

Abstract
Children born less than 30 weeks gestational age (GA) are at high risk for neurodevelopmental delay compared to term peers. Prenatal risk factors and neonatal epigenetics could help identify preterm children at highest risk for poor cognitive outcomes. We aimed to understand the associations among cumulative prenatal risk, neonatal DNA methylation, and child cognitive ability at age 3 years, including whether DNA methylation mediates the association between prenatal risk and cognitive ability. We studied 379 neonates (54% male) born less than 30 weeks GA who had DNA methylation measured at NICU discharge along with 3-year follow up data. Cumulative prenatal risk was calculated from 24 risk factors obtained from maternal report and medical record and epigenome-wide neonatal DNA methylation was assayed from buccal swabs. At 3 year follow up, child cognitive ability was assessed using the Bayley Scales of Infant and Toddler Development (3rd edition). Cumulative prenatal risk and DNA methylation at two CpGs were uniquely associated with child cognitive ability. Using high-dimensional mediation analysis, we also identified differential methylation of 309 CpGs that mediated the association between cumulative prenatal risk and child cognitive ability. Many of the associated CpGs were located in genes (TNS3, TRAPPC4, MAD1L1, APBB2, DIP2C, TRAPPC9, DRD2) that have previously been associated with prenatal exposures and/or neurodevelopmental phenotypes. Our findings suggest a role for both prenatal risk factors and DNA methylation in explaining outcomes for children born preterm and suggest we should further study DNA methylation as a potential mechanism underlying the association between prenatal risk and child neurodevelopment.
Keywords: preterm, prenatal risk, DNA methylation, epigenetics, cognitive ability, neurodevelopment, mediation
Children born preterm are at increased risk of poor cognitive and behavioral outcomes compared to peers born at term. Children born < 30 weeks gestational age (GA) are at especially high risk for delays or deficits in neuromotor, cognitive, and behavioral development (Aarnoudse-Moens et al., 2009; Allen, 2008; Aylward, 2014; Doyle et al., 2010; Hack et al., 2004, 2009; Hille et al., 2008; Stephens & Vohr, 2009; Vohr et al., 2012). Despite this overall trend, there is considerable heterogeneity in outcomes for this high-risk group. Longitudinal studies following contemporary cohorts of very preterm children into toddlerhood and beyond have shown that upwards of two-thirds of children score within normal limits on standardized assessments of cognitive, language, and/or motor development (Camerota et al., 2022). However, this means there are still a sizeable number of children with moderate to severe delays in one or more developmental domains. Understanding what factors predict long-term developmental outcomes in children born < 30 weeks GA is an important research priority, as it could aid in the early identification of infants who require more intensive follow-up, resources, and/or intervention. In the current study, we examined prenatal risk factors and neonatal epigenetics as two complementary influences on cognitive development among children born < 30 weeks GA. Epigenetic modifications, specifically studied via patterns of DNA methylation, have received increasing attention as a potential biological process that may explain the enduring impact of early life experiences on child developmental outcomes. Thus, in addition to examining the unique roles of prenatal risk factors and neonatal epigenetics in predicting long-term cognitive outcomes for preterm children, we also explore whether associations between prenatal risk and cognitive outcomes are mediated by neonatal epigenetic patterns.
Prenatal Risk in Children Born Very Preterm
Child characteristics (e.g., male sex) and neonatal medical morbidities (e.g., brain injury, bronchopulmonary dysplasia) are known contributors to cognitive outcomes among children born very preterm (Helderman et al., 2012; McGowan et al., 2022; O’Shea et al., 2008). More recently, inspired by the increasing popularity of prenatal programming frameworks (e.g., Developmental Origins of Health and Disease [DOHAD], fetal programming, and First 1000 Days; (Camerota & Willoughby, 2021), research has begun to investigate risk factors in the prenatal environment that increase the risk of poor cognitive outcome for children born preterm. Beyond contributing to the risk of preterm birth, risk factors in the prenatal environment may exacerbate the impact of preterm birth and associated illness/immaturity in this already vulnerable population. Recent studies have uncovered associations between prenatal maternal mental health conditions (e.g., depression and anxiety), socioeconomic factors (e.g., income, education), substance use (e.g., tobacco, alcohol), and other prenatal medical conditions (e.g., obesity, diabetes) and the health and developmental outcomes of children born preterm (Fung et al., 2014; Helderman et al., 2012; Hofheimer et al., 2020; Nosavan et al., 2022), resulting in a better understanding of the cumulative adversities preterm infants may face (adverse prenatal conditions combined with preterm birth and associated stressors during NICU stay). However, most research in preterm samples has investigated the impact of prenatal risk factors on health and/or neurodevelopment measured in the neonatal period, rather than investigating long-term associations. The lack of studies on prenatal risk factors and long-term neurodevelopmental outcomes in this population is a known gap in the literature, especially given evidence from term-born populations that prenatal programming has long-reaching associations with neurodevelopmental outcomes measured in infancy, childhood, and beyond (for a review, see Sandman et al., 2011). To the extent that infants born preterm are more susceptible to environmental conditions and at heightened risk for adverse neurodevelopmental outcomes, clarifying the magnitude and persistence of prenatal programming associations in this population is an important next step.
Prior studies investigating prenatal risk factors in relation to child cognitive development have also tended to investigate single risk factors in isolation, rather than assessing prenatal risk holistically. This approach has been useful so far in helping to identify specific risk factors (e.g., maternal depression, psychosocial stress, nutritional factors) that are independently associated with cognitive outcomes. However, to the extent that prenatal risk factors co-occur and/or have compounding impacts on development, an individual variable approach may not capture the full extent of prenatal programming effects. Cumulative risk approaches account for multiple risk factors simultaneously and may have greater power to detect associations with child outcomes as opposed to individual variable approaches (Burchinal et al., 2000; Evans et al., 2013). Cumulative risk approaches are also consistent with an allostatic load perspective that emphasizes additive effects of environmental risk factors on functioning of biological stress systems (McEwen, 1998). We have used a cumulative risk approach in our prior work to show associations between prenatal risk factors and age 3 executive function (Camerota & Willoughby, 2020) in a sample of children primarily born at term. We have also shown how high-risk prenatal phenotypes are related to worse neurodevelopmental and behavioral outcomes for toddlers born < 30 weeks GA (Camerota et al., 2023). However, the associations between cumulative prenatal risk and cognitive outcomes past age 2 have not yet been studied in children born very preterm.
The Role of Epigenetics
Given the well-documented associations between prenatal risk factors and outcomes for children born both preterm and term, an important next step is to better understand the potential mechanisms underlying these associations. One potential mechanism involves epigenetic modifications, or changes to gene expression that do not alter the genetic sequence itself. Detailed descriptions of epigenetic mechanisms and their application to child development have now been published (e.g., Lester et al., 2016). Here, we will specifically consider the role of DNA methylation, one of the most well-studied epigenetic mechanisms in human (and especially developmental) research. Briefly, DNA methylation involves the addition of a methyl group to a cytosine molecule that is typically followed by a guanine molecule at a so-called cytosine-phosphate-guanine (or CpG) site. The methyl group is an epigenetic mark added on top of DNA, so it does not change the sequence of DNA itself, but it can alter the expression of genes. DNA methylation is typically concentrated in the promoter regions of genes, and increased DNA methylation in this region is typically associated with decreased gene transcription. By increasing or decreasing DNA methylation, it is possible to change the expression of a gene, and subsequently alter cellular functioning. DNA methylation is a key mechanism involved in embryogenesis, explaining how undifferentiated (largely unmethylated) stem cells differentiate into specific cell types, via activation or silencing of specific genes.
Critically, DNA methylation is dynamic and is known to be influenced by environmental factors such as prenatal stress (Cao-Lei et al., 2020) and chemical exposures (Martin & Fry, 2018). Differential methylation of genes involved in the stress response system are the most well-studied in relation to prenatal stress and child outcomes. For example, a body of literature has investigated links between early life stress and DNA methylation of NR3C1, a gene that encodes for glucocorticoid receptors (GR) that bind with cortisol. First studied in animal models (Turecki & Meaney, 2016; Weaver et al., 2004) and then replicated in humans (Sosnowski et al., 2018), it has been shown that higher levels of early life stress are associated with increased methylation of NR3C1 and higher stress reactivity in offspring. Epigenome-wide association studies (assessing associations between exposures and DNA methylation across the genome) have largely confirmed the results of candidate gene studies, showing associations among prenatal risk factors (e.g., stress, mood disorders, obesity) and DNA methylation of specific CpG sites in children (Non et al., 2014; Sharp et al., 2017). In turn, individual differences in DNA methylation have been linked to differences in neurodevelopment including associations with neurodevelopmental disorders such as attention-deficit hyperactivity disorder (Neumann et al., 2020; Walton et al., 2017) and autism spectrum disorders (Mordaunt et al., 2020). However, few studies have explicitly tested mediated pathways from prenatal risk to child neurodevelopmental outcomes, possibly because until recently, there were no methods for testing indirect effects in the context of high-throughput (epi)genomics studies. With the development of high-dimensional mediation methods in recent years (for a review, see Zeng et al., 2021), such studies are emerging. Two recent studies conducted in a South African birth cohort found that prenatal tobacco and alcohol exposure (Abrishamcar et al., 2022) and prenatal exposure to indoor air pollution (Feil et al., 2023) were related to infant and toddler cognitive development via their association with newborn DNA methylation at multiple CpGs sites, including methylation of genes and pathways implicated in fetal brain development, cognitive development, and neurodevelopmental delay. Thus, though this remains an emerging area of research, preliminary findings are promising and suggest the plausibility of DNA methylation as a potential molecular mediator linking prenatal risk factors to children’s cognitive development.
Epigenetics and Preterm Birth
Studies examining epigenetic processes in the context of preterm birth have generally supported the results arising from primarily term samples, with some differences. Preterm birth has been shown to leave an epigenetic signature that persists into adolescence and adulthood (Cruickshank et al., 2013; Tan et al., 2018). Prenatal risk, measured cumulatively and as distinct phenotypes, has been shown to predict DNA methylation at birth at multiple specific CpG sites across the epigenome in children born < 30 weeks (Camerota et al., 2021). In this study, prenatal risk was primarily associated with lower DNA methylation, and many of the identified CpGs were located in genes previously shown to be associated with physical and mental health outcomes as well as neurodevelopmental markers (Camerota et al., 2021). Further candidate gene and epigenome-wide findings in preterm children show associations between DNA methylation and bacterial sepsis (Tendl et al., 2013), pain-related stress (Chau et al., 2014; Provenzi et al., 2015), medical morbidities (Everson et al., 2020) and neonatal neurobehavior (Everson et al., 2019; Lester et al., 2015). Thus, while there is preliminary data to suggest that DNA methylation is related to both prenatal risk and neurobehavior in preterm samples, epigenetics has not yet been shown to predict longer-reaching outcomes, nor has it been tested as a mediator linking prenatal risk to cognitive outcomes.
