

Abstract
Remote hydrofunctionalizations of alkenes incorporate functional groups distal to existing carbon–carbon double bonds. While remote carbonylations are well-known, remote hydrofunctionalizations are most common for addition of relatively nonpolar B–H, Si–H, and C–H bonds with alkenes. We report a system for the remote hydroamination of disubstituted alkenes to functionalize an alkyl chain selectively at the subterminal, unactivated, methylene position. Critical to the high regioselectivity and reaction rates are the electronic properties of the substituent on the amine and the development of the ligand DIP-Ad-SEGPHOS by evaluating the steric and electronic effects of ligand modules on reactivity and selectivity. The remote hydroamination is compatible with a broad scope of alkenes and aminopyridines and enables the regioconvergent synthesis of amines from an isomeric mixture of alkenes. The products can be derivatized by nucleophilic aromatic substitution on the amino substituent with a variety of nucleophiles.
The selective functionalization at electronically and sterically similar methylene carbons distal to a reactive site has been a long-standing goal in organic synthesis but has been challenging to develop because of the similar reactivities of unactivated methylene groups.1–5 The remote hydrofunctionalization of alkenes, which places a hydrogen on the alkene and a functional group distal to the alkene, is a potential approach to this synthetic goal (Scheme 1A). Remote hydrosilylation6–12 by chain walking is a classic reaction, and remote hydrofunctionalization with boranes,13–22 as well as arenes or heteroarenes,23–26 is known. Nickel-catalyzed formal remote hydroarylation27–39 and hydroalkylation40–42 has also been reported, but the remote hydrofunctionalization of alkenes with more polar X–H bonds, especially those of amines,43,44 remains underdeveloped. The few reported transformations occur in a formal sense by combining silanes as the hydride donor and esters of hydroxylamine or dioxazolone as the aminating reagents (Scheme 1B).45–48
Scheme 1.
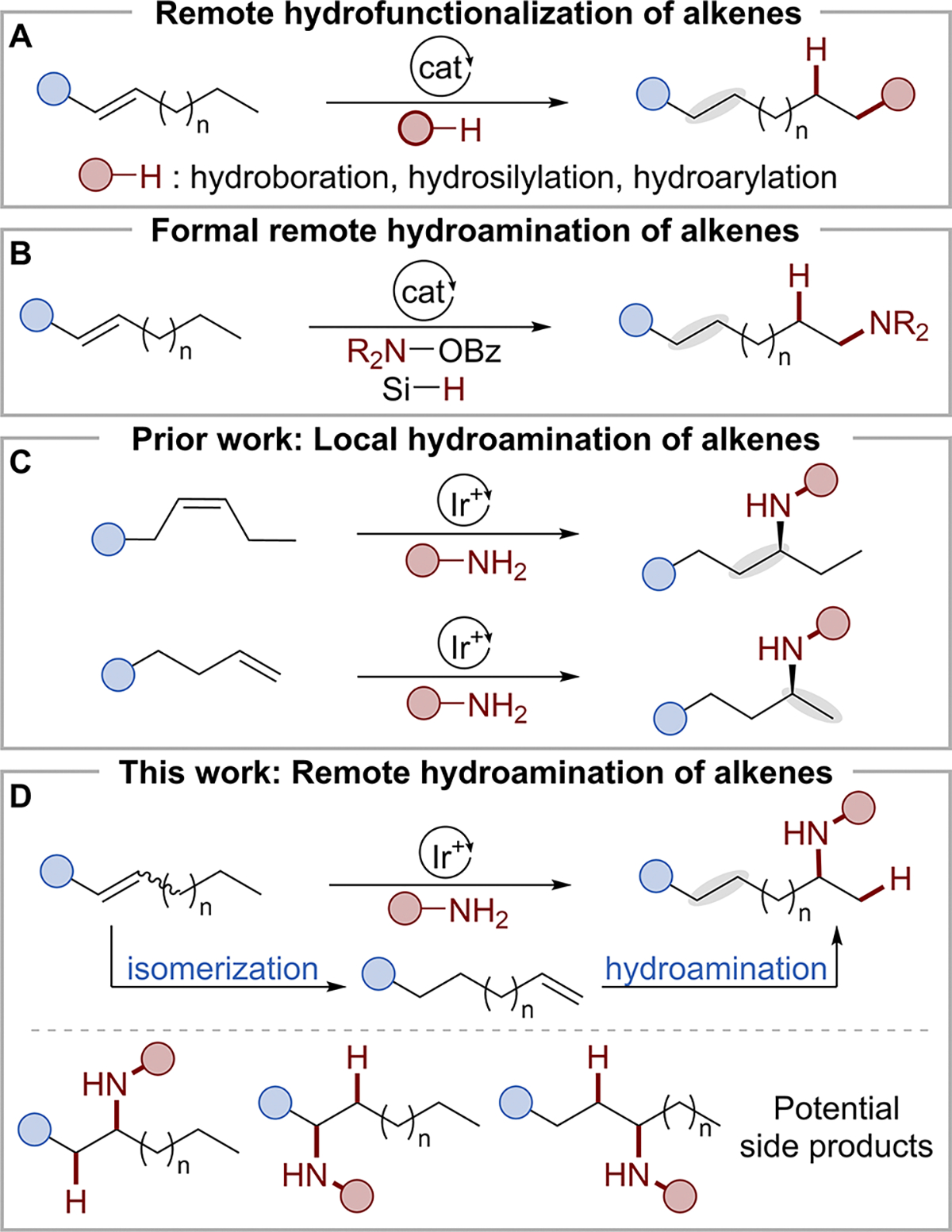
