

Abstract
New particle formation (NPF) in the tropical upper troposphere is a globally important source of atmospheric aerosols1–4. It is known to occur over the Amazon basin, but the nucleation mechanism and chemical precursors have yet to be identified2. Here we present comprehensive in situ aircraft measurements showing that extremely low-volatile oxidation products of isoprene, particularly certain organonitrates, drive NPF in the Amazonian upper troposphere. The organonitrates originate from OH-initiated oxidation of isoprene from forest emissions in the presence of nitrogen oxides from lightning. Nucleation bursts start about 2 h after sunrise in the outflow of nocturnal deep convection, producing high aerosol concentrations of more than 50,000 particles cm−3. We report measurements of characteristic diurnal cycles of precursor gases and particles. Our observations show that the interplay between biogenic isoprene, deep tropical convection with associated lightning, oxidation photochemistry and the low ambient temperature uniquely promotes NPF. The particles grow over time, undergo long-range transport and descend through subsidence to the lower troposphere, in which they can serve as cloud condensation nuclei (CCN) that influence the Earth’s hydrological cycle, radiation budget and climate1,4–8.
Subject terms: Atmospheric chemistry, Climate sciences, Atmospheric chemistry
Aircraft measurements over the Amazon show that new particle formation in the upper troposphere emerges when isoprene, emitted by forests, undergoes oxidation in the presence of nitrogen oxides produced by lightning.
Main
Isoprene is emitted in large quantities by vegetation such as broad-leafed trees and it has the largest source strength of all biogenic volatile organic compounds (VOCs). Globally, emissions amount to about 500–600 Tg a−1, representing more than half of the total biogenic VOC emissions, with tropical South America contributing an estimated 163 Tg a−1 (refs. 9,10). Isoprene is primarily emitted into the atmosphere during daylight hours and is typically converted within 1–2 h into oxygenated VOCs, mostly through reaction with hydroxyl radicals (OH)11,12. Isoprene mixing ratios in the continental boundary layer of the Amazon basin range between 1 and 20 ppbv, with a diurnal cycle peaking in the afternoon13. Although isoprene-derived oxygenated organic molecules (IP-OOMs) are known to contribute substantially to the production of secondary organic aerosol mass by means of condensation onto pre-existing particles14,15, they are unable to form new particles in the boundary layer and are even thought to inhibit NPF from monoterpenes16,17.
Deep convection can transport isoprene from the boundary layer to the upper troposphere within 1–2 h (refs. 18,19). Palmer et al.20 demonstrated that nocturnal deep convection delivers isoprene to the tropical upper troposphere, leading to secondary organic aerosol production20. Deep convective clouds are also associated with lightning activity21 and thus represent a main source of nitrogen oxides (NOx)22 that play a central role in OH formation (recycling) in the upper troposphere23. Furthermore, pre-existing aerosol particles are efficiently scavenged from boundary-layer air that ascends in precipitating deep convective clouds2,24. As a result, the outflow of nocturnal deep convection contains increased levels of isoprene and NOx, with a weak condensation sink (CS) for condensation of vapours onto pre-existing aerosols. The low temperatures of the upper troposphere and intense solar (actinic) radiation during the daytime create favourable conditions for NPF, that is, aerosol nucleation and initial growth up to detectable particle size in the convective outflow1,2,7,25.
Although high number concentrations of freshly formed particles have frequently been observed in the upper troposphere1–3,7,25–28, the conditions and chemical processes underlying NPF have remained elusive. Kulmala et al.29 proposed that insoluble organics are transported to the upper troposphere by deep convection to produce new particles and Ekman et al.30 suggested that isoprene itself could be the precursor for upper tropospheric NPF over the Amazon. A study of the aerosol composition in the upper troposphere over the Amazon, focusing on larger particles (dp > 50 nm), found isoprene-derived secondary organic aerosols and organic nitrates to be the main components31. Recent measurements at the Chacaltaya mountain site in Bolivia (5,240 m a.m.s.l.) revealed isoprene oxidation products, including organonitrates in the gas and aerosol phases in air masses from the free troposphere over the Amazon basin32. However, the precursors that drive particle nucleation and early growth were not identified. Although NPF supplies about half the CCN globally4,33, in the continental and marine boundary layer in the tropics it is rare34 and typical NPF events have not been observed over the Amazon rainforest, mostly because vapours that are condensable at cold conditions remain volatile at high temperatures35–38. However, several studies have demonstrated that particles form in the cold upper troposphere and are subsequently transported to lower altitudes, at which they act as CCN1,2,4,6,7,39,40.
Observation of NPF
The Chemistry of the Atmosphere: Field Experiment in Brazil (CAFE-Brazil) took place from December 2022 to January 2023 (Extended Data Fig. 1). It studied NPF by conducting several measurement flights in the upper troposphere up to 13.8 km altitude over the Amazon basin (Methods). High concentrations of freshly formed aerosol particles (dp < 5 nm) were frequently detected at altitudes above 8 km. Several condensation nuclei (CN)-counter channels were operated to measure the total particle number concentrations N2, N3, N4 and N5 larger than the lower detection cut-off diameters of 2, 3, 4 and 5 nm, respectively, and the differences, for example, N2–5, denote the concentration of freshly formed particles in the range 2–5 nm (Methods). In situ observations of the chemical transformation of precursor gases into extremely low-volatility organic compounds (ELVOCs) and the onset of rapid and strong NPF in air masses influenced by recent outflow from a nocturnal mesoscale convective system are shown in Fig. 1. By repeatedly examining the same air masses while flying a holding pattern centred on the outflow, we followed the photochemical development during the night–day transition (Extended Data Fig. 2). Measurements before sunrise were used to characterize air masses at an altitude of 13 km. Higher isoprene mixing ratios of 400–850 pptv (equivalent to pmol mol−1) were found up to 3 h after cloud outflow. Trajectory calculations indicate that the air had been in contact with an electrically active mesoscale convective system that had developed south of the flight path during night-time (Fig. 2), which was sampled frequently (T1–T9 in Figs. 1 and 2). The outflow also comprised high levels of NOx. Photochemical production of OH radicals was observed to start at sunrise and increased with solar elevation, reaching concentrations of about 6 × 105 cm−3 (corresponding to 0.1 pptv) during T4. During this period, radiation reflected from cirrus layers below the sampled air masses resulted in an increase in actinic flux and enhanced rates of photochemical processing, with 300–500 pptv of NO formed from NO2 photolysis after sunrise (Fig. 1 and Extended Data Fig. 2). The reaction of isoprene with OH initiates the formation of isoprene hydroxy peroxy radicals (ISOPOO).
Extended Data Fig. 1. Overview of flight tracks, NPF events and schematic of the isoprene-driven NPF.

a, HALO aircraft flight tracks during CAFE-Brazil. RF 19, described in detail in this study, is highlighted in orange. b, NPF events over the Amazon basin and adjacent areas. Flight path segments are shown as black lines for flight altitudes >8 km for all research flights RF 05 to RF 20 in Brazil (except RF 16, and for RF 17, channel N2 is replaced by channel N3). NPF data points are coloured according to the local time of measurement. c, Transport and chemistry of isoprene-driven NPF. Emission of isoprene by vegetation, transport to the upper troposphere by nocturnal deep convection, photochemical formation of first-generation and second-generation oxidation products owing to reactions involving OH and NO at upper tropospheric temperatures, NPF from isoprene-derived organonitrates, zonal and downward transport, aerosol growth to CCN size and activation of CCN into cloud droplets. Satellite picture data in a and b from https://wvs.earthdata.nasa.gov.
Fig. 1. Time series of radicals, trace gases and aerosol particles.

Measurements were performed on 23 January 2023 over the Amazon basin north of Manaus (RF 19). a, OH and HO2. b, Isoprene, sum of the first-generation isoprene oxidation products methyl vinyl ketone (MVK), methacrolein (MACR) and isoprene hydroxy hydroperoxide (ISOPOOH) and NO. c, Isoprene-derived organonitrates C5H11O4(ONO2) and C5H10O2(ONO2)2. d, Particle concentrations N2–4 and N4–5, as well as pressure altitude. The light-red bar (T9) indicates the period between 10:05 and 10:10 local time, during which the strongest NPF was detected. Grey bars (T1–T8) represent previous sampling periods of the air mass examined at T9, traced by backward trajectories originating from the position of the aircraft during that period. The ambient temperature was about −58 °C at a flight altitude of 12.3 km during T1–T9. During the 4–5 min of the NPF event encounters, the aircraft travels more than 50 km.
Extended Data Fig. 2. Extended dataset for RF 19.

a, Actinic flux j(O1D) resulting from total spectral actinic flux densities (downward + upward). Note that the high reflectivity from a cirrus cloud deck below the aircraft increased the total flux considerably. b, OH and HO2. c, Ozone and CO. d, isoprene, sum of methyl vinyl ketone (MVK), methacrolein (MACR) and isoprene hydroxyhydroperoxide (ISOPOOH), monoterpenes. e, NO, NO2 (upper limit) and NO2 calculated from photostationary state using NO, O3 and j(NO2). f, Organonitrates C5H11O4(ONO2) and C5H10O2(ONO2)2. g, Gaseous sulfuric acid and MSA (upper limit). Note that a fraction of the signals (especially for methane sulfonic acid) is probably because of evaporation of pre-existing particles in the inlet line. h, Particle number concentrations N2, N3, N4 and N5. i, Aerosol compounds sulfate, nitrate and organics for particles >50 nm (AMS measurements). Note the high correlation between organics and nitrate also for the particles >50 nm. j, Particle concentration for particle sizes 0.25–1 µm and 1–40 µm (OPC measurements) and estimated CS for different assumptions of particle size. k, Water vapour and flight altitude. l, Latitude and longitude of aircraft position. m, Ambient temperature and pressure. n, Wind velocity and direction. For details on uncertainties and detection limits, see Methods.
Fig. 2. Flight path and air-mass back trajectories.

a, Flight path for RF 19 (grey). The diamond-shaped pattern ABCD was flown repeatedly and the NPF air mass was encountered several times (thick-coloured sections T4–T9) also before the NPF event started (T1–T3). Coloured sections T1–T8 are each slightly shifted to the right to prevent overlap. Thin yellow lines with 1-h markers indicate 3 h of back trajectories from the NPF period T9 used to calculate and identify periods T1–T8. b, Back trajectories from the locations of the aircraft between 08:05 and 08:15 local time (black and red markers) to 140 min before (yellow-centred markers) and infrared satellite image (GOES-16, band 13: 10.3 µm; https://ftp.cptec.inpe.br/goes/goes16/retangular/ch13/2023/01/) indicating the approximate cloud-top temperatures at 05:50 local time. Temperatures below −50 °C are coloured. The back-trajectory calculations used the horizontal wind speed and direction measured by the research aircraft. The air parcels in which NPF was detected (red circles) trace back to the convective cloud and were in contact with convection more recently (relative to the sampling time) than those in which NPF was not detected (black circles). Satellite picture data in a obtained from https://wvs.earthdata.nasa.gov, NASA Worldview Snapshots.
