiomics2Targets (CC BY-NC) | Deng et al.1 |
https://github.com/MaayanLab/Multiomics2Targets https://doi.org/10.6084/m9.figshare.26199887 |
| The source code for the eXpression2Kinases analysis (CC BY-NC) | Chen et al.4; Clarke et al.5 | https://github.com/MaayanLab/x2k-pathway-analysis |
| The Multiomics2Targets website (CC BY-NC) | Deng et al.1 | https://multiomics2targets.maayanlab.cloud/ |
| KEA3 (CC BY-NC) | Kuleshov et al.6 | https://maayanlab.cloud/kea3/ |
| ChEA3 (CC BY-NC) | Keenan et al.7 | https://maayanlab.cloud/chea3/ |
| Enrichr (CC BY-NC) | Chen et al.8; Kuleshov et al.9; Xie et al.10 | https://maayanlab.cloud/Enrichr/ |
| TargetRanger (CC BY-NC) | Marino et al.11 | https://targetranger.maayanlab.cloud/ |
| Geneshot (Apache License 2.0) | Lachmann et al.12 | https://maayanlab.cloud/geneshot/ |
Materials and equipment
The Multiomics2Targets platform is a freely accessible webserver available at https://multiomics2targets.maayanlab.cloud/. Multiomics2Targets was developed utilizing the Appyter framework13 which enables the creation of interactive input forms and the automatic execution and visualization of Jupyter Notebooks. The backend and accompanying code used for the analysis was developed in Python 3.9, and the UI is styled and created with Jinja2 and Svelte components.
Step-by-step method details
Preparing the data and uploading it to Multiomics2Targets
Timing: <5 min
Uploaded data matrices must conform to a specified format. Only properly formatted datasets should be uploaded to the Multiomics2Targets site for analysis.
-
1.
Prepare data matrices in the format where samples are the columns, and gene/proteins are the rows. Such data matrices can represent transcriptomics, proteomics, and/or phosphoproteomics collected from matching patients.
Note: For the CPTAC3 data, all 10 cancer types have canned examples available on the Multiomics2Targets site. The CPTAC3 data3 is available from https://pdc.cancer.gov/pdc/cptac-pancancer. The prepared data matrices serve as examples for how to prepare your data for upload to Multiomics2Targets.
-
2.
Access the Multiomics2Targets site from https://multiomics2targets.maayanlab.cloud/. Troubleshooting 1.
-
3.
Before uploading the data, ensure that the file format follows the format specified in the tooltips (Figure 1A) next to each data type.
For the transcriptomics data matrices, ensure each row contains a gene that maps to an NCBI gene symbol or ENSEMBL ID, and the expression level of that gene. Check that the columns contain unique sample IDs.Note: NCBI synonyms are mapped to official symbols.Note: The pipeline performs normalization, so it expects un-normalized gene counts.-
a.For the uploaded proteomics data matrices, ensure that each row contains a gene or a protein name, and each column corresponds to a unique sample ID, matching those same IDs of the other uploaded data and metadata matrices.Note: If some samples are missing for one or more of the data modalities, the pipeline excludes these samples from the analysis when it is applicable.
-
b.For the uploaded phosphoproteomics data matrices, ensure phosphosites or protein names are on each row, and each column contains a unique sample ID, matching those same IDs as the other uploaded data and metadata matrices.
-
c.For uploaded metadata, check that each row contains a sample ID matching those in the uploaded data matrices type(s), and each column contains a metadata attribute that is either categorial (text strings) or numerical.
-
a.
-
4.
Ensure that the files you prepare complete uploading. Alternatively, click on any of the upload areas to open a native file explorer where users can select their data or metadata files from the local file system. Troubleshooting 2.
Note: A progress bar appears once a file is dragged into any of the colored areas (Figure 1B).
Figure 1.

Screenshots from the upload form of Multiomics2Targets
(A) By hovering over the question mark icon, users are provided with an example that explains how to format the input data files.
