

Abstract
Abstract
Thermophilic xylanases catalyzing the cleavage of β-1,4-glycosidic bonds in xylan have applications in food, feed, biorefinery, and pulp industries. In this study, a hyperthermophilic endo-xylanase was obtained by further enhancement of thermal tolerance of a thermophilic GH11 xylanase originated from metagenome of bagasse pile based on rational design. Introducing N13F and Q34L to the previously reported X11P enzyme shifted the optimal working temperature to 85 °C and led to 20.7-fold improvement in thermostability at 90 °C along with a marked increase in Tm to 93.3 °C. X11PNQ enzyme converted xylan to prebiotic xylooligosaccharides with high specificity on xylobiose to xylohexaose and high operational stability at 85 °C, resulting in 10.3-folds yield improvement compared to the parental enzyme. Molecular dynamic simulation and quantum mechanical analysis revealed improved H-bonding networks within GH11 xylanase principal domains and greater dynamic cross-correlations. A novel thermostabilization mechanism by π-amide interaction with slightly lower interaction energy than the native H-bond, but compensated by increased occurrence frequency was firstly demonstrated for thermophilic enzymes. The enzyme represents one of the most thermostable xylanases ever reported with biotechnological potential.
Key points
• Hyperthemophilic xylanase X11PNQ was obtained by rational design engineering.
• X11PNQ showed specificity to prebiotic xylooligosaccharides (XOS) at 85 °C with improved t1/2 at 90 °C.
• Novel thermostabilization by π-amide interaction was demonstrated by MD/QM.
Supplementary information
The online version contains supplementary material available at 10.1007/s00253-024-13356-3.
Keywords: Hyperthermophilicity, Xylanase, Rational design, Molecular dynamic simulation, Oligosaccharides
Introduction
Endo-1,4-β-xylanase (EC 3.2.1.8) is a key enzyme cleaving xylan, the major hemicellulose in herbaceous plants. Xylanase represents one of the most commercially important enzymes with a wide range of applications, particularly as an animal feed additive and as catalysts for food processing and biobleaching step in the pulp industry (Alokika 2019). An increasing demand for xylanases has been witnessed with the emerging use in biomass saccharification and production of xylooligosaccharide (XOS) prebiotic from agricultural residues (Yegin 2023). A variety of endo-xylanases classified in different glycosyl hydrolase families originated from different microbes, including from bacterial (Bacillus, Geobacillus, Paenibacillus, and Streptomyces), and fungal origins (Aspergillus, Trichoderma, and Paecillomyces), and from environmental metagenomes have been characterized for variations in their structures, catalytic properties, and specificities (Li et al. 2022; Verma and Satyanarayana 2020). Most of them work optimally under mesophilic to thermophilic conditions (40–70 °C) under slightly acidic to neutral conditions (pH 4–7). Searching for xylanases with optimal activity and stability in an extended range is thus of great interest for improving their performance in existing applications and expanding their use in other fields of industries.
Hyperthermophilic enzymes work optimally at an extremely high-temperature range, above 80 °C with stability under the operating conditions (Atalah et al. 2019). Compared to their non-thermophilic homologs, they possess advantages on allowing catalysis of reactions at higher temperatures, increasing operational lifetime under processing conditions, and extending their shelf-life at room temperature. Hyperthermophilic xylanases are desirable for various applications, for examples, in biofuel production, pulp and paper processing, food and feed industries, and textiles, and biomass processing in biorefineries where enzyme with stability under high-temperature operational or processing conditions are desirable (Ajeje et al. 2021). Searching for xylanases with high-operating temperatures has been achieved by exploring enzymes from thermophilic or geothermal sources by culture-dependent microbial isolation and culture-independent metagenomic strategy (Verma and Satyanarayana 2020) and through enzyme engineering (Sürmeli and Şanlı-Mohamed 2023). Xylanases from different microbial sources were engineered for improving its optimal catalytic temperature and thermostability by rational design (Miao et al. 2022; Paës and O’Donohue 2006; Qian et al. 2015; Tian et al. 2017; Wang et al. 2017; Wu et al. 2023) and directed evolution (Kumar et al. 2018b). These works reported a number of xylanases with optimal activity in the thermophilic range with only few examples for hyperthermophilic enzymes. However, developing a high-performance xylanase optimally working in the extreme temperature range with high stability under the operational conditions for long reaction time with defined product specificity is still a challenge.
Molecular dynamic (MD) simulations utilize classical atomistic models to emulate biomolecular systems and have been extensively used to complement experimental methods of protein design. Application of quantum mechanics (QM) calculation on MD simulations (MD) has been shown as a highly efficient tool to further enhance analysis of MD trajectories to identify chemical interactions in complex environments, like biological systems (van der Kamp and Mulholland 2013). The combinatorial MD/QM analyses allow for conformation scanning over time as well as representative geometries for interaction energy determination relevant to protein stabilization (Khan et al. 2016a, b; Wei et al. 2022). It offers a powerful tool for identifying non-covalent interaction (NCI) behaviors contributing to thermostabilizing mechanisms by allowing quantification of hydrogen-bonding environments, examination of network-based statistical approaches, and various other inter-molecular interactions. Analyses of the global or regional dynamics present in a protein can further reveal the conformational preferences they possess (Bauer et al. 2019). In recent years, investigation into π-interactions, i.e., those involving π-electron clouds of an aromatic ring, has revealed near-hydrogen bond levels of strength (Krone et al. 2020). So far, studies on influence of π-interactions on stabilization of biological structures have been rather limited. Exploration on roles of π-interactions on stabilizing thermophilic enzymes is thus of great interest.
In this work, a hyperthermophilic xylanase X11PNQ has been constructed based on the reported glycosyl family 11 xylanase from metagenome representing uncultured microbes in a sugarcane bagasse collecting pile (Kanokratana et al. 2015; Mhuantong et al. 2015) which was previously engineered by random mutagenesis (Boonyapakron et al. 2021). The enzyme was further improved by targeted site-directed mutagenesis and its 3-dimensional structure has been elucidated by X-ray crystallography. The basis of its hyperthermophilicity was then explained by molecular dynamic simulation through application of quantum mechanics. The enzyme represents one of the most hyperthermophilic xylanases ever reported with a novel mechanism on structural thermostability demonstrated.
Materials and methods
Materials
The xylanase X11P gene was obtained from X11 gene, a metagenome-derived gene from sugarcane bagasse collection site engineered by random mutagenesis reported previously (Boonyapakron et al. 2021) (accession number AGN70389 for the original X11 sequence and QOV09177 for the X11P sequence). Expression vector pET28a( +) was from Novagen (Darmstadt, Germany). Escherichia coli DH5α (Invitrogen, Carlsbad, CA, USA) was used as the host strain for DNA cloning. Escherichia coli Rosetta (DE3) (Novagen, Darmstadt, Germany) was used for expression of recombinant enzymes. All chemicals and reagents are analytical or molecular biology grades and purchased from major chemical suppliers (Sigma-Aldrich, St. Louis, Mo, USA; Fluka, Buchs, Switzerland; and Merck, Kenilworth, NJ, USA).
Construction of mutant enzymes
X11P mutations were generated through site-directed mutagenesis using PCR method. Briefly, a 50 μL PCR reaction containing 10-ng plasmid DNA template, 0.2 μM forward and reverse primers (Supplemental Table S1), 0.2 mM each dNTPs, and 1 U of Phusion High-Fidelity DNA polymerase (Thermo Scientific, Scientific, Waltham, MA, USA) were run at one cycle of 5 min at 95 °C, 18 cycles with 30 s at 95 °C, 30 s at 62 °C and 7 min at 68 °C, then 10 min at 68 °C. The amplified products were digested with 20 U of DpnI at 37 °C for 2 h. The constructs were confirmed by nucleotide sequence analysis (ATGC sequencing, Patumthani, Thailand). The sequences were submitted to GenBank as X11PN (GenBank accession no: OR790445), X11PQ (GenBank accession no: OR790446), and X11PNQ (GenBank accession no: OR790447).