Further reactions lead to forming first-generation and second-generation products, including various organonitrates. High concentrations of highly oxidized isoprene alkyl nitrates were observed for the first time at 08:10–08:14 local time (T4), coincident with bursts of N2–5 particles (Fig. 1 and Extended Data Figs. 2–5). The chemical ionization atmospheric pressure interface ime-of-flight (CI-APi-TOF) mass spectrometer can detect highly oxidized ELVOCs as well as inorganic species such as sulfuric acid41–43, which cause NPF (Methods). The CI-APi-TOF data were strongly dominated by the mononitrate C5H11O4(ONO2) and the dinitrate C5H10O2(ONO2)2 of isoprene (Fig. 3), causing nucleation and the early growth of the freshly formed particles (Methods and Extended Data Fig. 3). These molecular properties have been confirmed by laboratory observations of isoprene nitrates in charged molecular clusters and neutral particles at upper-troposphere conditions44. The CI-APi-TOF measurements represent a combination of gas phase and nanoaerosol composition owing to ram-pressure-induced adiabatic heating and, thus, partial evaporation of molecular clusters and nanoparticles in the inlet system (Methods).
Extended Data Fig. 5. Time series and mass-defect plots for CI-APi-TOF data before and during NPF events of RF 19.

a, Time series of the most prominent mononitrates and dinitrates C5H11O4(ONO2) and C5H10O2(ONO2)2, representative IP0N compound C5H12O6 and MSA. b, Signals strongly shift between an air parcel that is not influenced by the fresh convection (which contains larger amounts of pre-existing particles that probably contribute to the MSA signal owing to particle evaporation in the inlet) and signals during the first (c) and second (d) encounters of the strong NPF event (T4 and T5). High concentrations of the mononitrates and dinitrates are rapidly forming.
Fig. 3. Molecular composition detected with the CI-APi-TOF mass spectrometer during the strongest NPF event of RF 19 (T9, 10:06–10:10 local time).

a, Mass defect versus mass-to-charge ratio with labelling of the molecular composition for prominent peaks. For clarity, the nitrate reagent ion (NO3−, m/z = 62) has been omitted from all labels. Circle size indicates peak intensity and colour indicates isoprene oxidation products without a nitrate group (IP0N, green), mononitrates (IP1N, magenta), dinitrates (IP2N, blue) and MSA signals (orange). b, Saturation vapour pressure C* of isoprene and its oxidation products as calculated with the SIMPOL model49. The compounds C5H12O6, C5H11O4(ONO2) and C5H10O2(ONO2)2 are ELVOCs at −58 °C. c, Reaction scheme leading to the formation of C5H12O6, C5H11O4(ONO2) and C5H10O2(ONO2)2. The molecular structures shown are exemplary. IVOCs, intermediate-volatility organic compounds; LVOCs, low-volatility organic compounds; SVOCs, semivolatile organic compounds.
Extended Data Fig. 3. Details of NPF event during RF 19.

a, IP0N, IP1N and IP2N measured with the CI-APi-TOF mass spectrometer as well as IP0-2N = IP0N + IP1N + IP2N and MSA. b, Calculated saturation ratio for IP0N, IP1N and IP2N, as well as estimated saturation ratio necessary to overcome the Kelvin barrier for particle diameters of 1.0, 1.1 and 1.2 nm, respectively (Methods). c, Particle concentrations N2, N3, N4 and N5 as measured by four channels of the FASD instrument. d, Estimated maximum of the CS when assuming all particles N5 to have maximum sizes of 20, 50 or 100 nm. During T3, when NPF is first observed by the initial increase of N2, the CS is less than 2 × 10−4 s−1.
The air mass in which the first NPF burst was observed at 08:10–08:14 local time (T4) initially contained a low number of pre-existing particles (N2 < 3,000 cm−3) and hence a low CS, estimated at CS < 1 × 10−3 s−1 (T2 and T3; Methods and Extended Data Fig. 3). Although no N2–5 particles were detected in T2, several hundred more particles were detected in the 2-nm CN-counter channel during period T3, indicating the onset of NPF (Extended Data Fig. 3). Within the 20 min between T3 and T4, more than 25,000 cm−3 N2–5 particles were formed, which corresponds to a formation rate J2 for particles >2 nm of about 20 cm−3 s−1. For this first NPF burst, particle concentrations measured in all CN-counter channels increased by at least an order of magnitude, except for the largest channel (>5 nm), which only increased slightly over previous background values. This implies a growth rate of about 9 nm h−1 from an initial cluster size of around 1 nm up to 4 nm. At the subsequent encounter of the air mass, another 20 min later (T5), the 5-nm channel shows an increase by one order of magnitude, indicating growth of the 2–5-nm particles to sizes beyond 5 nm. The growth rate represents an estimate, limited by the uncertainties of cut-off diameters of the CN-counter channels and air-mass inhomogeneities. Later in the day, N2–5 particles were ubiquitous throughout the upper troposphere, indicating that NPF events are widespread, and nucleation plumes disperse and mix with background air. At that time, the numbers of N2–5 particles did not reach the same levels as during the early morning, indicating reduced availability of precursors and particle abundance owing to coagulation and mixing. During the later flight sections, we could not repeat quasi-Lagrangian air-mass encounters, thus formation and growth rates cannot be estimated. Nevertheless, the CN-counter measurements confirm the presence of N2–5 particles.
Formation of organonitrates
The highest concentrations of nitrates and N2–5 particles were detected at 10:06–10:10 local time (T9), for which CI-APi-TOF data are shown in Fig. 3. Isoprene oxygenated organic molecules comprise about 97% of the detected reaction products, with two compounds dominating, C5H11O4(ONO2) and C5H10O2(ONO2)2. We categorized all isoprene oxidation products detected by the CI-APi-TOF mass spectrometer into non-nitrogen-containing, mononitrates and dinitrates (IP0-2N = IP0N + IP1N + IP2N). The CI-APi-TOF mass spectrometer only detects oxidized isoprene products with number of oxygen atoms ≥ 4 and with increasing sensitivity with the number of oxygen atoms and functional groups41 (Methods). Therefore, these measurements represent lower limits of the ambient concentrations of compounds that contain 4–9 oxygen atoms and, although species such as C5H8O4 are detected, they may be underestimated.
The two main compounds C5H11O4(ONO2) and C5H10O2(ONO2)2 are the likely products of second-generation OH oxidation in which the intermediate RO2 radicals react twice (sequentially) with NO to form the dinitrate C5H10O2(ONO2)2 or with HO2 and NO to form the mononitrate C5H11O4(ONO2), respectively (Fig. 3c). The corresponding first-generation intermediate closed-shell products are isoprene hydroxyl peroxide (ISOPOOH, C5H10O3), formed when ISOPOO reacts with HO2, and isoprene hydroxy nitrate (IHN, C5H9O(ONO2)) in the case of NO reaction. The isomeric structures of the first-generation and second-generation products shown in Fig. 3c are exemplary; other formation pathways may exist and further reaction pathways (for example, forming peroxy nitrates RO2NO2 from reaction with NO2) complement the scheme. Although isoprene oxidation chemistry has been investigated in detail, including under high-NOx conditions11,45–48, studies at low-temperature and low-pressure conditions in the upper troposphere are lacking. The reaction of NO with the RO2 peroxy radical can produce either nitrates or alkoxy radicals and NO2. The branching ratio for this reaction is an intricate function of temperature, pressure, molecular size and structure. Still, at the low-temperature conditions in the upper troposphere, the branching ratio probably shifts towards the termolecular channel, which produces organic nitrates11.
Further isoprene nitrate compounds include, for example, C5H11NO8 and C5H10N2O9, which are probably peroxynitrates formed from the reaction of the RO2 radicals with NO2. These compounds were also detected, but at 10–40 times lower abundance compared with the respective alkyl nitrates (Fig. 3). The lower abundance is explained by the much lower concentration of NO2 (as derived from the photostationary state approximation) compared with NO (Extended Data Fig. 2).
We used the SIMPOL model49 to estimate the volatility and the supersaturation ratio for C5H12O6, C5H11O4(ONO2) and C5H10O2(ONO2)2 (Methods). At −60 °C in the upper troposphere, all three compounds have a saturation vapour pressure, C*, of about 3 × 10−7 µg m−3, which is in the volatility class of ELVOCs50. At T9, a total supersaturation ratio of 0.5–1.0 × 105 is calculated. For these conditions, C5H11O4(ONO2) and C5H10O2(ONO2)2 are expected to participate in particle nucleation and early growth. It can be expected that the nucleation is boosted by the involvement of IP0N, as well as small amounts of inorganic species, such as sulfuric acid44. The influence of H2SO4 on the nucleation cannot be directly inferred from our observations as the sulfuric acid concentration was low, below the detection limit of about 2.0 × 106 molecules cm−3 (Methods).
Role of monoterpenes and ions
Highly oxidized organic molecules (HOMs) generated from the oxidation of monoterpenes such as α-pinene have been suggested to cause NPF in the tropical upper troposphere owing to their extremely low or ultralow volatility42,43. Our observations at high altitudes show no evidence of monoterpene-derived HOMs (neither nitrates nor non-nitrates) and therefore do not support this hypothesis. We only observed typical monoterpene-derived HOMs in the Amazon boundary layer. Even in fresh convective outflow, the monoterpene mixing ratio in the upper troposphere was typically well below 60 pptv (Extended Data Fig. 2), providing insufficient precursor concentrations to explain our NPF data. Furthermore, HOM formation in the upper troposphere is too slow43 for monoterpenes to play an important role in NPF or early particle growth, as the HOM yield decreases with decreasing temperature.
Ion-induced nucleation51,52 could play a notable role in NPF, in which ions produced from galactic cosmic rays may promote IP-OOMs nucleation44. This nucleation pathway is limited by the ion pair production rate in the tropical upper troposphere, which may reach 40 cm−3 s−1 (ref. 51). A NPF rate of about 20 cm−3 s−1, as determined by our observations, is thus within this range. Nevertheless, our observations show that the limiting factor for NPF is not the presence of ions (which are ubiquitous) but, rather, is determined by the photochemical generation of ELVOCs from isoprene.
