

Abstract
The potential applications afforded by the generation and reactivity of artificial metalloenzymes (ArMs) in microorganisms are vast. We show that a non-pathogenic E. coli strain, Nissle 1917 (EcN), is a suitable host for the creation of ArMs from cytochrome P450s and artificial heme cofactors. An outer-membrane receptor in EcN transports an iridium porphyrin into the cell, and the Ir-CYP119 (CYP119 containing iridium porphyrin) assembled in vivo catalyzes carbene insertions into benzylic C–H bonds enantioselectively and site-selectively. The application of EcN as a whole-cell screening platform eliminates the need for laborious processing procedures, drastically increases the ease and throughput of screening, and accelerates the development of Ir-CYP119 with improved catalytic properties. Studies to identify the transport machinery suggest that a transporter different from the previously assumed ChuA receptor serves to usher the iridium porphyrin into the cytoplasm.
Graphical Abstract
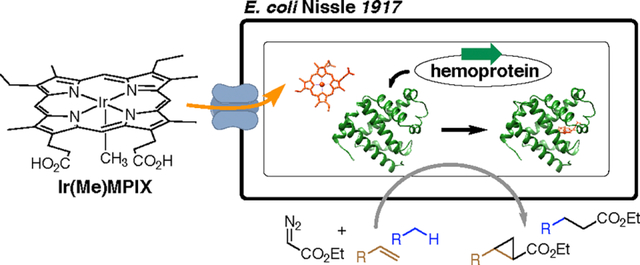
INTRODUCTION
Artificial metalloenzymes (ArMs) lie at the interface of homogeneous catalysis and biocatalysis and are created by the incorporation of a synthetic metal complex into a protein scaffold. By doing so, the versatile reactivity of transition-metal catalysts can be merged with the unique selectivity provided by the confined, shape-selective space of an enzyme active site.1 While the directed evolution of protein structures and the synthesis of new organometallic complexes have made possible the creation of the unnatural catalytic properties of ArMs,2 the reactivity of such enzymes in living cells is limited, due to the multitude of chemical functional groups present in the cell. In particular, efficient cofactor uptake, specific in vivo assembly, and catalytic reactivity without inhibition by cellular components are challenging to achieve with ArMs.3
Nature has achieved site-selective and enantioselective C–H bond functionalization in cells ranging from bacteria to humans through billions of years of evolution,4 and our group and others have sought to mimic this site-selectivity with unnatural metalloenzymes.5,6 Such reactions of native systems are illustrated by site-selective oxidation of C–H bonds in complex natural products catalyzed by cytochrome P450 enzymes7,8 and the directed evolution of such enzymes for reactions on non-native substrates,9 with altered regioselectivity,10 or with reagents for group transfer not known to be part of nature.11–13
Our group reported the first examples of enzyme-catalyzed intramolecular and intermolecular insertions of carbenes into sp3 C–H bonds14 and, most recently, showed that evolution of the CYP119 scaffold containing an iridium porphyrin in the active site allows site-selective reactions at one of two nearly equivalent methylene groups of a substituted phthalan.15 Although these studies with ArMs have shown that site-selective functionalization of C–H bonds can be achieved, the methods by which the ArMs are generated have limited the rate of evolution of such enzymes. Moreover, the incorporation of such unnatural C–H bond functionalization into artificial biosynthetic pathways akin to those reported recently16 would require that the enzymes assemble and react in whole cells.
This long-term goal of expanding natural metabolism involving ArMs generated in vivo16 and the shorter-term goal of facilitating directed evolution of ArMs would benefit from and even require assembly and reactivity of ArMs in cells without the need for in vitro reconstitution steps.17 Previously, we created artificial hemoproteins with iron substituted by an abiological metal in the porphyrin scaffold by generating apoprotein and incorporating the artificial cofactor into this structure. While this general workflow generated active catalysts, rapid directed evolution was hampered by the need to purify the protein14,18 or to generate cell lysates prior to reconstitution with synthetic metalloporphyrins.15,19
Recently, Ward and co-workers overcame these hurdles by compartmentalizing streptavidin–biotin-based ArMs on the outer membrane20 and in the periplasm21,22 of E. coli. These alternative expression platforms exploit a propitious environment for the assembly and catalysis by ArMs because the protein matrix is readily accessible and the interference from intracellular components is lower than that in the cytoplasm. However, the potential problem of incorrect folding and incomplete secretion, the absence of cofactors, and the low concentration of biosynthetic intermediates in the periplasm or cell surface limits the applicability of catalysis in these locations. Thus, we sought to develop a platform for the recombinant production of Ir-CYP119 in the cytoplasm and use of such enzymes for non-natural reactions in E. coli. More specifically, we sought to conduct stereoselective and site-selective functionalization of C–H bonds with ArMs generated and reacting in such whole cells.
Our goal of assembling active artificial metalloenzymes for site-selective and enantioselective C–H bond functionalization in E. coli requires that we identify an approach to introduce the abiotic cofactor, Ir(Me)MPIX (MPIX for mesoporphyrin IX), at the stage of protein expression. Several strategies have been reported to transport heme analogues into cells. Marletta and co-workers reported the expression of H-NOX (heme nitric oxide/oxygen binding protein) substituted with Ru(CO)MPIX in the E. coli strain RP523 that is heme-permeable and hemB defective (Figure 1a).23 The co-expression of an outer-membrane heme transporter ChuA also has been reported to incorporate synthetic heme derivatives into the hemoproteins of interest (Figure 1b).24–26 However, the cell line in the former method requires anaerobic cultivation to reach a sufficient cell density, and the overexpressed transporters on limited membrane space in the latter case can disrupt the normal functions of native membrane proteins and receptors.
Figure 1.