Protein expression and purification
Protein expression was modified from BENNY protocol (Alessa et al. 2019). X11P and its variants were grown in 2xTY medium supplemented with 50 μg/mL kanamycin and 34 μg/mL chloramphenicol at 30 °C for 24 h. The cultures were used as the inoculum at 2% (v/v) for recombinant protein expression in TY-based auto-induction medium (AIM; 16 g/L tryptone, 10 g/L yeast extract, 3.3 g/L (NH4)2SO4, 6.8 g/L KH2PO4, 7.1 g/L Na2HPO4, 0.5 g/L glucose, 2.0 g/L α-lactose, and 0.15 g/L MgSO4) supplemented with the same antibiotics. After cultivation at 30 °C for 24 h, cells were harvested by centrifugation at 8600 × g for 10 min. Recombinant proteins were extracted by sonication in lysis buffer (100 mM sodium phosphate buffer, 300 mM NaCl, 20 mM imidazole pH 8.0) using 10 mL cell culture/mL lysis buffer. Lysates were clarified by centrifugation at 17,400 × g for 60 min twice. Supernatants were purified by Ni2+-NTA affinity chromatography using the HisPrep FF 16/10 column (GE Healthcare, Uppsala, Sweden) equilibrated with lysis buffer. The His-tagged proteins were eluted using an imidazole gradient of 20–200 mM in 100 mM sodium phosphate buffer, 300 mM NaCl, and pH 8.0. The eluent fraction containing the target protein were pooled, buffer-exchanged with 20 mM sodium phosphate buffer, 100 mM NaCl, 10% glycerol, pH 7.4, and then concentrated using Amicon Ultra-15 Centrifugal Filter Units with a 10-kDa cutoff (Millipore, Darmstadt, Germany). Protein concentration was measured by NanoDrop spectrophotometer (Nanodrop OneC, Thermo Fisher Scientific, Waltham, MA, USA) at UV spectrum 280 nm using mass extinction coefficient of 77,470 M−1 cm−1 determined by quantitative amino acid analysis from ExPASY (https://web.expasy.org/protparam/). The protein profile was analyzed by SDS-PAGE.
Xylanase activity assay
Xylanase activity was determined based on the amount of liberated reducing sugar using the 3,5-dinitrosalicylic acid (DNS) method (Miller 1959). The assay reaction contained 2.58 nM enzyme and 1.0% w/v beechwood xylan (Megazyme, Wicklow, Ireland) in 100 mM sodium phosphate buffer (pH 6.0) and incubated at 85 °C for 10 min. The effect of temperature was investigated at different values ranging from 50 to 100 °C at pH 6.0. The influence of pH was determined at 85 °C through the xylanase standard assay in the range 4.0–7.0 using the following buffer (100 mM): sodium acetate buffer (pH 4.0, 5.0, 5.6), sodium citrate buffer pH 6.0, and sodium phosphate buffer (pH 6.0, 7.0). The reaction was performed as previous reported (Boonyapakron et al. 2017).
Effect of surfactants and divalent ions
The effect of surfactants was investigated by pre-incubating 2.58 nM of the enzyme at 90 °C for 10 min in the presence of different surfactants with polyethylene glycol (PEG) 4000, PEG 8000, Tween 20, Tween 80, Triton X-100, Triton X-305, and Tergiton NP-40 at the final concentration of 1% v/v in sodium phosphate buffer pH 6.0. The effect of divalent ions (CaCl2, MgCl2, MnCl2, and CoCl2) were determined at 1.0 mM at 90 °C for 10 min. Residual activity was determined under the standard condition (85 °C, 10 min). The relative activity was defined as a percentage of the released reducing sugar relative to the maximal amount released in the absence of pre-incubation (t0).
Thermal stability assay
Temperature stability was determined by incubating 25.8 nM of enzyme at optimal pH 6.0 from 50 to 90 °C with varying incubation times at the selected temperature. After pre-incubation, enzyme activity was determined by DNS method using 2.58 nM of enzyme at the optimal temperature (85 °C) for 10 min as described above. The kinetic parameters for thermal inactivation were obtained according to Eq. 1.
| 1 |
where A0 is the initial activity obtained without pre-incubation, A is the residual activity after heat treatment, kd is thermal inactivation rate constant, and t is the exposure time.
The half-life of the xylanase (t1/2) was determined from the relationship in Eq. (2).
| 2 |
Kinetic analysis
The enzyme kinetic parameters of X11P and its variants were determined using beechwood xylan as the substrate under optimal reaction conditions. Reaction assays were performed in triplicate by incubating 320 μL of beechwood xylan pH 6.0 at different concentrations (0.1–2.0% w/v) and 20 μL of 22 nM of enzyme. The assay reaction was incubated at 85 °C for 0–150 min. The reaction was terminated by adding 680 μL of the DNS reagent. The reducing sugar was developed by boiling at 100 °C for 10 min and measuring A540. The Michaelis–Menten constant (Km) and maximum velocity (Vmax) values were calculated according to the Michaelis–Menten equation using Sigma-Plot 14.0 program (Systat Software, San Jose, CA, USA).
XOS production from beechwood xylan
Enzymatic hydrolysis was performed in 1 mL reactions containing 2.19 nM of enzyme and 4% (w/v) beechwood xylan in 50 mM sodium acetate solution pH 6.0. The reactions were incubated at 85 °C for 1–72 h with shaking at 900 rpm in a ThermoMixer (Eppendorf, Hamburg, Germany). After incubation, the reaction was terminated by boiling for 10 min and centrifuged at 12,500 × g for 10 min. The digested products from hydrolysates were determined by high-performance anion-exchange chromatography with pulsed amperometric detector (HPAEC-PAD) using a Dionex ICS-5000+ SP (Thermo Fisher Scientific, Waltham, MA). XOS (10 μL aliquots) were separated on a Dionex CarboPac™ 100 analytical-exchange column with 0.25 mL/min flow rate using a two-stage binary gradient elution of NaOAc-NaOH solution. XOS was detected with the Dionex ED50 electrochemical detector. The product profile and yield were determined by calibrating with the standard mono (X1) and oligosaccharides (X2–X6).
Differential scanning fluorimetry (DSF)
Thermodynamic stability of the enzyme was conducted by differential scanning fluorimetry with Sypro Orange dye (Sigma-Aldrich, St. Louis, Mo, USA) following the standard protocol (Niesen et al. 2007). The sample mixture contained 4.34 nM of enzyme and 12.5 × Sypro Orange in 100 mM sodium phosphate buffer pH 6.0. Samples were heated from 20 to 100 °C at a constant heating rate of 1 °C/1 min. The fluorescence intensities for the thermal unfolding profile were quantified using the FRET filter (excitation 450–290 nm, emission 560–580 nm) with the CFX real-time PCR machine (Bio-Rad, Hercules, CA).
Statistical analysis
The differences between the parental X11P and the engineered variants were analyzed by one-way ANOVA using Tukey’s post hoc test with the confidence level of 95%. Asterisks indicate statistically significant differences between the wild type and variants with p value < 0.05. All experiments were performed in triplicate and results are presented as mean standard ± deviation. All experiments were performed in triplicate. The differences between groups were analyzed by one-way ANOVA using Tukey’ s post hoc test. The corrected p value < 0.05 was considered statistically significant.
Protein crystal preparation and structure determination
The structures of the X11P and X11PNQ were elucidated by X-ray crystallography. Protein crystallization was setup using a microbatch technique (Chitnumsub et al. 2004). Crystallization of both proteins in apo form was initially screened with various crystallization solutions for best crystal growth selection (Supplemental Fig. S1). The crystal was harvested in liquid nitrogen without cryo-protectant solution. Native diffraction datasets were collected using SC XRD series: D8 venture (Bruker, Billerica, MA, USA) at NSTDA Characterization and Testing Service Centre (NCTC, Patumthani, Thailand). Data were processed using HKL2000 package (Otwinowski and Minor 1997). Structural phase was solved by molecular replacement using PHASER (McCoy et al. 2007) in the CCP4 suite. Structure refinement and adjustment were performed using REFMAC5 (Murshudov et al. 1997) and COOT (Emsley and Cowtan 2004). The structures were validated in PROCHECK (Laskowski et al. 1993). Two protein coordinates were deposited in the protein data bank. The PDB codes are 8X65 (X11P) and 8X66 (X11PNQ).
Molecular dynamic simulation
The crystal structures of enzyme variants were protonated using the H + + Webserver for the experimental pH of 6.0 (Anandakrishnan et al. 2012). Complexes were loaded into Amber22 by parameterization through the native tleap program. Proteins were solvated using the conventional TIP3P water box, and the AMBERff14SB force field was used for dynamic motions (Case et al. 2023; Jorgensen et al. 1983; Maier et al. 2015). The systems were tested for their thermostability by examining 0.2 µs trajectories at the respective ambient, elevated, and hyper temperatures of 300 K, 350 K, and 400 K.