(B) Once a well-formatted file is selected or dragged to the designated area, a progress bar provides feedback about the status of the upload. The progress bar turns green once the upload is complete.
Tuning the pipeline parameters
Adjusting the available parameter settings on the Multiomics2Targets landing page may improve the results and provide the user with greater control in how the pipeline is executed.
-
5.Optional: Inspect the various parameter options and adjust as necessary (Figure 2):
-
a.Specify a column in the uploaded metadata file to serve as cluster annotations. These annotations replace automatic clustering computed by the default pipeline.
-
b.Choose whether to prioritize membrane proteins for target identification.Note: Prioritizing membrane genes/proteins relates to identifying cell-surface proteins highly expressed in the uploaded cancer samples while lowly expressed across many normal cells and tissues as reported in several healthy tissue atlases. It is recommended to keep this option enabled.
-
c.Choose whether to perform background normalization for target identification.Note: Normalization to a background also relates to the identification of aberrantly expressed genes in the cancer samples. This step aligns the uploaded data into the same distribution of the healthy background atlases. It is recommended to keep this option enabled.
-
d.Choose whether to impute protein expression.Note: Imputation of protein and phosphoprotein expression levels is achieved by filling missing values in the uploaded data matrices with averages. If the proteomics or phosphoproteomics matrices were previously imputed with a different method, this option should be disabled. The thresholds for the imputation procedure were tuned with the CPTAC3 data but can be adjusted based on the user’s needs.
-
e.Choose the number of differentially expressed genes (DEGs) and differentially phosphorylated proteins (DPPs) to utilize in the pipeline.Note: The number of DEGs and DPPs is set by default to 500. The user may decrease or increase these values depending on the desired breadth of coverage. This is most relevant to the transcriptomics and phosphoproteomics data matrices.
-
f.Set UMAP parameters.Note: UMAP parameters determine how the pipeline determines sample assignment to clusters. Such assignment to clusters is based on the normalized transcriptomics data. If the points on the UMAP appear to belong to one cluster, try to adjust these parameters to potentially reveal subtypes.
-
a.
-
6.
Optional: Select a method for adding intermediates to the enriched transcription factors for the Expression2Kinases part of the pipeline. These options include gene-gene co-expression correlations from Geneshot12 and protein-protein co-expression correlations derived from the Cancer Cell Line Encyclopedia (CCLE).14
-
7.
Optional: Select a cancer type to utilize for the DepMap15 cancer cell-line screening analysis.
Note: The most logical selection here is selecting the cancer type of the uploaded data. The analysis checks how the identified cell surface targets and driver kinases behave in the DepMap dataset. Use CMD (Mac) or CTRL key (Windows) to select multiple cancer types to use for validation with DepMap.
Figure 2.

Screenshot of the Adjustable Parameters for the Multiomics2Targets pipeline
Submit the pipeline for execution
Submission of the uploaded files initiates the execution of the pipeline. Results can be viewed as they are computed, and the full version of the analysis report can be exported upon completion of the pipeline execution.
-
8.
Selecting Submit at the bottom of the input form executes the pipeline with the uploaded data matrices and selected parameters.
Note: The Appyter framework renders the results cell by cell in a Jupyter Notebook style. The user may inspect and download the results while the pipeline computes in sequence. Troubleshooting 3.
-
9.
Save the URL of the report for future use and retrieval, and for sharing the results with collaborators.
Note: Each run of the pipeline receives a unique and persistent identifier that is embedded in the URL. Hence, you can share the URL, or return to the results page later. By pasting the URL in the browser URL text box, you are directed the same executed result page. Troubleshooting 4.
-
10.
Optional: To view the code executed in each step of the analysis, click on the “toggle code” button placed directly below the abstract.
-
11.
Optional: The user may export the results as a PDF, HTML, and a ZIP bundle (including all the figures, tables and executed cells) by clicking on the “Download as” dropdown menu button (Figure 3).
Note: The selected output type is automatically downloaded to your default downloads directory.
Figure 3.

