

Abstract
New synthetic methods for the catalytic functionalization of C─H bonds have the potential to revolutionize the synthesis of complex molecules1-4. However, the realization of this synthetic potential requires the ability to functionalize selectively one C─H bond in a compound containing many such bonds and an array of functional groups. The site-selective functionalization of aliphatic C─H bonds is one of the greatest challenges that must be met for C─H bond functionalization to be used widely in complex-molecule synthesis1,3,5,6, and processes catalysed by transition-metals provide the opportunity to control selectivity7,8. Current methods for catalytic, aliphatic C─H bond functionalization typically rely on the presence of one inherently reactive C─H bond9,10, or on installation and subsequent removal of directing groups that are not components of the desired molecule8. To overcome these limitations, we sought catalysts and reagents that would facilitate aliphatic C─H bond functionalization at a single site, with chemoselectivity derived from the properties of the catalyst and site-selectivity directed by common functional groups11 contained in both the reactant and the desired product. Here we show that the combination of an iridium-phenanthroline catalyst and a dihydridosilane reagent leads to the site-selective γ-functionalization of primary C─H bonds controlled by a hydroxyl group, the most common functional group in natural products12. The scope of the reaction encompasses alcohols and ketones bearing many substitution patterns and auxiliary functional groups; this broad scope suggests that this methodology will be suitable for the site-selective and diastereoselective functionalization of complex natural products.
Our strategy for the functionalization of aliphatic C─H bonds directed by hydroxyl groups is outlined below:
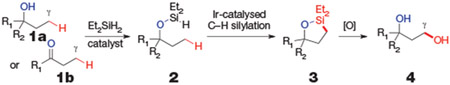
In this approach, a single dihydridosilane reagent has two roles. First, the silane attaches to the oxygen atom of an alcohol (1a) or ketone (1b) by forming a (hydrido)silyl ether (2) to direct the C─H bond functionalization. The (hydrido)silyl ether is formed by dehydrogenative coupling with the alcohol or by hydrosilylation of the ketone. Second, the Si─H unit of the silyl ether undergoes dehydrogenative functionalization of a primary C─H bond without isolation of the intermediate (hydrido)silyl ether. Tamao-Fleming oxidation of the oxasilolane (3) formed by this process then yields a 1,3-diol (4) from a net 1,4-hydroxyl-directed, aliphatic C─H bond functionalization. On the basis of our recently developed, iridium-catalysed silylation of aromatic C─H bonds13, we anticipated that both silylation reactions could be catalysed by iridium complexes13,14. However, successful implementation of this strategy would require the identification of a catalyst that is reactive for the silylation of unactivated aliphatic C─H bonds, not just the silylation of typically more reactive aromatic C─H bonds. It would also require that the directing effect be strong enough to ensure that intermolecular functionalization of aromatic C─H bonds would not interfere with the desired directed functionalization of an aliphatic C─H bond.
We began by evaluating iridium complexes containing a series of bipyridine ligands (L) as catalysts for the dehydrogenative cyclization of (hydrido)silyl ether (5), generated in situ by an iridium-catalysed dehydrogenative coupling of diethylsilane (Et2SiH2) with tetrahydrolinalool. The reaction is outlined below:
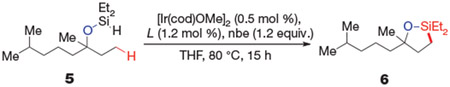
In the presence of 1.0 mol % of a combination of [Ir(cod)OMe]2 (cod, 1,5-cyclooctadiene; Me, methyl group) and 1,10-phenanthroline (phen), with norbornene (nbe) as an H2 acceptor13, we observed 77% conversion of 5 to form oxasilolane (6) in 68% yield (Table 1a). The same reaction conducted with 4,4’-di-tert-butylbipyridine (dtbpy), the most commonly used ligand for iridium-catalysed arene borylation15, occurred with only slightly higher conversion and yield (Table 1b). To probe systematically the effect of the electronic properties of ancillary ligands on iridium-catalysed aliphatic C─H bond silylation, we conducted reactions with catalysts generated from a series of 4,7-disubstituted phenanthroline derivatives (Table 1c-g). Reactions conducted with the more electron-donating ligands occurred to higher relative conversions and formed the silylation product in higher yields than did reactions conducted with the less electron-donating ligands16. Ultimately, we obtained the highest yield of 6 with a catalyst containing 3,4,7,8-tetramethyl-1,10-phenanthroline (Me4phen, Table 1h), the most electron-donating phenanthroline derivative in this series16.
Table 1 ∣.