The Current Study
Given the gaps in the literature, the current study had three main aims. First, we aimed to conduct an epigenome-wide association study (EWAS) to understand whether DNA methylation during the neonatal period predicted the cognitive outcome of preschoolers born very preterm. We hypothesized that we would find associations between DNA methylation of specific CpGs and child cognitive ability, based on prior evidence showing associations between neonatal DNA methylation and neonatal neurodevelopment in preterm samples (Everson et al., 2019). Second, using CpGs identified as significant predictors in the EWAS, we aimed to test the unique and joint contributions of cumulative prenatal risk and DNA methylation to child cognitive ability. Given research from primarily term samples, and preliminary evidence showing similar patterns in preterm samples, we hypothesized that higher levels of cumulative prenatal risk would be associated with worse cognitive outcomes at age 3 years. We also expected that cumulative prenatal risk and DNA methylation would uniquely predict cognitive outcomes. Finally, we aimed to test whether DNA methylation mediated the pathway between prenatal risk and cognitive ability, using a combination of high-dimensional and causal mediation analyses. In line with emerging evidence (Abrishamcar et al., 2022; Feil et al., 2023) we hypothesized that we would find significant mediated pathways through one or more CpG site(s).
Methods
Population and Procedures
Participants included mothers and infants from the Neonatal Neurobehavioral Outcomes in Very Preterm Infants (NOVI) study, a multisite, longitudinal study of infants born < 30 weeks GA. The NOVI study recruited infants from April 2014 through June 2016 at nine university affiliated NICUs across the United States. Inclusion criteria included: (1) infant birth at less than 30 weeks GA; (2) parental ability to read and speak English or Spanish; and (3) residence within 3 hours of the NICU and follow up clinic. Exclusion criteria included: (1) maternal age < 18 years; (2) maternal cognitive impairment (precluding ability to obtain informed consent); (3) maternal or infant death; and (4) infant major congenital anomalies. Enrollment and consent procedures were approved by local institutional review boards and all mothers provided written informed consent for themselves and their infants’ participation.
At the time of enrollment, NOVI-trained staff collected maternal and infant demographic and medical information via maternal interview and medical record abstraction. Neonatal medical information was collected using a standardized data collection procedure using the Vermont-Oxford Network protocols (Vermont Oxford Network, 2018). Infant buccal cells were collected for epigenetic analyses during the week of NICU discharge. Pertinent to this analysis, children returned to the clinic for a 3 year follow up visit. Trained research staff administered standardized neurodevelopmental assessments and parents completed questionnaires.
Of the 704 infants enrolled in the NOVI Study, 494 were seen at the 3 year follow up visit. There were no differences in demographic or medical characteristics for children seen versus not seen at age 3 follow up. Of the 494 seen at age 3, 379 had both neonatal DNA methylation and age 3 cognitive assessment data and were included in the current analysis. Figure 1 shows participant flow and Table 1 shows demographic and medical characteristics of included participants. This study follows STROBE guidelines for observational studies.
Figure 1.
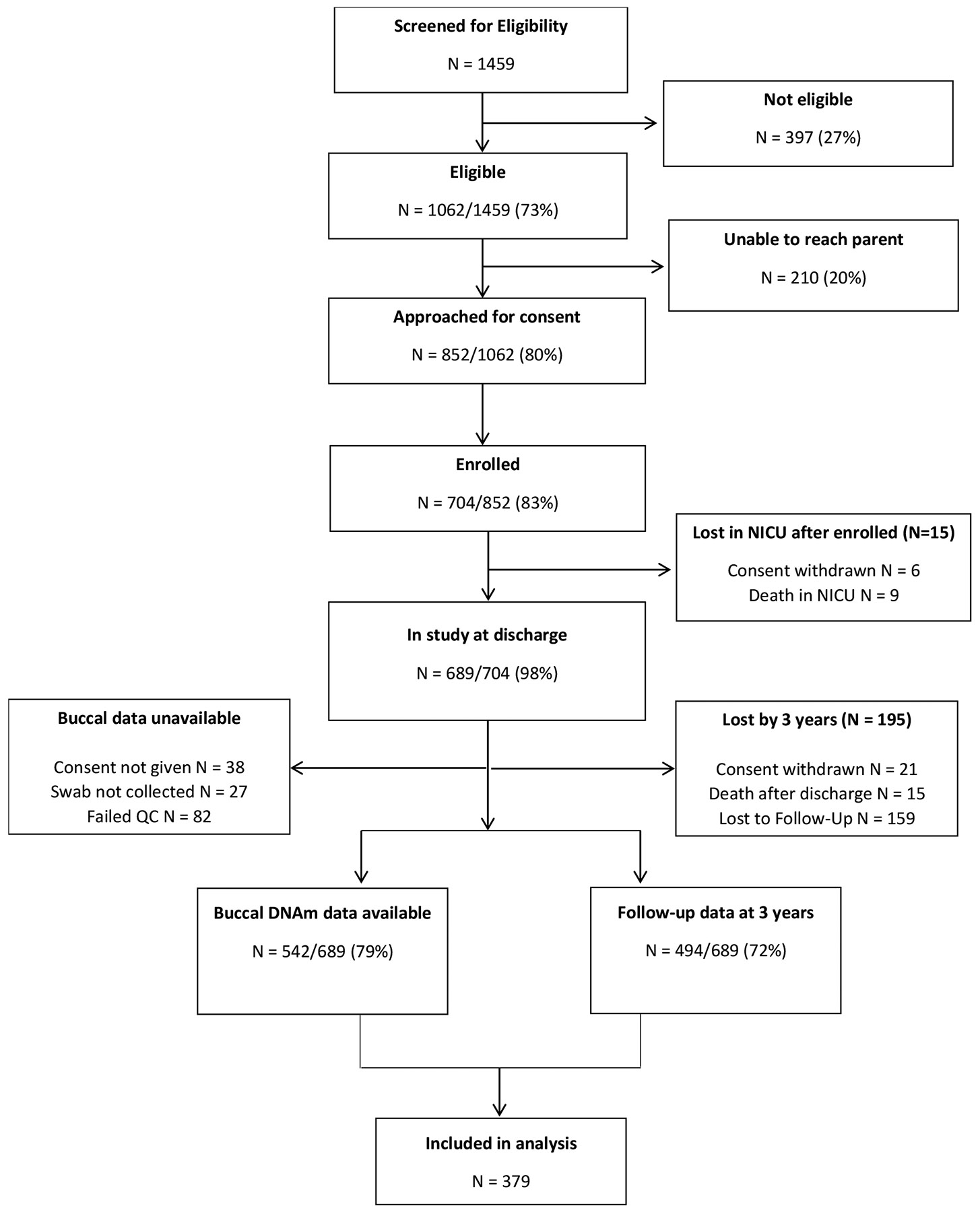
Study flowchart.
Table 1.
Maternal and infant characteristics
| Maternal characteristics (N = 335) | M (SD) or % (n) |
|---|---|
| Minoritized race or ethnicity | 55% (183/333) |
| American Indian / Alaska Native race | 0.3% (1/335) |
| Asian race | 3.9% (13/335) |
| Black or African American race | 20% (68/335) |
| Native Hawaiian or Other Pacific Islander race | 1.5% (5/335) |
| White race | 47% (156/335) |
| More than one race | 19% (64/335) |
| Unknown/not reported race | 8.4% (28/335) |
| Hispanic/Latino/a ethnicity | 22% (74/335) |
| Low SES: Hollingshead level 5 | 8.4% (28/333) |
| Maternal education: < HS/GED | 14% (45/332) |
| No partner | 26% (86/333) |
| Infant characteristics (N = 379) | M (SD) or % (n) |
| Sex = Male | 54% (203/379) |
| Multiple gestation | 28% (104/378) |
| Vaginal delivery | 29% (109/378) |
| Severe retinopathy of prematurity (ROP) | 5.6% (21/378) |
| Necrotizing enterocolitis/sepsis | 20% (76/378) |
| Bronchopulmonary dysplasia (BPD) | 49% (184/378) |
| Serious brain injury | 11% (41/377) |
| GA at birth (weeks) | 27.04 (1.88) |
| Head circumference (cm) | 24.45 (2.41) |
| GA at NICU discharge (weeks) | 39.93 (4.56) |
| Length of NICU stay (days) | 89.72 (37.8) |
| Birth weight (g) | 949.4 (272) |
| Weight at discharge (g) | 2945 (812) |
Note. There were 379 infants born to 335 mothers included in the current analyses. The number of infants and mothers are different due to multiple births. Minority race or ethnicity was defined as any non-White race (e.g., Black, Asian) or ethnicity (e.g., Hispanic and/or Latino/a). Serious brain injury included parenchymal echodensity, periventricular leukomalacia, or ventricular dilation diagnosed via cranial ultrasound. SES, socioeconomic status; HS, high school; GED, General Equivalency Diploma; GA, gestational age; NICU, neonatal intensive care unit.
Measures
Prenatal Risk Factors
We assessed 24 pre- and perinatal risk factors using a combination of parent self-report and medical record abstraction (Table 2). These variables included demographic (maternal age > 35 years, low SES, < high school degree, minority race/ethnicity, no relationship partner), physical health (underweight, obese, high gestational weight gain, hypertension, pre-eclampsia, diabetes, STI/HIV, infection), mental health (depression, anxiety, pregnancy moods and feelings), and substance use (tobacco, alcohol, marijuana, illicit drugs) factors. We created a cumulative prenatal risk index by summing each of the risk factors and dividing by the total number of non-missing items. The majority of women (>95%) had data for all items. Additional information regarding this approach has been published previously (Camerota et al., 2021).
Table 2.