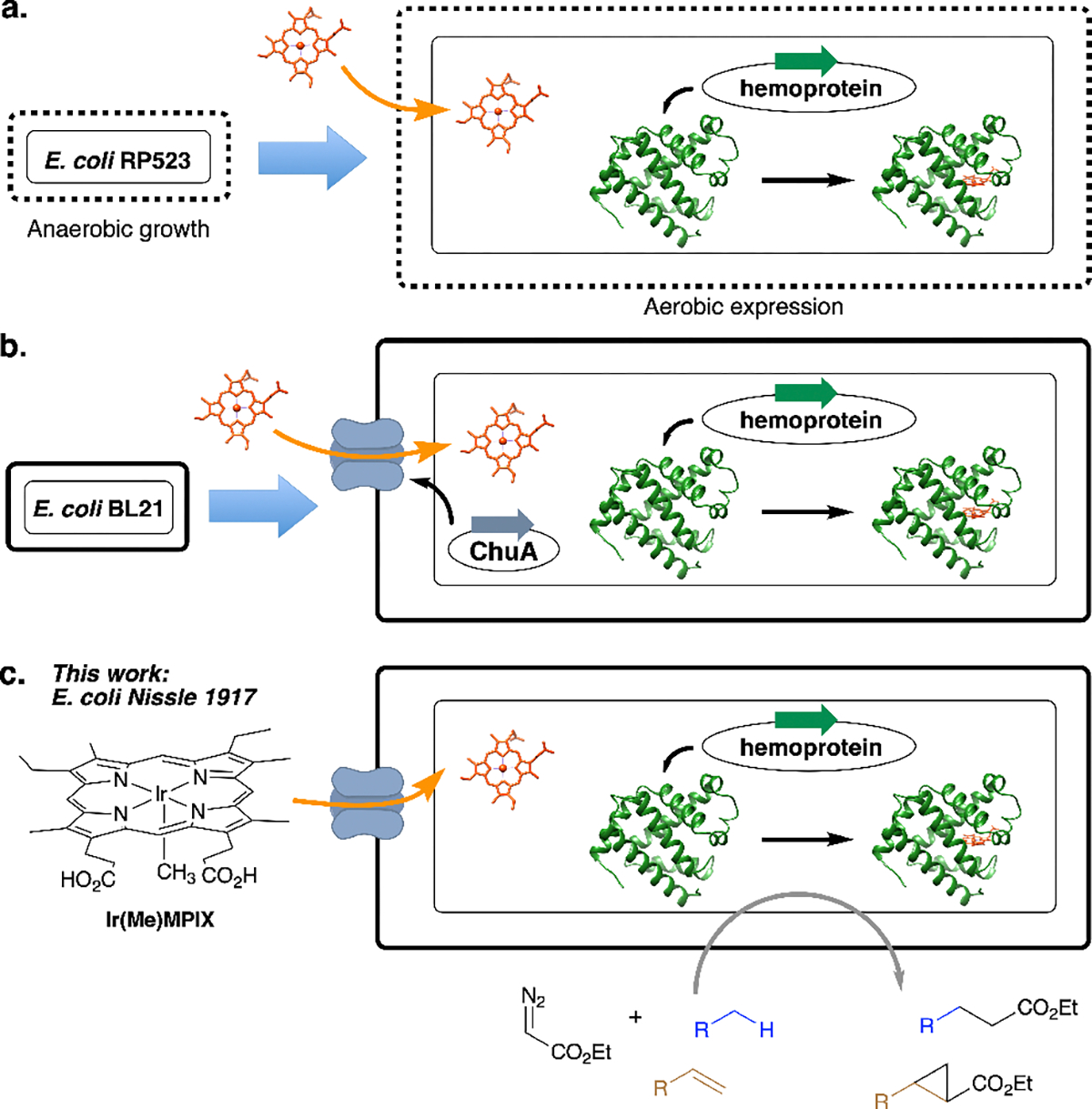
Methods to introduce artificial metalloporphyrins at the level of protein expression. (a) The use of an E. coli strain RP523 with a mutation that renders the membrane heme-permeable. (b) Co-expression of an outer-membrane transporter ChuA with the hemoprotein of interest. (c) This work: in vivo assembly of Ir-CYP119 within E. coli Nissle 1917 that contains a native uptake system for metalloporphyrins.
Here, we report the assembly of Ir-CYP119 enzymes in an E. coli strain (EcN) containing a chromosomally encoded outer-membrane transporter and subsequent stereoselective and site-selective intermolecular C–H bond functionalization in whole cells. In addition to achieving organometallic C–H bond functionalization with artificial metalloenzymes in E. coli, our investigation of the outer-membrane transporters on EcN identified a new transporter that facilitates the uptake of iridium porphyrins to the cytoplasm with efficiency that is higher than that of the previously assumed principal heme transporter ChuA.
RESULTS AND DISCUSSION
Enhanced Iridium Uptake by Nissle 1917.
Recently, the E. coli strain Nissle 1917 (EcN), which is a probiotic reagent used in the treatment of various gastroenterological diseases27 and contains a chromosomal copy encoding the heme acquisition system chu, has been shown to enhance the incorporation of heme during the expression of hemoproteins.28 We envisioned that the intrinsic cofactor uptake system in this E. coli strain would avoid the need to introduce a second plasmid encoding the transporter in a co-expression strategy and enable the in vivo assembly and whole-cell catalysis of Ir-CYP119 (Figure 1c). If so, directed evolution of artificial metalloenzymes should be accelerated.
Because metalloporphyrins containing metals other than iron are known to have antibacterial activities, especially against microorganisms that express a heme transport system,30 we first investigated whether Ir(Me)MPIX is toxic to EcN cells (Figure 2a). EcN cells were transformed with a CYP119-expression plasmid, and the kinetics of cell growth in the presence of 0.1, 0.5, and 1 ppm of Ir(Me)MPIX were monitored over time. Compared with the cell culture that was incubated in the absence of Ir(Me)MPIX, the growth of the cells with 0.1–1 ppm of iridium porphyrin added to the medium were barely impacted. These data show that the cell growth in the media with concentrations of Ir(Me)MPIX near or below 1 ppm should be suitable for generating ArMs.
Figure 2.
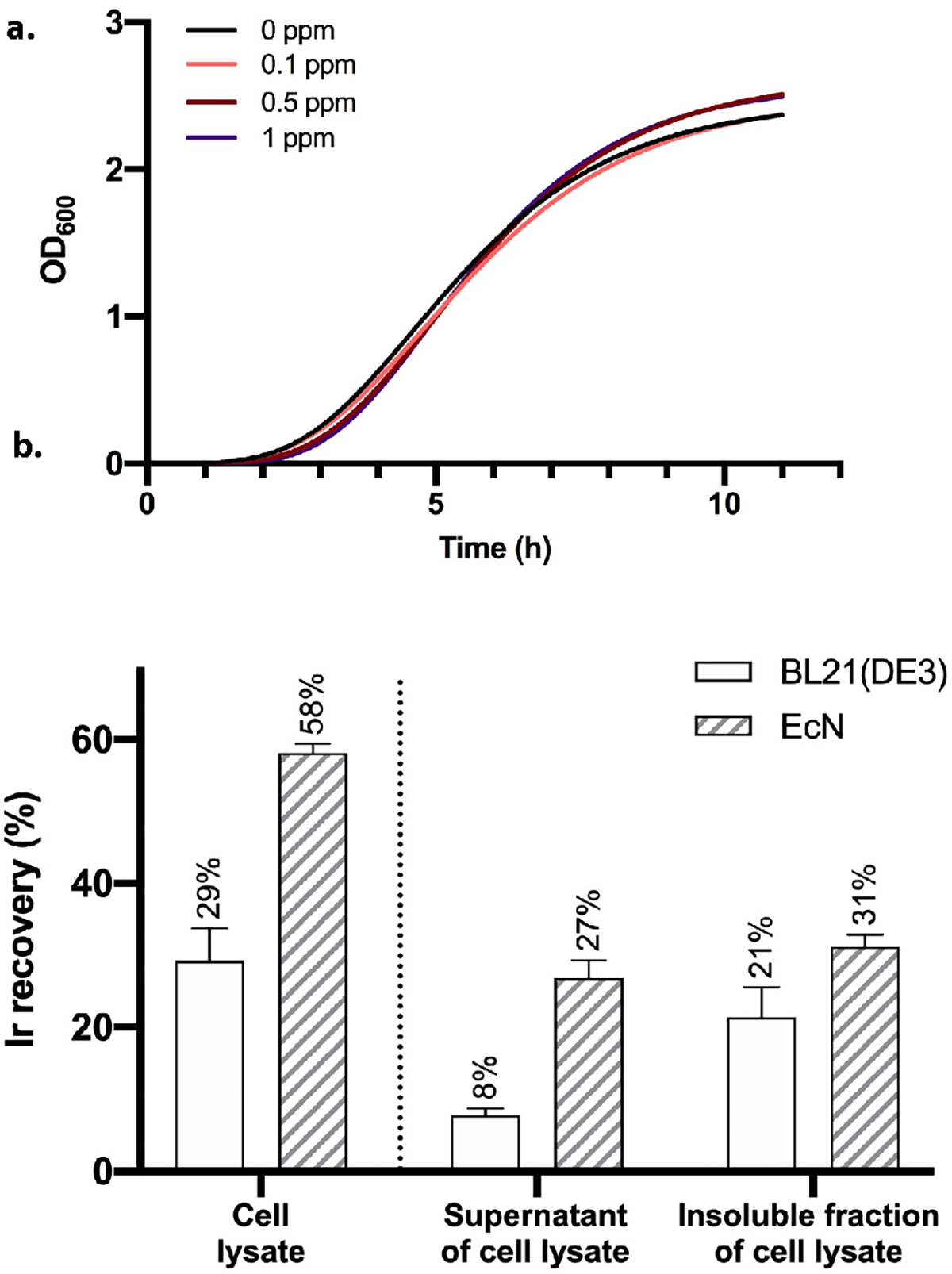
(a) The cell growth curves of EcN in the presence of 0, 0.1, 0.5, and 1 ppm of Ir(Me)MPIX. 1 ppm = 1 mg/L. The curves were fitted to a Gompertz growth model using Prism 8.29 (b) The percentages of iridium added to the growth medium that were recovered in the total cell lysates and the percent within the supernatants of cell lysates and the insoluble fractions of cell lysates from the two E. coli strains expressing CYP119. The data are shown as the average from three biological replicates, with error bars indicating 1 standard deviation.