Examination of bipyridine ligands for aliphatic C─H bond silylation (5→6)
| Ligand, L | Conversion | Yield | |
|---|---|---|---|
| a | phen | 77% | 68% |
| b | dtbpy | 83% | 76% |
| c | 4,7-Cl2phen | 13% | <2% |
| d | 4,7-(HO)2phen | 33% | 19% |
| e | 4,7-Ph2phen | 82% | 72% |
| f | 4,7-Me2phen | 94% | 85% |
| g | 4,7-(MeO)2phen | 94% | 87% |
| h | 3,4,7,8-Me4phen | 100% | 99% |
Corrected, overall gas chromatography yields (0.25-mmol scale) with dodecane as an internal standard. Reagentsand conditions: tetrahydrolinalool (1.0 equiv.), Et2SiH2 (1.2 equiv.), [Ir(cod)OMe]2 (0.05 mol %), THF, room temperature, 17 h; removal of volatiles, then [Ir(cod)OMe]2(0.5 mol %), ligand (1.2 mol %), nbe (1.2 equiv.), THF, 80 °C, 15 h. For full experimental details, see the Supplementary Information.
After identifying a suitable catalyst for aliphatic C─H bond silylation, we developed a telescoped procedure for transformation of a starting alcohol (7) or ketone (8) to a 1,3-diol (Fig. 1). Following iridium-catalysed aliphatic C─H silylation, we obtained the diol simply by adding MeOH, KHCO3 and aqueous H2O2 to the crude silylation reaction mixture and heating at 50 °C. To provide compounds that were easily purified by silica-gel chromatography, we acylated the diols thus obtained before isolation. Figure 1 shows a series of products prepared by this sequence of C─H silylation and oxidation. Both tertiary (9a) and secondary acyclic alcohols (9b, c) underwent aliphatic C─H bond oxygenation with comparable efficiency. Under most conditions, oxygenation of a primary C─H bond in the presence of a secondary alcohol would be challenging because of competitive oxidation of the secondary alcohol to the corresponding ketone. Our strategy allows an overall oxidation of a primary C─H bond in the presence of a secondary alcohol because it uses a mildly reducing silane as the functionalizing reagent.
Figure 1 ∣. Hydroxyl-directed γ-oxygenation of secondary and tertiary alcohols and ketones.
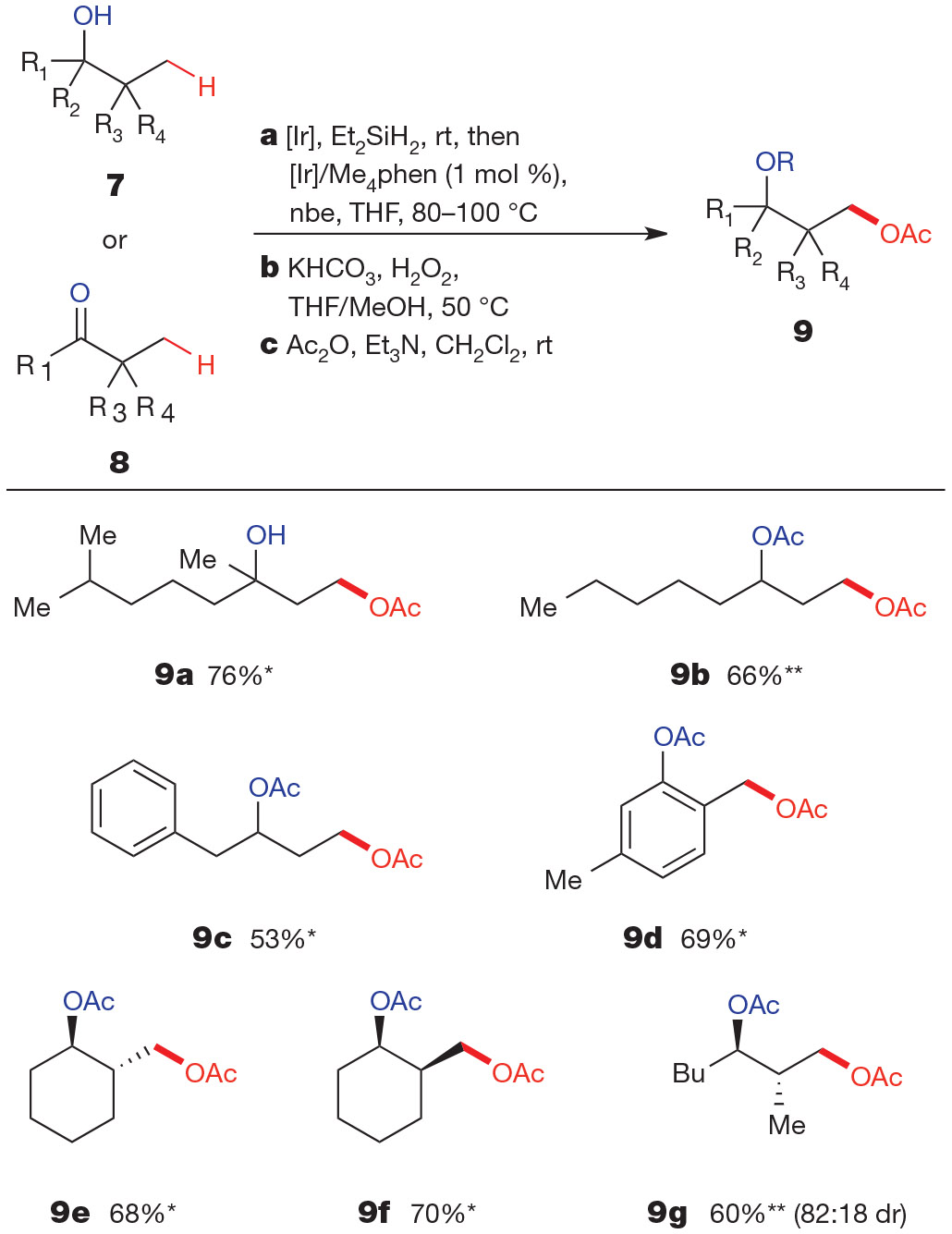