Prevalence of individual prenatal risk factors
| Prenatal risk factors | % (n) |
|---|---|
| Maternal age > 35 ^ | 20% (65/328) |
| Low SES: Hollingshead level 5 ^ | 8.4% (28/333) |
| Maternal education: < HS/GED ^ | 14% (45/332) |
| Minoritized race or ethnicity ^ | 55% (183/333) |
| No partner ^ | 26% (86/333) |
| No prenatal care + | 0.9% (3/333) |
| Underweight ^ | 3.4% (11/327) |
| Obese ^ | 38% (124/327) |
| Too much weight gained ^ | 18% (60/327) |
| Hypertension + | 27% (89/332) |
| Pre-eclampsia + | 21% (69/334) |
| Diabetes + | 6.3% (21/334) |
| STI/HIV + | 6.6% (22/334) |
| Infection + | 11% (38/334) |
| Tobacco + | 14% (46/333) |
| Alcohol + | 3.0% (10/334) |
| Marijuana + | 8.4% (28/334) |
| Illegal substances + | 5.4% (18/334) |
| Depression ^ + | 13% (44/335) |
| Anxiety ^ + | 13% (45/335) |
| Pregnancy "Hard Time" ^ | 11% (35/324) |
| Pregnancy "Felt Down" ^ | 8.6% (28/327) |
| Pregnancy "Felt Hopeless" ^ | 2.5% (8/327) |
| Pregnancy "Felt Slow" ^ | 21% (67/327) |
Note. Minority race or ethnicity was defined as any non-White race (e.g., Black, Asian) or ethnicity (e.g., Hispanic and/or Latino/a). SES, socioeconomic status; HS, high school; GED, General Equivalency Diploma; STI, sexually transmitted infection; HIV, Human immunodeficiency virus. +denotes variable obtained from medical record; ^denotes variable obtained from maternal report.
Neonatal DNA Methylation
Study staff collected buccal swabs within three days of NICU discharge. Neonatal DNA methylation data were generated following a processing pipeline detailed elsewhere (Everson et al., 2020). Briefly, DNA was extracted from buccal swab samples, underwent bisulfite modification, and was assayed for genome-wide DNA methylation using the Illumina MethylationEPIC Beadchip array. Array data were normalized (Aryee et al., 2014; Liu & Siegmund, 2016), standardized across Type-I and Type-II probe designs (Pidsley et al., 2013; Teschendorff et al., 2013), and underwent quality control measures such as the exclusion of samples with poor detection p-values or sex-mismatch and exclusion of probes with low detection p-values, those on the X or Y chromosome, those with single nucleotide polymorphisms within the binding region, and those that could cross-hybridize to other regions of the genome. To reduce multiple testing burden and increase our power to detect meaningful associations, we collapsed data from highly correlated, proximal CpG sites into co-methylated regions (CMRs) using a procedure described previously (Gatev et al., 2020). Also consistent with our prior work, we excluded CpGs or CMRs with low variability (SD < 0.02) and we recoded outliers (defined as values falling 3 interquartile ranges [IQR] below the 25th percentile or 3 IQR above the 75th percentile) to missing.
Following these pre-processing steps, 452,453 methylation loci were available from 542 samples (83% of 651 with buccal swab consent; 77% of entire NOVI cohort; see Figure 1).
Child cognitive ability
Child cognitive ability was assessed at the 3 year follow up visit using the Bayley Scales of Infant and Toddler Development – 3rd edition (BSID-III; Bayley, 2006). The Bayley Scales are a widely used measure of cognitive ability for children up to age 42 months and assesses skills such as exploration and manipulation, memory, object relatedness, habituation, visual preference, and pretend play. Our primary outcome variable was the cognitive composite score, an age-normed standardized score with population mean of 100 and standard deviation of 15.
Analysis Plan
Independent effects of prenatal risk and DNA methylation on child cognitive ability
We conducted an epigenome-wide association study (EWAS) to test associations between DNA methylation at birth and 3-year cognitive ability. We used generalized estimating equation (GEE) models to test associations between DNA methylation at ~450,000 CpG sites (independent variables) and child cognitive ability (dependent variable), accounting for nesting of children within families (i.e., multiple births) and controlling for child sex, study site, neonatal morbidities, GA at birth, and age at buccal collection as well as biological and technical covariates (i.e., cellular heterogeneity and sample plate). We adjusted for cellular heterogeneity by controlling for the proportion of epithelial cells, determined using previously developed reference methylomes (Zheng et al., 2018). P-values were adjusted for multiple testing using the Benjamini-Hochberg false discovery rate (FDR; Benjamini & Hochberg, 1995). We used an FDR threshold of 10% for interpreting epigenome-wide significant findings.
Using the CpGs significantly associated with child cognitive ability in the EWAS, we conducted secondary analyses to determine the unique contributions of DNA methylation and cumulative prenatal risk to child cognitive ability. We did this by estimating three models testing (1) DNA methylation alone, (2) cumulative prenatal risk alone, and (3) DNA methylation and cumulative prenatal risk together as predictors of child cognitive ability. This set of analyses helped us understand whether the association between DNA methylation and cognitive ability is confounded by prenatal risk, whether the association between prenatal risk and cognitive ability is explained by DNA methylation, or whether the two are independently associated. We ran these analyses as GEE models accounting for nesting of children within families and controlling for child sex, study site, neonatal morbidities, GA at birth, and maternal postnatal psychological distress, measured by the Global Severity Index of the Brief Symptom Inventory (Derogatis & Melisaratos, 1983) and averaged across the time period from birth to 3 years. Models that included DNA methylation as an independent variable additionally controlled for important technical and biological factors including age at buccal collection, proportion of epithelial cells, and sample plate.
DNA methylation as a mediator linking prenatal risk to child cognitive ability
Analyses described so far have aimed to (1) identify CpGs that are strongly associated with child cognitive ability, and (2) assess the unique contributions of DNA methylation and cumulative prenatal risk to child cognitive ability. Next, our goal was to formally test whether DNA methylation mediates the association between prenatal risk and child cognitive ability. To address this goal, we considered several methods.
First, we tested for mediation using the CpGs that were significantly associated with child cognitive ability in the EWAS described above. We implemented causal mediation analysis (CMA) using the mediation R package to obtain estimates of average causal mediated effect (ACME), average direct effect (ADE), total effect (TE), and the proportion mediated (PM) in the population (Tingley et al., 2014).
Next, we conducted high-dimensional mediation analyses (HDMA) to test whether DNA methylation across the epigenome mediated the association between cumulative prenatal risk and child cognitive ability. HDMA was preferred over traditional mediation frameworks for several reasons. A traditional mediation approach takes the results of an EWAS conducted on either the exposure (i.e., prenatal risk) or the outcome (i.e., child cognitive ability) and tests whether any significantly associated CpGs identified in the EWAS mediates the association between exposure and outcome. However, each EWAS can only account for one side of the exposure (E)-mediator (M)-outcome (O) pathway (i.e., only the E-M or the M-O path). HDMA methods have specifically been developed to address the large number of composite null hypotheses (i.e., E-M and M-O paths tested simultaneously) that must be tested when investigating epigenome-wide DNA methylation data.
Thus, to supplement the CMA described above, we conducted HDMA using two methods, implemented using the HIMA2 and DACT R packages. HIMA2 uses a penalized regression approach and involves three steps (Perera et al., 2022). First, dimension reduction is handled using a sure independence screening (SIS) method (Fan & Lv, 2008). Then, regression parameters are estimated on this reduced set of mediators using a de-biased Lasso procedure. Finally, the results are adjusted for multiple testing using an FDR correction.
The divide-aggregate-composite null test (DACT) method is an alternative HDMA approach that has been shown to be more highly powered than the HIMA method (Liu et al., 2022). It utilizes the Efron empirical null framework to estimate proportions of the three sub-null hypotheses that make up the composite hypothesis (i.e., E-M path = 0 but M-O path ≠ 0; E-M path ≠ 0 but M-O path = 0; both E-M path and M-O path = 0) across all epigenetic mediators. Then it conducts the DACT for the composite null hypothesis of no mediation effect in the three sub-null cases. Finally, it calculates a DACT p-value that is a weighted sum of all three p values under the three sub-null hypotheses. In order to implement the DACT, we first conducted an EWAS to obtain coefficients and p values for the E-M path. That is, we conducted a series of GEE models testing the association between cumulative prenatal risk and DNA methylation at each CpG site. We already obtained coefficients and p values for the M-O path (association between DNA methylation and child cognitive ability) in the first step of this analysis plan. Consistent with methods described elsewhere (Abrishamcar et al., 2022; Feil et al., 2023), we then filtered CpG sites from both the E-M and M-O paths using a p value threshold of 0.05. We additionally filtered coefficients based on their direction (i.e., positive or negative) to ensure an overall negative total effect, given our hypothesis that increased prenatal risk would be associated with poorer child cognitive ability. We conducted DACT on this reduced set of CpGs and adjusted the resulting DACT p values using FDR correction.
To formally test the magnitude and significance of the indirect pathway between cumulative prenatal risk, DNA methylation, and child cognitive ability, we conducted CMA using significant CpGs identified from HDMA. As described previously, we obtained estimates of the ACME, ADE, TE, and PM in the population along with their 95% confidence intervals.
All HDMA and CMA models controlled for the same covariates described previously (child sex, study site, neonatal morbidities, GA at birth, GA at buccal collection, cellular heterogeneity, sample plate, and maternal postnatal psychological distress [for M-O paths]). All CMA models were run using cluster-robust standard errors to account for nesting of children in families.
Data for these analyses are accessible through NCBI Gene Expression Omnibus (GEO) via accession series GSE128821. This study was not preregistered.
Results
Descriptive Statistics
There were 379 children (54% male) born to 335 mothers in the current analyses (Table 1). The mean GA at birth was 27.0 weeks (SD = 1.88 weeks) and 72% of the sample were singleton births. Of the families with multiple births (n = 39 families), the majority (n = 36 families) were twin births with a smaller number of triplets (n = 1 family) and quadruplets (n = 2 families). The sample was demographically diverse: 55% identified as a minoritized race or ethnicity (< 1% American Indian/Alaska Native, 3.9% Asian, 1.5% Black, 20% Native Hawaiian or Other Pacific Islander, 47% White, 19% multiracial, 8.4% unknown race, 22% Hispanic and/or Latino/a). The majority of mothers in the sample had a high school degree or higher (86%) and had a relationship partner (74%). Overall, there were few demographic and medical characteristics that differed between children included versus excluded from the current analyses (Supplemental Table 1). Notably, children included in the current analysis were more likely to be White (47% versus 37%, p = .02) and to have shorter NICU stays (89.7 versus 99.4 days, p = .005).
The mean cumulative prenatal risk score in this sample was 0.15 (SD = .10), corresponding to approximately 3.5 individual risk factors (SD = 2.3). The most common prenatal risk factors (Table 2) were minoritized race or ethnicity (55%), obesity (38%) and hypertension (27%). Approximately 13% of mothers had depression and 13% had anxiety. The mean BSID-III cognitive composite score was 93.6 (SD = 13.5) with 17% meeting the cutoff for mild delay (composite score < 85) and 5.8% meeting the cutoff for severe delay (composite score < 70). There were no differences in prenatal risk scores or BSID-III cognitive scores for participants included versus excluded in the current analyses (all p > .05).