Diel cycles of NPF
Our aircraft measurements, aggregated from 11 flights, show a pronounced diel cycle for the concentration of N2–5 particles, NPF events and isoprene oxidation products in the tropical upper troposphere (Fig. 4 and Extended Data Figs. 6 and 7). Here NPF events are defined conservatively from the differences of the 2-nm and 5-nm CN-counter channels by 0.7N2 − 1.3N5 > 0 (Methods). The concentration of N2–5 particles at night and in the early morning was generally small and no NPF events were detected before 08:00 local time. About 2 h after sunrise, we frequently detected high concentrations of N2–5 particles, dominating the total particle number concentration. Although N2–5 particle concentrations were negligible before the events, N2–5 was frequently greater than 10,000 scm−3 (converted to standard conditions according to the International Union of Pure and Applied Chemistry (IUPAC): 273.15 K and 1,000 hPa) during the events. NPF events were similar to those observed near the Earth’s surface in other environments in extratropical regions34, that is, in terms of abrupt occurrence of particles, strong initial growth, the role of photochemical production of the nucleating and condensable species and the large spatial extent of the events34. During a dedicated flight, the high N2–5 particle and organonitrate concentrations were observed in an area extending more than 300 km across and the two vertical layers in which the NPF was detected each extended over more than 0.5 km (Extended Data Fig. 8). During all flights, NPF events were associated with the occurrence of isoprene nitrates IP1N and IP2N (Fig. 4 and Extended Data Fig. 4). Shen et al.44 studied isoprene-driven NPF for upper-troposphere conditions around −30 and −50 °C with and without NOx at the CLOUD chamber facility. For isoprene + NOx conditions, both the gas-phase IP-OOMs spectrum as well as the resulting nucleation rates are in good agreement with our field study (see discussion in Methods, Extended Data Fig. 9 and Extended Data Table 1). Furthermore, Shen et al.44 show with APi-TOF measurements that isoprene organonitrates participate directly in the initial cluster formation. Although they found that, without NOx, IP-OOMs nucleate even stronger than in the presence of NOx, nitrate-dominated IP-OOMs still lead to substantial nucleation rates, which are in agreement with our findings. During CAFE-Brazil, for all observed NPF events, the CI-APi-TOF spectra were dominated by nitrate IP-OOMs, IP1N and IP2N, as the presence of isoprene in the tropical upper troposphere is intrinsically coupled with high NOx conditions through the NOx production from lightning during the deep convective transport. We found strong NPF events during all five morning flights in the upper troposphere. Therefore, it seems likely that, at least at the beginning of the wet season (December to January), such events occur every morning over a large part of the Amazon, affected by the outflow of night-time deep convection accompanied by lightning.
Fig. 4. Diurnal cycle and altitude dependence of NPF events, N2–5 particles and precursor gases.
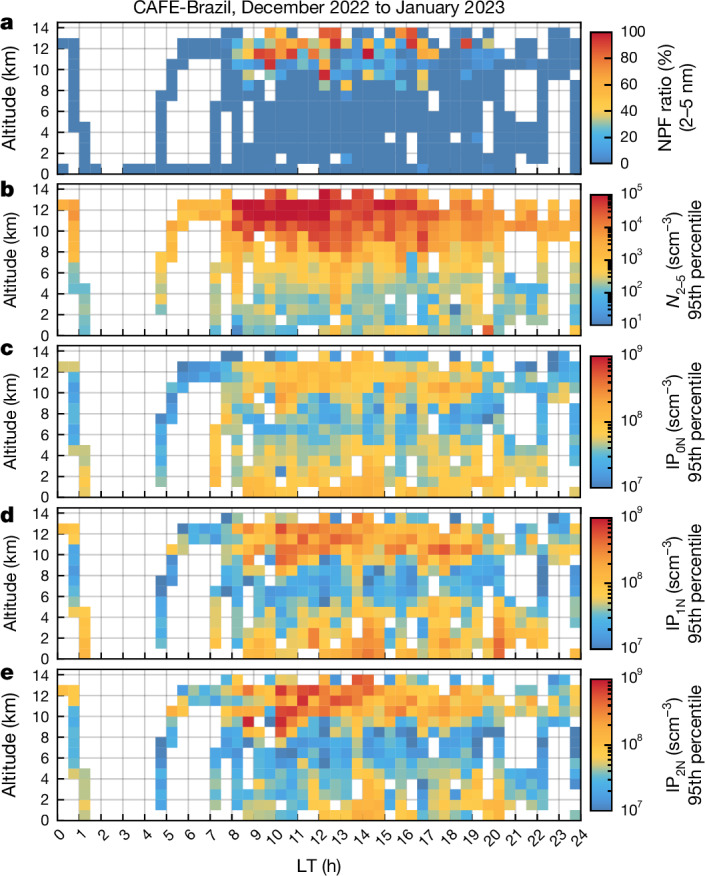
Pixels are calculated as bins of 1 km altitude and 30 min time and combine data from 11 research flights over the Amazon basin. a, Frequency of occurrence of NPF events, defined as the ratio of NPF events to the total number of measurements for each pixel. b, Concentrations of N2–5 particles. c–e, Isoprene organonitrates IP0N (c), mononitrates IP1N (d) and dinitrates IP2N (e). Panels b–e show the 95th percentile of the concentration, that is, 5% of the data contributing to a pixel are even higher than the value indicated by the colour. In the upper troposphere, high concentrations of organonitrates and N2–5 particles occurred frequently, whereas concentrations were low in the lower and middle tropospheres and during the night. Data influenced by the Manaus plume and by biomass burning have been removed. For direct comparability, all concentration data are normalized to standard conditions (273.15 K, 1,000 hPa).
Extended Data Fig. 6. Altitude profiles for N2–5 particles and isoprene oxidation products IP0,1,2N measured by the CI-APi-TOF mass spectrometer.

All data were converted to standard conditions (273.15 K, 1,000 hPa) for altitude-independent comparability and comparability with previous studies1,2. Converted to standard conditions, maximum particle concentrations N2–5 for upper tropospheric NPF events were frequently larger than 100,000 scm−3. Data for flights RF 09 to RF 20, excluding RF 16 and using N3–5 for RF 17. Data influenced by biomass burning and by the Manaus plume have been removed. Grey lines denote average concentrations and shaded areas give 5th to 95th percentiles.
Extended Data Fig. 7. Frequency of NPF events detected during RF 09 to RF 20.

a, Data from Fig. 4 are binned vertically into boundary layer (0–2 km), lower and middle free troposphere (2–8 km) and the upper troposphere (>8 km). Time bins with respect to day-time when NPF is observed (08:00–18:00 LT) as well as night-time and early morning (before 08:00 LT and after 18:00 LT). b, The number of measurements per pixel for the data shown in Fig. 4. The number in each pixel denotes the number of contributing aircraft flights. Numbers in red denote pixels with at least three flights and 20 data points per flight. The colours denote the total number of data points obtained, with each data point representing a 1-min average value during a flight. Data from RF 16 are excluded; N2–5 is replaced by N3–5 for RF 17.
Extended Data Fig. 8. Spatial extent of the NPF event observed during RF 17, measured in the morning of 18 January 2023.

a, Continuously high concentrations of N3–5 particles are detected in an area with an extent of more than 300 km in two layers between 10.6–11.5 and 12.0–12.6 km. A cirrus cloud deck was present at altitudes below 10.8 km. b, High concentrations of dinitrates IP2N were also detected. c, Vertical profile of the number concentration of N3–5 along the latitudinal cross-section, indicated by colour. d, Vertical profile of the number concentration of IP2N along the latitudinal cross-section, indicated by colour. Satellite picture data in a and b obtained from https://wvs.earthdata.nasa.gov.
Extended Data Fig. 4. Correlation of N2–5 particle concentration with trace gases.

Correlation of N2–5 with IP0N (a), IP1N (b), IP2N (c), MSA (d), sulfuric acid (e) and relative humidity (f). Coloured data represent research flights RF 09 to RF 20, excluding RF 16, and for RF 17, channel N2 is replaced by N3 for technical reasons. Only data during NPF events measured between 08:00 and 18:00 local time (LT) and for altitudes >8 km are shown (see Fig. 4 and Extended Data Fig. 8). Data points with black circles represent RF 19. Correlation coefficients represent the entire dataset.
Extended Data Fig. 9. Comparison of isoprene-driven NPF of CAFE-Brazil RF 19 with the CLOUD isoprene + NOx experiment.

Mass-defect plots for NO3−-CIMS spectra from CAFE-Brazil, RF 19, T9, at −58 °C (same data as in Fig. 3a) (a) and Shen et al.44, adopted from their Fig. 3d, measured at the CLOUD chamber at −48 °C (b). Note that the original Fig. 3d in Shen et al. also contains the signals from NH4+-CIMS and Br−-CIMS instruments, which extends the range towards lower-oxygen-content molecules. Note also that their Fig. 3d shows the signals with the mass and mass defect of the reagent ion removed. We have added the effect of the NO3− clustering here to make the data directly comparable with Fig. 3a of our study. Marker size is linearly scaled to concentration. The CLOUD NO3−-CIMS has a lower limit of detection (2 × 104 cm−3 NO3−-CIMS at CLOUD versus 1 × 106 cm−3 for the NO3−-CI-APi-TOF aircraft instrument at CAFE-Brazil) and thus can detect many smaller peaks. During RF 19, T9, the overall signal intensity of IP0-2N was higher than in the CLOUD experiment, so the marker size in the lower panel has been scaled down by a factor of 5 for comparability. The red edge indicates C5H10O2(ONO2)2 clustered to a NO3− reagent ion, which is the highest peak in RF 19, T9. Panels c–j show the conditions during time intervals T1–T9 from RF 19 (this study) and from the CLOUD isoprene + NOx experiment44. c, Concentrations of IP0N, IP1N and IP2N. d, Relative fractions of IP0N, IP1N and IP2N. e, Ratio of NO to HO2. f, Mixing ratios of isoprene, NO and NO2. g, Rate coefficients for RO2 + HO2 and for RO2 + NO reactions. h, Rate coefficients kRO2+HO2 and kRO2+NO. i, Organonitrate yield α. j, Ratio of (kNO+RO2*[NO]*α)/(kHO2+RO2*[HO2]).
Extended Data Table 1.
Comparison of NPF conditions during CAFE-Brazil RF 19 with the CLOUD experiment

Median concentrations, rate constants and meteorological parameters for each time interval (T1–T9) of RF 19, as well as data from Shen et al.44, for an isoprene + NOx experiment at the CLOUD chamber (same experiment as shown in Fig. 3d in ref. 44). For retrieving the values within each T interval, we selected a time frame corresponding to measurements from a consistent air mass, as determined by the distinct enhancements in our experimental data. Note that, for determining IP0,1,2N for CLOUD Fig. 3d (last column) and their fractions, only data from the NO3−-CI-APi-TOF is used.
The aggregated concentration measurements of the IP0N, IP1N and IP2N compounds show a similar diel cycle and altitude dependence as the N2–5 particles (Fig. 4). To be representative of pristine conditions, data that were measured during two encounters with biomass-burning plumes and data influenced by pollution emissions from the city of Manaus have been removed. Note that all data of Fig. 4 (as opposed to Figs. 1–3) were converted to standard temperature and pressure conditions for altitude-independent comparability and comparability with previous studies1,2. Overall, we observed NPF events during nearly 30% of measurements conducted at altitudes between 8 and 14 km and between 08:00 and 18:00 local time (Extended Data Fig. 7). At altitudes <8 km at any time and at >8 km during the night and in the early morning before 08:00 local time, NPF events represented less than 1%. Notable amounts of IP0N, IP1N and IP2N are also observed at altitudes <8 km, particularly in the boundary layer at midday, but the compounds are too volatile at these temperatures for NPF to occur (Figs. 3 and 4). We note that our measurements do not represent random averages, as the measurement flights had specific objectives, including the identification of NPF events in the upper troposphere.
Atmospheric implications
The newly formed particles in the upper troposphere can be transported over large distances well beyond the Amazon region27. During transport, the aerosol size distribution changes owing to coagulation and continued condensation of low-volatile species. A fraction of the newly formed particles is expected to be transported downwards38,40, affecting the abundance of CCN at low altitudes in the tropics1,6. The modelling study in ref. 4 suggests that 35% of the low-altitude CCN were initially formed by nucleation in the free and upper troposphere4. Williamson et al.1 demonstrated that descending dry air in subsidence over the oceans contains more CCN-sized particles than moist air at low altitudes, indicating that NPF in tropical convective regions can increase the CCN concentrations of the lower troposphere. Because higher CCN concentrations can substantially influence the cloud microphysical and optical properties8, the process probably affects the atmospheric radiation budget and the climate5.