Overall isolated yields for reactions conducted on a 1.0-mmol scale following purification by silica-gel chromatography. Reagents and conditions: a, 7 (to give products marked *) or 8 (products marked **) (1.0 equiv.), Et2SiH2 (1.2 equiv.), [Ir(cod)OMe]2 (0.05 mol %), THF, room temperature (rt); removal of volatiles, then [Ir(cod)OMe]2 (0.5 mol %), Me4phen (1.2 mol %), nbe (1.2 equiv.), THF, 80–100 °C. b, KHCO3 (2.5 equiv.), 30% aqueous H2O2 (10 equiv.), THF/MeOH, 50 °C. c, Ac2O (1.5–3.0 equiv.), DMAP (0–0.05 equiv.), CH2Cl2/Et3N, room temperature. Ac2O, acetic anhydride; DMAP, 4-dimethylaminopyridine; Et3N, triethylamine. For full experimental details, see the Supplementary Information.
Reactions of phenols (9d) and cyclic aliphatic alcohols (9e, f) also occurred under these conditions in good yields. The C─H functionalization of cyclic alcohols proved insensitive to the stereochemistry of the ring fusion in the bicyclic oxasilolane intermediate; trans- and cis-2-methylcyclohexanol underwent this process in similar yields (9e, f). However, the reaction of an acyclic substrate bearing diastereotopic methyl groups formed 9g as the major product with significant diastereoselectivity (82:18 dr), resulting from C─H bond silylation to form the less strained trans-disubstituted oxasilolane intermediate.
The products in Fig. 2 illustrate the range of functional groups that are tolerated by the aliphatic C─H bond oxygenation sequence. Aryl halides (9h, i), an aryl trifluoromethyl group (9j), and an internal carbon─carbon double bond (9k) were all compatible with the reaction conditions. The sequence also tolerated phenols protected as the corresponding benzyl or silyl ethers (9l–n); protected catechols (9o); and aliphatic alcohols protected with trialkylsilyl groups (9p, q). Although aldehydes were not compatible, the C─H functionalization occurred in high yield when this functionality was masked as an acetal (9r). None of the examples in Fig. 2 suffered from competing intra- or intermolecular aromatic C─H bond functionalization, an observation that is particularly striking for 9l, which contains three unhindered aromatic C─H bonds. Because the catalyst for the present aliphatic C─H silylation is similar to that used most commonly for aromatic C─H borylation15, the absence of products from aromatic functionalization can be attributed to the powerful directing ability of the (hydrido)silyl group to promote selective functionalization of a δ─C─H bond.
Figure 2 ∣. Functional-group tolerance of hydroxyl-directed γ-oxygenation.