Neonatal DNA methylation and child cognitive ability
After correction for multiple testing, there were two CpGs with significant associations with child cognitive ability within a 10% FDR (Table 3). These two CpGs were cg20276927 and cg22358121, annotating to the TNS3 and TRAPPC4 gene, respectively. Both CpGs were negatively associated with child cognitive ability. The magnitude of the coefficients indicated that an increase of DNA methylation from the 25th to the 75th percentile was associated with an expected decrease in Bayley cognitive composite scores of 4.24 (cg20276927) and 3.52 (cg22358121) points, equivalent to approximately 0.31 and 0.26 SD, respectively.
Table 3.
Epigenome-wide association study results for CpG sites significantly associated with child cognitive ability (FDR < 10%)
| CpG | Location | Gene annotation | Coefficient |
p value (raw) |
p value (FDR) |
|---|---|---|---|---|---|
| cg20276927 | chr7: 47497808 | TNS3 (5’UTR) | –4.24 | 3.76E-07 | 0.085 |
| cg22358121 | chr11: 118890977 | TRAPPC4 (Body) | –3.52 | 2.66E-07 | 0.085 |
Note. The coefficient represents the expected change in Bayley-III cognitive composite scores associated with an increase of DNAm from the 25th to 75th percentile.
Contributions of DNA methylation and cumulative prenatal risk to child cognitive ability
To examine whether associations identified in the EWAS could be explained by prenatal risk, or if DNA methylation and prenatal risk were independently associated with child cognitive ability, we performed a series of secondary analyses. We estimated three models to test the associations of (1) cumulative prenatal risk alone, (2) DNA methylation (of the two CpGs identified in the EWAS) alone, and (3) cumulative prenatal risk and DNA methylation together on child cognitive ability and compared coefficients and confidence intervals across these three models. Standardized model coefficients are summarized in Figure 2. A full table of regression coefficients appear in Supplemental Table 2. DNA methylation and cumulative prenatal risk were each independently associated with child cognitive ability and the magnitude of the associations were similar when they were modeled together in a single model (Figure 2; red lines) versus in separate models (Figure 2; blue and green lines). In the joint model, a 1-SD increase in cumulative prenatal risk was associated with a 2.9 point (0.21 SD) decrease in BSID-III cognitive scores, whereas a 1-SD increase in DNA methylation of cg20276927 and cg22358121 was associated with a 3.2 point (0.23 SD) and 2.7 point (0.20 SD) decrease in BSID-III cognitive scores, respectively (all p < .001).
Figure 2.
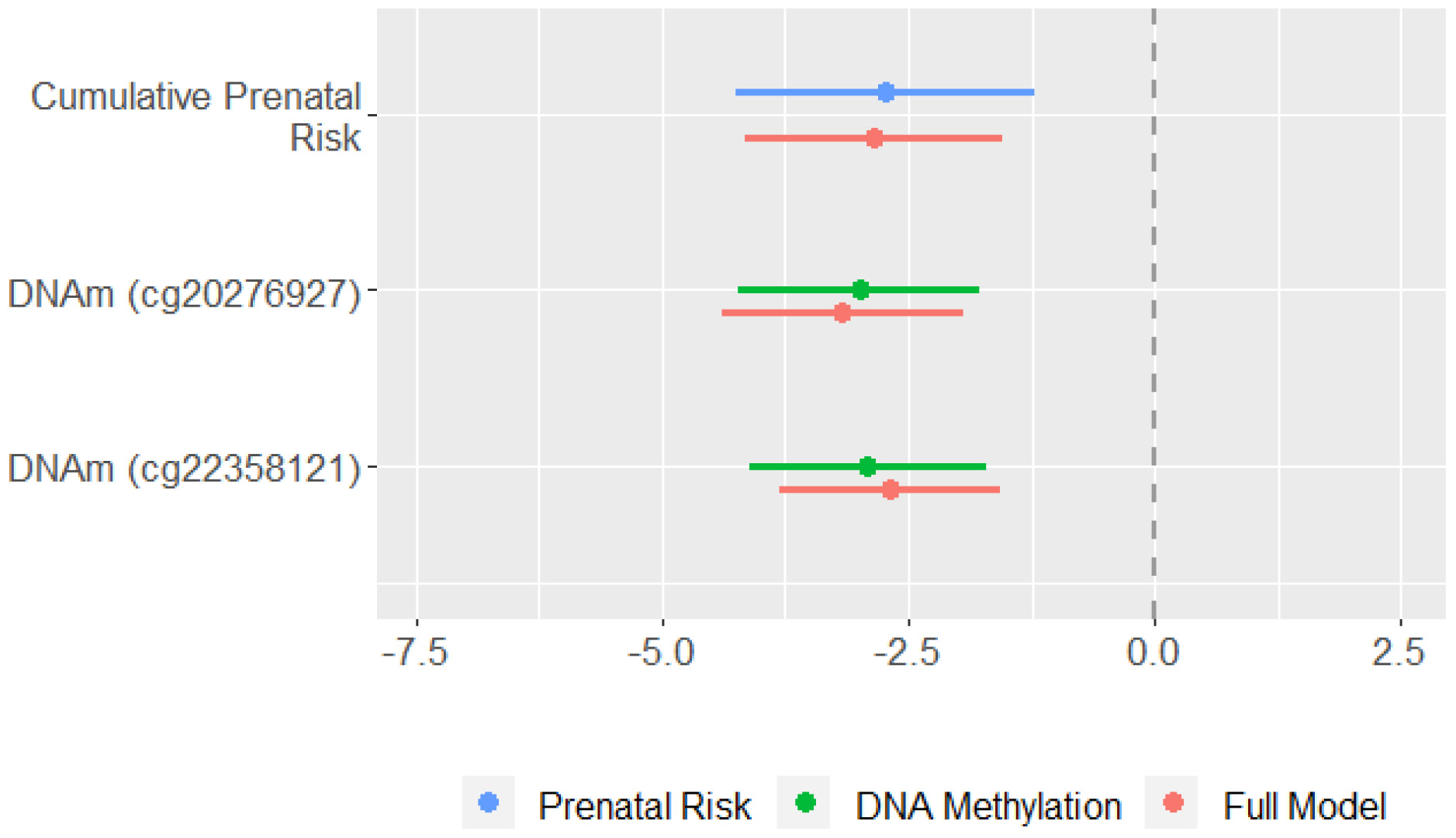
Comparison of model coefficients and 95% confidence intervals from GEE models testing cumulative prenatal risk (blue), DNA methylation of two CpG sites (green), and both cumulative prenatal risk and DNA methylation together (red). Coefficient represents the expected change in Bayley cognitive composite scores associated with a 1-SD increase in the independent variable. All models accounted for nesting of children in families and controlled for child sex, study site, neonatal morbidities, GA at birth, and maternal postnatal psychological distress from birth to age 3 years. Models including DNA methylation as a predictor additionally controlled for age at buccal swab, proportion of epithelial cells, and sample plate. DNAm = DNA methylation; GEE = generalized estimating equations.
DNA methylation as a mediator
We first applied CMA to the two CpGs identified in our EWAS, to test whether DNA methylation of these two CpGs were considered significant mediators between cumulative prenatal risk and BSID-III cognitive scores. The ADE and TE for the impact of cumulative prenatal risk on cognitive ability were significant in both models and ranged from −3.61 to −3.82 (all p < .001). For cg20276927, the ACME was 0.12 (95% CI = −0.38 to 0.66; p = 0.62). For cg22358121, the ACME was −0.13 (95% CI = −0.61 to 0.30; p = 0.54). Thus, results of the CMA did not support a mediated pathway from cumulative prenatal risk to child cognitive ability via either of these two CpGs, which aligns with our above findings that DNA methylation of these CpGs and cumulative prenatal risk have independent associations with cognitive ability.
Given the advantages of HDMA described above, we next used HIMA2 and DACT to test whether DNA methylation of other CpGs may be mediators of the association between cumulative prenatal risk and child cognitive ability. Using HIMA2, we did not identify any CpG sites as significant mediators. However, using DACT, we identified 362 potential mediators. Following CMA, this list was reduced to 309 CpGs with significant indirect effects. Acknowledging that the results from HIMA2 may be an underestimate of the true mediation effect, whereas the results from DACT may be an overestimate, we chose to interpret the top 25 CpG sites with the smallest p-values following CMA (all p ≤ .002). The results for all 309 CpGs are listed in Supplemental Table 3.
As shown in Table 4, the ACME for the top 25 CpGs ranged from −0.38 to −0.68, whereas the ADE and TE ranged from −3.05 to −3.55 and −3.62 to −4.14, respectively (all p ≤ .002). The proportion of the total effect mediated through DNA methylation ranged from .10 to .18.
Table 4.