The Multiomics2Targets Reports
Screenshot from the top of the notebook. Export options become available upon completion of the pipeline execution.
Expected outcomes
Multiomics2Targets is a freely accessible online platform and a workflow engine that performs integrative analysis on uploaded transcriptomics, proteomics, and phosphoproteomics data matrices created from profiling cohorts of cancer patients. The platform expands previous work by calibrating the Expression2Kinases (X2K) workflow.4,5 By ranking protein kinases from enriched transcription factors inferred from the transcriptomics data together with the rankings of kinases based on the phosphoproteomics data, Multiomics2Targets compares the workflows for agreement. The Multiomics2Targets pipeline produces figures and tables that pinpoint to novel cell signaling pathways driven by specific protein kinases. Additionally, the pipeline utilizes the TargetRanger11 algorithm to identify genes that encode for cell-surface proteins that are highly expressed in cancer subtypes and lowly expressed across most human cells and tissues. These targets may become subjects for developing antibody drug conjugate (ADC) and CAR T cell therapies. The pipeline further confirms the identified protein kinases and the cell-surface targets by inspecting their expression at the protein level. The produced reports from Multiomics2Targets contain all the analysis in a research publication-style report that can be downloaded and shared.
Quantification and statistical analysis
The Multiomics2Targets pipeline utilizes multiple tools to produce many figures and tables. The reports contain interactive visualizations and links to external tools for further analysis and visualization of the uploaded data. The reports also contain automatically generated descriptions of the results produced with the OpenAI LLM service GPT-4o. The results visualized in Figure 4 are representative figures produced from the analysis of the glioblastoma (GBM) CPTAC3 cohort. Users can load this dataset directly from the Multiomics2Targets site as the default example. To summarize the results dynamically, we instruct the GPT-4o model to strictly describe and compare the results across the identified clusters. All GPT generated text is encapsulated in yellow boxes. More details about the prompting of GPT-4o to produce these descriptions are provided in the original manuscript.
Figure 4.

Representative results from the glioblastoma (GBM) CPTAC3 cohort
(A) Labeled automatic clustering performed on the uploaded transcriptomics data matrix.
(B) Diagrams of pathways that are up regulated in cluster 3 identified with the X2K analysis.
(C) Heatmap displaying the enrichment ranks of transcription factors returned from a ChEA3 analysis applied to the down-regulated genes of each cluster along with tracks of accompanying metadata.
(D) Cell-surface targets identified to be upregulated in one or more of the GBM clusters compared to multiple healthy tissue and cell-type backgrounds, annotated with Z-scored protein expression.
(E) Recovery of protein kinases inferred with X2K compared to protein kinases directly inferred from the phosphoproteomics.
(F) Recovered kinases Z-scores in individual samples compared to their average across the cohort.
If transcriptomics data was uploaded, the pipeline conducts clustering of the samples based on the log2 quantile normalized expression of the 5,000 most variable genes. It then visualizes these samples and clusters as a UMAP.16 After clustering, the pipeline performs Enrichr8,9,10 enrichment analysis with gene set libraries created from Wikipathways17 (Wikipathways_2023_Human), GO Biological Processes18 (GO_Biological_Processes_2023) and MGI Mammalian Phenotype19 (MGI_Mammalian_Phenotype_Level_4_2021) to characterize the collective biological processes that are most prominent in each cluster. The large language model (LLM) GPT-4o provides consensus labels for each cluster (the full prompt design can be seen here: https://github.com/MaayanLab/Multiomics2Targets/blob/262ce5e17b8403bd015b5f5378a6d663b92b7424/helpers.py#L213). The details about prompting GPT-4o to produce these summaries are discussed in the original manuscript1 (Figure 4A). Additional downstream analyses of the identified clusters include:
-
1.
The Expression2Kinases (X2K) workflow4,5 that is applied to analyze the samples in each cluster. The workflow identifies pathways that contain enriched transcription factors, inferred intermediates that connect these transcription factors, and enriched kinases that phosphorylate the transcription factors and the intermediates. The workflow visualizes the identified pathways as ball-and-stick diagrams for pathway members identified in at least 25% of the samples in each cluster (Figure 4B). These pathways may represent driver pathways that could be targeted to alter the oncogenic phenotype in a particular subtype.