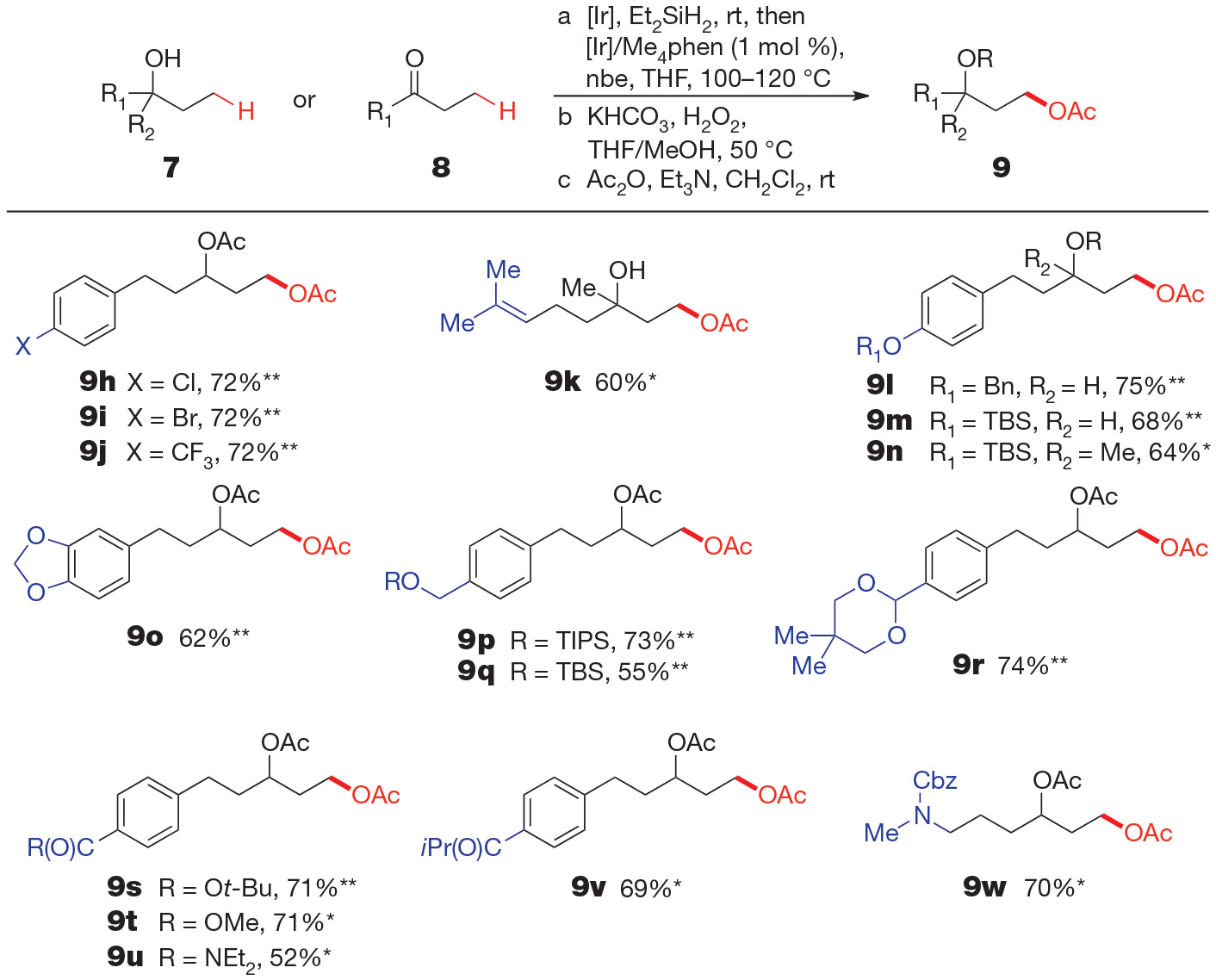
Overall isolated yields for reactions conducted on a 1.0-mmol scale following purification by silica-gel chromatography. Reagents and conditions: a, 7 (to give products marked *)or 8 (products marked **) (1.0 equiv.), Et2SiH2 (1.2 equiv.), [Ir(cod)OMe]2 (0.05 mol %), THF, room temperature; removal of volatiles, then [Ir(cod)OMe]2 (0.5 mol %), Me4phen (1.2 mol %), nbe (1.2 equiv.), THF, 100–120 °C. b, KHCO3 (2.5 equiv.), 30% aqueous H2O2 (10 equiv.), THF/MeOH, 50 °C. c, Ac2O (1.5–3.0 equiv.), DMAP (0–0.05 equiv.), CH2Cl2/Et3N, room temperature. For compounds 9t, 9u, 9v and 9w, step a (first half) used Ru(PPh3)3Cl2 (0.2 mol %) in benzene or toluene at 50 °C. For compound 9u, step a (second half) used 2 mol % [Ir]/Me4phen. For full experimental details, see the Supplementary Information.