Top 25 CpGs identified as mediators of the relationship between prenatal risk and child cognitive ability using DACT followed by CMA
| CpG | Location | Gene annotation | ACME (95% CI) |
ADE (95% CI) |
Total Effect (95% CI) |
Prop Med (95% CI) |
|---|---|---|---|---|---|---|
| cg14986172 | chr19: 59097644 | -- | −0.44 (−1.01, −0.08) | −3.40 (−5.25, −1.49) | −3.85 (−5.68, −1.92) | 0.11 (0.02, 0.28) |
| cg22633096 | chr10: 64451393 | -- | −0.43 (−0.87, −0.11) | −3.33 (−5.25, −1.36) | −3.76 (−5.59, −1.90) | 0.11 (0.03, 0.28) |
| cg23674189 | chr19: 5785241 | PRR22 (TSS1500); DUS3L (Body) | −0.50 (−0.96, −0.13) | −3.24 (−5.07, −1.38) | −3.74 (−5.56, −1.84) | 0.13 (0.03, 0.33) |
| cg25679431 | chr3: 32189008 | GPD1L (Body) | −0.49 (−0.97, −0.14) | −3.33 (−5.28, −1.43) | −3.82 (−5.65, −1.88) | 0.12 (0.04, 0.30) |
| cg26236192 | chr20: 59379733 | -- | −0.61 (−1.15, −0.19) | −3.20 (−4.98, −1.38) | −3.82 (−5.65, −1.91) | 0.16 (0.06, 0.35) |
| cg26817877 | chr2: 173539621 | -- | −0.55 (−1.05, −0.17) | −3.18 (−4.99, −1.34) | −3.73 (−5.55, −1.87) | 0.15 (0.04, 0.35) |
| cg00056002 | chr16: 88933996 | PABPN1L (TSS1500) | −0.46 (−0.97, −0.11) | −3.51 (−5.43, −1.61) | −3.97 (−5.79, −2.04) | 0.11 (0.03, 0.27) |
| cg02458632 | chr2: 62678081 | -- | −0.52 (−1.01, −0.14) | −3.31 (−5.16, −1.47) | −3.83 (−5.74, −1.88) | 0.13 (0.04, 0.30) |
| cg03112480 | chr13: 25317909 | -- | −0.39 (−0.81, −0.08) | −3.37 (−5.20, −1.51) | −3.76 (−5.63, −1.87) | 0.10 (0.02, 0.26) |
| cg03409278 | chr18: 24004595 | LINC01543 (Body) | −0.69 (−1.36, −0.18) | −3.45 (−5.45, −1.40) | −4.14 (−6.09, −2.07) | 0.16 (0.04, 0.38) |
| cg03737367 | chr19: 50005045 | MIR150 (TSS1500) | −0.42 (−0.88, −0.09) | −3.32 (−5.16, −1.44) | −3.74 (−5.60, −1.83) | 0.11 (0.03, 0.28) |
| cg04996645 | chr2: 87820882 | LINC00152 (Body) | −0.51 (−1.02, −0.12) | −3.22 (−5.05, −1.31) | −3.72 (−5.59, −1.80) | 0.13 (0.03, 0.34) |
| cg06176876 | chr20: 5159289 | CDS2 (Body) | −0.56 (−1.13, −0.14) | −3.21 (−4.97, −1.37) | −3.77 (−5.60, −1.90) | 0.14 (0.04, 0.34) |
| cg08190384 | chr3: 128734868 | EFCC1 (Body) | −0.57 (−1.15, −0.15) | −3.14 (−5.01, −1.30) | −3.71 (−5.63, −1.72) | 0.15 (0.04, 0.36) |
| cg08673194 | chr17: 17302863 | -- | −0.44 (−0.86, −0.09) | −3.19 (−4.99, −1.34) | −3.62 (−5.49, −1.74) | 0.12 (0.03, 0.29) |
| cg09125754 | chr2: 130886714 | POTEF (1st Exon; 5’UTR) | −0.53 (−1.07, −0.13) | −3.23 (−5.05, −1.41) | −3.76 (−5.66, −1.83) | 0.14 (0.04, 0.33) |
| cg09166630 | chr15: 57887006 | GCOM1 (Body); MYZAP (Body) | −0.38 (−0.83, −0.07) | −3.32 (−5.14, −1.42) | −3.70 (−5.51, −1.78) | 0.10 (0.02, 0.25) |
| cg11139633 | chr19: 34282474 | -- | −0.59 (−1.12, −0.17) | −3.10 (−4.90, −1.25) | −3.69 (−5.54, −1.75) | 0.15 (0.04, 0.36) |
| cg12984408 | chr10: 47106705 | LINC00842 (Body) | −0.67 (−1.29, −0.19) | −3.05 (−4.88, −1.22) | −3.72 (−5.64, −1.73) | 0.18 (0.06, 0.39) |
| cg13319235 | chr8: 96364247 | C8orf37-AS1 (Body) | −0.46 (−0.97, −0.10) | −3.24 (−5.02, −1.40) | −3.70 (−5.56, −1.75) | 0.12 (0.03, 0.30) |
| cg15156941 | chr15: 76578733 | ETFA (Body) | −0.41 (−0.85, −0.09) | −3.55 (−5.31, −1.76) | −3.96 (−5.82, −2.05) | 0.10 (0.02, 0.24) |
| cg19789753 | chr2: 242805962 | -- | −0.49 (−0.95, −0.13) | −3.24 (−5.04, −1.39) | −3.73 (−5.60, −1.80) | 0.13 (0.03, 0.32) |
| cg23830414 | chr21: 44491247 | CBS (Body) | −0.68 (−1.30, −0.21) | −3.08 (−4.98, −1.13) | −3.76 (−5.67, −1.81) | 0.18 (0.05, 0.42) |
| cg25061914 | chr22: 37447468 | KCTD17 (TSS1500) | −0.43 (−0.88, −0.09) | −3.45 (−5.33, −1.52) | −3.88 (−5.83, −1.93) | 0.11 (0.02, 0.27) |
| cg25426208 | chr6: 27198646 | -- | −0.54 (−1.11, −0.14) | −3.19 (−5.14, −1.26) | −3.73 (−5.58, −1.77) | 0.14 (0.04, 0.35) |
Note. CpGs not annotated to a gene are intergenic. ACME = average causal mediated effect; ADE = average direct effect; CMA = causal mediation analysis; DACT = divide-aggregate-composite null test; Prop Med = proportion mediated. All mediated pathways were significant at p < .002.
To better understand the possible biological mechanisms explaining the DNA methylation mediation results, we annotated the significant CpGs from the HDMA to their associated genes and then conducted gene enrichment analyses using two databases (Gene Ontology [GO] and Kyoto Encyclopedia of Genes and Genomes [KEGG]). This analysis helps us understand whether the genes identified in our analysis are disproportionately representative of certain cellular functions or biological processes. However, after adjustment for multiple testing, there were no significantly enriched pathways found in either the GO or KEGG databases.
Post-Hoc Analysis
To better understand the clinical relevance of our findings, we conducted post-hoc analyses to understand whether cumulative prenatal risk and DNA methylation of the two CpGs identified in the EWAS predicted whether a child scored in the clinical range on the Bayley (cognitive composite score < 85, indicative of mild delay). We found that cumulative prenatal risk and DNA methylation of both CpGs significantly predicted odds of scoring in the mild delay range. A 1-SD increase in cumulative prenatal risk was associated with a 1.47 increase in odds of mild delay, whereas a 1-SD increase in DNA methylation of cg20276927 and cg22358121 was associated with 1.50 and 1.53 increase in odds of mild delay, respectively (all p ≤ .01). Full model results are shown in Supplementary Table 4.
Discussion
The aim of this study was to test whether prenatal risk factors and DNA methylation predict cognitive outcomes for children born less than 30 weeks GA. A second goal was to understand whether DNA methylation mediates the association between cumulative prenatal risk and child cognitive ability. Results from our EWAS pointed to two CpG sites that were associated with child cognitive ability at age 3. Further analysis showed that DNA methylation of these two CpG sites and cumulative prenatal risk were independently associated with child cognitive ability, though these specific CpG sites did not mediate the relationship between cumulative prenatal risk and child cognitive ability. Results from HDMA suggested that DNA methylation of other CpG sites may mediate the relationship between prenatal risk and child cognitive ability, with DNA methylation of over 300 CpG sites potentially playing a role. However, differences in the results of two HDMA approaches suggest a need for further study and replication. Together these results lend support for neonatal DNA methylation as an early indicator of cognitive development for children born preterm, and suggest it should be further explored as a possible biological mechanism that may explain the long-lasting impact of prenatal risk factors on developmental outcomes in this population.
The two CpGs that were associated with child cognitive ability in our EWAS annotated to the TNS3 and TRAPPC4 genes. The TNS3 gene is thought to be involved in dephosphorylation and intracellular signal transduction. We have previously found that DNA methylation of the TNS3 gene (albeit at different CpG sites) is related to the number of neonatal morbidities experienced by infants in the NOVI study (Everson et al., 2020). In the prior study, more morbidities were associated with greater methylation of TNS3, whereas in the current study, greater methylation of TNS3 was associated with poorer cognitive ability at age 3 years. Whereas the current study controlled for neonatal medical morbidities, a known contributor to developmental delay, a future study might assess the role of TNS3 (and related cellular signaling mechanisms) in explaining some of the relationship between neonatal medical problems and downstream developmental outcomes in preterm children. Alternatively, it would be interesting to investigate whether neonatal medical problems are part of the mediated pathway from prenatal risk to child cognitive development. Prior GWAS studies have also found that genetic variation in the TNS3 gene is linked to cognitive phenotypes, including adults’ general cognitive functioning (Davies et al., 2018).
The TRAPPC4 gene encodes for a trafficking protein particle complex that regulates vesicle-mediated transport (i.e., transport of substances within membrane-bound vesicles). Unlike the TNS3 gene, prior studies have not implicated epigenetic or genetic variation in TRAPPC3 in any cognitive phenotypes. However, a splicing variant of TRAPPC4 that results in decreased expression has recently been associated with early-infantile-onset neurodegenerative syndrome in a small number of patients (Ghosh et al., 2021). Further investigation of this specific gene and/or related biological pathways is therefore warranted to understand its implication for infant and child cognitive development.
Cumulative prenatal risk and DNA methylation of the two CpG sites identified in the EWAS were both independently associated with child cognitive scores at age 3. These results imply that the impact of prenatal risk on cognitive outcomes is not totally due to DNA methylation of these CpGs, nor is the relationship between DNA methylation and cognitive outcomes confounded by prenatal risk. Our finding that cumulative prenatal risk is a unique predictor of cognitive ability in this high-risk group is novel, as most studies with preterm infants have not investigated multiple prenatal risk factors simultaneously, nor have they followed children into the preschool period. Our findings suggest that it could be important for clinicians to consider prenatal risk factors as an indicator of which neonates may require more extensive follow-up or early intervention following very preterm birth. Moreover, this studyadds to existing work examining either prenatal risk factors (e.g., Nosavan et al., 2022) or DNA methylation (e.g., Everson et al., 2019) as predictors of outcomes in the context of preterm birth and suggests the utility of better understanding both domains in tandem in order to enhance risk stratification.
Our results showed that a one standard deviation change in either prenatal risk or DNA methylation of each CpG site was associated with approximately a 3 point change in BSID-III cognitive composite scores, equivalent to about 0.2 SD. Although each individual association might be of small magnitude, it is notable that experiencing higher levels of prenatal risk in tandem with increased DNA methylation of one or both CpGs could be associated with larger, compounded associations of 0.5 SD or more. Additionally, given that children born < 30 weeks GA are already at risk for poorer cognitive outcomes, these small to moderate effect sizes could have enhanced clinical importance, as these risk factors may be more likely to push children’s scores into the range of ‘mild’ (composite score < 85) or ‘moderate’ (composite score < 70) delay. Our post-hoc analyses showed just this: children with elevated prenatal risk and/or greater DNA methylation of one or both CpGs were more likely to score in range of mild cognitive delay. Thus, what may be considered a small shift in the absolute value of scores could constitute a meaningful shift in the number of preterm children classified as having a developmental delay or disability (Carey et al., 2023) and who therefore would qualify for additional services or interventions. Thus, small but additive effects are especially important to understand in the context of this high-risk group.