The Multiomics2Targets workflow visualizes the enrichment results from ChEA,7 KEA,6 and Z-scored protein and phosphoprotein expression as heatmaps (Figure 4C). Observing patterns in these heatmaps can help identify modules of kinases, transcription factors, and other proteins that may contribute to specific phenotypes in the identified subtypes. Along with metadata tracks at the top of these heatmaps, users can interactively further investigate the clusters with Clutergrammer.20 Multiomics2Targets provides a link to view the results with Clustergrammer above each heatmap. In Clustergrammer, the user can identify the samples and corresponding proteins/genes in each cluster by zooming and panning. Users can also download and export the visualized matrices in Clustergrammer (Troubleshooting 5).
The workflow additionally annotates cell surface targets that are highly expressed in each cluster, and lowly expressed across most healthy tissues and cell types with Z-scored proteomics expression, which the pipeline visualizes as a heatmap (Figure 4D). The analysis compares the transcriptomics expression levels in each cluster to healthy tissue and cell type expression levels obtained from the ARCHS4,21 GTEx,22 and Tabula Sapiens23 using limma voom.24 The differentially expressed genes for each cluster are also filtered for genes that encode for cell-surface proteins. The list of cell surface proteins is determined by combining information from COMPARTMENTS25 and the Human Protein Atlas (HPA).26 The set of membrane proteins was previously published in an article describing TargetRanger.11 Targets appearing at the top of the heatmap, and with lighter colors across the cells, represent genes/proteins that are up regulated in many or all the clusters. These identified targets are relevant for multiple subtypes. The targets appearing at the bottom of the heatmap are typically those that are subtype specific. Additionally, if the user selects a cancer type in Step 7, the source cell lines corresponding to that cancer type from DepMap15 are identified (Table 1). Additionally, Multiomics2Targets queries PubMed to identify the number of articles that mentioned the target with and without the term “cancer” and the specific cancer type, to assess the novelty of the identified targets.
-
2.
Utilizing the matched phosphoproteomics data, the Multiomics2Targets pipeline additionally assesses the recovery of protein kinases identified through the X2K algorithm4,5 compared to those enriched directly from the differentially phosphorylated proteins with KEA3. The pipeline provides an assessment of the level of agreement between the X2K and KEA3 results per cluster at the individual sample level. The Multiomics2Targets workflow denotes the samples with greater than expected recovery level with an asterisk (Figure 4E). For the samples that exhibit greater than expected recovery, if a matching proteomics data matrix is uploaded and available, the pipeline visualizes the Z-scored protein expression of the identified kinases, comparing the expression across the cohort to the expression in the sample (Figure 4F). Additionally, Multiomics2Targets perform similar validation with DepMap15 as described above in relation to the cell-surface targets. Overall, the most dysregulated kinases for individual samples could become personalized targets.
Table 1.
Example from a target validation table utilizing DepMap’s cancer cell line gene effect scores, and novelty assessed by PubMed citations
| Target | Ranked top 10 in (cluster, background) | Mean gene effect | Literature co-mentions with cancer | Literature co-mentions with diffuse glioma |
|---|---|---|---|---|
| ADCYAP1R1 | (3, ARCHS4) (1, ARCHS4) |
0.068 | 7 | 0 |
| APOC2 | (6, ARCHS4) | 0.0509 | 22 | 0 |
| AQP4 | (5, ARCHS4) (4, ARCHS4) (4, TS) (2, ARCHS4) (2, TS) (0, ARCHS4) (0, TS) (3, ARCHS4) (3, TS) (1, ARCHS4) (1, TS) |
0.1402 | 115 | 0 |
| ATP13A4 | (4, ARCHS4) (2, ARCHS4) (3, ARCHS4) |
0.0099 | 4 | 0 |
| BCAN | (5, ARCHS4) (5, TS) (4, ARCHS4) (4, TS) (3, ARCHS4) (3, TS) (1, ARCHS4) (1, TS) |
−0.1101 | 36 | 0 |
Limitations
There are several limitations to the Multiomics2Targets platform. The Multiomics2Targets workflow identifies clusters from uploaded cohorts utilizing only the transcriptomics data type. A more integrative multi-omics strategy to identify clusters may be more robust. Additionally, Multiomics2Targets is intended for analysis of large cohorts. Smaller collections of samples may lack the statistical power for the various analyses provided by the platform. Since producing such large-scale datasets is a major undertaking, the application of Multiomics2Targets can be applied to few projects. The Multiomics2Targets webserver is implemented as a Kubernetes job with 5Gb of memory allocated. However, for large cohorts this may not be sufficient, and the analysis would need to be run locally.