Whereas a tert-butyl ester was tolerated by our standard conditions (9s), methyl ester, amide, ketone and carbamate functional groups underwent competitive hydrosilylation when forming the silyl ether in the presence of the iridium catalyst. However, diethyl(hydrido)silyl ethers formed from the coupling of Et2SiH2 with alcohols containing auxiliary methyl ester, ketone and carbamate moieties without competitive reduction of the carbonyl functionalities in the presence of 0.2 mol % of the ruthenium complex Ru(PPh3)3Cl2 (ref. 17) in which Ph is a phenyl group. Moreover, the carbonyl groups in these compounds were compatible with the C─H silylation process and subsequent Tamao-Fleming oxidation; thus, we obtained diol derivatives 9t, 9v and 9w in good overall yield from the starting alcohols. A small amount of carbonyl hydrosilylation occurred during ruthenium-catalysed silylation of the alcohol containing an amide functional group, but we still obtained diol product 9u in 52% overall yield.
Because the iridium-catalysed C─H functionalization tolerates auxiliary functionality and is highly selective for reaction at a single C─H bond, it allows site-selective γ-functionalization of natural products bearing hydroxyl and carbonyl groups (Fig. 3). The reaction of (+)-fenchol (10) under our standard conditions led to selective functionalization of the C9 methyl group, directed by the C2 hydroxyl group, to generate 9-hydroxyfenchol diacetate (11a) in 66% yield (Fig. 3a). Previously, 11a was accessed indirectly from other terpene skeletons by multi-step rearrangement sequences18; direct oxidation of (+)-fenchol at the C9 position by fermentation formed diol 11b in only 4.0% yield and was accompanied by three further diol isomers19. Exo-selective hydrosilylation of (+)-camphor (12) catalysed by 0.05 mol % [Ir(coe)2Cl]2, in which coe is cyclooctene (92:8 exo/endo), followed by directed C─H functionalization at the C10 methyl group, gave 13a in 57% yield. The previous synthesis of exo-2,10-camphanediol (13b) began with the significantly more expensive (+)-ketopinic acid20.
Figure 3 ∣. Directed aliphatic C─H functionalization of natural products.
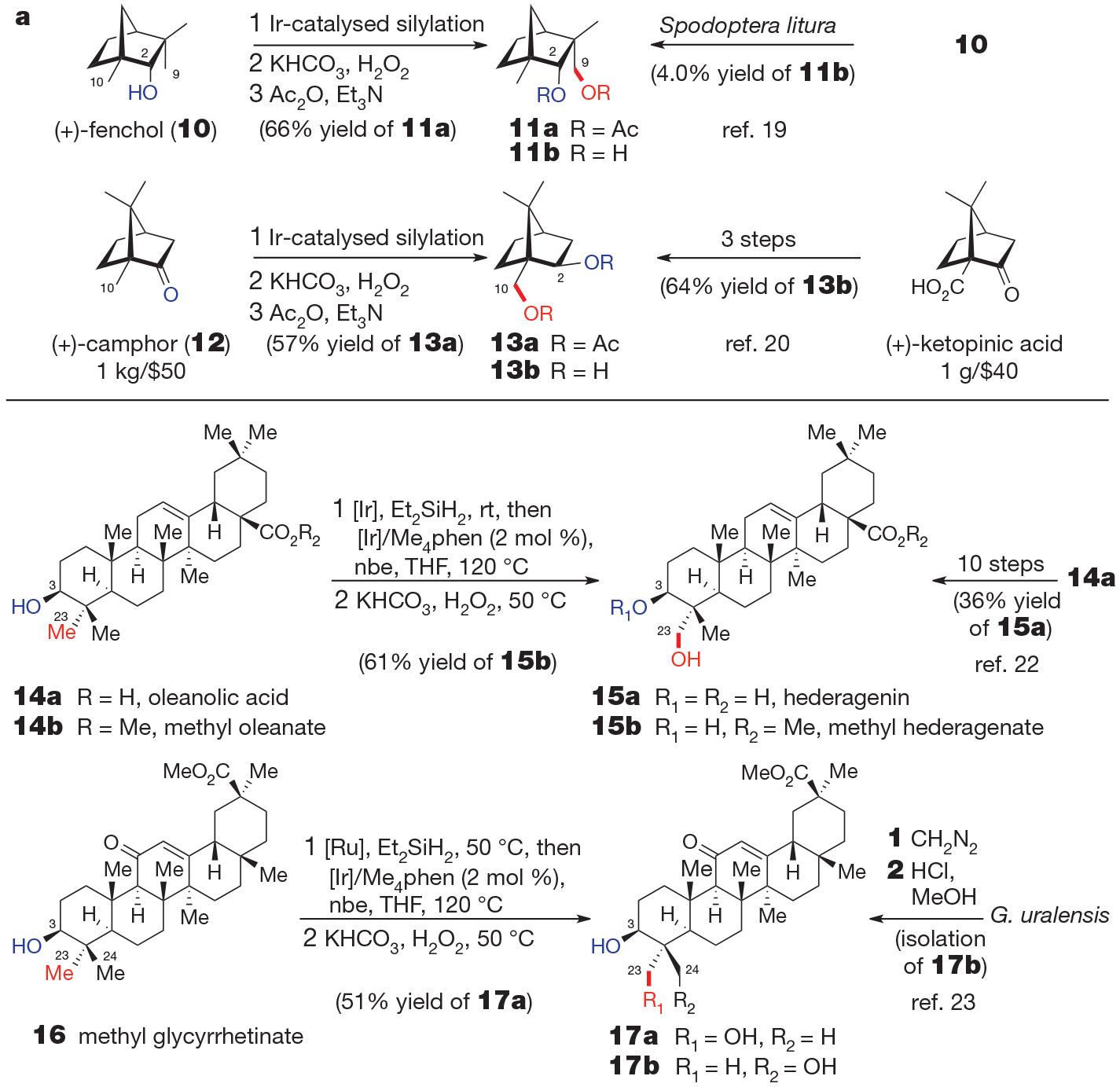
a, Directed γ-functionalization of the monoterpenes fenchol (10) and camphor (12). b, Hydroxyl-directed γ-functionalization of the triterpenoid saponins methyl oleanate (14b) and methyl glycyrrhetinate (16). For full experimental details, see the Supplementary Information.
Finally, we conducted the iridium-catalysed selective functionalization of a pair of triterpenoid saponin aglycons. Triterpenoid saponins (for example, 15, in which R3 = sugar, Fig. 3b) exhibit a range of biological activities, including anti-inflammatory, anti-fungal, anti-leishmanial and anti-tumour properties21. Together with the carbohydrate portions of saponins, the aglycon moiety is important in modulating biological activity; for example, C23-hydroxylated saponins possess strong haemolytic activity21. Because the extraction of saponins from natural sources can be time-consuming and low-yielding, laboratory synthesis from the corresponding aglycon is often the preferred method to gain material for