(A) Remote Hydrofunctionalization of Alkenes. (B) Formal Remote Hydroamination of Alkenes. (C) Prior Work: Direct Hydroamination of Alkenes. (D) This Work: Remote Hydroamination of Disubstituted Alkenes
We envisioned a reaction in which a single reagent containing an N–H bond could trigger alkene isomerization and deliver an amino group to form the product. While synthetically appealing, this reaction could generate a mixture of products with the amino group at multiple internal sites.49 Our group recently reported catalytic hydroamination of internal and terminal alkenes with a 2-aminopyridine (Scheme 1C)50–52 that suppressed isomerization and led to direct hydroamination. We envisioned that appropriate modifications to the amine and catalyst could increase the rate of isomerization over that of hydroamination at internal sites thereby creating a remote hydroamination of internal alkenes by selective N–H addition to the terminal alkene (Scheme 1D). Herein, we report the remote hydroamination of unactivated, internal alkenes to place the amino group at the unactivated subterminal carbon of an alkyl chain. This process was enabled by tuning the electronic properties of the N–H donor to retard the rate of migratory insertion and tuning the backbone and aryl fragment of the bisphosphine ligand to promote the desired remote hydroamination regioselectively.
To achieve remote hydroamination of internal alkenes, we first sought to identify an aminopyridine that would change the selectivity from the hydroamination we reported previously. To do so, we conducted the reactions of a model alkene, cis-4-octene (1a) with a series of 2-aminopyridines in combination with our previously developed cationic iridium catalyst.50,51 The reaction with the electron-rich 6-methyl-2-aminopyridine (2a) led to slow isomerization of cis-4-octene, the formation of the 2-aminoalkane (3aa) with low selectivity (30%), and the 4-aminoalkane from direct addition of the N–H bond to the internal alkene as the major product (Scheme 2A, entry 1).
Scheme 2. Investigation of the Reaction Conditions for the Remote Hydroamination of cis-4-Octene.