Troubleshooting
Problem 1
Unable to access the web server at the specified URL (related to Step 2).
Potential solution
Post an issue on GitHub to inquire about the status of the web server if it is inaccessible at the specified URL. Users can run the pipeline locally with Docker. The site will be accessible at localhost:5000 after running the following command:
> docker run --device /dev/fuse --cap-add SYS_ADMIN --security-opt apparmor:unconfined -p 5000:5000 -it maayanlab/x2ktr:0.1.05
Users may optionally provide an OpenAI API key to receive automatically generated descriptions of the results throughout the report:
> docker run --device /dev/fuse --cap-add SYS_ADMIN --security-opt apparmor:unconfined -p 5000:5000 -e OPENAI_API_KEY=sk-… -it maayanlab/x2ktr:0.1.05
Please refer to the GitHub repository at https://github.com/MaayanLab/Multiomics2Targets for the latest Docker image version.
If the user is unable to run the pipeline locally, please post an issue on GitHub at: https://github.com/MaayanLab/Multiomics2Targets/issues/new.
Problem 2
The file upload is not finishing and appears to be stuck in “progressing” (related to Step 4).
Potential solution
The speed of the file upload is dependent on the user’s upload speed. For large files, this speed may not be sufficient to complete. To bypass this issue, the user can provide a Passthrough URI which can be in the form of a GA4GH DRS, AWS S3, Google Cloud GS, FTP, HTTP, or HTTP link. The passthrough option can be selected for any of the file upload sections.
Problem 3
An error occurs in the execution of a specific Jupyter Notebook cell when executing the pipeline.
Potential solution
An informative error message should be displayed below the cell block where the error occurred. Please ensure that the file format conforms to the provided guidelines. For example, the gene or protein identifiers should conform to the standards specified on the input page. If the issue persists, please post an issue on GitHub with a link to the specific workflow instance in which the error occurred, as well as a screenshot of the section of the report with the error. If the issue persist and it is clear that it is not due to wrong file formatting, the issue should be posted on the associated Multiomics2Target repo on GitHub at: https://github.com/MaayanLab/Multiomics2Targets/issues/new.
Problem 4
An error occurs indicating the Kernel Died (related to Step 8).
Potential solution
This error indicates that the Kubernetes job did not have sufficient memory to run the pipeline with the uploaded files. To mitigate this issue, you can run the pipeline locally. Docker needs to be installed. The application should be accessible at localhost:5000 after running the following command.
> docker run --device /dev/fuse --cap-add SYS_ADMIN --security-opt apparmor:unconfined -p 5000:5000 -it maayanlab/x2ktr:0.1.05
Users may optionally provide an OpenAI API key to receive automatically generated descriptions of the results throughout the report.
> docker run --device /dev/fuse --cap-add SYS_ADMIN --security-opt apparmor:unconfined -p 5000:5000 -e OPENAI_API_KEY=sk-… -it maayanlab/x2ktr:0.1.05
Please refer to the GitHub repository at: https://github.com/MaayanLab/Multiomics2Targets for the latest Docker image version.
If the user is unable to run the pipeline locally, please post an issue on GitHub at: https://github.com/MaayanLab/Multiomics2Targets/issues/new.