To put this into perspective, more than 7,000 mesoscale convective systems occur over the Amazon each year, covering a surface area of more than 40,000 km2 per system, typically lasting for 4 h (refs. 53,54) and with high lightning activity21,55. Therefore, NPF from the oxidation of biogenic isoprene combined with lightning NOx may represent a notable mechanism of particle production in the tropical upper troposphere. It seems likely that this also occurs in the upper troposphere over other tropical forests, that is, in Central Africa, Southeast Asia and Northern Australia, that are also characterized by strong isoprene emissions9,10 and ubiquitous deep convection associated with lightning.
Methods
The CAFE-Brazil mission took place in Brazil between 30 November 2022 and 29 January 2023, which covers the end of the dry-to-wet season transition and the beginning of the wet season in the Amazon. The German High Altitude and Long Range Research Aircraft (HALO) was stationed at Manaus International Airport, conducting a total of 16 local research flights (143 flight hours), as well as the transfer flights between Germany and Brazil (26 flight hours). An overview of the flight tracks is shown in Extended Data Fig. 1a. The research flights, at altitudes between 0.3 and 13.8 km, covered the region from 11° 33′ S to 4° 40′ N and from 72° 33′ W to 33° 50′ W. The aircraft was equipped with instrumentation to measure the in situ concentration of trace gases, radicals, aerosol number concentration and several aerosol physical and chemical properties. Descriptions of the methods used for the detection of the gas compounds and aerosol properties are given here.
CI-APi-TOF mass spectrometer
The CI-APi-TOF mass spectrometer56,57 measures gaseous molecules that can contribute to NPF (that is, aerosol nucleation and initial growth up to detectable particle size). Gases such as isoprene oxidation products (IP0,1,2N), methanesulfonic acid (MSA) and sulfuric acid are detected. The instrument was specifically designed and certified for aircraft use and it is optimized for low inlet losses, a constant ionization pressure and minimal internal gas consumption.
The CI-API-TOF mass spectrometer uses a trace-gas inlet initially developed to measure OH radicals. It is almost identical to the inlet used for the HydrOxyl Radical measurement Unit based on fluorescence Spectroscopy (HORUS) instrument. The sampling location is placed at a sufficient distance from the fuselage to avoid the influence of the aircraft’s boundary layer. The inlet uses a set of shrouds and a flow restrictor to decelerate the air by a factor of 10 to around 25 m s−1 before sampling into the inlet line to reduce turbulence58. The flow restrictor, however, also causes a ram-pressure effect and thus adiabatic heating of the sample air depending on ambient pressure and air velocity, which will be discussed below in more detail.
The sampling line consists of a 20.5-mm inner diameter 1.8-m-long stainless-steel tube with an 8-mm orifice at the beginning (located in the centre part of the inlet). The sampling line has two bends with radii of about 120 and 500 mm, respectively, to enable installation close to the cabin wall. The inlet line is thermally insulated and has a temperature sensor 300 mm downstream of the sampling position inside the inlet. The sample flow is kept constant at 25.0 slpm for all altitudes to reduce wall losses, while still being in a laminar flow regime.
At the end of the sampling tube, the air reaches the SCORPION (Switchable Corona Powered Ion Source), which consists of a pressure-control and an ionization stage. The pressure-control stage comprises two sequentially placed and conically shaped orifices with 1.4 mm inner diameter each. Between these two orifices, a PID-controlled solenoid regulation valve allows for variable pumping, which provides a constant pressure of 200 hPa in the ionization region. The ionization region is located directly after the second orifice. It consists of a 20.5-mm inner diameter stainless-steel tube and two orthogonally attached ion source units, of which only one is used at any given time. The ion source uses a corona discharge to produce (HNO3)0,1,2NO3− reagent ions from gaseous HNO3. Because a corona discharge also produces OH radicals, which could alter the chemical composition of the sample air, we implemented a counter-flow regime, such that the nitrate reagent ions are pushed towards the sample air by means of an electric field, whereas the gas flow, which also carries OH radicals, is directed away from the sample air towards the exhaust.
The nitrate reagent ions are mixed with the sample air and travel along the main drift tube (130 mm length) with a reaction time between 180 and 350 ms, depending on altitude. Although the ionization pressure is kept constant at 200 hPa, the flow along the drift region varies between 2.7 slpm at ground level and 1.4 slpm at 12 km altitude, causing different reaction times. The higher flow at lower altitudes is needed to maintain the ionization pressure at 200 hPa with the given orifice diameters. Although the reaction times are around 3–5 times longer than in an Eisele–Tanner-type ion source59 (about 50 ms), the ionization pressure is also lower by a factor of 5, leading to a roughly comparable number of collisions between reagent ions and sample gas. However, because of the extra orifices in the pressure stage and a relatively long inlet line, the overall sensitivity of SCORPION is lower by about one order of magnitude compared with a nitrate reagent ion long time-of-flight (LTOF) mass spectrometer instrument as deployed, for example, at the CLOUD chamber42,43. The detection limit for SCORPION is between 5 × 105 and 5 × 106 cm−3.
At the end of the SCORPION drift tube, the ions enter the Tofwerk time-of-flight mass spectrometer by means of a 350-µm inner diameter orifice. We use an HTOF with a resolution of around 4,000 at the m/z range 250–300 amu. The data are recorded at 1 Hz; however, it is averaged to 10 s resolution before post-processing and high-resolution peak fitting in Tofware (version 3.2.5, Aerodyne).
To derive the ambient concentrations of IP0,1,2N, several pressure-dependent and temperature-dependent correction factors have to be applied to the fitted peak intensities as follows:
Here (IP0,1,2N)cps represents the fitted peak intensities in counts per second. Tr(m/z) is the m/z-dependent correction factor for the relative change in instrument transmission efficiency60. For the IP0,1,2N m/z range, this correction is about 10–20%, depending on the exact m/z ratio.
A pressure-dependent calibration factor is applied to the normalized signal (at 12.2 km altitude, C230hPa = 6.5 × 1010 cm−3, whereas at ground level, C1000hPa = 2.1 × 1011 cm−3). This calibration factor was experimentally estimated for gaseous sulfuric acid by generating a known amount of gaseous sulfuric acid by means of ultraviolet (UV)-induced OH production (from photolysis of H2O) and subsequent oxidation of sulfur dioxide61 to sulfuric acid62. We constructed a dedicated calibration rack for the CI-APi-TOF mass spectrometer, similar to that described in ref. 62. The calibration unit can be operated at pressures between 200 and 1,000 hPa.
After applying the calibration factor, a background correction of the signal is performed (BG). Background measurements were performed in flight by overflowing SCORPION with synthetic air from the internal gas bottle. This can, however, only be done above 9 km altitude, as at lower altitudes more gas would be needed to overflow the ion source than provided by the internal gas bottle. Typically, 2–3 background measurements are performed per flight, each lasting 10 min.
The factor I(pi, Ti, Fi) represents the correction for losses to the wall in the 1.8-m-long sampling line and depends on inlet pressure pi, inlet temperature Ti and inlet flow Fi. It is based on the parametrization for straight tube losses and thus ignores the two curves in our inlet tube. The inlet loss estimation uses the experimentally determined diffusion coefficient for gaseous sulfuric acid and its pressure-dependent and temperature-dependent parametrizations61. The inlet correction factor is about 1.65 for a typical high-altitude flight scenario (12 km altitude).
The correction factor A(pa, pi, Ta, Ti) scales the concentration levels from inlet temperature Ti and pressure pi conditions to ambient temperature Ta and pressure pa conditions. This scaling is necessary as the ram pressure caused by the flow restrictor increases the inlet pressure compared with ambient conditions (at 12.2 km altitude, at which most of research flight (RF) 19 was flown from 188 hPa ambient pressure to 268 hPa inlet pressure). This pressure increase also induces an adiabatic heating of the sampling air. Further heating comes from the limited thermal insulation of the inlet line. At 12.2 km altitude, we measure a temperature increase from −58 °C (ambient) to −13 °C (inlet), so ΔT = 45 °C. Although the inlet residence time at these conditions is rather short (0.38 s), this temperature increase could lead to the evaporation of molecules from the aerosol to the gas phase, which could enhance our measured gas-phase signals. At −58 °C, the main IP0,1,2N products are ELVOCs, whereas they shift to the low-volatility organic compounds range at −13 °C (Fig. 3). Especially for freshly nucleated particles with diameters in the size range in which the Kelvin effect has a role, this could cause evaporation. Therefore, the measurements are considered to be a combination of pure gas-phase concentration and potentially re-evaporated aerosol-phase molecules. However, even if a large fraction of our signal would originate from evaporation of freshly nucleated particles, this still highlights the crucial role of isoprene-derived oxidation products for the NPF process in the upper troposphere over the Amazon.
The measurement of sulfuric acid was affected by a sulfur contamination inside the stainless-steel vessel containing the liquid HNO3 supply of the ion source. This led to an increased instrumental background for sulfuric acid of about 2 × 106 cm−3 for our measurements at altitudes above 8 km.
The overall uncertainty of the CI-APi-TOF measurements is ±62%. This consists mainly of the uncertainty of the sulfuric acid calibration factor of 55%, as well as the uncertainty of the mass-dependent transmission correction (20%) and the inlet loss correction (20%). No calibration standards are available for the measurement of ELVOCs from isoprene or monoterpenes. Therefore, we use the same calibration factor as determined for the sulfuric acid measurements. The sensitivity of the CI-APi-TOF instrument is high for the detection of highly oxidized organics with many functional groups, for example, HOMs from monoterpenes. For those, this approach is well established42, but the CI-APi-TOF instrument is expected to be less sensitive for the smaller IP-OOMs, especially when the molecules contain few oxygen atoms. This general dependence is confirmed by the comparison of nitrate reagent ions with, for example, bromide or iodide reagent ions41. The concentrations of the isoprene oxidation products detected by the CI-APi-TOF mass spectrometer should therefore be considered as lower limits. Compared with the IP0N, IP1N and IP2N reported in ref. 44, the CI-APi-TOF instrument will detect smaller fractions of these compound classes, as more mass spectrometers using other reagent ions are used in ref. 44 to complement the IP0,1,2N measurements. Nevertheless, the relative changes of individual compounds over time, as shown, for example, in Extended Data Fig. 5, are not affected by this effect.
We also apply the calibration factor estimated for sulfuric acid to derive the concentration of IP0-2N, as a direct calibration for IP0-2N is not possible. It was shown that HOMs with sufficient oxygen content, that is, a sufficient number of functional groups to cluster with NO3−, are charged with nitrate reagent ions at the kinetic limit, similar to sulfuric acid41,63. Riva et al.41 conducted a detailed comparison of various reagent ions and their respective sensitivities towards pure organic as well as nitrate HOMs derived from monoterpene oxidation. They found that both nitrate and non-nitrate HOMs can be charged by NO3− reagent ions at the kinetic limit as long as HOMs contain more than six oxygen atoms in the non-nitrate case and more than seven oxygen atoms in the nitrate case. Assuming that these results are transferable to IP0-2N, this means that some of our reported IP0-2N could be charged below the unit charging efficiency of the kinetic limit. This leads to an increase in calibration factor for these species and our reported concentrations represent a lower limit estimation, especially for low-oxygen-content IP0-2N.