Input parameterization was followed by energy minimization over 10,000 cycles using a cutoff distance of 10.0 Å. The timestep for all steps was 2 fs; coordinates were saved every 500 and 5000 steps for pre-production and production, respectively. Emulating a canonical ensemble for calibration, key components were held constant, denoted via: number of particles (N), volume (V), temperature (T), and pressure (P). A heating ensemble of NVT followed minimization for 60 ps, raising the temperature of the system from 0 K to the target temperature using the Langevin thermostat and a coupling constant of 2 ps. Equilibration under NPT (isothermal-isobaric ensemble) was then performed for 1 ns (500,000 steps) to reach a target density of 1 g/mol (Qausain et al. 2020). Tetra-replicate 200 ns production runs were carried out for X11P and X11PNQ using 4 sequential 50 ns micro-trajectories.
Root mean square deviation (RMSD) and radius of gyration (Rg) information were acquired through backbone atom calculations, while root mean square fluctuation (RMSF) was calculated using carbon-alpha positions, encapsulating the average deviation of the protein’s backbone atomic set from a reference structure (first frame of the production run), the overall compactness of this set, and the residue-dependent average deviation at the carbon-alpha, respectively; data was transformed via the R package Bio3D and plotted with ggplot2 (Grant et al. 2021). Visualizations were performed with VMD and PyMol (Version 2.0 Schrödinger, LLC; Humphrey et al. 1996).
Input parameterization was followed by energy minimization over 10,000 cycles using a cutoff distance of 10.0 Å. The timestep for all steps was 2 fs; coordinates were saved every 500 and 5000 steps for pre-production and production, respectively. An NVT heating ensemble followed the minimization for 60 ps, raising the temperature of the system from 0 K to the target temperature using the Langevin thermostat and a coupling constant of 2 ps. Equilibration under NPT was then performed for 1 ns (500,000 steps) to reach a target density of 1 g/mol. Tetra-replicate 200 ns production runs were carried out for X11P and X11PNQ using 4 sequential 50-ns micro-trajectories.
Solvent-accessible surface area (SASA), residue-residue distances, and hydrogen-bonding were computed with Amber’s CPPTRAJ program for the active site and mutated residue regions (Roe and Cheatham 2013). A Python script was used for isolating hydrophobic residue positions as indices in the protein sequence for feeding into CPPTRAJ to determine hydrophobicity. All data were saved both as time series over the length of the trajectories and as averages. Hydrogen-bond data were likewise accessed through CPPTRAJ program tools.
Free energy landscapes (FEL) were generated in a temperature-dependent manner with 200 bins for a 200 × 200 matrix representing the 20,000 frames of the 200 ns simulation, determined by the following equation:
| 3 |
where Gα is the free energy, T is the temperature, P(qα) is a specific state, and Pmax(q) is the most likely conformation; the overall process follows a Boltzmann distribution, utilizing the constant k. These free energies are plotted atop the first two principal components and are representative of the overall trajectory of protein motion (Papaleo et al. 2009).
Dynamic cross-correlation matrices (DCCM) were constructed to assess the coordinated movements between residue regions. Matrices were generated for atomic position relationships between residue Cα’s following the formula:
| 4 |
The equation quantifies the relationships between Cα atom positions, i and j, in terms of their cross-products and obtaining the covariance (Tahir Khan et al. 2023). DCCM was co-opted with ensemble network analysis for the identification of residue neighborhoods. The network maps were exported into VMD for visualization.
For the calculation of the potential π-amide interaction, an in-house script was derived in R. Hydrogen-free trajectories were analyzed using CPPTRAJ. Center of masses (COM) for the N13F phenyl ring and the Y17/F18 amide region was defined. Interactions within 4.0 angstroms between these centers were assessed based on a 40° angle threshold between a point on the amide’s normal plane, COMamide, and COMaromatic. Frames were scored based on meeting both distance and angle criteria.
Utilizing the data frame constructed for π-amide analysis, ideal instances of the π-amide occurrence were reviewed with emphasis at a 300 K, “mild-state” conformation of the protein. Two systems were generated for the N13/N13F region: one in which the H-bonding partner Q55 was present with the amide residues Y17 and F18 (system 1), and one in which Q55 was not present (system 2) in effort to reduce computational loads of many atom systems during the differential energy screening.
The cluster model approach was used to extract and constrain residues N13F, Y17, and F18, as well as N13(F), Y17, F18, and Q56, from a frame with interaction potential. N13(F) and Q56 were stripped of backbone atoms and end-capped with a constrained methyl group. Y17/F18 was kept as a single fragment end-capped at nitrogen and oxygen termini with methyl groups; all backbone carbon and hydrogens were constrained with the central amide carbon as an exception.
Resultant systems were QM optimized in Gaussian 16 revision B.01 with DFT M062X-D3/6–311 + G(d,p), and interaction energies were obtained and calculated using M062X-D3/ma-TZVP (Frisch et al. 2016; Zhao and Truhlar 2011; Zheng et al. 2010). Optimizations and energy calculations were performed with SMD (solvation model density) implicit solvent modeling (Marenich et al. 2009). Interaction energies were deduced as in Eq. (5) through complex and monomer single point energy (SPE) calculations using coordinates of the optimized complex geometry and corrected via basis set superposition error obtained with counterpoise from the gas phase SPE (Mentel and Baerends 2014).
| 5 |
The independent gradient model (IGM) with Hirshfeld partitioning (IGMH) was employed to visualize the weak interactions present at the N13(F) substitution site with Multiwfn wavefunction analyzer (Lu and Chen 2012, 2022). The isosurfaces obtained for the systems were plotted on the standard scatterplot for ; visualizations of the molecular system were extracted from VMD.
Results
Rational design of hyperthermophilic mutants
The recombinant X11P xylanase (Boonyapakron et al. 2021) containing a single P71T mutation was previously obtained by removal of the substrate binding domain and random mutagenesis of X11 xylanase originally isolated from sugarcane bagasse pile metagenome which shared 74% homology to a GH11 xylanase from uncultured bacteria (Kanokratana et al. 2015). In this present work, X11P was further increased for thermostability and prolonged lifetime of the enzyme at extremely high temperature by rational design, focusing on increasing interactions in the two-layered β-sheets. Additional mutations were introduced to X11P based on sequence alignment with EvXyn11 (Dumon et al. 2008), a GH11 xylanase from uncultured bacteria with 76% identity to X11P. EvXyn11 was then subsequently engineered for improved thermostability to EvXyn11TS, which containing 7 mutations (S9P, T13F, N14H, Y18F, Q34L, S35E, and S71T). Compared to EvXyn11TS, 3 out of the 7 mutations already exist in X11P (Supplemental Fig. S2). The effect on introducing single mutation (S9P, N13F, Q34L, and N35E) on thermostability of X11P was then investigated. The pre-screening result showed that N13F and Q34L enhanced thermostability of X11P, while no significant improvement was observed for S9P and N35E (data not shown).
Characterization of enzyme variants
The enzyme X11P was further engineered by targeted sequence alteration for N13F and Q34L as single and combined mutations. All enzyme variants were expressed as soluble forms in E. coli and purified to > 95% homogeneity using Ni-affinity chromatography (Supplemental Fig. S3). Catalytic activity and thermostability of the enzyme variants (X11P, X11PN, X11PQ, and X11PNQ) were compared. Increased optimal working temperature from 80 to 85 °C were observed for X11PQ and X11PNQ (Fig. 1A). All enzymes showed no changes in their optimal pH at 6.0 (Fig. 1B). Improvement in specific activity was observed for X11PN (27%) compared to X11P while no significant change was shown for X11PNQ but with 30% lower activity for X11PQ (Table 1). This reflected in increased Vmax and kcat of X11PN while marked decreases were shown for X11PQ. A slight decrease in Km was also observed for X11PN while no significant change was shown for X11PQ and X11PNQ compared to X11P. This led to the overall kcat/Km in the order X11PN > X11P > X11PNQ > X11PQ.
Fig. 1.