While the results from our EWAS suggest that prenatal risk and DNA methylation of two neurocognitive-associated genes may uniquely contribute to risk for poor cognitive outcomes, we also aimed to investigate whether DNA methylation operated as a biological intermediary linking prenatal risk to cognitive outcomes in preterm children. We did not find evidence that either of the two CpGs linked to cognitive ability in our EWAS were significant mediators. However, when utilizing HDMA methods, particularly the DACT approach, we found over 300 potential epigenetic mediators underlying the association between cumulative prenatal risk and child cognitive outcomes. This discrepancy in results illustrates the difficulty of studying putative mechanistic pathways in development with high-dimensional mediator data. Although our EWAS was conducted to identify CpG sites that were strongly associated with child cognitive ability, with stringent adjustment for multiple testing, it only considered one side of the mediated pathway (the M-O relationship). It is therefore not surprising that these same CpGs were not identified as being significantly related to the exposure variable (i.e., cumulative prenatal risk). HDMA methods have been developed to account for the fact that individual EWAS approaches are not sufficient for identifying statistical mediators. Rather, approaches are needed that incorporate preliminary screening or dimension reduction techniques, with less stringent multiple testing correction at preliminary analysis stages.
To date there have been few studies using HDMA methods in the context of child development studies. The majority of studies examining epigenetics as a mediator of environmental risk have used candidate gene approaches (for a review, see Barker et al., 2018). Two recent studies used HDMA to test whether DNA methylation mediated the associations between prenatal tobacco and alcohol exposure (Abrishamcar et al., 2022) and indoor air pollution (Feil et al., 2023) and child neurodevelopmental outcomes in a South African birth cohort. Similar to our findings, both prior studies identified significant epigenetic mediators, but only with the DACT (as opposed to HIMA) approaches. Additionally, we found several epigenetic mediators in common with these prior studies when considering our full list of 309 CpGs and their associated genes. Differential methylation within the MAD1L1 and ABCB4 genes were previously been shown to mediate the association between prenatal tobacco exposure and child cognitive ability at 6 months of age (Abrishamcar et al., 2022). The MAD1L1 (mitotic arrest deficient 1 like 1) gene plays a key role in cell division. Multiple studies now show it is differentially methylated as a function of prenatal smoking (Abrishamcar et al., 2022; Joubert et al., 2016). In turn, differential methylation of MAD1L1 in cortical tissue has been linked to autism spectrum disorder (Nardone et al., 2014). Thus, evidence is growing that differential methylation of MAD1L1 may be a mechanism linking prenatal risk factors to child neurodevelopment.
We also found overlap in methylation of genes (though not specific CpGs) previously shown to mediate the association between indoor air pollution and child neurodevelopmental outcomes (Feil et al., 2023). These overlapping genes include APBB2, DIP2C, NFATC1, PTPRN2, and TRAPPC9. DNA methylation of some of these genes has previously been associated with disorders such as schizophrenia (APBB2, DIP2C, TRAPPC9) and attention-deficit hyperactivity disorder (TRAPPC9) in child and adult samples (Hannon et al., 2016; Li et al., 2021; Montano et al., 2016; Neumann et al., 2020). Thus, these genes and their associated functions may also play a role in explaining associations between prenatal risk factors and/or exposures and downstream neurodevelopmental outcomes. Also of note, one of the CpGs identified as a significant mediator in our analysis annotated to DRD2, a dopamine receptor gene that is part of a family of genes thought to be important in reward processing and the development of psychopathology, particularly substance use disorders (Noble, 2003). On the whole, we found evidence that epigenetic mechanisms, including methylation of neurodevelopmental-associated genes, may mediate the association between cumulative prenatal risk and child cognitive outcomes. It is noteworthy that some of our results were consistent with prior studies, especially since prior studies have not specifically tested epigenetic mechanisms in children born preterm.
Strengths and Limitations
The examination of epigenetic mediation in a diverse sample of children born < 30 weeks GA and followed prospectively since birth is a key strength of our study. Many of our key variables were measured objectively, including medical record reports of maternal prenatal medical variables and assessment of child cognitive outcomes. However, our findings should be considered in light of several study limitations. There were some differences in the characteristics of children included in this analysis versus those lost to follow-up. Children included were more likely to be White and to have shorter NICU stays compared to those excluded, though there were not differences in cumulative prenatal risk based on attrition. Our use of a cumulative risk index could be considered too crude of a marker of prenatal risk, as it equates the severity of each individual risk factor and lacks specificity about the types of risk factors different children were exposed to. On the other hand, cumulative prenatal risk indices have been used before to examine associations with DNA methylation (Rijlaarsdam et al., 2016) and child cognitive outcomes (Camerota & Willoughby, 2020), and have the benefit of stronger prediction of outcomes compared to individual variable approaches (Burchinal et al., 2000). Alternative statistical techniques, such as person-centered approaches, could be considered in future studies to improve the precision of measurement of prenatal risk, though the application of HDMA approaches to multinomial data and the clinical utility of such models (i.e., ability to use person-centered approaches in clinical settings) are pertinent considerations.
We measured DNA methylation in buccal cells, a peripheral tissue that is easily accessible and arises from the same germ layer as the brain. While studies have shown specific advantages of buccal swabs over other peripheral tissues in epigenetic research (Lowe et al., 2013), we cannot assume that our findings are indicative of processes occurring in the brain; rather, they may be more likely to represent systemic biological processes. We specifically used an EWAS approach because there have been no prior studies investigating DNA methylation and age 3 cognitive outcomes for children born very preterm. A downside to this approach is that we did not have specific a priori hypotheses about genes or biological processes that are mechanistically linked to prenatal risk or cognitive development. Thus our findings should be confirmed via replication. Additionally, we chose to remove outliers in DNA methylation data prior to conducting our EWAS as a way reduce the likelihood of identifying false positive associations (Mansell et al., 2019). We acknowledge that this may have led to increased likelihood of Type II error, but we considered this preferable to increased risk of Type I error. Like several prior studies, we only found significant mediation using one of two HDMA approaches. As others have pointed out, DACT may be better powered to detect significant mediators compared to HIMA (Abrishamcar et al., 2022; Feil et al., 2023). Alternatively, the increased power of the DACT method may be accompanied by a greater likelihood of false positive findings. Thus, our results should be interpreted cautiously until we learn more about the robustness of different HDMA methods (particularly in behavioral epigenetic studies). Finally, DNA methylation is influenced both by environmental factors, such as the prenatal risk factors that we studied here, and by genetics. Since we do not have genetic data in this cohort, we cannot disentangle whether some of the associations we report are driven by genetic variants (or genetic by epigenetic interactions), though this is an important future direction.
Conclusion
In sum, we report independent and mediated associations of cumulative prenatal risk, neonatal DNA methylation, and child cognitive ability at age 3 years, in a sample of children born < 30 weeks GA. Understanding the pre- and perinatal factors that are associated with better or worse outcomes in this high-risk group, as well as the mechanisms underlying the transmission of these risk factors, could be useful in both identifying and mitigating risk. Clinicians could use information about children’s prenatal risk factors in combination with their DNA methylation profiles to identify children at highest risk for cognitive delay and provide additional supports to disrupt the negative developmental sequelae. Future research might investigate whether there are factors in the postnatal environment that buffer preterm children from poor outcomes, especially those who have experienced a multitude of prenatal risk factors.
Supplementary Material
Public significance statement:
Understanding what factors predict long-term developmental outcomes in children born less than 30 weeks gestation could help inform prevention and intervention efforts for this high-risk group. This study shows that prenatal risk factors and neonatal DNA methylation could help identify preterm children at high risk for poor cognitive outcomes at age 3 years. DNA methylation may also help explain some of the long-term effects of prenatal risk on child outcomes.
FUNDING STATEMENT
Research reported in this publication was supported by the National Institutes of Health under award numbers R01HD072267, R01HD084515, and UG3OD023347. Dr. Camerota was additionally supported by a career development award from the National Institute of Mental Health (NIMH), grant K01MH129510. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
CONSENT STATEMENT
This study was approved by local Institutional Review Boards, and participants gave informed consent.
DATA AVAILABILITY STATEMENT
Data used in this publication are accessible through NCBI Gene Expression Omnibus (GEO) via accession series GSE128821.