Problem 5
The Clustergrammer link does not open the specified matrix, does not load, or displays an error (related to quantification and statistical analysis).
Potential solution
Clustergrammer has a limit on the size of matrices that it can visualize. If the uploaded cohort is large, some Clustergrammer links may not become unavailable. However, the matrices can be downloaded using the links below the heatmap figures as CSVs and investigated further locally with alternative tools. In addition, when large data matrices are loaded, some of the features of Clustergrammer are disabled. To explore specific regions in the Clustergrammer heatmaps, try to zoom in by using the mouse wheel, or by clicking the gray trapezoids. Zooming in will also uncover the gene and sample names.
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Avi Ma’ayan (avi.maayan@mssm.edu).
Technical contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Avi Ma’ayan (avi.maayan@mssm.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
No new data was generated in this study. CPTAC3 data was analyzed. This dataset is available for download from: https://pdc.cancer.gov/pdc/cptac-pancancer. The source code used to create Multiomics2Targets is available from: https://github.com/MaayanLab/Multiomics2Targets.
A snapshot of the code with a DOI is available from Zenodo at https://zenodo.org/records/13948700; https://doi.org/10.5281/zenodo.13948700.
Acknowledgments
This project is funded by NIH grants U24CA271114, U24CA264250, OT2OD030160, OT2OD036435, and R01DK131525.
Author contributions
G.B.M. wrote the paper, and G.B.M. and E.Z.D. developed the Multiomics2Targets website, performed the TargetRanger and X2K analyses, and produced the figures. D.J.B.C. developed the Appyter and performed some of the initial analyses. I.D. created the protein co-expression data matrix from CCLE. A.C.R. and P.W. contributed ideas and participated in discussions. W.M. assisted in preparing the inputs to the ChEA3 and KEA3 analyses. A.M. initiated the project, provided direction and ideas, managed the project, and wrote the paper.
Declaration of interests
The authors declare no competing interests.
References
- 1.Deng E.Z., Marino G.B., Clarke D.J.B., Diamant I., Resnick A.C., Ma W., Wang P., Ma'ayan A. Multiomics2Targets identifies targets from cancer cohorts profiled with transcriptomics, proteomics, and phosphoproteomics. Cell Rep. Methods. 2024;4 doi: 10.1016/j.crmeth.2024.100839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellis M.J., Gillette M., Carr S.A., Paulovich A.G., Smith R.D., Rodland K.K., Townsend R.R., Kinsinger C., Mesri M., Rodriguez H., et al. Connecting genomic alterations to cancer biology with proteomics: the NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov. 2013;3:1108–1112. doi: 10.1158/2159-8290.CD-13-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y., Dou Y., Da Veiga Leprevost F., Geffen Y., Calinawan A.P., Aguet F., Akiyama Y., Anand S., Birger C., Cao S., et al. Proteogenomic data and resources for pan-cancer analysis. Cancer Cell. 2023;41:1397–1406. doi: 10.1016/j.ccell.2023.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen E.Y., Xu H., Gordonov S., Lim M.P., Perkins M.H., Ma'ayan A. Expression2Kinases: mRNA profiling linked to multiple upstream regulatory layers. Bioinformatics. 2012;28:105–111. doi: 10.1093/bioinformatics/btr625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke D.J.B., Kuleshov M.V., Schilder B.M., Torre D., Duffy M.E., Keenan A.B., Lachmann A., Feldmann A.S., Gundersen G.W., Silverstein M.C., et al. eXpression2Kinases (X2K) Web: linking expression signatures to upstream cell signaling networks. Nucleic Acids Res. 2018;46:W171–W179. doi: 10.1093/nar/gky458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuleshov M.V., Xie Z., London A.B.K., Yang J., Evangelista J.E., Lachmann A., Shu I., Torre D., Ma'ayan A. KEA3: improved kinase enrichment analysis via data integration. Nucleic Acids Res. 2021;49:W304–W316. doi: 10.1093/nar/gkab359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keenan A.B., Torre D., Lachmann A., Leong A.K., Wojciechowicz M.L., Utti V., Jagodnik K.M., Kropiwnicki E., Wang Z., Ma'ayan A. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019;47:W212–W224. doi: 10.1093/nar/gkz446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma'ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie Z., Bailey A., Kuleshov M.V., Clarke D.J.B., Evangelista J.E., Jenkins S.L., Lachmann A., Wojciechowicz M.L., Kropiwnicki E., Jagodnik K.M., et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021;1 doi: 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marino G.B., Ngai M., Clarke D.J.B., Fleishman R.H., Deng E.Z., Xie Z., Ahmed N., Ma'ayan A. GeneRanger and TargetRanger: processed gene and protein expression levels across cells and tissues for target discovery. Nucleic Acids Res. 2023;51:W213–W224. doi: 10.1093/nar/gkad399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lachmann A., Schilder B.M., Wojciechowicz M.L., Torre D., Kuleshov M.V., Keenan A.B., Ma'ayan A. Geneshot: search engine for ranking genes from arbitrary text queries. Nucleic Acids Res. 2019;47:W571–W577. doi: 10.1093/nar/gkz393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarke D.J.B., Jeon M., Stein D.J., Moiseyev N., Kropiwnicki E., Dai C., Xie Z., Wojciechowicz M.L., Litz S., Hom J., et al. Appyters: Turning Jupyter Notebooks into data-driven web apps. Patterns (N Y) 2021;2 doi: 10.1016/j.patter.2021.100213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghandi M., Huang F.W., Jané-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R., 3rd, Barretina J., Gelfand E.T., Bielski C.M., Li H., et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsherniak A., Vazquez F., Montgomery P.G., Weir B.A., Kryukov G., Cowley G.S., Gill S., Harrington W.F., Pantel S., Krill-Burger J.M., et al. Defining a Cancer Dependency Map. Cell. 2017;170:564–576.e16. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leland M.H.,J., Saul N., GroBberger L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018;3:861. doi: 10.21105/joss.00861. [DOI] [Google Scholar]
- 17.Slenter D.N., Kutmon M., Hanspers K., Riutta A., Windsor J., Nunes N., Mélius J., Cirillo E., Coort S.L., Digles D., et al. WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018;46:D661–D667. doi: 10.1093/nar/gkx1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eppig J.T., Blake J.A., Bult C.J., Kadin J.A., Richardson J.E., Mouse Genome Database Group The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res. 2015;43:D726–D736. doi: 10.1093/nar/gku967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez N.F., Gundersen G.W., Rahman A., Grimes M.L., Rikova K., Hornbeck P., Ma'ayan A. Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data. Sci. Data. 2017;4 doi: 10.1038/sdata.2017.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lachmann A., Torre D., Keenan A.B., Jagodnik K.M., Lee H.J., Wang L., Silverstein M.C., Ma'ayan A. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun. 2018;9:1366. doi: 10.1038/s41467-018-03751-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.GTEx Consortium The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tabula Sapiens Consortium∗. Jones R.C., Karkanias J., Krasnow M.A., Pisco A.O., Quake S.R., Salzman J., Yosef N., Bulthaup B., Brown P., et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022;376 doi: 10.1126/science.abl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Binder J.X., Pletscher-Frankild S., Tsafou K., Stolte C., O'Donoghue S.I., Schneider R., Jensen L.J. COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database. 2014;2014 doi: 10.1093/database/bau012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thul P.J., Lindskog C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018;27:233–244. doi: 10.1002/pro.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data was generated in this study. CPTAC3 data was analyzed. This dataset is available for download from: https://pdc.cancer.gov/pdc/cptac-pancancer. The source code used to create Multiomics2Targets is available from: https://github.com/MaayanLab/Multiomics2Targets.
A snapshot of the code with a DOI is available from Zenodo at https://zenodo.org/records/13948700; https://doi.org/10.5281/zenodo.13948700.