Aerosol number concentration and nucleation-mode particles
The Fast Aerosol Size Distribution (FASD) instrument is a compact multichannel system to detect newly formed particles. It was developed specifically for operation aboard the HALO aircraft. The system combines the concept of well-established commercial ultrafine condensation particle counters64 (CPCs) with central temperature management, central butanol vapour supply, central pressure control and a new type of flow system. The prototype, designed and built at the Max Planck Institute for Chemistry (MPIC), simultaneously measures aerosol concentration in ten channels every second. The ten-channel device is about the size of two commercial ultrafine CPCs. Similar aircraft-based instruments use several CPCs65–67.
For each CPC channel, the sample air is fed by means of a short (6 mm) capillary into a heated mixing chamber, surrounded by particle-free butanol-rich sheath air and fed into the cold condensation region. The supersaturated butanol condenses on the aerosol particles, causing them to grow sufficiently to be detected in a downstream optical particle counter. To minimize diffusional losses, all channels share a central sample line with a distance of only about 38 mm between adjacent inlets and the inlet capillaries protrude into the core sample flow.
The FASD instrument gradually reduces the butanol content of each sheath flow in a dilution chamber between the saturator and the channels. The exhaust air from each channel is collected and cleaned of particles by a HEPA filter and returned to the saturator and dilution stage by a central pump. There is a total of four pressure stages in the sheath flow loop, with the highest pressure in the saturator and the butanol dry air upstream of the dilution. Two valves control the saturated and dry air flow ratio into the dilution chamber. Although the saturated air is introduced at a single point, the dry air flow is split to replenish the air flowing to each channel. The mixing, condensation and detection areas of each channel are close to the ambient pressure level in the inlet and outlet lines. Finally, the lowest pressure is upstream of the sheath flow pump and also drives the removal of butanol that condenses on the walls of each condenser. This butanol is collected in a reservoir and can be reused as normally no water vapour condenses.
A valve controls the total sample flow. This valve allows some air to leave the sheath flow loop from the dilution stage to the exhaust line. In total, an equal amount of air enters through the short inlet capillary of each channel. Another valve sets the desired flow through the dilution stage and allows some air to flow directly from the end of the dilution stage to the sheath flow pump. Flow uniformity is achieved by carefully selecting the flow resistances, which results in pressure differentials between stages in the range 10–20 hPa. The condenser sections are cooled to a target temperature of 10 °C by a water cooling system. The waste heat is transferred from the cold water cycle to the hot water cycle through Peltier elements, in which the heat is eventually released to the ambient air through an external radiator. To avoid unwanted butanol condensation, it is essential that the dilution chamber and the individual sheath flow transfer lines and mixing chambers of each channel maintain temperatures above the saturator temperature. This is achieved by heating the saturator indirectly, while keeping the other parts actively warm, resulting in a permanent temperature gradient that prevents unwanted condensation.
The particle activation is determined by the temperature difference between condensation and saturation, the pressure differentials and the dilution factor. By measuring the activation curves under given conditions and comparing them with theoretical calculations, the activation behaviour can be derived for fluctuating measurement conditions65. Initial measurements of selected particle sizes in the range 2–30 nm show that the performance of the first channel of the prototype is comparable with a commercial ultrafine CPC, whereas the measured values of the other channels fit to an effective dilution to around 75% per channel.
Although diffusional losses between the channels are not apparent, the final calibration requires further measurements65,67. Therefore, for this study, the theoretical cut-off diameters are obtained using Kelvin’s equation68 with a dilution of 75% per stage. For each measurement time step, the corresponding particle activation diameters can be calculated from the actual temperatures and pressure differences and are typically in the range 2–6 nm. Measurement periods without NPF events can be used to determine systematic deviations of sample flows owing to slight variations in flow resistances. To keep the measurement conditions of the channels stable, regardless of changes in ambient pressure, the FASD instrument is operated at a constant pressure of typically 200 hPa. The pressure control is achieved by continuously calculating the net flows in and out of the FASD instrument based on flow controller and pressure sensor data. The calculation results are used to drive two PID controllers that regulate the outflow by using a mass flow controller and the inflow by using a custom-made size-changing orifice69.
Proton transfer reaction time-of-flight mass spectrometry
A proton transfer reaction time-of-flight mass spectrometer46,70,71 (PTR-TOF-MS 8000, Ionicon Analytik) was used for the fast high-mass-resolution airborne measurements of VOCs (m/z < 500 amu). Isoprene, its oxidation products including methyl vinyl ketone, methacrolein and isoprene hydroxyhydroperoxide, and total monoterpenes reported in this study were measured at m/z = 69.069, 71.049 and 137.132 amu, respectively. In this technique, hydronium ions (H3O+) are used as a reagent to ionize molecules in air that have a higher proton affinity than water (693 kJ mol−1). The instrument was operated with a drift pressure of 2.2 mbar and an E/N of 137 Td. Air was drawn from outside the aircraft to the instrument through a fuselage-mounted inlet housing, in a heated 2-m-long, 0.64-cm outer diameter Teflon inlet line. Quantification of the detected compounds was performed by frequent in-flight background determinations with zero air and several ground-based calibrations using a gravimetrically prepared gas standard containing isoprene, methyl vinyl ketone and α-pinene (Apel-Riemer Environmental). At a time resolution of 1 min, the detection limit (3σ) was calculated to be 100, 41 and 25 pptv for isoprene, its oxidation products and total monoterpenes, respectively. An ozone correction for isoprene was applied on the basis of laboratory experiments and comparison with the gas chromatography–mass spectrometry data. The total uncertainty of measurement was usually below 25%. Further detailed information about the proton transfer reaction time-of-flight mass spectrometry used is given ref. 72, its response to atmospheric ozone in ref. 73 and the configuration in the aircraft is described in ref. 74.
Gas chromatography–mass spectrometry
VOCs were measured in situ using a customized gas chromatograph coupled to a commercial quadrupole mass spectrometer (Agilent Technologies 5973 MSD). The system has been described in detail previously75 and its configuration in the aircraft payload for the CAFE-Brazil campaign is given elsewhere74. In brief, ambient air is drawn at 200 scm3 through the Trace Gas Inlet (TGI, Enviscope) to the instrument by means of a heated 2-m-long Teflon line (0.6 cm outer diameter) equipped with a sodium thiosulfate ozone scrubber73. Within a series of traps in a liquid-nitrogen-cooled cryo-concentrator, the sampled air is dried (−10 °C), enriched for VOCs (−160 °C) for 1 min and then concentrated into a small volume (−160 °C) before being rapidly heated to inject the sample into the gas chromatograph. The compounds are separated by a DB-624 UI, 10 m, 0.25 mm, 1.4 µm capillary column (Agilent Technologies). The temperature programme of the custom-built gas chromatograph oven is as follows: 30 °C for 50 s, then 30 °C to 200 °C at 1.8 °C s−1 and constant 200 °C for the rest of the chromatogram. After separation, the compounds are electronically ionized (70 eV) and detected by the mass spectrometer in the selected ion mode. In the configuration used for the CAFE-Brazil campaign, more than 35 compounds could be resolved and quantified in a 2.4-min chromatogram, the overall measurement frequency being 3 min. Calibration was achieved using a gravimetrically prepared multicomponent pressurized standard (Apel-Riemer Environmental), with a stated accuracy of 5%, with calibrations being performed before, during and after each flight. Isoprene was detected at m/z = 67 amu and a retention time of 0.7 min with a detection limit of 5 pptv and an uncertainty of about 10%.
NOx measurements
Nitrogen oxides were measured through photolysis chemiluminescence with the two-channel instrument Nitrogen Oxides Analyzer for HALO (NOAH). In one channel, nitric oxide (NO) is converted to excited-state nitrogen dioxide (NO2*) by reaction with excess amounts of ozone (O3). A photon is emitted during de-excitation of NO2*, which is detected by a photomultiplier tube. The signal is converted to ambient NO mixing ratios using normal on-ground calibrations between the flights. The second channel is identical except for using a photolytic converter, in which NO2 is photolysed to NO at a wavelength of around 395 nm before the addition of O3. The conversion efficiency of the converter (the fractional conversion of NO2 to NO) was 29% during the CAFE-Brazil campaign. Owing to enhanced temperatures in the instrument and the photolytic converter, NO2 reservoir species, mostly methyl peroxy nitrate, can release NO2. Therefore, the NO2 measurement represents a sum of NO2 and thermally labile nitrates and represents an upper limit of the NO2 mixing ratios at night-time. For the day-time data, we use NO2 derived from the photostationary state (inferred from NO, O3 and j(NO2)) in this study instead (Extended Data Fig. 2). The 1 Hz detection limit for NO is 8 pptv and the overall measurement uncertainty is 5%. A detailed description of the instrument is presented in refs. 76,77. Details on the thermal decomposition of NO2 reservoir species in photolytic converters and resulting interferences can be found, for example, in refs. 77,78,79.
HOx measurements
The airborne HORUS instrument is based on the Fluorescence Assay by Gas Expansion–Laser-Induced Fluorescence of OH (FAGE-LIF) instrument as described in detail in ref. 80. It was developed specifically for operation on the HALO research aircraft and combines an external inlet shroud with an in-flight calibration system, OH and HO2 detection axes, a laser system and a vacuum system. The OH is drawn into the detection axis through a critical orifice at a pressure range of 300–1,300 Pa, depending on ambient pressure. It is selectively excited on the Q1(2) transition line (A2Σ +−X2Π, ν′ = 0, ν″ = 0) by a 3-kHz pulsed UV laser light around 308 nm. The UV laser emission wavelength is periodically tuned on and off resonance of the OH Q1(2) transition to quantify the fluorescence background. An inlet pre-injector (IPI) system is installed to remove atmospheric OH to measure the chemical OH background signal. The airborne IPI system has been redesigned to fit within the inlet shroud system, while maintaining similar operational features as the on-ground IPI installation81.
HO2 is measured indirectly through the quantitative conversion of atmospheric HO2 to OH by injection of NO within HORUS.
| 1 |
With excess amounts of NO used in the conversion of HO2 to OH, subsequent HONO formation has to be taken into account.
| 2 |
The losses resulting from internal HONO formation are dependent on pressure and amount to less than 1% above 10 km and less than 2% at ground level.
We account for the reaction of RO2 with NO leading to HO2 formation, which then generates OH in the presence of NO. We reduce the NO addition to limit the contribution of RO2 to the detected OH levels. The instrument operates in two modes during flights: one with a low NO addition, creating an internal NO concentration of 1.5 ± 0.1 × 1013 cm−3 and achieving a conversion rate of 20–40% depending on altitude, and another with a high NO addition of 7 ± 0.1 × 1013 cm−3, reaching a conversion rate of more than 95%. Furthermore, NO titrations are conducted at different altitudes to ensure accurate measurement of the HO2 contribution. The HORUS instrument 1σ accuracy is ±22.6% for OH and ±22.1% for HO2. Precision depends on ambient pressure and instrument performance.