To assess our hypothesis that iridium porphyrins could be transferred efficiently into EcN, quantification of the cellular iridium in EcN harboring Ir-CYP119 was conducted with ICP-MS (inductively coupled plasma-mass spectrometry). As a control, a common laboratory E. coli strain without genetically encoded heme transporter, BL21(DE3), was cultivated under the same conditions. The expression of Ir-CYP119 was induced by adding 0.2 ppm of anhydrotetracycline, and 0.1 ppm of iridium porphyrin was added upon induction. The amount of iridium in the cell lysates was measured after 18 h of incubation. The percentage of the total added iridium that was recovered in lysed EcN cells (58%) was measurably higher than that in BL21(DE3) (29%) (Figure 2b). Further, the amounts of iridium in the supernatants of the two cell lysates, which contain mostly soluble proteins, were much different. The iridium content in the supernatant of EcN cell lysates was 3 times (27%) higher than that of the BL21(DE3) cells containing no known heme transporters (8%).
Most importantly, the UV–vis absorbance spectrum of the Ir-CYP119 contained in the supernatant of EcN cell lysates was indistinguishable from that of Ir-CYP119 reconstituted in vitro (Figure S1). The UV–vis spectrum of the enzyme purified from the EcN cell lysate contained a characteristic Soret peak at 400 nm, and ICP analysis of the same protein indicated that 19% of the protein contained the artificial cofactor (Table S1). In contrast, the Soret peak is almost undetectable in the UV–vis spectrum of the CYP119 purified from BL21(DE3) cells lacking a transporter (Figure S1), and ICP results showed that little (3%) of the CYP119 purified from BL21(DE3) contained the cofactor, even when 0.5 ppm of Ir(Me)MPIX was supplemented to the cell culture during growth (Table S1). These data show that EcN is a suitable platform for assembly of the Ir-CYP119 in vivo, but they do not speak to the identity of the system that transports the iridium-containing cofactor. Studies to identify potential transporters in this cell line are presented in the final section of this paper.
C–H Bond Functionalization in Nissle 1917.
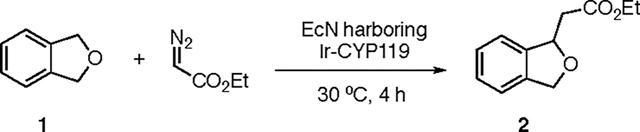
Having demonstrated the enhancement of iridium uptake and assembly of Ir-CYP119 in EcN, the carbene insertion into benzylic C–H bonds was conducted with EcN cells harboring a CYP119 variant that was previously shown to catalyze the enantioselective C–H activation of phthalan 1.15 No product from carbene insertion was observed with EcN cultivated in the absence of iridium cofactor. However, the reaction proceeded with 70:30 enantiomeric ratio (er) and 91 TON (Table 1, entry 1) when the EcN culture was supplemented with 1 ppm of Ir(Me)MPIX at the same time as the induction of protein expression.
Table 1.
Evaluation of Carbene Insertion into Phthalan 1 Catalyzed by Ir-CYP119 in Nissle 1917 Cellsa
| |||
|---|---|---|---|
| entry | Ir(Me)MPIXb(ppm) | erc | TONd |
| 1 | 1 | 70:30 | 91 ± 11 |
| 2 | 0.5 | 87:13 | 273 ± 12 |
| 3 | 0.2 | 92:8 | 789 ± 16 |
| 4 | 0.1 | 92:8 | 1024 ± 29 |
| 5e | 0.1 | 92:8 | 1314 ± 56 |
| 6f | 0.1 | 50:50 | 56 ± 7 |
Reaction conditions: EcN expressing CYP119 mutant (C317G, A152L, T213G, P252S, V254A), OD600 ~30, phthalan (2 μmol), EDA (16 μmol), DMSO (20 μl), M9-N (300 μL), 30 °C, 4 h.
The concentrations of Ir(Me)MPIX added to EcN cells during protein expression.
The er (enantiomeric ratio) was determined by chiral HPLC.
The TON (turnover number) was calculated by the number of substrate molecules converted to the product per iridium cofactor added to the cell culture.
OD600 ~15.
The reaction was performed with EcN cells containing an empty vector pASK-IBA3C under otherwise identical conditions. All data are shown as the average from three biological replicates.
To increase the er of the reactions in whole cells, the concentration of Ir(Me)MPIX in cell cultures was decreased from 1 to 0.1 ppm (Table 1, entries 1–4). The er increased to 92:8, and the biocatalyst afforded the C–H bond functionalization product in E. coli with a TON of 1024 when the loading of iridium cofactor in the culture medium was as low as 0.1 ppm (Table 1, entry 4). This selectivity and activity is comparable to that of the catalysts generated in vitro by addition of cofactor to purified apo protein.15 With a cell suspension of lower density (OD600 ~15), the reaction in E. coli still proceeded with a high enantioselectivity (Table 1, entry 5). In contrast, the reaction of phthalan with EDA (ethyl diazoacetate) in the presence of EcN cells containing an empty vector lacking the CYP119 genes, under otherwise identical growth and reaction conditions, gave only trace amounts of product, and the product was racemic (Table 1, entry 6). Without the coordination environment provided by the CYP119 cavity, the unbound iridium porphyrin catalyzed the C–H activation with low reactivity and no enantioselectivity.
Together, these data imply that the C–H bond functionalization occurring in the presence of EcN cells results from the reactivity of a combination of unbound cofactor and assembled Ir-CYP119. A decrease in the iridium loading suppresses the racemic background reaction catalyzed by unbound iridium porphyrins and leads to higher enantioselectivity from the reactions predominantly catalyzed by Ir-CYP119 assembled in EcN cells.
Having established the conditions for enantioselective carbene insertion into C–H bonds with Ir-CYP119 in E. coli cells, we evaluated the site-selectivity of C–H bond functionalization with a panel of 4-substituted phthalans catalyzed by EcN containing the assembled Ir-CYP119 (Scheme 1). CYP119-ML1, CYP119-PL1, and CYP119-PL2, which are mutants developed in our prior work, preferentially reacted at one of the two benzylic positions in the substituted phthalans over the other.15 With 4-bromophthalan 3a as substrate, EcN cells expressing Ir-CYP119-PL1 selectively activated the C–H bond in the methylene group para to bromine and generated the carbene insertion product with a 3.1:1 ratio of constitutional isomers. Cells expressing Ir-CYP119-ML1 preferentially formed the isomer resulting from reaction at the methylene group meta to bromine with a ratio of 1:3.2. These ratios are measurably higher than those obtained with the reactions catalyzed by Ir-CYP119-PL1 and Ir-CYP119-ML1 formed in vitro from the purified apoprotein and iridium porphyrin complex (1.7,1 and 1:2.2, respectively) (Table S2). The reactions of 4-chlorophthalan 3b in EcN also formed either isomer with good site-selectivity catalyzed by the two Ir-CYP119 mutants assembled in vivo, and the product ratios were higher than those from the same enzymes generated in vitro. Finally, the reactions of substrates containing electron-donating (3c) and bulky alkyl (3d) substituents both gave the product from insertion into the methylene para to the substituent, like the analogous reactions in vitro. These higher or similar selectivities show that the reactions in EcN are catalyzed by the intact Ir-CYP119. The free cofactor reacts to give a nearly 1:1 ratio of constitutional isomers.15
Scheme 1. Site-Selective C–H Activation Catalyzed by EcN Cells Harboring Ir-CYP119a.