References
- Aarnoudse-Moens CSH, Weisglas-Kuperus N, van Goudoever JB, & Oosterlaan J (2009). Meta-Analysis of Neurobehavioral Outcomes in Very Preterm and/or Very Low Birth Weight Children. Pediatrics, 124(2), 717–728. 10.1542/peds.2008-2816 [DOI] [PubMed] [Google Scholar]
- Abrishamcar S, Chen J, Feil D, Kilanowski A, Koen N, Vanker A, Wedderburn CJ, Donald KA, Zar HJ, Stein DJ, & Hüls A (2022). DNA methylation as a potential mediator of the association between prenatal tobacco and alcohol exposure and child neurodevelopment in a South African birth cohort. Translational Psychiatry, 12(1), 418. 10.1038/s41398-022-02195-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MC (2008). Neurodevelopmental outcomes of preterm infants. Current Opinion in Neurology, 21(2), 123–128. 10.1097/WCO.0b013e3282f88bb4 [DOI] [PubMed] [Google Scholar]
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, & Irizarry RA (2014). Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics, 30(10), 1363–1369. 10.1093/bioinformatics/btu049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward GP (2014). Neurodevelopmental Outcomes of Infants Born Prematurely. Journal of Developmental & Behavioral Pediatrics, 35(6), 394–407. 10.1097/01.DBP.0000452240.39511.d4 [DOI] [PubMed] [Google Scholar]
- Barker ED, Walton E, & Cecil CAM (2018). Annual Research Review: DNA methylation as a mediator in the association between risk exposure and child and adolescent psychopathology. Journal of Child Psychology and Psychiatry, 59(4), 303–322. 10.1111/jcpp.12782 [DOI] [PubMed] [Google Scholar]
- Bayley N (2006). Bayley Scales of Infant and Toddler Development—Third Edition. Harcourt Assessment. [Google Scholar]
- Benjamini Y, & Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological), 57(1), 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Burchinal MR, Roberts JE, Hooper S, & Zeisel SA (2000). Cumulative risk and early cognitive development: A comparison of statistical risk models. Developmental Psychology, 36(6), 793–807. 10.1037/0012-1649.36.6.793 [DOI] [PubMed] [Google Scholar]
- Camerota M, Graw S, Everson TM, McGowan EC, Hofheimer JA, O’Shea TM, Carter BS, Helderman JB, Check J, Neal CR, Pastyrnak SL, Smith LM, Dansereau LM, DellaGrotta SA, Marsit CJ, & Lester BM (2021). Prenatal risk factors and neonatal DNA methylation in very preterm infants. Clinical Epigenetics, 13(1), 171. 10.1186/s13148-021-01164-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camerota M, McGowan EC, Carter BS, Check J, Dansereau LM, DellaGrotta SA, Helderman JB, Hofheimer JA, Neal CR, O’Shea TM, Pastyrnak SL, Smith LM, & Lester BM (2023). Maternal Prenatal Risk Phenotypes and Neurobehavioral Outcomes Among Infants Born Very Preterm. The Journal of Pediatrics, 113521. 10.1016/j.jpeds.2023.113521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camerota M, McGowan EC, Hofheimer JA, O’Shea TM, Carter BS, Helderman JB, Check J, Neal CR, Pastyrnak SL, Smith LM, Loncar CM, Sheinkopf SJ, Dansereau LM, DellaGrotta SA, & Lester BM (2022). Neurodevelopmental profiles of infants born <30 weeks gestation at 2 years of age. Pediatric Research, 91(6), 1579–1586. 10.1038/s41390-021-01871-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camerota M, & Willoughby MT (2020). Prenatal Risk Predicts Preschooler Executive Function: A Cascade Model. Child Development, 91(3), e682–e700. 10.1111/cdev.13271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camerota M, & Willoughby MT (2021). Applying Interdisciplinary Frameworks to Study Prenatal Influences on Child Development. Child Development Perspectives, 15(1), 24–30. 10.1111/cdep.12395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao-Lei L, de Rooij SR, King S, Matthews SG, Metz G. a. S., Roseboom TJ, & Szyf M (2020). Prenatal stress and epigenetics. Neuroscience and Biobehavioral Reviews, 117, 198–210. 10.1016/j.neubiorev.2017.05.016 [DOI] [PubMed] [Google Scholar]
- Carey EG, Ridler I, Ford TJ, & Stringaris A (2023). Editorial Perspective: When is a ‘small effect’ actually large and impactful? Journal of Child Psychology and Psychiatry, jcpp.13817. 10.1111/jcpp.13817 [DOI] [PubMed] [Google Scholar]
- Chau CMY, Ranger M, Sulistyoningrum D, Devlin AM, Oberlander TF, & Grunau RE (2014). Neonatal pain and COMT Val158Met genotype in relation to serotonin transporter (SLC6A4) promoter methylation in very preterm children at school age. Frontiers in Behavioral Neuroscience, 8(DEC), 409. 10.3389/fnbeh.2014.00409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshank MN, Oshlack A, Theda C, Davis PG, Martino D, Sheehan P, Dai Y, Saffery R, Doyle LW, & Craig JM (2013). Analysis of epigenetic changes in survivors of preterm birth reveals the effect of gestational age and evidence for a long term legacy. Genome Medicine, 5(10), 96. 10.1186/gm500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G, Lam M, Harris SE, Trampush JW, Luciano M, Hill WD, Hagenaars SP, Ritchie SJ, Marioni RE, Fawns-Ritchie C, Liewald DCM, Okely JA, Ahola-Olli AV, Barnes CLK, Bertram L, Bis JC, Burdick KE, Christoforou A, DeRosse P, … Deary IJ (2018). Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nature Communications, 9(1), 2098. 10.1038/s41467-018-04362-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derogatis LR, & Melisaratos N (1983). The Brief Symptom Inventory: An introductory report. Psychological Medicine, 13(3), 595–605. 10.1017/S0033291700048017 [DOI] [PubMed] [Google Scholar]
- Doyle LW, Roberts G, & Anderson PJ (2010). Outcomes at Age 2 Years of Infants < 28 Weeks’ Gestational Age Born in Victoria in 2005. The Journal of Pediatrics, 156(1), 49–53.e1. 10.1016/j.jpeds.2009.07.013 [DOI] [PubMed] [Google Scholar]
- Evans GW, Li D, & Whipple SS (2013). Cumulative risk and child development. Psychological Bulletin, 139(6), 1342–1396. 10.1037/a0031808 [DOI] [PubMed] [Google Scholar]
- Everson TM, Marsit CJ, Michael O’Shea T, Burt A, Hermetz K, Carter BS, Helderman J, Hofheimer JA, McGowan EC, Neal CR, Pastyrnak SL, Smith LM, Soliman A, DellaGrotta SA, Dansereau LM, Padbury JF, & Lester BM (2019). Epigenome-wide Analysis Identifies Genes and Pathways Linked to Neurobehavioral Variation in Preterm Infants. Scientific Reports, 9(1), 1–13. 10.1038/s41598-019-42654-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everson TM, O’Shea TM, Burt A, Hermetz K, Carter BS, Helderman J, Hofheimer JA, McGowan EC, Neal CR, Pastyrnak SL, Smith LM, Soliman A, DellaGrotta SA, Dansereau LM, Padbury JF, Lester BM, & Marsit CJ (2020). Serious neonatal morbidities are associated with differences in DNA methylation among very preterm infants. Clinical Epigenetics, 12(1), 1–15. 10.1186/s13148-020-00942-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, & Lv J (2008). Sure Independence Screening for Ultrahigh Dimensional Feature Space. Journal of the Royal Statistical Society Series B: Statistical Methodology, 70(5), 849–911. 10.1111/j.1467-9868.2008.00674.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil D, Abrishamcar S, Christensen GM, Vanker A, Koen N, Kilanowski A, Hoffman N, Wedderburn CJ, Donald KA, Kobor MS, Zar HJ, Stein DJ, & Hüls A (2023). DNA methylation as a potential mediator of the association between indoor air pollution and neurodevelopmental delay in a South African birth cohort. Clinical Epigenetics, 15(1), 31. 10.1186/s13148-023-01444-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung GPG, Chan LM, Ho YC, To WK, Chan HB, & Lao TT (2014). Does gestational diabetes mellitus affect respiratory outcome in late-preterm infants? Early Human Development, 90(9), 527–530. 10.1016/j.earlhumdev.2014.04.006 [DOI] [PubMed] [Google Scholar]
- Gatev E, Gladish N, Mostafavi S, & Kobor MS (2020). CoMeBack: DNA methylation array data analysis for co-methylated regions. Bioinformatics (Oxford, England), 36(9), 2675–2683. 10.1093/bioinformatics/btaa049 [DOI] [PubMed] [Google Scholar]
- Ghosh SG, Scala M, Beetz C, Helman G, Stanley V, Yang X, Breuss MW, Mazaheri N, Selim L, Hadipour F, Pais L, Stutterd CA, Karageorgou V, Begtrup A, Crunk A, Juusola J, Willaert R, Flore LA, Kennelly K, … Gleeson JG (2021). A relatively common homozygous TRAPPC4 splicing variant is associated with an early-infantile neurodegenerative syndrome. European Journal of Human Genetics: EJHG, 29(2), 271–279. 10.1038/s41431-020-00717-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack M, Taylor HG, Schluchter M, Andreias L, Drotar D, & Klein N (2009). Behavioral outcomes of extremely low birth weight children at age 8 years. Journal of Developmental and Behavioral Pediatrics, 30(2), 122–130. 10.1097/DBP.0b013e31819e6a16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack M, Youngstrom EA, Cartar L, Schluchter M, Taylor HG, Flannery D, Klein N, & Borawski E (2004). Behavioral outcomes and evidence of psychopathology among very low birth weight infants at age 20 years. Pediatrics, 114(4), 932–940. 10.1542/peds.2003-1017-L [DOI] [PubMed] [Google Scholar]
- Hannon E, Dempster E, Viana J, Burrage J, Smith AR, Macdonald R, St Clair D, Mustard C, Breen G, Therman S, Kaprio J, Toulopoulou T, Hulshoff Pol HE, Bohlken MM, Kahn RS, Nenadic I, Hultman CM, Murray RM, Collier DA, … Mill J (2016). An integrated genetic-epigenetic analysis of schizophrenia: Evidence for co-localization of genetic associations and differential DNA methylation. Genome Biology, 17(1), 176. 10.1186/s13059-016-1041-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helderman JB, O’Shea TM, Kuban KCK, Allred EN, Hecht JL, Dammann O, Paneth N, McElrath TF, Onderdonk A, & Leviton A (2012). Antenatal Antecedents of Cognitive Impairment at 24 Months In Extremely Low Gestational Age Newborns. PEDIATRICS, 129(3), 494–502. 10.1542/peds.2011-1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille ETM, Dorrepaal C, Perenboom R, Gravenhorst JB, Brand R, & Verloove-Vanhorick SP (2008). Social Lifestyle, Risk-taking Behavior, and Psychopathology in Young Adults Born Very Preterm or with a Very Low Birthweight. The Journal of Pediatrics, 152(6), 793–800.e4. 10.1016/j.jpeds.2007.11.041 [DOI] [PubMed] [Google Scholar]
- Hofheimer JA, Smith LM, McGowan EC, O’Shea TM, Carter BS, Neal CR, Helderman JB, Pastyrnak SL, Soliman A, Dansereau LM, DellaGrotta SA, & Lester BM (2020). Psychosocial and medical adversity associated with neonatal neurobehavior in infants born before 30 weeks gestation. Pediatric Research, 87(4), 721–729. 10.1038/s41390-019-0607-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, Reese SE, Markunas CA, Richmond RC, Xu C-J, Küpers LK, Oh SS, Hoyo C, Gruzieva O, Söderhäll C, Salas LA, Baïz N, Zhang H, Lepeule J, … London SJ (2016). DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. American Journal of Human Genetics, 98(4), 680–696. 10.1016/j.ajhg.2016.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester BM, Conradt E, & Marsit C (2016). Introduction to the Special Section on Epigenetics. Child Development, 87(1), 29–37. 10.1111/cdev.12489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester BM, Marsit CJ, Giarraputo J, Hawes K, LaGasse LL, & Padbury JF (2015). Neurobehavior related to epigenetic differences in preterm infants. Epigenomics, 7(7), 1123–1136. 10.2217/epi.15.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Li Y, Qin H, Tubbs JD, Li M, Qiao C, Lin J, Li Q, Fan F, Gou M, Huang J, Tong J, Yang F, Tan Y, & Yao Y (2021). Genome-wide DNA methylation analysis of peripheral blood cells derived from patients with first-episode schizophrenia in the Chinese Han population. Molecular Psychiatry, 26(8), 4475–4485. 10.1038/s41380-020-00968-0 [DOI] [PubMed] [Google Scholar]