Aerosol mass spectrometer
The composition of non-refractory aerosol particles in the diameter range approximately 50–800 nm was measured using a compact time-of-flight aerosol mass spectrometer (C-ToF-AMS)82,83. The instrument samples the aerosol particles by means of a constant pressure inlet and an aerodynamic lens into the vacuum system. The particles are flash vaporized on a 600 °C surface and the resultant gas-phase molecules are ionized by electron ionization. The ions are analysed by a time-of-flight mass spectrometer. The C-ToF-AMS has been operated on HALO since the ACRIDICON-CHUVA campaign in 2014, which also took place over the Amazon rainforest31.
Other measurements
Carbon monoxide was measured with the quantum cascade laser absorption spectrometer TRISTAR with a mean total measurement uncertainty of 3.5% (refs. 84,85).
Upward and downward spectral actinic flux densities in the range 280–650 nm are measured by combinations of two CCD spectroradiometers86,87.
The Fast AIRborne Ozone (FAIRO) instrument measures ozone with high temporal resolution (10 Hz). It combines two independent techniques, UV photometry and chemiluminescence detection88.
Data from the Basic HALO Measurement and Sensor System (BAHAMAS) are used for determination of the aircraft position, wind velocity and direction, humidity, temperature and pressure89.
Trajectory calculations
Quasi-Lagrangian sampling periods during HALO flights were identified in Fig. 1 (marked by grey shading) using the flight measurements in combination with backward trajectories as described in the following.
The sampling period with the highest number of N2–5 particles (light-red shading in Fig. 1 (T9)) was chosen as the reference period, with its limits defined as 10:05:46 and 10:10:30 local time. During this sampling period, HALO covered a distance of 62.2 km, which becomes relevant during the final computational step determining the quasi-Lagrangian sampling periods. To identify time intervals earlier during the flight in which approximately the same air mass as during the reference period was examined, backward trajectories were calculated for a set of ten parcels. These were initialized at HALO’s instantaneous location every 30 s (at 0 s and 30 s times) during the reference period. We used a simple Euler scheme with a 30-s time step to calculate the trajectories. We further assumed a constant wind velocity for each trajectory. The wind speed and direction were based on HALO measurements at the parcel initialization time. Vertical movements were not considered. These computations result in an m × 10 matrix containing the ten position vectors of the parcels initialized at HALO’s location during the reference period (p1, p2,…, p10) at each historical time (ti, ti − Δt,…, ti − (m − 1)Δt). We then calculated the Euclidean distance from each parcel to HALO’s location (in km), at each location along the trajectories. This distance is always computed with respect to HALO’s instantaneous position at the equivalent time. Finally, the quasi-Lagrangian intervals highlighted by the grey shading in Fig. 1 (T1–T8) were determined by selecting time periods along the backward trajectories for which the mean distance from the parcels to HALO was less than the sampling distance of the reference period (that is, 62.2 km).
The trajectories shown in Fig. 2 were computed using the above described method, but instead initializing 1-min-spaced parcels between 08:05 and 08:15 local time.
It should be noted that the calculated trajectories involve uncertainties, mainly because of the assumption of constant wind speed and the neglect of vertical velocity. However, tests, in which further backward trajectories were computed from each semi-Lagrangian time interval itself (not shown), indicate that the time-dependent changes in wind speed from one phase of the flight pattern loop to another did not substantially affect the estimates of the air-mass location. This indicates that we may use the approximation of constant horizontal wind speed on the spatio-temporal scales analysed for this specific flight. Also, convective clouds typically induce atmospheric gravity waves in their surroundings. Consequently, air parcels expelled by convective outflows may oscillate in the vertical dimension during horizontal displacements, remaining close to the outflow level for a few hours. Thus, vertical errors in parcel location are expected to be within a range smaller than the spatial extent of the air mass in which NPF was identified (about 62 km). We thus argue that neglecting the vertical velocity component serves as a reasonable proxy for the air-mass locations before the measurements in the near outflow proximity, in the absence of sufficiently accurate time-dependent vertical wind-speed data. This assumption also immediately breaks down when the backward trajectories come in contact with active convection.
Given the absence of a true value to evaluate parcel trajectories at this scale (note that atmospheric models provide time-dependent three-dimensional wind velocities, but these are highly sensitive to the representation of the location and structure of convective storms, making them unsuitable for this type of high-resolution analysis), the final validation should come from the tracer measurements themselves. The strong agreement between this trajectory analysis and the in situ measurements is reassuring and provides some validation of the conclusions presented here. Note that, during T8, only small amounts of N2–5 particles and IP0-2N were found, indicating that, at this time, the centre of the NPF air mass had probably moved north of section BC of the flight track (Fig. 2).
Identification of NPF
To identify a NPF event, we conservatively assumed a measurement uncertainty of 30% for each channel of the FASD instrument (including statistical uncertainty, drifts in the flows and other systematic uncertainties). The difference between N2 and N5 is classified as a NPF event if 0.7N2 − 1.3N5 > 0 cm−3 (refs. 28,66).
Condensation sink
The condensation of a vapour to aerosol particles is described by the CS (ref. 90). For vapours of ultralow or extremely low volatility, the condensation to pre-existing large particles competes with the NPF process. The size distribution of the aerosol is crucial for determining the CS. An Ultra-High Sensitivity Aerosol Spectrometer (UHSAS) was generally used in combination with the FASD CPC measurements to determine the aerosol size distribution in the size range 60–1,000 nm, but for RF 19, the UHSAS was not operational. Therefore, we performed a rough estimation of the range of the upper limit of the CS by assuming that all aerosol particles measured by the 5-nm FASD channel N5 have a size of 20, 50 or 100 nm. These three estimates are given in Extended Data Figs. 2 and 3. Before and outside the NPF events, the 50-nm or 100-nm assumptions give a reasonable range (compared with the other research flights when the UHSAS was operational), whereas during the NPF events, the <20-nm assumption is more likely to be correct.
Saturation vapour pressure
The SIMPOL model is used to obtain an estimate of the temperature-dependent saturation vapour pressures Ci* for the isoprene oxidation products49 (Fig. 3). SIMPOL is based on the group-contribution method, in which the number of functional groups of an organic molecule determines its saturation vapour pressure. For the SIMPOL-derived saturation vapour pressures, an uncertainty of one order of magnitude in the volatility distribution is assumed43. The saturation ratio Si* of a compound can be determined by calculating Si* = [ci]mi/(NACi*), with [ci] denoting the concentration of compound ci and mi its molecular mass. The saturation ratio, Si*, of a compound can then be compared with estimates of the Kelvin diameters beyond which condensation is favoured over evaporation (Extended Data Fig. 3).
Comparison of CAFE-Brazil results to the CLOUD laboratory measurements of Shen et al.
Shen et al.44 report laboratory measurements from the CLOUD chamber that investigate the role of isoprene for NPF at cold upper troposphere conditions (around −30 and −50 °C) with and without NOx. For isoprene + NOx conditions at −48 °C, they report an IP-OOMs distribution (Fig. 3d in ref. 44) that is similar to the one we report for our atmospheric measurements (Fig. 3a and Extended Data Fig. 9). Note that the figure in ref. 44 shows not only IP-OOMS data from a NO3− reagent ion CIMS similar to the CI-APi-TOF used in our study, but, in addition, data from IP-OOMs measured by further mass spectrometers using NH4+ and Br− reagent ions. This extends the range of detected compounds towards higher volatility compounds with lower oxygen content41,44. These extra compounds are not expected to drive nucleation on their own but may contribute to the growth of newly formed particles after they have reached the respective Kelvin diameter. Comparing only the NO3− reagent ion data, the CLOUD data match our mass-defect plot (Extended Data Fig. 9). Nitrates, especially dinitrates, dominate the range in both cases. In ref. 44, the NO3−-CIMS measurement shows an even stronger dominance of nitrate IP-OOMs than our study. The highest peak in our study (C5H10N2O8) is the second highest peak in ref. 44, whereas the highest peak found by Shen et al. is the closely related C5H10N2O9, which can be formed by low-temperature RO2 reaction with NO2 instead of NO (ref. 44). This is more likely in CLOUD as it was operating at a higher NO2/NO ratio owing to lower NO2 photolysis rates in CLOUD compared with the upper troposphere over the Amazon during daytime (CLOUD: NO2/NO ≈ 3.1; CAFE-Brazil, RF 19, T9, NO2/NO ≈ 1.1; Extended Data Fig. 9 and Extended Data Table 1). Nevertheless, there is good overall agreement between the spectra recorded in CLOUD and our study.
Shen et al.44 report that non-nitrate IP-OOMs are more effective for nucleation than nitrate IP-OOMs, which can be seen from lower nucleation rates at comparable concentration (extended data figure 5 in ref. 44). Even with the lower nucleation efficiency for isoprene nitrates, the authors still measured notable nucleation rates at −48 °C in a nitrate-IP-OOMs-dominated experiment (J1.7 ≈ 4 cm−3 s−1 for 2 × 108 cm−3 of IP1-2N) with a gas-phase IP-OOMs spectrum similar to the one we report, as discussed above. Furthermore, figure 3c in ref. 44 confirms that nitrate-IP-OOMs do participate in initial cluster formation. The authors also state that the role of nitrates for nucleation could increase at colder temperatures, as encountered during our flights (−58 °C for RF 19). They report a more than 100-fold increase in nucleation rate for isoprene + NOx conditions when the temperature is reduced from about −30 to −50 °C. Given the observation that, at −50 °C, nitrate-IP-OOMs are weaker nucleators than non-nitrate-IP-OOMs, which means that they do not nucleate at the kinetic limit at −50 °C, it is plausible that the nitrate-IP-OOMs-driven nucleation rate increases when the temperature is reduced from −50 °C to −58 °C. Although the CLOUD experiment could only measure as cold as −50 °C, the expected increase in nucleation rate at colder temperatures leads to a good agreement with the NPF rate of 20 cm−3 s−1 for a concentration of 2.7 × 108 cm−3 of IP1-2N reported here. We note that, even if nitrates dominate the IP-OOMs spectra, we do not rule out an important contribution of non-nitrates to the initial steps of cluster formation and nucleation.
Overall, the results of the Shen et al.44 laboratory study agree with the nitrate-dominated IP-OOMs spectra that were measured in the upper troposphere over the Amazon. Taking into account the lower temperatures during our flights, the formation rates measured in the laboratory for isoprene + NOx conditions are comparable with those estimated in our study.
The probable reason for why nitrate IP-OOMs are much more prevalent than non-nitrate-IP-OOMs is that the presence of isoprene and NOx in the upper troposphere over the Amazon region is intrinsically coupled through deep convection, as isoprene is transported rapidly from the boundary layer to the upper troposphere, and NOx is produced by lightning. Both isoprene and NOx accumulate during the night. After sunrise, photolysis leads to the production of OH and NO (with NO presence enhancing OH recycling as well). OH and NO then trigger isoprene oxidation and the corresponding RO2 termination reactions that lead to the reported nitrate-dominated IP-OOMs spectra and NPF events.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-024-08192-4.