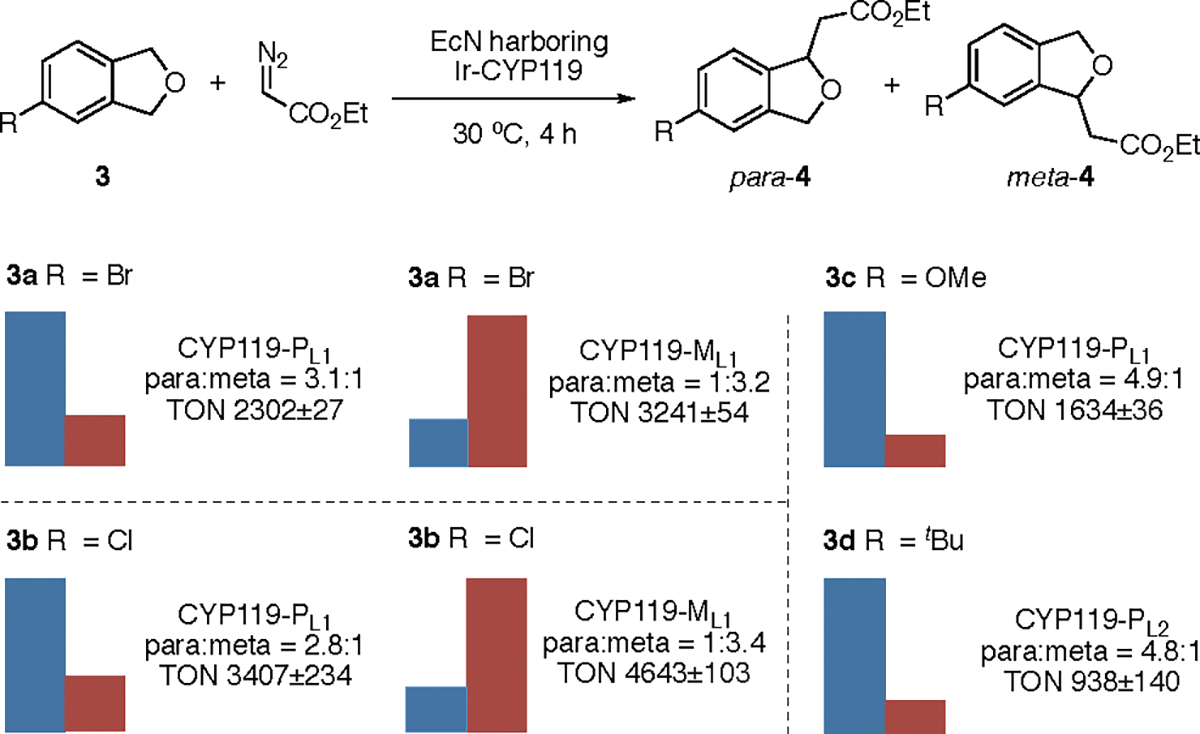
aReaction conditions: EcN expressing CYP119 mutant, OD600 ~30, phthalan derivatives (2 μmol), EDA (16 μmol), DMSO (20 μL), M9-N (300 μL), 30 °C, 4 h. The cells were cultivated with 0.1 ppm of Ir(Me)MPIX during protein expression. The heights of bars correspond to the relative amounts of para (blue) and meta (red) products. All data are shown as the average from three biological replicates.
The TONs obtained for the reactions in EcN cells were much higher than those for the reactions with the artificial metalloenzymes generated and used in vitro (Table S2). For example, the TONs of reactions of halophthalans 3a and 3b conducted with whole cells were 10–20 times higher than those for reactions in vitro. Moreover, the reactions of substrates 3c and 3d in EcN both proceeded with TONs 8– 9 times higher than those of the reactions catalyzed by the same enzymes reconstituted in vitro.
One could imagine that the catalytic C–H bond functionalization occurs by a process catalyzed by Ir-CYP119 that is released by partial cell lysis, rather than a reaction catalyzed within intact E. coli. To distinguish between these scenarios, we first measured the increase in the formation of functionalized product 2 versus time and found that the number of turnovers leveled off after around 4 h (Figure 3a). To assess whether the active catalyst was present during this time in cells or had been released into the medium, the supernatant and cells were separated from the reaction mixture by centrifugation at time intervals differing by 1 h over this 4 h period, and each set of cells was resuspended in M9-N reaction buffer. This suspension and the supernatant were separately fed a new batch of substrates (4-chlorophthalan 3b and EDA) and allowed to react for an additional 2 h. Additional production of insertion product 4b was consistently observed in the reactions containing the resuspended cell pellets (Figure 3b), and the reactivity of the resuspended cell pellets decreased over time. The loss of the reactivity is attributed to the potential toxicity of EDA (Figure S2), rather than leakage of Ir-CYP119 into the medium, because no additional formation of 4b was observed from the reactions with isolated supernatants. These data clearly show that the activity of the whole-cell catalysts originated from the Ir-CYP119 contained within the intact cells.
Figure 3.
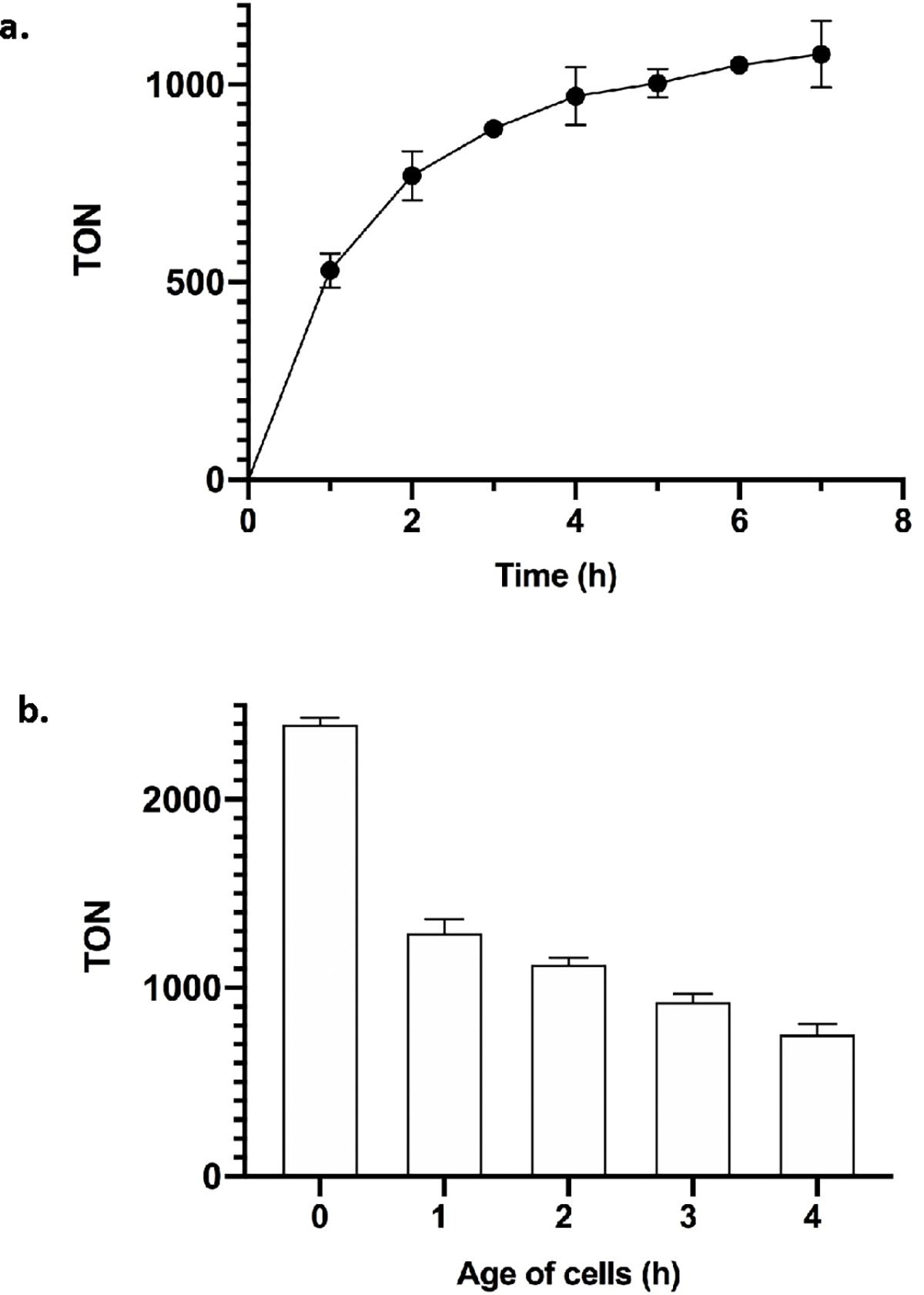
(a) The turnover numbers for the reaction of phthalan 1 with EDA vs time (h) in whole cells. Reaction conditions: EcN expressing CYP119 mutant, OD600 ~30, phthalan 1 (2 μmol), EDA (16 μmol), DMSO (20 μL), M9-N (300 μL), 30 °C, 1–7 h. The cells were cultivated with 0.1 ppm of Ir(Me)MPIX during protein expression. (b) The C–H activation of 4-chlorophthalan 3b catalyzed by resuspended pellets. The age of cells refers to the length of time for which the cells had been used for catalysis before resuspension. Reaction conditions: resuspended EcN cell pellets, OD600 ~30, 4-chlorophthalan 3b (1 μmol), EDA (8 μmol), DMSO (10 μL), M9-N (300 μL), 30 °C, 2 h. All data are shown as the average from three biological replicates, with error bars indicating 1 standard deviation.