- Liu J, & Siegmund KD (2016). An evaluation of processing methods for HumanMethylation450 BeadChip data. BMC Genomics, 17(1), 469. 10.1186/s12864-016-2819-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Shen J, Barfield R, Schwartz J, Baccarelli AA, & Lin X (2022). Large-Scale Hypothesis Testing for Causal Mediation Effects with Applications in Genome-wide Epigenetic Studies. Journal of the American Statistical Association, 117(537), 67–81. 10.1080/01621459.2021.1914634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe R, Gemma C, Beyan H, Hawa MI, Bazeos A, Leslie RD, Montpetit A, Rakyan VK, & Ramagopalan SV (2013). Buccals are likely to be a more informative surrogate tissue than blood for epigenome-wide association studies. Epigenetics, 8(4), 445–454. 10.4161/epi.24362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansell G, Gorrie-Stone TJ, Bao Y, Kumari M, Schalkwyk LS, Mill J, & Hannon E (2019). Guidance for DNA methylation studies: Statistical insights from the Illumina EPIC array. BMC Genomics, 20(1), 1–15. 10.1186/s12864-019-5761-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EM, & Fry RC (2018). Environmental Influences on the Epigenome: Exposure- Associated DNA Methylation in Human Populations. Annual Review of Public Health, 39(1), 309–333. 10.1146/annurev-publhealth-040617-014629 [DOI] [PubMed] [Google Scholar]
- McEwen BS (1998). Stress, adaptation, and disease. Allostasis and allostatic load. Annals of the New York Academy of Sciences, 840, 33–44. 10.1111/j.1749-6632.1998.tb09546.x [DOI] [PubMed] [Google Scholar]
- McGowan EC, Hofheimer JA, O’Shea TM, Kilbride H, Carter BS, Check J, Helderman J, Neal CR, Pastyrnak S, Smith LM, Camerota M, Dansereau LM, Della Grotta SA, & Lester BM (2022). Analysis of Neonatal Neurobehavior and Developmental Outcomes Among Preterm Infants. JAMA Network Open, 5(7), e2222249. 10.1001/jamanetworkopen.2022.22249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montano C, Taub MA, Jaffe A, Briem E, Feinberg JI, Trygvadottir R, Idrizi A, Runarsson A, Berndsen B, Gur RC, Moore TM, Perry RT, Fugman D, Sabunciyan S, Yolken RH, Hyde TM, Kleinman JE, Sobell JL, Pato CN, … Feinberg AP (2016). Association of DNA Methylation Differences With Schizophrenia in an Epigenome-Wide Association Study. JAMA Psychiatry, 73(5), 506–514. 10.1001/jamapsychiatry.2016.0144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordaunt CE, Jianu JM, Laufer BI, Zhu Y, Hwang H, Dunaway KW, Bakulski KM, Feinberg JI, Volk HE, Lyall K, Croen LA, Newschaffer CJ, Ozonoff S, Hertz-Picciotto I, Fallin MD, Schmidt RJ, & LaSalle JM (2020). Cord blood DNA methylome in newborns later diagnosed with autism spectrum disorder reflects early dysregulation of neurodevelopmental and X-linked genes. Genome Medicine, 12(1), 88. 10.1186/s13073-020-00785-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardone S, Sams DS, Reuveni E, Getselter D, Oron O, Karpuj M, & Elliott E (2014). DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Translational Psychiatry, 4(9), e433. 10.1038/tp.2014.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann A, Walton E, Alemany S, Cecil C, González JR, Jima DD, Lahti J, Tuominen ST, Barker ED, Binder E, Caramaschi D, Carracedo Á, Czamara D, Evandt J, Felix JF, Fuemmeler BF, Gutzkow KB, Hoyo C, Julvez J, … Tiemeier H (2020). Association between DNA methylation and ADHD symptoms from birth to school age: A prospective meta-analysis. Translational Psychiatry, 10(1). 10.1038/s41398-020-01058-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble EP (2003). D2 dopamine receptor gene in psychiatric and neurologic disorders and its phenotypes. American Journal of Medical Genetics, 116B(1), 103–125. 10.1002/ajmg.b.10005 [DOI] [PubMed] [Google Scholar]
- Non AL, Binder AM, Kubzansky LD, & Michels KB (2014). Genome-wide DNA methylation in neonates exposed to maternal depression, anxiety, or SSRI medication during pregnancy. Epigenetics, 9(7), 964–972. 10.4161/epi.28853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosavan NP, Smith LM, Dansereau LM, Roberts MB, Hofheimer JA, Carter BS, Helderman JB, McGowan EC, Neal CR, Pastyrnak S, Della Grotta SA, O’Shea TM, & Lester BM (2022). Associations between maternal pre-pregnancy body mass index and neonatal neurobehavior in infants born before 30 weeks gestation. Journal of Perinatology, 42(4), 483–490. 10.1038/s41372-021-01308-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea TM, Kuban KCK, Allred EN, Paneth N, Pagano M, Dammann O, Bostic L, Brooklier K, Butler S, Goldstein DJ, Hounshell G, Keller C, McQuiston S, Miller A, Pasternak S, Plesha-Troyke S, Price J, Romano E, Solomon KM, … for the Extremely Low Gestational Age Newborns Study Investigators. (2008). Neonatal Cranial Ultrasound Lesions and Developmental Delays at 2 Years of Age Among Extremely Low Gestational Age Children. Pediatrics, 122(3), e662–e669. 10.1542/peds.2008-0594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera C, Zhang H, Zheng Y, Hou L, Qu A, Zheng C, Xie K, & Liu L (2022). HIMA2: High-dimensional mediation analysis and its application in epigenome-wide DNA methylation data. BMC Bioinformatics, 23(1), 296. 10.1186/s12859-022-04748-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, & Schalkwyk LC (2013). A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics, 14(1), 293. 10.1186/1471-2164-14-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzi L, Fumagalli M, Sirgiovanni I, Giorda R, Pozzoli U, Morandi F, Beri S, Menozzi G, Mosca F, Borgatti R, & Montirosso R (2015). Pain-related stress during the Neonatal Intensive Care Unit stay and SLC6A4 methylation in very preterm infants. Frontiers in Behavioral Neuroscience, 9(APR), 99. 10.3389/fnbeh.2015.00099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijlaarsdam J, Pappa I, Walton E, Bakermans-Kranenburg MJ, Mileva-Seitz VR, Rippe RCA, Roza SJ, Jaddoe VWV, Verhulst FC, Felix JF, Cecil CAM, Relton CL, Gaunt TR, McArdle W, Mill J, Barker ED, Tiemeier H, & van IJzendoorn MH (2016). An epigenome-wide association meta-analysis of prenatal maternal stress in neonates: A model approach for replication. Epigenetics, 11(2), 140–149. 10.1080/15592294.2016.1145329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandman CA, Davis EP, Buss C, & Glynn LM (2011). Prenatal programming of human neurological function. International Journal of Peptides, 2011. 10.1155/2011/837596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp GC, Salas LA, Monnereau C, Allard C, Yousefi P, Everson TM, Bohlin J, Xu Z, Huang R-C, Reese SE, Xu C-J, Baïz N, Hoyo C, Agha G, Roy R, Holloway JW, Ghantous A, Merid SK, Bakulski KM, … Relton CL (2017). Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: Findings from the pregnancy and childhood epigenetics (PACE) consortium. Human Molecular Genetics, 26(20), 4067–4085. 10.1093/hmg/ddx290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosnowski DW, Booth C, York TP, Amstadter AB, & Kliewer W (2018). Maternal prenatal stress and infant DNA methylation: A systematic review. Developmental Psychobiology, 60(2), 127–139. 10.1002/dev.21604 [DOI] [PubMed] [Google Scholar]
- Stephens BE, & Vohr BR (2009). Neurodevelopmental Outcome of the Premature Infant. Pediatric Clinics of North America, 56(3), 631–646. 10.1016/j.pcl.2009.03.005 [DOI] [PubMed] [Google Scholar]
- Tan Q, Li S, Frost M, Nygaard M, Soerensen M, Larsen M, Christensen K, & Christiansen L (2018). Epigenetic signature of preterm birth in adult twins. Clinical Epigenetics, 10(1), 87. 10.1186/s13148-018-0518-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tendl KA, Schulz SMF, Mechtler TP, Bohn A, Metz T, Greber-Platzer S, Kasper DC, Herkner KR, & Item CB (2013). DNA methylation pattern of CALCA in preterm neonates with bacterial sepsis as a putative epigenetic biomarker. Epigenetics, 8(12), 1261–1267. 10.4161/epi.26645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, & Beck S (2013). A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics, 29(2), 189–196. 10.1093/bioinformatics/bts680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingley D, Yamamoto T, Hirose K, Keele L, & Imai K (2014). mediation: R Package for Causal Mediation Analysis. Journal of Statistical Software, 59(5). 10.18637/jss.v059.i05 [DOI] [Google Scholar]
- Turecki G, & Meaney MJ (2016). Effects of the Social Environment and Stress on Glucocorticoid Receptor Gene Methylation: A Systematic Review. Biological Psychiatry, 79(2), 87–96. 10.1016/j.biopsych.2014.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermont Oxford Network. Manual of operations: Part 2. Data definitions and infant data forms. (2018). Vermont Oxford Network. [Google Scholar]
- Vohr BR, Stephens BE, Higgins RD, Bann CM, Hintz SR, Das A, Newman JE, Peralta-Carcelen M, Yolton K, Dusick AM, Evans PW, Goldstein RF, Ehrenkranz RA, Pappas A, Adams-Chapman I, Wilson-Costello DE, Bauer CR, Bodnar A, Heyne RJ, … Fuller J (2012). Are Outcomes of Extremely Preterm Infants Improving? Impact of Bayley Assessment on Outcomes. The Journal of Pediatrics, 161(2), 222–228.e3. 10.1016/j.jpeds.2012.01.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton E, Pingault J-B, Cecil CAM, Gaunt TR, Relton CL, Mill J, & Barker ED (2017). Epigenetic profiling of ADHD symptoms trajectories: A prospective, methylome-wide study. Molecular Psychiatry, 22(2), 250–256. 10.1038/mp.2016.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver ICG, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, & Meaney MJ (2004). Epigenetic programming by maternal behavior. Nature Neuroscience, 7(8), 847–854. 10.1038/nn1276 [DOI] [PubMed] [Google Scholar]
- Zeng P, Shao Z, & Zhou X (2021). Statistical methods for mediation analysis in the era of high-throughput genomics: Current successes and future challenges. Computational and Structural Biotechnology Journal, 19, 3209–3224. 10.1016/j.csbj.2021.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SC, Webster AP, Dong D, Feber A, Graham DG, Sullivan R, Jevons S, Lovat LB, Beck S, Widschwendter M, & Teschendorff AE (2018). A novel cell-type deconvolution algorithm reveals substantial contamination by immune cells in saliva, buccal and cervix. Epigenomics, 10(7), 925–940. 10.2217/epi-2018-0037 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data used in this publication are accessible through NCBI Gene Expression Omnibus (GEO) via accession series GSE128821.