Characteristics of the enzyme variants and production of XOS. A Temperature activity profile of enzymes measured at 50 to 100 °C; B pH activity profile at 85 °C in buffers ranging from pH 4.0 to 7.0; C reducing sugar was assessed using 4% xylan at 85 °C for 1–72 h; D production of xylose and its oligomers as mg/g of xylan (bar graph) and g/mg of enzyme (closed circles) at 85 °C for 24 h. *p-value < 0.05
Table 1.
Kinetic parameters of X11P and its mutants at pH 6.0 and 85 °C
| Enzyme | Protein concentration (nM) | Km (mg/mL) | Vmax (nmole/min) | kcat (min−1) | kcat/Km (mL/mg/min) | Specific activity (μmole/min/mg) | Tm (°C) |
|---|---|---|---|---|---|---|---|
| X11P | 1.29 | 5.40 ± 1.0 | 79.3 ± 5.8 | 181,050 | 33,562 | 6690 ± 172 | 84.7 ± 0.6 |
| X11PN | 1.29 | 4.56 ± 0.5 | 102.9 ± 4.1 | 234,932 | 51,513 | 8490 ± 249 | 90.0 ± 0.0 |
| X11PQ | 1.29 | 5.65 ± 0.9 | 59.0 ± 3.6 | 134,703 | 23,839 | 4654 ± 238 | 87.7 ± 0.6 |
| X11PNQ | 1.29 | 5.36 ± 0.5 | 71.7 ± 2.5 | 163,699 | 30,521 | 6516 ± 322 | 93.3 ± 0.6 |
Incorporation of mutations led to increasing Tm of the purified enzymes from 84.7 °C of X11P to 93.3 °C of X11PNQ (Table 1). The triple mutant X11PNQ showed the highest thermostability followed by X11PQ, X11PN, and X11P as demonstrated by t1/2. An increase in t1/2 was observed for X11PNQ compared to the other mutants at 70–90 °C. However, higher thermostability was shown for X11PQ at the lower temperature range from 50 to 60 °C. In the purified form with the absence of substrate, X11PNQ showed t1/2 of 14.5, 46.4, and 66.3 min at 90, 85, and 80 °C, respectively and markedly extended to 14.3 h at 70 °C and 45.2 days at 50 °C (Table 2). The result showed a more pronounced increase in t1/2 up to 20 folds at the temperature above 80 °C compared to at the lower temperature range. Addition of external additives (PEG polyols and surfactants) further increased the activity by 1.5–1.6-fold at 90 °C. This led to the resultant increase in the relative activity of 83–94% compared to that in the absence of additive with no pre-incubation (Table 3). A slight improvement in thermostability was observed in the presence of divalent metal icons (Ca2+, Mg2+, Mn2+, and Co2+) (Supplemental Table S2).
Table 2.
Half-life of enzymes without coexisting of substrate at varying temperatures
| Temperature (°C) | t1/2 | |||
|---|---|---|---|---|
| X11P | X11PN | X11PQ | X11PNQ | |
| 50 | 30.0 ± 0.5a | 31.8 ± 0.5a | 58.6 ± 1.0a | 45.2 ± 1.4a |
| 60 | 14.7 ± 1.2a | 22.8 ± 1.1a | 26.6 ± 0.5a | 22.1 ± 0.4a |
| 70 | 8.4 ± 0.3b | 8.3 ± 0.5b | 10.7 ± 0.2b | 14.3 ± 0.8b |
| 80 | 4.4 ± 0.7c | 24.9 ± 2.4c | 11.7 ± 1.2c | 66.3 ± 2.9c |
| 85 | 2.4 ± 0.1c | 15.6 ± 0.0c | 9.9 ± 1.0c | 46.4 ± 1.5c |
| 90 | 0.7 ± 0.0c | 5.5 ± 0.1c | 2.3 ± 0.2c | 14.5 ± 2.1c |
aPresented as day, bpresented as hour, cpresented as min
Table 3.
Effect of surfactants on X11PNQ activity
| Surfactants | Specific activity (U/mg) | Relative activity (%) | ||
|---|---|---|---|---|
| t0 | t10 | t*0 | t**10 | |
| w/o surfactant | 5519 ± 162 | 2434 ± 100 | 100 | 44 |
| PEG 4000 | 8998 ± 70 | 5407 ± 143 | 163 | 98 |
| PEG 8000 | 8968 ± 129 | 5233 ± 137 | 163 | 95 |
| Tween 20 | 8561 ± 117 | 4757 ± 71 | 155 | 86 |
| Tween 80 | 8628 ± 63 | 5241 ± 180 | 156 | 95 |
| TritonX-100 | 8425 ± 168 | 4557 ± 158 | 153 | 83 |
| TritonX-305 | 8358 ± 271 | 4625 ± 119 | 151 | 84 |
| Tergiton NP-40 | 8708 ± 145 | 4746 ± 102 | 158 | 86 |
Activity was compared between with no pre-incubation
(t0) and with pre-incubation at 90 °C for 10 min (t10)
*Relative activity at t0 compared to without surfactant at t0
**Relative activity at t10 compared to without surfactant at t0
To explore the potential of X11P variants for XOS production at high temperature, we studied hydrolysis of beechwood xylan at 4% w/v at 85 °C with varied times (1–72 h). Analysis of the reducing sugar hydrolyzed by X11P variants showed the increasing of reducing sugar over the time with the maximum yield at 24 h (Fig. 1C). X11PNQ exhibited the highest reducing sugar for 9.36 mg/mL, equivalent to threefold increase over X11P. Product distribution from hydrolysis of beechwood xylan using X11P and its variants at 85 °C is shown in Fig. 1D. X11P showed product specificities toward longer chain oligosaccharides in the order of X3 > X4 > X5 > X6 > X2 with the minor presence of X1. Increasing product yield up to 110.37 ± 0.55 mg/g of xylan, equivalent to 10.4 × , 2.7 × , and 2.6 × was obtained using X11PNQ, followed by X11PN and X11PQ, compared to X11P, respectively. The highest product of XOS was observed from X11PQ catalysis yielding 86 g product/mg of enzyme, equivalent to 10.4 folds increase compared to X11P. The alternation in product specificities reflected the thermal stability of the enzyme variants as shown by their t1/2, which showed marked improvement in its t1/2.
Crystal structure determination and molecular dynamic simulations through quantum mechanics
To elucidate the mechanism of N13F and Q34L effect on enzyme structure, crystal structures of X11P and X11PNQ were elucidated. The crystal structures of X11P and X11PNQ were determined by molecular replacement using a GH11 xylanase from an uncultured bacterium (pdb code 2VUJ with 77% sequence identity) as a template which 1.7 Å and 2.3 Å resolution, respectively. Data collection and refinement statistics are listed in Supplemental Table S3. The overall fold of X11P displays the right-handed β-jelly roll fold typical for the GH11 family of xylanases (Fig. 2). The overall core structures of X11P and X11PNQ are not different. MD simulation was further employed to explain stabilizing interactions in the hyperthermophilic mutants based on their elucidated crystal structures.
Fig. 2.
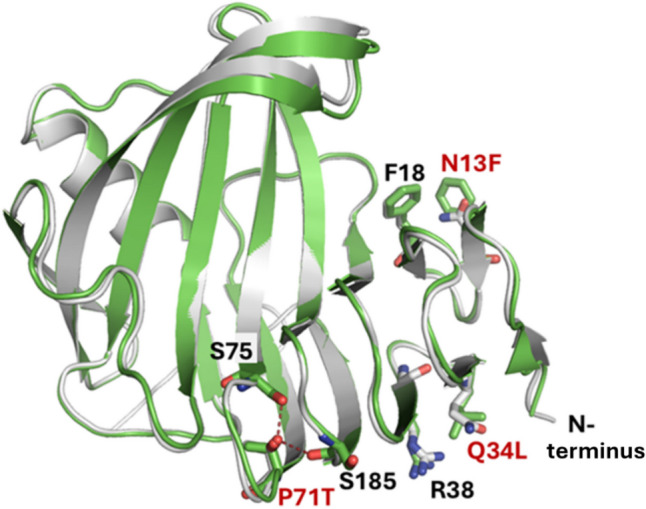
Overlaid structures of X11P and X11PNQ. Ribbon structures of X11P (white color) and X11PNQ (green) show the engineered residues of N13F, Q34L, and P71T, as well as residues F18, R38, S75, and S185, interacting with the engineered residues
From MD simulation, X11P and X11PNQ both showed strong stability at ambient and elevated temperatures (Fig. 3); however, X11PNQ showed improved thermostability at the extreme temperature of 400 K (Fig. 3, right). In addition, while the RMSD trend for X11PNQ at lower temperatures is often elevated to X11P, the overall distribution is narrower. This observation may be due to X11PNQ requiring more movement from the crystal structure to obtain an equilibrium conformation. Once this equilibrium is attained, it appears that X11PNQ remains more compact. The thumb-loop region through residues 127–137 shows the most deviation in each mutant. X11PNQ has a mild increased level of this movement at ambient and elevated temperatures, while this is the region of primary destabilization in X11P at 400 K. As this region is associated with the binding of substrate, it is unclear if this movement is a feature or a defect in the lower temperature groups. X11P and X11PNQ showed varying levels of both residue-residue and residue-water-residue (bridging) hydrogen bonding (Fig. 4). A proton channel exists between D24-N48 and N48-E180. Hydrogen bond levels are comparable for both proteins for the D24-N48 interaction, while the bridged interaction shows a mild increase in interaction for X11PNQ; however, the N48-E180 interaction is a strict bridge interaction and shows considerable increased occupancy in X11P over X11PNQ. Despite this, a greater variety of active site (palm, outer palm, and edge-of-finger regions) residues show improved H-bond interactions in X11PNQ for both residue-residue and bridged mechanisms.