Directed Evolution of Ir-CYP119 in Whole Cells.
Having shown that the assembly and reactivity of Ir-CYP119 can be achieved in EcN cells, we sought to exploit this capability to develop a streamlined protocol for expressing and screening Ir-CYP119 mutants in EcN with 96-well microtiter plates (Figure S3). Our group has previously evolved an Ir-CYP119 mutant that catalyzed diastereoselective cyclopropanations of monoterpenes, but the directed evolution process was impeded by the need to purify the proteins prior to measuring the selectivity.18
To demonstrate the potential to evolve ArMs by conducting the screening with intact EcN cells, we sought to evolve Ir-CYP119 to catalyze diastereoselective cyclopropanation of a sesquiterpenoid, (+)-nootkatone. (+)-Nootkatone 5 contains an unconjugated 1,1-disubstituted alkene possessing one secondary alkyl group, making the stereoselectivity challenging to achieve. One of the CYP119 mutants (CYP119-P) in the pool of mutants studied for C–H activation15 was chosen as the starting host from which to start directed evolution. This Ir-CYP119 variant catalyzed the cyclopropanation of (+)-nootkatone in EcN with a dr (diastereomeric ratio) of 1.0:4.4:1.8:1.5, whereas the free cofactor afforded the products with a distinct ratio of 1.1:1.0:2.7:2.4 (Figure 4).
Figure 4.
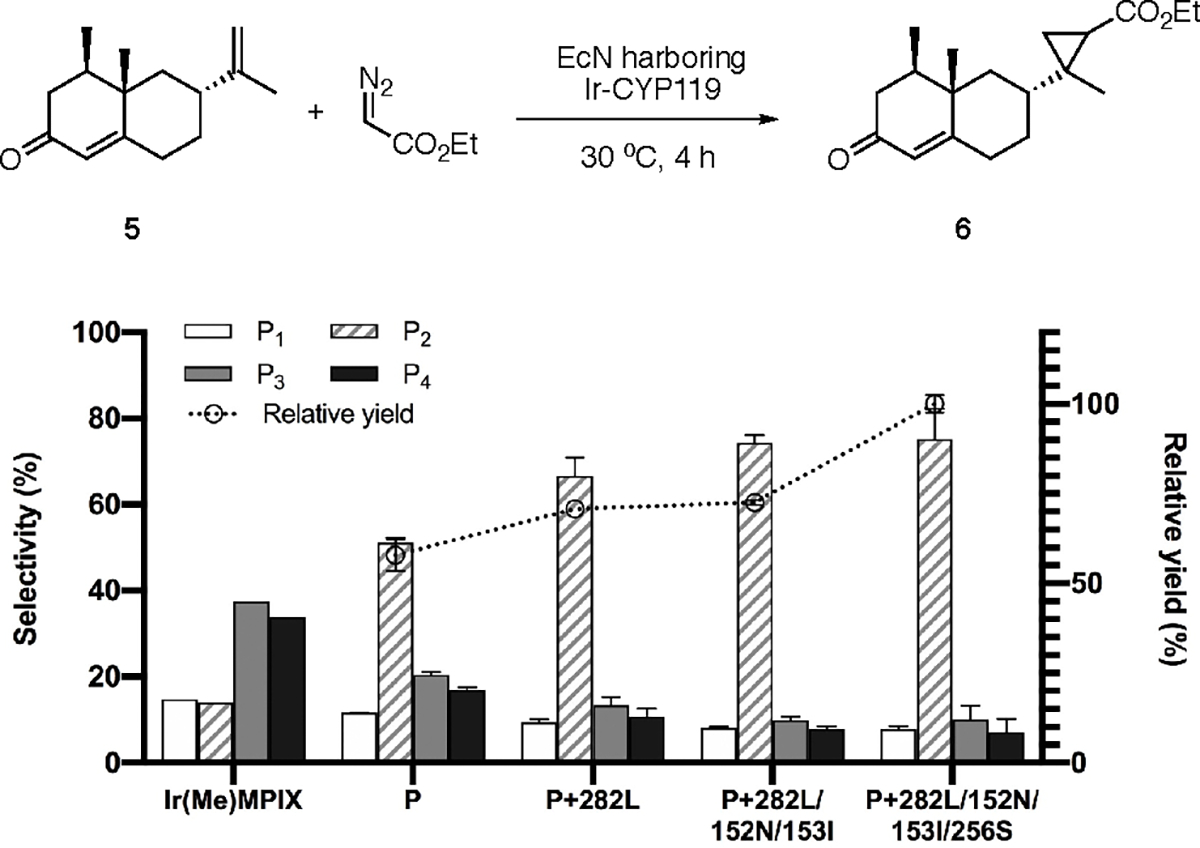
Directed evolution of Ir-CYP119 for diastereoselective cyclopropanation of (+)-nootkatone 5 with EcN as a screening platform. Reaction conditions: EcN expressing CYP119 mutant, OD600 ~30, 5 (2 μmol), EDA (16 μmol), DMSO (20 μL), M9-N (300 μL), 30 °C, 4 h. The cells were cultivated with 0.1 ppm of Ir(Me)MPIX during protein expression. P1–P4 are the four diastereomeric products numbered in the order of elution by gas chromatography. The selectivity of each diastereomer corresponds to its percent in the total four diastereomeric products. The relative yield is calculated by normalizing the yield of the reaction catalyzed by the final mutant as 100%. All data from the reactions in EcN are shown as the average from three biological replicates, with error bars indicating 1 standard deviation.