biological studies21, and syntheses of both natural and non-natural saponins starting from methyl hederagenate (15b) have been described21. The potent biological activity of saponins bearing a C23-hydroxylated aglycon, coupled with the broader availability of the C23-unfunctionalized materials, suggest that a general method for chemoselective C23-oxygenation (for example, 14→15) would provide a short synthesis of saponins that would otherwise be challenging to access. An earlier synthesis of hederagenin (15a) from oleanolic acid (14a) required ten steps and proceeded with 36% overall yield22.
As shown in Fig. 3b, our iridium-catalysed silylation of aliphatic C─H bonds converted methyl oleanate (14b) to methyl hederagenate (15b) in three steps and 61% overall yield, without isolation of any intermediates. Similarly, selective oxygenation of the C23 methyl group of methyl glycyrrhetinate (16) provided diol 17a in 51% overall yield. C24-hydroxylated glycyrrhetinate derivative 17b has previously been obtained from Glycyrrhiza uralensis23, but the C23-hydroxylated diastereomer 17a has not been reported. On the basis of these results, we anticipate that this iridium-catalysed C─H bond functionalization should be suitable for the hydroxyl-directed C23-functionalization of a series of triterpenoids, enabling the preparation of both natural and unnatural derivatives.
The hydroxyl-directed functionalization of primary C─H bonds that we have developed overcomes several limitations and complements the scope of current methods for aliphatic C─H bond functionalization. The regioselectivity is distinct from that of classical, stoichiometric methods for 1,5-functionalization24, which involve radical intermediates and often require strongly electrophilic reagents; the selectivity for reactions of primary C─H bonds distinguishes our method from more recent alcohol-directed 1,4-functionalizations, which occur only with weaker benzylic and tertiary C─H bonds25,26. Furthermore, by our method, a single reagent docks at the existing functional group and effects the C─H functionalization process. Previous methods for catalytic, directed functionalization of aliphatic C─H bonds, which inspired the design of the current system, have led to regioselective functionalization of neighbouring C─H bonds but require installation and removal of a separate directing group before and after the C─H bond functionalization and purification of the intermediates27,28. Finally, the iridium catalyst reacts with primary C─H bonds in preference to secondary C─H bonds, and this selectivity complements that of non-directed oxidations that typically occur at the weaker and more electron-rich C─H bonds9,10,29. Considering the high selectivity of this process, the tolerance for auxiliary functionality, and the complementarity with radical-based reactions and direct oxidations, this method is suitable for both de novo synthesis of complex organic molecules and derivatization of natural products. It should inspire the development of related strategies for selective C─H bond functionalization with reagents that both dock at existing functional groups and deliver functionality to a single C─H bond30.