Fig. 3.

Trajectory analyses for X11P and X11PNQ.cSuperimposed plots for RMSD, RMSF, and Rg for X11P (black) and X11PNQ (green) at the simulated temperatures: 300 K, 350 K, and 400 K. Notable peaks are highlighted at the cord and thumb-loop regions
Fig. 4.

Hydrogen bonding patterns of xylanase domains. Notable residue pairs showing improved interactions are plotted in histograms (bottom) for conventional and bridged hydrogen bonds. Corresponding domains are emphasized as facet wrapped labels. Potential substitution responsible for the effects are noted. Exemplary hydrogen bonds are visualized over the protein model for the four main effected regions: fingers, outer palm, palm, and the β6–β7 loop domains
Hydrogen bonds were lost between the native contacts Q34-Q2 and N13-Q56; however, this loss was correlated with improved H-bond networks near each site. Q34L saw a larger network down the β-sheet scaffold improving T40-S188, S72-S184, and S30-S41 interactions (fingers domain); the flexible β6-β7 loop housing Q56 saw fortified associations with R61-G59/T57 and T57-Q56 interactions, as well as improving relations between the loop and the palm regions via G58-N173 (and W82-N173 within the palm). Some of the largest changes came in the form of side chain to backbone H-bonding between T40 and S188 and T57 and R61 which saw increases from 22 to 58% and 15 to 20.2%, respectively.
Global conformational changes of both proteins were examined through free energy landscapes (FEL); additionally, the indicative substitution with hydrophobic residues prompted hydrophobicity assessment (Fig. 5). Blue point regions depict energy minima in the FEL, while more saturated red colors emphasize energetic conformations. Large variations or patches depict global changes in conformation and may relate to domain changes. X11P traverses a dispersed path between − 10 and 10 on both PCAs (principal component analysis) while X11PNQ is localized at a minimum at 5 on PCA 1 and 0 for PCA 2 with a brief conformational pitstop at the inverse. Assessment of global hydrophobic residue surface area of each protein highlighted a marked increase in hydrophobicity for X11PNQ.
Fig. 5.

Free energy landscapes compared with hydrophobicity. A Population-based plot of hydrophobic residue surface area of X11P (black) and X11PNQ (green); B cartoon rendition of X11PNQ colored by its all-residue network matrix, and several residues within the 1–60 region are presented for aid; C free energy plots are presented at the peak for each protein followed by all-residue DCCM and truncated residue 2–60 analyses with corresponding networks mapped to the axes. All representations were generated using data from the 350 K simulations
Per-residue cross-correlation matrices were constructed to further probe alterations in conformational preferences; DCCM plots are colored by their correlations with positive associations as blue and anti-correlations colored pink (Fig. 6). While both proteins show similar cross-correlations for the length of the peptide, they contrast within the substitution-containing region of residues 1–60. Here, X11PNQ shows larger, positive correlations between all residues with a mild anti-correlated behavior within the first three residues before the proximal disulfide bond. The relation of this behavior was not correlated with hydrogen bonding improvements in this region.
Fig. 6.

All-residue DCCM (top) and truncated residue 2–60 (bottom) analyses with corresponding networks mapped to the axes are shown for X11P and X11PNQ; networks for axes vary between all-residue and 2–60 subsets due to the selection-dependent nature of the DCCM calculation. All plots were generated using data from the 350 K simulations
Identification of novel π-amide interaction
The presence of π-interaction in X11PNQ was further studied through application of quantum mechanics. The N13F substitution disrupts a hydrogen bond that exists between the original asparagine residue and a glutamine, Q56, on a neighboring loop in a range of 7–17% of total simulation time. Additional non-covalent interactions were examined at the substitution site, and the potential for a π-amide interaction between N13F and the backbone atoms of Y17/F18 was identified in lieu of the hydrogen bond (Fig. 7 top). System 1 was used to identify the energetic differences between the π-amide interaction and the hydrogen bond. The optimized H-bond structure showcased bonding behaviors between the nitrogen donor of N13 and the carbonyl oxygen of Q56. At a distance of 3.10 angstroms, the solvated interaction energy was − 8.0 kcal/mol. Meanwhile, the COM of the aromatic ring was 3.80 angstroms above the COM of the Y17/F18 backbone amide at an angle of 8.49°, and the solvated interaction energy was determined to be − 7.2 kcal/mol. System 2 was useful for determining various attributes of the calculation. For one, the solvent influence was determined by identifying the gas phase and solvent-phase energies. The optimized geometry had a COMN13F-COMamide distance of 3.75 angstroms and a corresponding angle of 9.34°. The smaller system yielded a gas-interaction energy of − 8.6 kcal/mol and a solvent-optimized energy of − 5.8, attributing the loss in energy to the strong dielectric constant of water. These results confirmed direct π-amide interactions comparable to other π-interactions and conventional hydrogen bonds (Krone et al. 2020). A one-dimensional potential energy scan was used to determine the interaction energy of the π-amide system at an extended distance of 4.5 angstroms and revealed an energy of − 4.7 kcal/mol, placing the system just below conventional hydrogen bonding cutoffs of − 5.0 kcal/mol (Supplemental Fig. S4).
Fig. 7.

IGMH interpretation of optimized π-amide geometry from cluster. Top: location of the N13F substitution site with the π-amide site highlighted (orange: N13F, Y17, and F18). Q34L is depicted as pink, the catalytic E88 and E180 as red, the thumb-loop region as cyan, and Q56 is shown as yellow. Bottom: IGMH wavefunction analyses of the pi-amide and hydrogen bonds present in system 1 of substituted N13F and native N13, respectively. VMD visualizations lay above scatter plots of the inter-atomic energies; a.u. is an abbreviation for atomic units as formatted by Multiwfn
To further understand thermostabilizing effect of this interaction, the π-amide and hydrogen bond wavefunctions were analyzed in Multiwfn to aid in visualization of their interaction pairs (Fig. 7 bottom). The IGMH method was used to isolate the van der Waals (vdW) and electrostatic interaction patterns. The backbone region was not removed from system 1 for the analysis in order to preserve any steric clashes that might prevent the close approach of Q56 to N13. As a result, hydrogen bond scatter plot remains largely in the green regions which are reserved more for positively interacting vdW interactions. While the hydrogen bonding is stronger than the π-amide, it is perhaps unable to form close enough approaches to show large strengths over the π-amide. In addition, while the 4.0 angstrom and 40° cutoff for the geometric search of π-amides revealed roughly an 11–15% occupancy in simulation, extending the distance cutoff to 4.5 angstroms revealed a 30% occurrence. Therefore, while the relative strength of the π-amide decreases with distance, its existence is longer than that of the native hydrogen bond (6–14%). Overall, the MD/QM simulation identified the function of π-amide interactions as an important mechanism on thermostabilization of the enzyme.