A combinatorial library targeting 10 residues around the active site of CYP119 was generated with mutagenic primers that carry a degenerate NNK codon at the targeted positions (Figure S4).31 A screening of ~400 random clones showed that the mutation of I282L increased the diastereoselectivity, such that the fraction of the major diastereomeric product increased from 51% (1.0:4.4:1.8:1.5) to 67% (1.0:7.2:1.4:1.2 dr). This minor isomer (14%) formed from the reaction catalyzed by the free cofactor. Further randomization at sites 152 and 153 with primers containing degeneracy (NNK) at both residues led to an enzyme containing 152N/153I mutations with a higher diastereoselectivity of 74% (1.0, 9.6:1.8:1.0 dr). A third round of mutation, this time at residues 254 and 256, identified a quadruple mutant 282L/152N/153I/256S that formed the cyclopropanation product of (+)-nootkatone with a 28% increase in the yield and 75% selectivity for the major isomer (Figure 4). This evolved mutant reacted with a nearly identical diastereoselectivity (70%) for the major diastereomeric product and yield of 46% when the reaction was performed in vitro with purified proteins and slow addition of diazo reagents (Table S3). This rapid directed evolution to dramatically increase the diastereoselectivity for cyclopropanation clearly shows the value of evolving artificial metalloenzymes assembled and reacting in vivo.
Identification of the Potential Transporter of the Ir(Me)PIX Cofactor in Nissle 1917.
After demonstrating that EcN is a suitable screening platform for the directed evolution of Ir-CYP119 in whole cells, we sought to clarify whether the uptake of the artificial cofactor into EcN is assisted by the heme receptor ChuA, as assumed in prior literature,24–26 or by a different transport system. We did so because we found that the reaction catalyzed in BL21(DE3) cells expressing CYP119 without a known heme transporter proceeded with a substantial er (82:18), albeit with low TON (205). This result suggests that trace amounts of Ir(Me)MPIX can enter the cells via passive diffusion or other carrier-dependent mechanisms.
To understand the role of the chu operon in the transit of iridium cofactors, we performed gene knockouts in the EcN strain by Red/ET homologous recombination.32 We found that the deletions of outer-membrane receptor ChuA and inner membrane transporters ChuU/ChuV had little impact on the enantioselectivity and TON of C–H activation of phthalan 1 catalyzed by these mutant strains expressing Ir-CYP119 (Figure S5). These data show that cells lacking the ChuA receptor or ChuU/ChuV transporters remain capable of transporting Ir(Me)MPIX into the cytoplasm and assembling Ir-CYP119 in vivo and imply that other transporters are responsible for the uptake of artificial cofactors.
We hypothesized that the potential transporter for iridium porphyrin would still possess some similarity with ChuA, since both Ir(Me)MPIX and hemin (native ligand of ChuA) comprise a porphyrin scaffold. A homology search for the ChuA protein using the BLASTP algorithm against the proteome of EcN was performed, and the 12 top hits were selected for further testing (Table S4). These genes were cloned and expressed in E. coli BL21(DE3), together with the CYP119 mutant for enantioselective C–H activation, and the capabilities of the resulting cells to assemble Ir-CYP119 by transporting Ir(Me)MPIX to the cytoplasm were evaluated by measuring the yield and enantioselectivity of the C–H bond functionalization of unsubstituted phthalan 1 catalyzed by these cells (Figure 5). If the iridium cofactor were transported into the cell with high efficiency and the artificial enzyme were assembled, the product would be formed with high enantioselectivity and high TON. If the cells lack a mechanism for efficient incorporation of Ir(Me)MPIX into the CYP119 mutant, then the product would be formed with low enantioselectivity and low TON.
Figure 5.
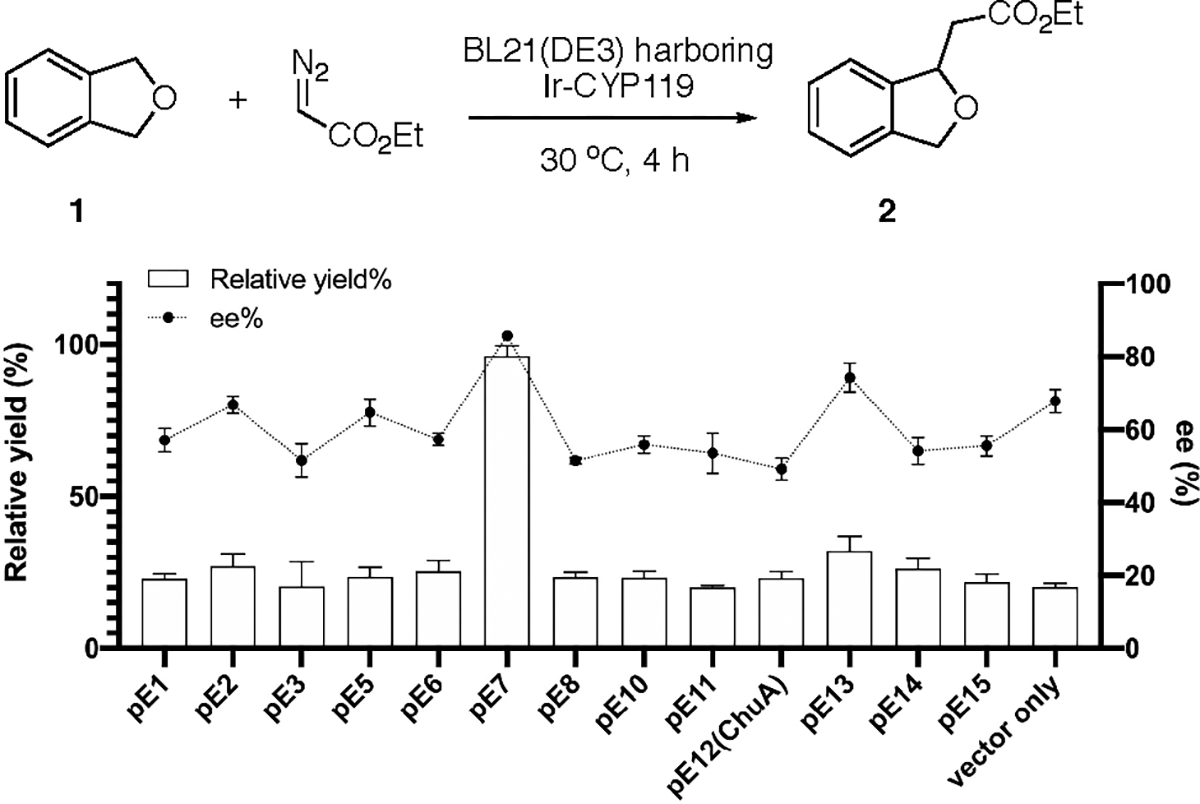
C–H bond functionalization of phthalan 1 catalyzed by BL21(DE3) cells co-expressing the potential transporter and CYP119 mutant. The cells were cultivated with 0.1 ppm of Ir(Me)MPIX during protein expression. Reaction conditions: BL21(DE3) expressing CYP119 mutant (C317G, A152L, T213G, P252S, V254A), OD600 ~20, phthalan (2 μmol), EDA (16 μmol), DMSO (20 μL), M9-N (300 μL), 30 °C, 4 h. The relative yield is calculated by normalizing the yield of the reaction in cells co-expressing the E7 transporter and CYP119 as 100%. All data are shown as the average from three biological replicates, with error bars indicating 1 standard deviation.
Consistent with our previous observation, BL21(DE3) cells co-transformed with a CYP119 plasmid and an empty vector lacking the genes for transporters catalyzed the enantioselective C–H activation with low TON (308) and 84:16 er (Figure 5, vector only). No increase in either the TON or er was observed when CYP119 was expressed with ChuA (encoded by pE12). However, the results of the reactions catalyzed by BL21(DE3) cells co-expressing CYP119 mutants with the series of potential transporters showed that the cells expressing the gene on pE7 yielded the product with high er (93:7) and a TON of 1463.