Source data
Acknowledgements
This work was conducted under authorization 002785/2019-51 and 1.043/2022 of the Brazilian National Council for Scientific and Technological Development (CNPq). We thank the German Aerospace Center Flight Experiments (DLR-FX) facility for operating HALO, specifically, the pilots, engineers, technicians and the operations team. We thank the CLOUD team for providing the intercomparison data and discussion. We acknowledge the use of imagery from the Worldview Snapshots application (https://wvs.earthdata.nasa.gov), part of the Earth Observing System Data and Information System (EOSDIS). This research has received support from the Max Planck Society and the German Research Foundation (DFG) within the SPP 1294 HALO (316646266, 442647232, 461448963, 461450470, 461450583), the TRR 301 TP-Change (428312742, projects A03, A04 and C06), the São Paulo Research Foundation (FAPESP) (grants 2017-17047-0 and 2023-04358-9), the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (438638/2018-2, 313355/2021-5 and 440171/2022-9) and the Dr. Hans Messer Foundation. N.T. received support from the Alexander von Humboldt Foundation. M.Z.-W. received support from the Heinrich Böll Foundation. G.R.U. was supported by FAPESP grants 2021/03547-7 and 2022/01780-9. M.A.C. was supported by FAPESP grant 2022/13257-9 and by the Serrapilheira Institute (Serra – 2211-41823). R.B. received financing from the Knut and Alice Wallenberg Foundation (project 2021.0169).
Extended data figures and tables
Author contributions
J.C., M.H., L.J.B., M.L.P., N.T., B.E.K., P.H., C.M.N., L.H.P., T.Kl., K.B., A.E., A.G., A.R., A.T., A.Z., B.B., B.A.H., C.Ma., C.Mo., C.N., D.D., D.M., F.R., G.R.U., I.D.V., J.-P.C., K.K., L.O., M.A.C., M.M., M.S., M.Z.-W., N.W., O.O.K., P.J., R.D., S.R., S.S.R., S.S., T.D., T.Ke., R.I.A., M.O.A., P.A., H.F., H.H., D.L.H., L.A.T.M., C.P., U.P., J.S., J.W. and J.L. prepared the measurement instruments or participated in scientific planning and forecasting. M.H., L.J.B., M.L.P., N.T., B.E.K., P.H., C.M.N., T.Kl., K.B., S.T.A., A.E., A.G., A.H., A.R., A.T., B.B., C.Ma., C.Mo., C.N., D.D., G.R.U., K.K., L.O., M.M., M.Z.-W., N.W., O.O.K., P.J., R.D., S.R., S.S.R., S.S., R.I.A., P.A., H.H., D.L.H. and C.P. collected the data. J.C., M.H., L.J.B., M.L.P., N.T., B.E.K., P.H., C.M.N., L.H.P., T.Kl., K.B., A.E., A.G., A.Z., B.B., C.N., H.K., K.K., M.Z.-W., N.W., S.R., R.I.A., H.H., C.P., U.P., J.S. and J.L. analysed the data. J.C., M.H., L.J.B., M.L.P., N.T., B.E.K., L.H.P., K.B., S.T.A., A.K., D.M., F.R., M.A.C., M.M., M.S., M.Z.-W., R.B., S.R., R.I.A., M.O.A., P.A., J.N.C., H.H., D.L.H., L.A.T.M., C.P., U.P., A.Pos., A.Poz., J.S., J.W. and J.L. contributed to the scientific discussion. J.C., M.H., L.J.B., L.H.P., T.Kl., K.B., F.R., R.I.A., M.O.A., P.A., H.H., U.P., A.Pos., A.Poz., J.W. and J.L. contributed to writing the manuscript.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Data availability
The full dataset shown in the figures is publicly available at 10.5281/zenodo.12527358 (ref. 91). Source data are provided with this paper.
Code availability
Data-analysis routines used to process data and generate plots are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Joachim Curtius, Martin Heinritzi
Extended data
is available for this paper at 10.1038/s41586-024-08192-4.
References
- 1.Williamson, C. J. et al. A large source of cloud condensation nuclei from new particle formation in the tropics. Nature574, 399–403 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Andreae, M. O. et al. Aerosol characteristics and particle production in the upper troposphere over the Amazon Basin. Atmos. Chem. Phys.18, 921–961 (2018). [Google Scholar]
- 3.Brock, C. A., Hamill, P., Wilson, J. C., Jonsson, H. H. & Chan, K. R. Particle formation in the upper tropical troposphere: a source of nuclei for the stratospheric aerosol. Science270, 1650–1653 (1995). [Google Scholar]
- 4.Merikanto, J., Spracklen, D. V., Mann, G. W., Pickering, S. J. & Carslaw, K. S. Impact of nucleation on global CCN. Atmos. Chem. Phys.9, 8601–8616 (2009). [Google Scholar]
- 5.Bellouin, N. et al. Bounding global aerosol radiative forcing of climate change. Rev. Geophys.58, e2019RG000660 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang, X., Gordon, H., Grosvenor, D. P., Andreae, M. O. & Carslaw, K. S. Contribution of regional aerosol nucleation to low-level CCN in an Amazonian deep convective environment: results from a regionally nested global model. Atmos. Chem. Phys.23, 4431–4461 (2023). [Google Scholar]
- 7.Clarke, A. D. et al. Particle production in the remote marine atmosphere: cloud outflow and subsidence during ACE 1. J. Geophys. Res. Atmos.103, 16397–16409 (1998). [Google Scholar]
- 8.Carslaw, K. S. et al. Large contribution of natural aerosols to uncertainty in indirect forcing. Nature503, 67–71 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Sindelarova, K. et al. Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years. Atmos. Chem. Phys.14, 9317–9341 (2014). [Google Scholar]
- 10.Guenther, A. B. et al. The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions. Geosci. Model Dev.5, 1471–1492 (2012). [Google Scholar]
- 11.Wennberg, P. O. et al. Gas-phase reactions of isoprene and its major oxidation products. Chem. Rev.118, 3337–3390 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Lelieveld, J. et al. Atmospheric oxidation capacity sustained by a tropical forest. Nature452, 737–740 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Yáñez-Serrano, A. M. et al. Diel and seasonal changes of biogenic volatile organic compounds within and above an Amazonian rainforest. Atmos. Chem. Phys.15, 3359–3378 (2015). [Google Scholar]
- 14.Claeys, M. et al. Formation of secondary organic aerosols through photooxidation of isoprene. Science303, 1173–1176 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Claeys, M. & Maenhaut, W. Secondary organic aerosol formation from isoprene: selected research, historic account and state of the art. Atmosphere12, 728 (2021). [Google Scholar]
- 16.Kiendler-Scharr, A. et al. New particle formation in forests inhibited by isoprene emissions. Nature461, 381–384 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Heinritzi, M. et al. Molecular understanding of the suppression of new-particle formation by isoprene. Atmos. Chem. Phys.20, 11809–11821 (2020). [Google Scholar]
- 18.Bardakov, R., Krejci, R., Riipinen, I. & Ekman, A. M. L. The role of convective up- and downdrafts in the transport of trace gases in the Amazon. J. Geophys. Res. Atmos.127, e2022JD037265 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang, D., Giangrande, S. E., Feng, Z., Hardin, J. C. & Prein, A. F. Updraft and downdraft core size and intensity as revealed by radar wind profilers: MCS observations and idealized model comparisons. J. Geophys. Res. Atmos.125, e2019JD031774 (2020). [Google Scholar]
- 20.Palmer, P. I., Marvin, M. R., Siddans, R., Kerridge, B. J. & Moore, D. P. Nocturnal survival of isoprene linked to formation of upper tropospheric organic aerosol. Science375, 562–566 (2022). [DOI] [PubMed] [Google Scholar]
- 21.Albrecht, R. I., Morales, C. A. & Silva Dia, M. A. F. Electrification of precipitating systems over the Amazon: physical processes of thunderstorm development. J. Geophys. Res. Atmos.116, D08209 (2011). [Google Scholar]
- 22.Schumann, U. & Huntrieser, H. The global lightning-induced nitrogen oxides source. Atmos. Chem. Phys.7, 3823–3907 (2007). [Google Scholar]
- 23.Lelieveld, J. et al. The South Asian monsoon—pollution pump and purifier. Science361, 270–273 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Yu, P. et al. Efficient in-cloud removal of aerosols by deep convection. Geophys. Res. Lett.46, 1061–1069 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clarke, A. D. & Kapustin, V. N. A Pacific aerosol survey. Part I: a decade of data on particle production, transport, evolution, and mixing in the troposphere. J. Atmos. Sci.59, 363–382 (2002). [Google Scholar]
- 26.Clarke, A. D. Atmospheric nuclei in the remote free-troposphere. J. Atmos. Chem.14, 479–488 (1992). [Google Scholar]
- 27.Krejci, R. et al. Evolution of aerosol properties over the rain forest in Surinam, South America, observed from aircraft during the LBA-CLAIRE 98 experiment. J. Geophys. Res. Atmos.108, D18 (2003). [Google Scholar]
- 28.Weigel, R. et al. In situ observations of new particle formation in the tropical upper troposphere: the role of clouds and the nucleation mechanism. Atmos. Chem. Phys.11, 9983–10010 (2011). [Google Scholar]
- 29.Kulmala, M. et al. Deep convective clouds as aerosol production engines: role of insoluble organics. J. Geophys. Res. Atmos.111, D17202 (2006). [Google Scholar]
- 30.Ekman, A. M. L. et al. Do organics contribute to small particle formation in the Amazonian upper troposphere?. Geophys. Res. Lett.35, L17810 (2008). [Google Scholar]
- 31.Schulz, C. et al. Aircraft-based observations of isoprene-epoxydiol-derived secondary organic aerosol (IEPOX-SOA) in the tropical upper troposphere over the Amazon region. Atmos. Chem. Phys.18, 14979–15001 (2018). [Google Scholar]
- 32.Zha, Q. et al. Oxidized organic molecules in the tropical free troposphere over Amazonia. Natl Sci. Rev.11, nwad138 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gordon, H. et al. Causes and importance of new particle formation in the present-day and preindustrial atmospheres. J. Geophys. Res. Atmos.122, 8739–8760 (2017). [Google Scholar]
- 34.Kerminen, V.-M. et al. Atmospheric new particle formation and growth: review of field observations. Environ. Res. Lett.13, 103003 (2018). [Google Scholar]
- 35.Wimmer, D. et al. Ground-based observation of clusters and nucleation-mode particles in the Amazon. Atmos. Chem. Phys.18, 13245–13264 (2018). [Google Scholar]
- 36.Varanda Rizzo, L. et al. Multi-year statistical and modeling analysis of submicrometer aerosol number size distributions at a rain forest site in Amazonia. Atmos. Chem. Phys.18, 10255–10274 (2018). [Google Scholar]
- 37.Martin, S. T. et al. Sources and properties of Amazonian aerosol particles. Rev. Geophys.48, RG2002 (2010). [Google Scholar]
- 38.Franco, M. A. et al. Occurrence and growth of sub-50 nm aerosol particles in the Amazonian boundary layer. Atmos. Chem. Phys.22, 3469–3492 (2022). [Google Scholar]