Discussion
In this study, a hyperthermophilic X11PNQ was obtained from further targeted engineering of the parental enzyme X11P (Boonyapakron et al. 2021) derived from X11, a GH11 xylanase originated from metagenomes of microbial community in sugarcane bagasse pile. Bagasse pile at sugar factory was reported as a potent source for lignocellulose-degrading enzymes in various glycosyl hydrolase and auxiliary enzyme families originated from diverse aerobic to anaerobic microbes as revealed by next-generation shotgun sequencing of its metagenome (Mhuantong et al. 2015; Bunterngsook et al. 2021). Thermophilic xylanases working under extremely high temperature has a wide range of applications with benefits on increased rate of reaction, prolonged operational lifetime, and reduced microbial contamination (Ajeje et al. 2021). To date, the number of reported thermophilic xylanases exhibiting functionality at elevated temperatures, particularly surpassing 85 °C, remains very limited (Supplemental Table S4). Compared to previously reported xylanases, the triple mutant X11PNQ is regarded as a highly thermostable enzyme capable of working at extremely high-temperature conditions. At 90 °C, X11PNQ displayed a comparable t1/2 to that of rMxyl, which is recognized as one of the most thermally stable xylanases (Verma et al. 2013). Nevertheless, X11PNQ exhibited a catalytic activity that is over 20 times higher than that of rMxyl. These characteristics of X11PNQ render it well-suited for a diverse range of applications. These include its application in the production of xylooligosaccharides, wherein its ability to operate at elevated temperatures facilitates enhanced substrate solubility with reduced viscosity in the reaction.
MD simulations provide an interrogative space for analyzing the unique functional and stability consequences of structural modifications across protein variants; the capacity to monitor macro and micro-molecular systems over time and under varying environmental conditions allows for the piecemeal deduction of stabilizing forces (Khan et al. 2016a, b). RMSD, RMSF, and Rg occupy the baseline equilibrium metrics for a simulated trajectory. Such measures have been established in the literature, including deleterious mutants corroborating lysosomal storage disorders and advantageous eukaryotic protein scaffolds permitting endosomal fusion (Khan et al. 2017; Khan et al. 2016a, b). Large-scale conformational changes across the trajectory can be identified and compared by projecting the free energy onto corresponding principal components; moreover, MD simulations offer opportunities for quantification of hydrogen bonds and other non-covalent interactions (Hassan et al. 2020; Khan et al. 2020a, b; Qausain et al. 2023). Combined, these methods provide a solid foundation for computationally exposing structural changes at both the global and local level of protein design.
While high-throughput mutation screening can cycle through variant scaffolds with high efficiency, computational simulations offer several benefits as an independent or in-tandem methodology. The in silico nature of these calculations can bridge the gap between theory and experiment in complex systems while providing kinetic information useful for optimizing mutagenesis strategies. Previous MD research on chitinase II identified maintenance of secondary structures in chitinase II up to 350 K (Khan et al. 2016a, b). Our work highlights the competitive advantage of X11PNQ using a similar temperature schema and posits secondary structure stability beyond 350 K for both simulated variants, in support of experimental findings.
Thermostabilization mechanism of X11PNQ was explored in this study using the MD/QM simulation based on its elucidated crystal structure. Overall, X11PNQ shows higher levels of compaction, conformational minimization, and hydrophobicity compared to its X11P parent, evidenced by a reduction in standard trajectory analysis data (RMSD, RMSF, and Rg) and free energy landscapes while increasing the global hydrophobic space of the xylanase by substitution with hydrophobic amino acids.
Both proteins persisted at elevated temperatures with a high degree of stability; however, the substitution improvements provided X11PNQ a greater degree of longevity in an extreme environment. While previous findings suggested an increase in arginine content can positively affect thermostability for modified xylanases, more recent work has shown increases in hydrophobicity can also improve xylanase thermostability (Stephens et al. 2014; Shi et al. 2023). The substitution of N13 and Q34 with hydrophobic residues increased the global hydrophobicity as well as improved the cross-correlation arrangements of residues in the vicinity of amino acids 1–60, despite the loss of hydrogen-bonding interactions between Q34-Q2 and N13-Q56. While the key proton channel residue interaction between N48 and catalytic E180 is not as occupied in bridged H-bonding in X11PNQ, the modified enzyme shows marked improvement in various active site residue-residue and bridged H-bonding interactions. Beyond the active site, both Q34L and N13F improved the local hydrogen-bonding of residues within their vicinity. The global conformation suggests incremental changes at multiple residues result in the cumulative improvement of X11PNQ’s thermostability profile.
In addition to well-known conventional non-covalent interactions, π-interactions have been introduced as another stabilization mechanism in biological systems. Unlike hydrogen-bonding patterns, π-interactions exhibit stacked, offset, and T-shaped orientations, with their cutoff distances, angles, and associated energies varying based on the identities of the interacting partners (Dalkas et al. 2014). Identification of novel π-interactions can thus aid in building a robust interaction portfolio for thermostable proteins.
According to MD/QM analysis, a π-amide interaction between N13F and the backbone amide region of Y17/F18 neighbors was identified as a hydrophobic consequence; this interaction’s relative strength and occupancy compared to hydrogen bonding provide X11PNQ with greater hydrophobicity without losing non-covalent interactions (NCI). This thermal stabilizing effect may also apply to the T13F mutation of EvXyn11, in which stabilizing mechanism was unclear in the previous report (Dumon et al. 2008). While π-π and cation-π interactions are well studied, documentation of π-amide interactions within biological systems remains scarce. Much of the investigation into these interactions is performed on ideal systems in the gas phase rather than on extracted conformations from peptide structures, with some examples involving ligand binding to active sites of enzymes (Stojanović et al. 2021). As a result, publications on their influence on biological structure stability are limited. The identification of π-amide interactions as a novel stabilization mechanism in thermophilic enzymes, first reported in this study, is therefore of great interest.
While these data offer a privileged insight into the global and local interaction environments and conformational preferences of X11P and X11PNQ across temperature ranges, the acquired MD simulation information is specific to unbound enzyme. Human programmed cell-death protein 1 (PD-1) has recently been examined for its behavior in the presence and absence of Pembrolizumab antibody, revealing loop stabilization and an increase in overall flexibility in the bound state. Contrastingly, the unbound state occupies a larger swathe of low-energy conformational space (Wang and Khan 2023). As a result, complex MD simulations or QM modeling of X11P and X11PNQ with xylan ligand may reveal additional stabilizing factors not witnessed in the unbound temperature scaffold.
In summary, a hyperthermophilic xylanase X11PNQ was reported in this study. The enzyme showed superior thermostability compared with most previously reported xylanases with specificities toward production of XOS at extremely high temperatures. MD simulation and QM analyses revealed thermostabilizing benefits of the PNQ variant through dynamic cross-correlations, network analyses, and FEL review. In addition to the improved hydrogen-bonding scaffolds in the principal finger, palm, and β6-β7 loop domains of the glycoside hydrolase family 11 (GH 11) xylanase, the π-amide interaction at N13F was firstly identified as a novel stabilization mechanism with a small reduction in interaction energy from the native H-bond, albeit with a larger frequency of occurrence. Application of the enzyme on catalyzing hydrolysis of xylan at high temperature in biorefinery is warranted.
Supplementary information
Below is the link to the electronic supplementary material.
Author contribution
KB: investigation, methodology, analysis, and writing—original draft preparation (enzyme engineering); BK: investigation, methodology, analysis, and writing—original draft preparation (computational analysis); KP: investigation; KA: investigation; NA: data analysis; AJ: data analysis; PC: data analysis; JYL: data analysis; TSW: conceptualization and writing—review and editing; XQZ: writing—review and editing; CGL: writing—review and editing; DQW: conceptualization, data analysis, supervision, and funding acquisition (computational analysis). VC: conceptualization, data analysis, supervision, writing—original draft preparation, project administration, and funding acquisition (enzyme engineering).
Funding
This work was supported by the NSRF via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation (Project “Artificial intelligence-guided synthetic biology for bio-based and biorefinery industry” B16F640052). Dong-Qing Wei is supported by grants from the National Science Foundation of China (Grant No. 32070662, 61832019, 32030063), Intergovernmental International Scientific and Technological Innovation and Cooperation Program of The National Key R&D Program (2023YFE0199200). The computations were partially performed at the Pengcheng Lab. and the Center for High-Performance Computing, Shanghai Jiao Tong University. Tuck Seng Wong was supported by the Visiting Professorship awarded by NSTDA, and the RAEng | Leverhulme Trust Senior Research Fellowship (LTSRF1819\15\21).
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information.
Declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
All authors declare no conflict of interest.