METHODS SUMMARY
We assembled iridium-catalysed silylation reactions under a nitrogen atmosphere using oven-dried glassware and dry, deoxygenated solvents. We assembled Tamao-Fleming oxidation reactions under air using commercial-grade solvents. We monitored reactions using gas chromatography (with a mass spectral detector or a flame ionization detector) or 1H nuclear magnetic resonance (NMR) spectroscopy.
General procedure for iridium-catalysed, directed aliphatic C─H bond functionalization.
We dissolved an alcohol or ketone substrate in tetrahydrofuran (THF) and treated it with a freshly prepared solution of [Ir(cod)OMe]2 (0.05 mol %) in THF and then with neat Et2SiH2 (1.2 equiv.). We stirred the resulting solution at room temperature (23 °C) until we observed complete conversion of the alcohol or ketone to the corresponding diethyl(hydrido)silyl ether. We then placed the reaction mixture under high vacuum for 1 h. We treated the concentrated diethyl(hydrido)silyl ether sequentially with freshly prepared solutions of nbe (1.2 equiv.) in THF and [Ir(cod)OMe]2 (0.5 mol %) in THF, and then with a slurry of Me4phen (1.25 mol %) in THF. We stirred the resulting solution at room temperature for 1 h and then heated it at 80–120 °C until we observed complete conversion to the corresponding oxasilolane. We then treated the crude reaction mixture containing the oxasilolane sequentially with MeOH, KHCO3 (2.5 equiv.) and H2O2 (30% solution in H2O, 10 equiv.), and stirred the resulting mixture overnight at 50 °C. We carefully quenched the reaction with aqueous NaHSO3, and extracted the resulting mixture with EtOAc. We washed the combined organic layers sequentially with 1 M HCl and saturated NaHCO3, and then dried them with MgSO4. We filtered the resulting organic layer through Celite and concentrated it to provide the crude diol, which we either purified directly or converted to the corresponding acetate derivative through treatment with Ac2O and Et3N. For full experimental details and characterization of all new compounds, along with copies of 1H and 13C NMR spectra, see Supplementary Information.
Supplementary Material
Acknowledgements
We thank the US National Science Foundation (CHE-0910641 to J.F.H.) and the US National Institutes of Health (GM087901 to E.M.S.) for funding this work, and Johnson Matthey for a gift of [Ir(cod)OMe]2.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of this article at www.nature.com/nature.
References
- 1.Gutekunst WR& Baran PS C─H functionalization logic in total synthesis. Chem. Soc. Rev 40,1976–1991 (2011). [DOI] [PubMed] [Google Scholar]
- 2.McMurray L, O’Hara F & Gaunt MJ Recent developments in natural product synthesis using metal-catalysed C─H bond functionalisation. Chem. Soc. Rev 40, 1885–1898 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Chen K& Baran PS Total synthesis of eudesmane terpenes by site-selective C─H oxidations. Nature 459, 824–828 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Godula K & Sames D C─H bond functionalization in complex organic synthesis. Science 312, 67–72 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Feng Y & Chen G Total synthesis of celogentin C by stereoselective C─H activation. Angew. Chem. Int. Ed 49, 958–961 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Giannis A, Heretsch P, Sarli V & Stößel A Synthesis of cyclopamine using a biomimetic and diastereoselective approach. Angew. Chem. Int. Ed 48, 7911–7914 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Yu J-Q & Shi Z in Topics in Current Chemistry Vol. 292 (Springer, 2010). [PubMed] [Google Scholar]