- 39.Wang, J. et al. Amazon boundary layer aerosol concentration sustained by vertical transport during rainfall. Nature539, 416–419 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Zhao, B. et al. High concentration of ultrafine particles in the Amazon free troposphere produced by organic new particle formation. Proc. Natl Acad. Sci.117, 25344–25351 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riva, M. et al. Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species. Atmos. Meas. Tech.12, 2403–2421 (2019). [Google Scholar]
- 42.Kirkby, J. et al. Ion-induced nucleation of pure biogenic particles. Nature533, 521–526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simon, M. et al. Molecular understanding of new-particle formation from α-pinene between −50 and +25 °C. Atmos. Chem. Phys.20, 9183–9207 (2020). [Google Scholar]
- 44.Shen, J. et al. New particle formation from isoprene under upper-tropospheric conditions. Nature10.1038/s41586-024-08196-0 (2024). [DOI] [PMC free article] [PubMed]
- 45.D’Ambro, E. L. et al. Molecular composition and volatility of isoprene photochemical oxidation secondary organic aerosol under low- and high-NOx conditions. Atmos. Chem. Phys.17, 159–174 (2017). [Google Scholar]
- 46.Liu, Y. et al. Isoprene photochemistry over the Amazon rainforest. Proc. Natl Acad. Sci.113, 6125–6130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thornton, J. A. et al. A near-explicit mechanistic evaluation of isoprene photochemical secondary organic aerosol formation and evolution: simulations of multiple chamber experiments with and without added NOx. ACS Earth Space Chem.4, 1161–1184 (2020). [Google Scholar]
- 48.Massoli, P. et al. Ambient measurements of highly oxidized gas-phase molecules during the Southern Oxidant and Aerosol Study (SOAS) 2013. ACS Earth Space Chem.2, 653–672 (2018). [Google Scholar]
- 49.Pankow, J. F. & Asher, W. E. SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds. Atmos. Chem. Phys.8, 2773–2796 (2008). [Google Scholar]
- 50.Schervish, M. & Donahue, N. M. Peroxy radical chemistry and the volatility basis set. Atmos. Chem. Phys.20, 1183–1199 (2020). [Google Scholar]
- 51.Kazil, J., Lovejoy, E. R., Barth, M. C. & O’Brien, K. Aerosol nucleation over oceans and the role of galactic cosmic rays. Atmos. Chem. Phys.6, 4905–4924 (2006). [Google Scholar]
- 52.Yu, F. Q. & Turco, R. P. Ultrafine aerosol formation via ion-mediated nucleation. Geophys. Res. Lett.27, 883–886 (2000). [Google Scholar]
- 53.Rehbein, A., Ambrizzi, T. & Mechoso, C. Mesoscale convective systems over the Amazon basin. Part I: climatological aspects. Int. J. Climatol.38, 215–229 (2018). [Google Scholar]
- 54.Anselmo, E. M., Machado, L. A. T., Schumacher, C. & Kiladis, G. N. Amazonian mesoscale convective systems: life cycle and propagation characteristics. Int. J. Climatol.41, 3968–3981 (2021). [Google Scholar]
- 55.Albrecht, R. I., Goodman, S. J., Buechler, D. E., Blakeslee, R. J. & Christian, H. J. Where are the lightning hotspots on Earth? Bull. Am. Meteorol. Soc.97, 2051–2068 (2016). [Google Scholar]
- 56.Jokinen, T. et al. Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF. Atmos. Chem. Phys.12, 4117–4125 (2012). [Google Scholar]
- 57.Kürten, A. et al. Neutral molecular cluster formation of sulfuric acid–dimethylamine observed in real time under atmospheric conditions. Proc. Natl Acad. Sci.111, 15019–15024 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Broch, S. Ein neues LIF-Instrument für flugzeug- und bodengebundene Messungen von OH- und HO2-Radikalen in der Troposphäre. Dissertation, Bergische Universität Wuppertal (2011).
- 59.Tanner, D. J., Jefferson, A. & Eisele, F. L. Selected ion chemical ionization mass spectrometric measurement of OH. J. Geophys. Res. Atmos.102, 6415–6425 (1997). [Google Scholar]
- 60.Heinritzi, M. et al. Characterization of the mass-dependent transmission efficiency of a CIMS. Atmos. Meas. Tech.9, 1449–1460 (2016). [Google Scholar]
- 61.Hanson, D. R. & Eisele, F. Diffusion of H2SO4 in humidified nitrogen: hydrated H2SO4. J. Phys. Chem. A104, 1715–1719 (2000). [Google Scholar]
- 62.Kurten, A., Rondo, L., Ehrhart, S. & Curtius, J. Calibration of a chemical ionization mass spectrometer for the measurement of gaseous sulfuric acid. J. Phys. Chem. A116, 6375–6386 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Ehn, M. et al. A large source of low-volatility secondary organic aerosol. Nature506, 476–479 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Kuang, C. & Ashish, S. Ultrafine Condensation Particle Counter Instrument Handbook10.2172/1245986 (US Department of Energy, 2024).
- 65.Brock, C. A. et al. Ultrafine particle size distributions measured in aircraft exhaust plumes. J. Geophys. Res. Atmos.105, 26555–26567 (2000). [Google Scholar]
- 66.Weigel, R. et al. Experimental characterization of the COndensation PArticle counting System for high altitude aircraft-borne application. Atmos. Meas. Tech.2, 243–258 (2009). [Google Scholar]
- 67.Williamson, C. et al. Fast time response measurements of particle size distributions in the 3–60 nm size range with the nucleation mode aerosol size spectrometer. Atmos. Meas. Tech.11, 3491–3509 (2018). [Google Scholar]
- 68.Thomson, W. LX. On the equilibrium of vapour at a curved surface of liquid. London Edinburgh Dublin Philos. Mag. J. Sci.42, 448–452 (1871). [Google Scholar]
- 69.Molleker, S. et al. Application of an O-ring pinch device as a constant-pressure inlet (CPI) for airborne sampling. Atmos. Meas. Tech.13, 3651–3660 (2020). [Google Scholar]
- 70.Jordan, A. et al. A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS). Int. J. Mass Spectrom.286, 122–128 (2009). [Google Scholar]
- 71.Rivera-Rios, J. C. et al. Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry. Geophys. Res. Lett.41, 8645–8651 (2014). [Google Scholar]
- 72.Wang, N. et al. Measurements of carbonyl compounds around the Arabian Peninsula: overview and model comparison. Atmos. Chem. Phys.20, 10807–10829 (2020). [Google Scholar]
- 73.Ernle, L., Ringsdorf, M. A. & Williams, J. Influence of ozone and humidity on PTR-MS and GC-MS VOC measurements with and without Na2S2O3 ozone scrubber. Atmos. Meas. Tech.16, 1179–1194 (2023). [Google Scholar]
- 74.Voigt, C. et al. Cleaner skies during the COVID-19 lockdown. Bull. Am. Meteorol. Soc.103, E1796–E1827 (2022). [Google Scholar]
- 75.Bourtsoukidis, E. et al. An aircraft gas chromatograph–mass spectrometer System for Organic Fast Identification Analysis (SOFIA): design, performance and a case study of Asian monsoon pollution outflow. Atmos. Meas. Tech.10, 5089–5105 (2017). [Google Scholar]
- 76.Tadic, I. et al. Net ozone production and its relationship to nitrogen oxides and volatile organic compounds in the marine boundary layer around the Arabian Peninsula. Atmos. Chem. Phys.20, 6769–6787 (2020). [Google Scholar]
- 77.Nussbaumer, C. M. et al. Modification of a conventional photolytic converter for improving aircraft measurements of NO2 via chemiluminescence. Atmos. Meas. Tech.14, 6759–6776 (2021). [Google Scholar]
- 78.Reed, C., Evans, M. J., Di Carlo, P., Lee, J. D. & Carpenter, L. J. Interferences in photolytic NO2 measurements: explanation for an apparent missing oxidant? Atmos. Chem. Phys.16, 4707–4724 (2016). [Google Scholar]
- 79.Jordan, N. et al. Potential interferences in photolytic nitrogen dioxide converters for ambient air monitoring: evaluation of a prototype. J. Air Waste Manage. Assoc.70, 753–764 (2020). [DOI] [PubMed] [Google Scholar]
- 80.Marno, D. et al. Calibration of an airborne HOx instrument using the All Pressure Altitude-based Calibrator for HOx Experimentation (APACHE). Atmos. Meas. Tech.13, 2711–2731 (2020). [Google Scholar]
- 81.Novelli, A. et al. Characterisation of an inlet pre-injector laser-induced fluorescence instrument for the measurement of atmospheric hydroxyl radicals. Atmos. Meas. Tech.7, 3413–3430 (2014). [Google Scholar]
- 82.Drewnick, F. et al. A new time-of-flight aerosol mass spectrometer (TOF-AMS)—instrument description and first field deployment. Aerosol Sci. Technol.39, 637–658 (2005). [Google Scholar]
- 83.Schmale, J. et al. Aerosol layers from the 2008 eruptions of Mount Okmok and Mount Kasatochi: in situ upper troposphere and lower stratosphere measurements of sulfate and organics over Europe. J. Geophys. Res. Atmos.115, D00L07 (2010). [Google Scholar]
- 84.Röder, L. L., Ort, L. M., Lelieveld, J. & Fischer, H. Quantitative analysis of temporal stability and instrument performance during field experiments of an airborne QCLAS via Allan-Werle-plots. Appl. Phys. B130, 118 (2024).
- 85.Tadic, I., Parchatka, U., Königstedt, R. & Fischer, H. In-flight stability of quantum cascade laser-based infrared absorption spectroscopy measurements of atmospheric carbon monoxide. Appl. Phys. B123, 146 (2017). [Google Scholar]
- 86.Bohn, B. & Lohse, I. Calibration and evaluation of CCD spectroradiometers for ground-based and airborne measurements of spectral actinic flux densities. Atmos. Meas. Tech.10, 3151–3174 (2017). [Google Scholar]
- 87.Bohn, B. & Lohse, I. Optical receiver characterizations and corrections for ground-based and airborne measurements of spectral actinic flux densities. Atmos. Meas. Tech.16, 209–233 (2023). [Google Scholar]
- 88.Zahn, A. et al. A fast and precise chemiluminescence ozone detector for eddy flux and airborne application. Atmos. Meas. Tech.5, 363–375 (2012). [Google Scholar]
- 89.Giez, A. et al. Determination of the Measurement Errors for the HALO Basic Data System BAHAMAS by Means of Error Propagation DLR-Forschungsbericht, DLR-FB-2022-27; 10.57676/5rdc-q708 (2023).
- 90.Tuovinen, S., Kontkanen, J., Cai, R. & Kulmala, M. Condensation sink of atmospheric vapors: the effect of vapor properties and the resulting uncertainties. Environ. Sci. Atmos.1, 543–557 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Curtius, J. et al. Isoprene nitrates drive new particle formationin Amazon’s upper troposphere. Zenodo10.5281/zenodo.12527358 (2024).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The full dataset shown in the figures is publicly available at 10.5281/zenodo.12527358 (ref. 91). Source data are provided with this paper.
Data-analysis routines used to process data and generate plots are available from the corresponding author on reasonable request.