Footnotes
Katewadee Boonyapakron and Braden Keiser are co-first authors.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dong-Qing Wei, Email: dqwei@sjtu.edu.cn.
Verawat Champreda, Email: verawat@biotec.or.th.
References
- Ajeje SB, Hu Y, Song G, Peter SB, Afful RG, Sun F, Asadollahi MA, Amiri H, Abdulkhani A, Sun H (2021) Thermostable cellulases / xylanases from thermophilic and hyperthermophilic microorganisms: current perspective. Front Bioeng Biotechnol 9:794304. 10.3389/fbioe.2021.794304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessa AHA, Tee KL, Gonzalez-Perez D, Omar Ali HEM, Evans CA, Trevaskis A, Xu J-H, Wong TS (2019) Accelerated directed evolution of dye-decolorizing peroxidase using a bacterial extracellular protein secretion system (BENNY). Bioresour Bioprocess 6(1):20. 10.1186/s40643-019-0255-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alokika SB (2019) Production, characteristics, and biotechnological applications of microbial xylanases. Appl Microbiol Biotechnol 103(21–22):8763–8784. 10.1007/s00253-019-10108-6 [DOI] [PubMed] [Google Scholar]
- Anandakrishnan R, Aguilar B, Onufriev AV (2012) H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res 40(Web Server issue):W537-41. 10.1093/nar/gks375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atalah J, Cáceres-Moreno P, Espina G, Blamey JM (2019) Thermophiles and the applications of their enzymes as new biocatalysts. Bioresour Technol 280:478–488. 10.1016/j.biortech.2019.02.008 [DOI] [PubMed] [Google Scholar]
- Bauer JA, Pavlovic J, Bauerova-Hlinkova V (2019) Normal mode analysis as a routine part of a structural investigation. Molecules 24(18):3293. 10.3390/molecules24183293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonyapakron K, Jaruwat A, Liwnaree B, Nimchua T, Champreda V, Chitnumsub P (2017) Structure-based protein engineering for thermostable and alkaliphilic enhancement of endo-β-1,4-xylanase for applications in pulp bleaching. J Biotechnol 259:95–102. 10.1016/j.jbiotec.2017.07.035 [DOI] [PubMed] [Google Scholar]
- Boonyapakron K, Chitnumsub P, Kanokratana P, Champreda V (2021) Enhancement of catalytic performance of a metagenome-derived thermophilic oligosaccharide-specific xylanase by binding module removal and random mutagenesis. J Biosci Bioeng 131(1):13–19. 10.1016/j.jbiosc.2020.09.008 [DOI] [PubMed] [Google Scholar]
- Bunterngsook B, Mhuantong W, Kanokratana P, Iseki Y, Watanabe T, Champreda V (2021) Identification and characterization of a novel AA9-type lytic polysaccharide monooxygenase from a bagasse metagenome. Appl Microbiol Biotechnol 105(1):197–210. 10.1007/s00253-020-11002-2 [DOI] [PubMed] [Google Scholar]
- Case DA, Aktulga HM, Belfon K, Ben-Shalom IY, Berryman JT, Brozell SR, Cerutti DS, Cheatham TE, Cisneros GA, Cruzeiro VWD, Darden TA, Forouzesh N, Giambaşu G, Giese T, Gilson MK, Gohlke H, Goetz AW, Harris J, Izadi S, Izmailov SA, Kasavajhala K, Kaymak MC, King E, Kovalenko A, Kurtzman T, Lee TS, Li P, Lin C, Liu J, Luchko T, Luo R, Machado M, Man V, Manathunga M, Merz KM, Miao Y, Mikhailovskii O, Monard G, Nguyen H, O’Hearn KA, Onufriev A, Pan F, Pantano S, Qi R, Rahnamoun A, Roe DR, Roitberg A, Sagui C, Schott-Verdugo S, Shajan A, Shen J, Simmerling CL, Skrynnikov NR, Smith J, Swails J, Walker RC, Wang J, Wang J, Wei H, Wu X, Wu Y, Xiong Y, Xue Y, York DM, Zhao S, Zhu Q, Kollman PA (2023) Amber22. UCSF, San Francisco [Google Scholar]
- Chitnumsub P, Yavaniyama J, Vanichtanankul J, Kamchonwongpaisan S, Walkinshaw MD, Yuthavong Y (2004) Characterization, crystallization and preliminary X-ray analysis of bifunctional dihydrofolate reductase-thymidylate synthase from Plasmodium falciparum. Acta Crystallogr D Biol Crystallogr 60(4):780–783. 10.1107/S0907444904001544 [DOI] [PubMed] [Google Scholar]
- Dalkas GA, Teheux F, Kwasigroch JM, Rooman M (2014) Cation–π, amino–π, π–π, and H-bond interactions stabilize antigen–antibody interfaces. Proteins: Struct Funct Bioinf 82(9):1734–1746. 10.1002/prot.24527 [DOI] [PubMed] [Google Scholar]
- Dumon C, Varvak A, Wall MA, Flint JE, Lewis RJ, Lakey JH, Morland C, Luginbühl P, Healey S, Todaro T, DeSantis G, Sun M, Parra-Gessert L, Tan X, Weiner DP, Gilbert HJ (2008) Engineering hyperthermostability into a GH11 xylanase is mediated by subtle changes to protein structure. J Biol Chem 283(33):22557–22564. 10.1074/jbc.M800936200 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60(12):2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR (2016) Gaussian 16, Revision B.01, Gaussian, Inc. Wallingford CT
- Grant BJ, Skjaerven L, Yao XQ (2021) The Bio3D packages for structural bioinformatics. Protein Sci 30(1):20–30. 10.1002/pro.3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan F, Khan FI, Song H, Lai D, Juan F (2020) Effects of reverse genetic mutations on the spectral and photochemical behavior of a photoactivatable fluorescent protein PAiRFP1. Spectrochim Acta A Mol Biomol Spectrosc 228:117807. 10.1016/j.saa.2019.117807 [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14(1):33–8. 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79(2):926–935. 10.1063/1.445869 [Google Scholar]
- Kanokratana P, Eurwilaichitr L, Pootanakit K, Champreda V (2015) Identification of glycosyl hydrolases from a metagenomic library of microflora in sugarcane bagasse collection site and their cooperative action on cellulose degradation. J Biosci Bioeng 119(4):384–391. 10.1016/j.jbiosc.2014.09.010 [DOI] [PubMed] [Google Scholar]
- Khan FI, Wei DQ, Gu KR, Hassan MI, Tabrez S (2016a) Current updates on computer aided protein modeling and designing. Int J Biol Macromol 85:48–62. 10.1016/j.ijbiomac.2015.12.072 [DOI] [PubMed] [Google Scholar]
- Khan FI, Shahbaaz M, Bisetty K, Waheed A, Sly WS, Ahmad F, Hassan MI (2016b) Large scale analysis of the mutational landscape in beta-glucuronidase: a major player of mucopolysaccharidosis type VII. Gene 576(1 Pt 1):36–44. 10.1016/j.gene.2015.09.062 [DOI] [PubMed] [Google Scholar]
- Khan FI, Aamir M, Wei D, Ahmad F, Hassan MI (2017) Molecular mechanism of Ras-related protein Rab-5A and effect of mutations in the catalytically active phosphate-binding loop. J Biomol Struct Dyn 35(1):105–118. 10.1080/07391102.2015.1134346 [DOI] [PubMed] [Google Scholar]
- Khan FI, Hassan F, Anwer R, Juan F, Lai D (2020) Comparative analysis of bacteriophytochrome Agp2 and its engineered photoactivatable NIR fluorescent proteins PAiRFP1 and PAiRFP2. Biomolecules 10(9):1286. 10.3390/biom10091286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan FI, Gupta P, Roy S, Azum N, Alamry KA, Asiri AM, Lai D, Hassan MI (2020b) Mechanistic insights into the urea-induced denaturation of human sphingosine kinase 1. Int J Biol Macromol 161:1496–1505. 10.1016/j.ijbiomac.2020.07.280 [DOI] [PubMed] [Google Scholar]
- Krone MW, Travis CR, Lee GY, Eckvahl HJ, Houk KN, Waters ML (2020) More than pi-pi-pi stacking: contribution of amide-pi and CH-pi interactions to crotonyllysine binding by the AF9 YEATS Domain. J Am Chem Soc 142(40):17048–17056. 10.1021/jacs.0c06568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Dangi AK, Shukla P (2018b) Engineering thermostable microbial xylanases toward its industrial applications. Mol Biotechnol 60(3):226–235. 10.1007/s12033-018-0059-6 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26(2):283–291. 10.1107/S0021889892009944 [Google Scholar]