- 8.Lyons TW & Sanford MS Palladium-catalyzed ligand-directed C─H functionalization reactions. Chem. Rev 110, 1147–1169 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen MS & White MC A predictably selective aliphatic C─H oxidation reaction for complex molecule synthesis. Science 318, 783–787 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Newhouse T & Baran PS If C─H bonds could talk: selective C─H bond oxidation. Angew. Chem. Int. Ed 50, 3362–3374 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu Y, Wang D-H, Engle KM & Yu J-Q Pd(II)-catalyzed hydroxyl-directed C─H olefination enabled by monoprotected amino acid ligands. J. Am. Chem. Soc 132, 5916–5921 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henkel T, Brunne RM, Müller H & Reichel F Statistical investigation into the structural complementarity of natural products and synthetic compounds. Angew. Chem. Int. Ed 38, 643–647 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Simmons EM& Hartwig JF Iridium-catalyzed arene ortho-silylation by formal hydroxyl-directed C─H activation. J. Am. Chem. Soc 132, 17092–17095 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boebel TA & Hartwig JF Silyl-directed, iridium-catalyzed ortho-borylation of arenes. A one-pot ortho-borylation of phenols, arylamines, and alkylarenes. J. Am. Chem. Soc 130, 7534–7535 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM & Hartwig JF C─H activation for the construction of C─B bonds. Chem. Rev 110, 890–931 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Crotti C. et al. Evaluation of the donor ability of phenanthrolines in iridium complexes by means of synchrotron radiation photoemission spectroscopy and DFT calculations. Dalton Trans. 133–142 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Corriu RJP & Moreau JJE Selective catalytic route to bifunctional silanes. Catalysis by rhodium and ruthenium complexes of the alcoholysis of diarylsilanes and the hydrosilylation of carbonyl compounds. J. Chem. Soc. Chem. Commun 38–39 (1973). [Google Scholar]
- 18.Bosworth N & Magnus PD Studies on terpenes. Part I. Rearrangement of 7-oxatricyclo[4,3,0,0]nonanes into 8-substituted 1,3,3-trimethylnorbornane derivatives. J. Chem. Soc. Perkin Trans I 943–948 (1972). [Google Scholar]
- 19.Miyazawa M & Miyamoto Y Biotransformation of (+)-(1R,2S)-fenchol by the larvae of common cutworm (Spodoptera litura). Tetrahedron 60, 3091–3096 (2004). [Google Scholar]
- 20.Deng JG, Jiang YZ, Liu GL, Wu LJ & Mi AQ A practical method for the synthesis of homochiral 2,10-camphanediols. Synthesis 963–965 (1991). [Google Scholar]
- 21.Plé K, Chwalek M & Voutquenne-Nazabadioko L Synthesis of α-hederin, δ-hederin, and related triterpenoid saponins. Eur.J. Org. Chem 2004, 1588–1603 (2004). [Google Scholar]
- 22.García-Granados A, López PE, Melguizo E, Parra A & Simeó Y Remote hydroxylation of methyl groups by regioselective cyclopalladation. Partial synthesis of hyptatic acid-A. J. Org. Chem 72, 3500–3509 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Kitagawa I, Hori K, Sakagami M, Zhou JL & Yoshikawa M Saponin and sapogenol. XLVIII. On the constituents of the roots of Glycyrrhiza uralensis Fischer from northeastern China. (2). Licorice-saponins D3, E2, F3, G2, H2, J2, and K2. Chem. Pharm. Bull. (Tokyo) 41, 1337–1345 (1993). [DOI] [PubMed] [Google Scholar]
- 24.Majetich G & Wheless K Remote intramolecular free radical functionalizations: an update. Tetrahedron 51, 7095–7129 (1995). [Google Scholar]
- 25.Chen K, Richter JM & Baran PS 1,3-diol synthesis via controlled, radical-mediated C─H functionalization. J. Am. Chem. Soc 130, 7247–7249 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Kasuya S, Kamijo S & Inoue M Direct construction of 1,3-diaxial diol derivatives by C─H hydroxylation. Org. Lett 11, 3630–3632 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Desai LV, Hull KL & Sanford MS Palladium-catalyzed oxygenation of unactivated sp3 C─H bonds. J. Am. Chem. Soc 126, 9542–9543 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Giri R. et al. Pd-catalyzed stereoselective oxidation of methyl groups by inexpensive oxidants under mild conditions: a dual role for carboxylic anhydrides in catalytic C─H bond oxidation. Angew. Chem. Int. Ed 44, 7420–7424 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Litvinas ND, Brodsky BH & Du Bois J C─H hydroxylation using a heterocyclic catalyst and aqueous H2O2. Angew. Chem. Int. Ed 48, 4513–4516 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Zalatan DN & Du Bois J in Topics in Current Chemistry Vol. 292 (eds Yu J-Q & Shi Z) 347–378 (Springer, 2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.