The high reactivity and enantioselectivity of whole cells containing the pE7 plasmid were corroborated by ICP data that revealed enhanced iridium uptake in these cells. More specifically, the percent of added iridium that was recovered from the BL21(DE3) cells co-expressing the protein on pE7 and CYP119 (59%) was almost the same as that of EcN cells (58%),33 while only 35% of the added iridium was recovered from the cells co-expressing ChuA (Figure S6). No increase in the iridium content in the supernatants of the lysates was observed in cells co-expressing CYP119 and ChuA over that from BL21(DE3) cells lacking a transporter (Ir recovery: 8% with ChuA vs 10% without transporter).34 In contrast, a much larger amount of iridium (30% of the added iridium) was recovered in the supernatant of lysed cells co-expressing the gene on pE7. Together with the reactivity of whole-cell catalysts in Figure 5, these results show that the protein encoded on pE7, among the potential transporters assessed in this study, led to the most efficient transport of the iridium cofactor into these E. coli cells.
Bioinformatic annotation suggests that the protein encoded on pE7 is a colicin I receptor (Table S5), which is a TonB-dependent outer-membrane transporter expressed by multiple E. coli strains (but not in BL21(DE3)), to bind and transport Fe3+ complexed to linear forms of catecholates.35 This result suggests that a siderophore receptor possessing a promiscuous ligand-binding site36,37 can be used to transport artificial cofactors into other Gram-negative bacteria.38
One could argue that the use of natural transport machinery to assemble artificial metalloenzymes in vivo will be limited to ArMs created from heme analogues as cofactors because natural heme transporters are well-known.3 The assembly of ArMs containing other types of cofactors should be more challenging, due to the absence of defined transporters that would act on synthetic cofactors more distantly related to natural ones. On the basis of this expectation, the requirement of cofactor transport has been circumvented by expressing the host protein with a tag for translocation to the periplasm or to express the protein on the cell surface.20,21 Our discovery of the promiscuous activity of the E7 protein expands the paradigm of the available cofactor transport machinery for the in vivo assembly of artificial hemoproteins. This finding suggests that the assembly of ArMs outside the hemoprotein family could be possible in whole cells and that the application of genetic, cellular, and bioinformatic tools could enable the identification of new transporters or the discovery of new functions of existing transporters to enable ArMs to be assembled in whole cells with synthetic cofactors that have little resemblance to natural ones.
CONCLUSIONS
In conclusion, we have shown that the probiotic E. coli strain Nissle 1917 (EcN) possessing a genetically encoded transport system is a suitable host for the efficient uptake of an iridium porphyrin complex and the in vivo assembly of Ir-CYP119. This strain enabled stereoselective and site-selective functionalization of C–H bonds by carbene insertion catalyzed by an ArM in whole cells and was shown to accelerate the directed evolution of Ir-CYP119 by enabling high-throughput screening of reactions with new substrates in whole cells. As part of these studies, a colicin I receptor in EcN was found to transport iridium porphyrins with high efficiency, and such transport by this receptor should be a valuable addition to expression-based strategies for assembling artificial hemoproteins, and enable the import of other types of artificial metallocofactors into E. coli.
Supplementary Material
ACKNOWLEDGMENTS
Initial experiments were supported by the Department of Energy, Laboratory Directed Research and Development funding, under contract DE-AC01-05CH11231, and subsequent studies were supported by National Science Foundation grant 2027943. Z.L. is an A*Star predoctoral fellow. Instruments in the College of Chemistry’s NMR facility are supported in part by the NIH (S10OD024998). A.M. is supported by the Joint Bioenergy Institute project, funded by the U.S. Department of Energy, Office of Science, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the U.S. Department of Energy. ICP-MS measurements were performed in the OHSU Elemental Analysis Core with partial support from NIH (S10RR025512).
Footnotes
The authors declare the following competing financial interest(s): J.D.K. has financial interests in Amyris, Ansa Biotechnologies, Apertor Pharma, Berkeley Yeast, Demetrix, Lygos, Napigen, ResVita Bio, and Zero Acre Farms.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c10975.
Description of materials and methods, Supplementary Figures S1–S10, and Supplementary Tables S1–S7 (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c10975
Contributor Information
Zhennan Liu, Department of Chemistry, University of California, Berkeley, California 94720, United States; Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States.
Jing Huang, Biological Systems and Engineering Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States; Joint BioEnergy Institute, Lawrence Berkeley National Laboratory, Emeryville, California 94608, United States.
Yang Gu, Department of Chemistry, University of California, Berkeley, California 94720, United States; Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States.
Douglas S. Clark, Department of Chemical and Biomolecular Engineering, University of California, Berkeley, California 94720, United States Molecular Biophysics and Integrated Bioimaging Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States.
Aindrila Mukhopadhyay, Department of Chemistry, University of California, Berkeley, California 94720, United States; Chemical Sciences Division and Environmental Genomics and Systems Biology Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States.
Jay D. Keasling, Biological Systems and Engineering Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States Joint BioEnergy Institute, Lawrence Berkeley National Laboratory, Emeryville, California 94608, United States; Department of Chemical and Biomolecular Engineering, University of California, Berkeley, California 94720, United States; Department of Bioengineering, University of California, Berkeley, California 94720, United States; Synthetic Biochemistry Center, Institute for Synthetic Biology, Shenzhen Institutes for Advanced Technologies, Shenzhen 518055, China; Center for Biosustainability, Danish Technical University, Lyngby 2800, Denmark.