- Li X, Dilokpimol A, Kabel MA, de Vries RP (2022) Fungal xylanolytic enzymes: diversity and applications. Bioresour Technol 344:126290. 10.1016/j.biortech.2021.126290 [DOI] [PubMed] [Google Scholar]
- Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33(5):580–592. 10.1002/jcc.22885 [DOI] [PubMed] [Google Scholar]
- Lu T, Chen Q (2022) Independent gradient model based on Hirshfeld partition: a new method for visual study of interactions in chemical systems. J Comput Chem 43(8):539–555. 10.1002/jcc.26812 [DOI] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C (2015) ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11(8):3696–3713. 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113(18):6378–6396. 10.1021/jp810292n [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40(4):658–674. 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentel LM, Baerends EJ (2014) Can the counterpoise correction for basis set superposition effect be justified? J Chem Theory Comput 10(1):252–267. 10.1021/ct400990u [DOI] [PubMed] [Google Scholar]
- Mhuantong W, Charoensawan V, Kanokratana P, Tangphatsornruang S, Champreda V (2015) Comparative analysis of sugarcane bagasse metagenome reveals unique and conserved biomass-degrading enzymes among lignocellulolytic microbial communities. Biotechnol Biofuels 8(1):16. 10.1186/s13068-015-0200-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H, Ma Y, Zhe Y, Tang X, Wu Q, Huang Z, Han N (2022) Improving the thermostability of a fungal GH11 xylanase via fusion of a submodule (C2) from hyperthermophilic CBM9_1-2. Int J Mol Sci 23(1):463. 10.3390/ijms23010463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31(3):426–428. 10.1021/ac60147a030 [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53(3):240–255. 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- Niesen FH, Berglund H, Vedadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2:2212. 10.1038/nprot.2007.321 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol 276:307–326 [DOI] [PubMed] [Google Scholar]
- Paës G, O’Donohue MJ (2006) Engineering increased thermostability in the thermostable GH-11 xylanase from Thermobacillus xylanilyticus. J Biotechnol 125(3):338–350. 10.1016/j.jbiotec.2006.03.025 [DOI] [PubMed] [Google Scholar]
- Papaleo E, Mereghetti P, Fantucci P, Grandori R, De Gioia L (2009) Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: the myoglobin case. J Mol Graph Model 27(8):889–899. 10.1016/j.jmgm.2009.01.006 [DOI] [PubMed] [Google Scholar]
- Qausain S, Khan FI, Lai D, Hassan MI, Basheeruddin M, Ahmed N, Khan MKA (2020) Mechanistic insights into the urea-induced denaturation of a non-seleno thiol specific antioxidant human peroxiredoxin 6. Int J Biol Macromol 161:1171–1180. 10.1016/j.ijbiomac.2020.05.168 [DOI] [PubMed] [Google Scholar]
- Qausain S, Khan FI, Khan MKA (2023) Conserved acidic second shell residue modulates the structure, stability and activity of non-seleno human peroxiredoxin 6. Int J Biol Macromol 242(Pt 3):124796. 10.1016/j.ijbiomac.2023.124796 [DOI] [PubMed] [Google Scholar]
- Qian C, Liu N, Yan X, Wang Q, Zhou Z, Wang Q (2015) Engineering a high-performance, metagenomic-derived novel xylanase with improved soluble protein yield and thermostability. Enzyme Microb Technol 70:35–41. 10.1016/j.enzmictec.2014.11.005 [DOI] [PubMed] [Google Scholar]
- Roe DR, Cheatham TE III (2013) PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 9(7):3084–3095. 10.1021/ct400341p [DOI] [PubMed] [Google Scholar]
- Shi C-Y, Zhan Y-F, Liu Y, Zhang Z-P, Shen X-Y, Wu C-K, Bai Z-Y, Zhang Z-A, Wang J (2023) Hydrophobic effects enhance xylooligosaccharides production from mulberry branch using xylanase-methacrylate conjugate-catalyzed hydrolysis. Biochem Eng J 193:108851. 10.1016/j.bej.2023.108851 [Google Scholar]
- Stephens DE, Khan FI, Singh P, Bisetty K, Singh S, Permaul K (2014) Creation of thermostable and alkaline stable xylanase variants by DNA shuffling. J Biotechnol 187:139–146. 10.1016/j.jbiotec.2014.07.446 [DOI] [PubMed] [Google Scholar]
- Stojanović S, Petrović Z, Zlatović M (2021) Amide–π interactions in active centers of superoxide dismutase. J Serb Chem Soc 86(9):781–793. 10.2298/JSC210321042S [Google Scholar]
- Sürmeli Y, Şanlı-Mohamed G (2023) Engineering of xylanases for the development of biotechnologically important characteristics. Biotechnol Bioeng 120(5):1171–1188. 10.1002/bit.28339 [DOI] [PubMed] [Google Scholar]
- Tahir Khan M, Dumont E, Chaudhry AR, Wei DQ (2023) Free energy landscape and thermodynamics properties of novel mutations in PncA of pyrazinamide resistance isolates of Mycobacterium tuberculosis. J Biomol Struct Dyn 1–12. 10.1080/07391102.2023.2268216 [DOI] [PubMed]
- Tian YS, Xu J, Chen L, Fu XY, Peng RH, Yao QH (2017) Improvement of the thermostability of xylanase from Thermobacillus composti through site-directed mutagenesis. J Microbiol Biotechnol 27(10):1783–1789. 10.4014/jmb.1705.05026 [DOI] [PubMed] [Google Scholar]
- van der Kamp MW, Mulholland AJ (2013) Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochem 52(16):2708–2728. 10.1021/bi400215w [DOI] [PubMed] [Google Scholar]
- Verma D, Satyanarayana T (2020) Xylanolytic extremozymes retrieved from environmental metagenomes: characteristics, genetic engineering, and applications. Front Microbiol 11:551109. 10.3389/fmicb.2020.551109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma D, Kawarabayasi Y, Miyazaki K, Satyanarayana T (2013) Cloning, expression and characteristics of a novel alkalistable and thermostable xylanase encoding gene (Mxyl) retrieved from compost-soil metagenome. PLoS ONE 8(1):e52459. 10.1371/journal.pone.0052459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Khan FI (2023) Investigation of molecular interactions mechanism of pembrolizumab and PD-1. Int J Mol Sci 24(13):10684. 10.3390/ijms241310684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ma R, Xie X, Liu W, Tu T, Zheng F, You S, Ge J, Xie H, Yao B, Luo H (2017) Thermostability improvement of a Talaromyces leycettanus xylanase by rational protein engineering. Sci Rep 7(1):15287. 10.1038/s41598-017-12659-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Liu M, Zhang K, Li J, Ouyang X (2022) Heterologous expression of family GH11 Aspergillus niger xylanase B (AnXylB11) in Pichia pastoris and the competitive inhibition by riceXIP: an experimental and simulation study. Colloids Surf B Biointerfaces 220:112907. 10.1016/j.colsurfb.2022.112907 [DOI] [PubMed] [Google Scholar]
- Wu Q, Zhang C, Zhu W, Lu H, Li X, Yang Y, Xu Y, Li W (2023) Improved thermostability, acid tolerance as well as catalytic efficiency of Streptomyces rameus L2001 GH11 xylanase by N-terminal replacement. Enzyme Microb Technol 162:110143. 10.1016/j.enzmictec.2022.110143 [DOI] [PubMed] [Google Scholar]
- Yegin S (2023) Microbial xylanases in xylooligosaccharide production from lignocellulosic feedstocks. Biomass Convers Biorefinery 13(5):3619–3658. 10.1007/s13399-022-03190-w [Google Scholar]
- Zhao Y, Truhlar DG (2011) Applications and validations of the Minnesota density functionals. Chem Phys Lett 502(1–3):1–13. 10.1016/j.cplett.2010.11.060 [Google Scholar]
- Zheng J, Xu X, Truhlar DG (2010) Minimally augmented Karlsruhe basis sets. Theor Chem Acc 128(3):295–305. 10.1007/s00214-010-0846-z [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and its Supplementary Information.