John F. Hartwig, Department of Chemistry, University of California, Berkeley, California 94720, United States; Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States
REFERENCES
- (1).Davis HJ; Ward TR Artificial Metalloenzymes: Challenges and Opportunities. ACS Cent. Sci. 2019, 5 (7), 1120–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Schwizer F; Okamoto Y; Heinisch T; Gu Y; Pellizzoni MM; Lebrun V; Reuter R; Köhler V; Lewis JC; Ward TR Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118 (1), 142–231. [DOI] [PubMed] [Google Scholar]
- (3).Jeschek M; Panke S; Ward TR Artificial Metalloenzymes on the Verge of New-to-Nature Metabolism. Trends Biotechnol. 2018, 36 (1), 60–72. [DOI] [PubMed] [Google Scholar]
- (4).Lewis JC; Coelho PS; Arnold FH Enzymatic functionalization of carbon–hydrogen bonds. Chem. Soc. Rev. 2011, 40 (4), 2003–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Perez-Rizquez C; Rodriguez-Otero A; Palomo JM Combining enzymes and organometallic complexes: novel artificial metalloenzymes and hybrid systems for C–H activation chemistry. Org. Biomol. Chem. 2019, 17 (30), 7114–7123. [DOI] [PubMed] [Google Scholar]
- (6).Upp DM; Lewis JC Selective C–H bond functionalization using repurposed or artificial metalloenzymes. Curr. Opin. Chem. Biol. 2017, 37, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kaspera R; Croteau R Cytochrome P450 oxygenases of Taxol biosynthesis. Phytochem. Rev. 2006, 5 (2), 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Niwa T; Murayama N; Imagawa Y; Yamazaki H Regioselective hydroxylation of steroid hormones by human cytochromes P450. Drug Metab. Rev. 2015, 47 (2), 89–110. [DOI] [PubMed] [Google Scholar]
- (9).Jung ST; Lauchli R; Arnold FH Cytochrome P450: taming a wild type enzyme. Curr. Opin. Biotechnol. 2011, 22 (6), 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang X; Peng Y; Zhao J; Li Q; Yu X; Acevedo-Rocha CG; Li A Bacterial cytochrome P450-catalyzed regio- and stereoselective steroid hydroxylation enabled by directed evolution and rational design. Bioresour. Bioprocess. 2020, 7 (1), 2. [Google Scholar]
- (11).Prier CK; Zhang RK; Buller AR; Brinkmann-Chen S; Arnold FH Enantioselective, intermolecular benzylic C–H amination catalysed by an engineered iron-haem enzyme. Nat. Chem. 2017, 9 (7), 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang RK; Chen K; Huang X; Wohlschlager L; Renata H; Arnold FH Enzymatic assembly of carbon–carbon bonds via iron-catalysed sp3 C–H functionalization. Nature 2019, 565 (7737), 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhang RK; Huang X; Arnold FH Selective CH bond functionalization with engineered heme proteins: new tools to generate complexity. Curr. Opin. Chem. Biol. 2019, 49, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dydio P; Key HM; Nazarenko A; Rha JYE; Seyedkazemi V; Clark DS; Hartwig JF An Artificial Metalloenzyme with the Kinetics of Native Enzymes. Science 2016, 354, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gu Y; Natoli SN; Liu Z; Clark DS; Hartwig JF Site-Selective Functionalization of (sp3)C–H Bonds Catalyzed by Artificial Metalloenzymes Containing an Iridium-Porphyrin Cofactor. Angew. Chem., Int. Ed. 2019, 58 (39), 13954–13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Huang J; Liu Z; Bloomer BJ; Clark DS; Mukhopadhyay A; Keasling JD; Hartwig JF Unnatural biosynthesis by an engineered microorganism with heterologously expressed natural enzymes and an artificial metalloenzyme. Nat. Chem. 2021, 13 (12), 1186–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Markel U; Sauer DF; Schiffels J; Okuda J; Schwaneberg U Towards the Evolution of Artificial Metalloenzymes—A Protein Engineer’s Perspective. Angew. Chem., Int. Ed. 2019, 58 (14), 4454–4464. [DOI] [PubMed] [Google Scholar]
- (18).Key HM; Dydio P.ł; Liu Z; Rha JY-E; Nazarenko A; Seyedkazemi V; Clark DS; Hartwig JF. Beyond Iron: Iridium-Containing P450 Enzymes for Selective Cyclo-propanations of Structurally Diverse Alkenes. ACS Cent. Sci. 2017, 3 (4), 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Dydio P; Key HM; Hayashi H; Clark DS; Hartwig JF Chemoselective, Enzymatic C–H Bond Amination Catalyzed by a Cytochrome P450 Containing an Ir(Me)-PIX Cofactor. J. Am. Chem. Soc. 2017, 139 (5), 1750–1753. [DOI] [PubMed] [Google Scholar]
- (20).Heinisch T; Schwizer F; Garabedian B; Csibra E; Jeschek M; Vallapurackal J; Pinheiro VB; Marlière P; Panke S; Ward TRE coli surface display of streptavidin for directed evolution of an allylic deallylase. Chem. Sci. 2018, 9 (24), 5383–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jeschek M; Reuter R; Heinisch T; Trindler C; Klehr J; Panke S; Ward TR Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature 2016, 537 (7622), 661–665. [DOI] [PubMed] [Google Scholar]
- (22).Rebelein JG; Cotelle Y; Garabedian B; Ward TR Chemical Optimization of Whole-Cell Transfer Hydrogenation Using Carbonic Anhydrase as Host Protein. ACS Catal. 2019, 9 (5), 4173–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Winter MB; McLaurin EJ; Reece SY; Olea C; Nocera DG; Marletta MA Ru-Porphyrin Protein Scaffolds for Sensing O2. J. Am. Chem. Soc. 2010, 132 (16), 5582–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lelyveld VS; Brustad E; Arnold FH; Jasanoff A Metal-Substituted Protein MRI Contrast Agents Engineered for Enhanced Relaxivity and Ligand Sensitivity. J. Am. Chem. Soc. 2011, 133 (4), 649–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bordeaux M; Singh R; Fasan R Intramolecular C(sp3)H amination of arylsulfonyl azides with engineered and artificial myoglobin-based catalysts. Bioorg. Med. Chem. 2014, 22 (20), 5697–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Reynolds EW; Schwochert TD; McHenry MW; Watters JW; Brustad EM Orthogonal Expression of an Artificial Metalloenzyme for Abiotic Catalysis. ChemBioChem. 2017, 18 (24), 2380–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Grozdanov L; Raasch C; Schulze J; Sonnenborn U; Gottschalk G; Hacker J; Dobrindt U Analysis of the Genome Structure of the Nonpathogenic Probiotic Escherichia coli Strain Nissle 1917. J. Bacteriol. 2004, 186 (16), 5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Fiege K; Querebillo CJ; Hildebrandt P; Frankenberg-Dinkel N Improved Method for the Incorporation of Heme Cofactors into Recombinant Proteins Using Escherichia coli Nissle 1917. Biochemistry 2018, 57 (19), 2747–2755. [DOI] [PubMed] [Google Scholar]
- (29).Nonlinear growth of Nissle 1917 was fit to a Gompertz growth model. See Table S7 of the Supporting Information for details.
- (30).Stojiljkovic I; Kumar V; Srinivasan N Non-iron metalloporphyrins: potent antibacterial compounds that exploit haem/Hb uptake systems of pathogenic bacteria. Mol. Microbiol. 1999, 31 (2), 429–442. [DOI] [PubMed] [Google Scholar]
- (31).Belsare KD; Andorfer MC; Cardenas FS; Chael JR; Park HJ; Lewis JC A Simple Combinatorial Codon Mutagenesis Method for Targeted Protein Engineering. ACS Synth. Biol. 2017, 6 (3), 416–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Baba T; Ara T; Hasegawa M; Takai Y; Okumura Y; Baba M; Datsenko KA; Tomita M; Wanner BL; Mori H Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006, 2 (1), 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).For comparison with a related transport system reported in ref 16, BL21 cells containing the Hug transport system and CYP119 led harvested cells to contain 95% of the added iridium. Additional data are needed on the levels of protein expression to make a direct comparison, but the similar orders of magnitude show that the unoptimized activity of this newly identified transporter is comparable to the most efficient one for this ArM known, and studies to increase this activity are ongoing.
- (34).Although ChuA has been reported to assist the assembly of an artificial P450 containing Ir(Me)-deuteroporphyrin IX in cells in ref 26, its expression was controlled by a strong T7 promoter with concurrent addition of 10 μM iridium porphyrin and the background transport by cells expressing no transporter was not evaluated. Under our expression conditions, the ChuA expressed is capable of transporting hemin but does not transport Ir(Me)MIPX efficiently (Figure S7).
- (35).Buchanan SK; Lukacik P; Grizot S; Ghirlando R; Ali MMU; Barnard TJ; Jakes KS; Kienker PK; Esser L Structure of colicin I receptor bound to the R-domain of colicin Ia: implications for protein import. EMBO J. 2007, 26 (10), 2594–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Peuckert F; Ramos-Vega AL; Miethke M; Schworer CJ; Albrecht AG; Oberthur M; Marahiel MA The Siderophore Binding Protein FeuA Shows Limited Promiscuity toward Exogenous Triscatecholates. Chem. Biol. 2011, 18 (7), 907–919. [DOI] [PubMed] [Google Scholar]
- (37).Krewulak KD; Vogel HJ Structural biology of bacterial iron uptake. Biochim. Biophys. Acta Biomembr. 2008, 1778 (9), 1781–1804. [DOI] [PubMed] [Google Scholar]
- (38).The uncovering of the E7 receptor was fortuitous. We cannot exclude the possibility that additional outer-membrane receptors can transport iridium porphyrins into EcN because this E. coli strain is known to possess a high number of iron acquisition systems.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.