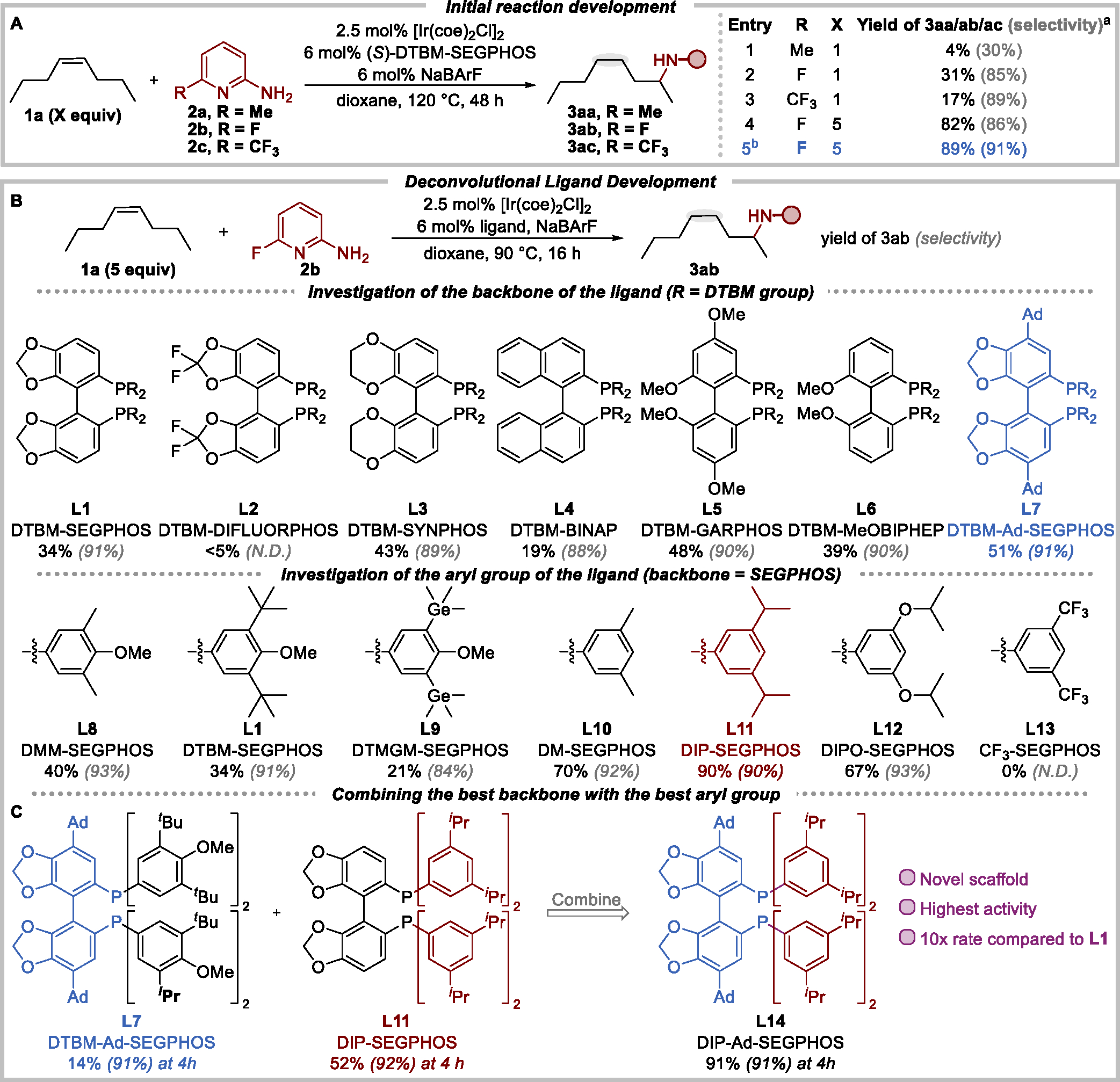
aYield represents the yield of 2-amine with respect to 2-aminopyridine as the limiting reagent; selectivity = 2-amine/all amines. bThe reaction was performed at 90 °C for 72 h.
We hypothesized that the selectivity for the 2-aminoalkane would increase if the rate of hydroamination could be retarded to enable more complete isomerization of the alkene. Building on the precedent that migratory insertions of alkenes into late transition-metal amido bonds are faster when the amido group is more electron donating,53,54 we examined the remote hydroamination with 2-aminopyridines bearing electron-withdrawing substituents. Rapid isomerization occurred, and the desired 2-amine was observed as the major product with the electron-deficient 6-fluoro- and 6-trifluoromethyl-2-aminopyridines (2b and 2c) as the N–H donor (Scheme 2A, entries 2 and 3). 6-Fluoro-2-aminopyridine (2b) was selected as the amine for further reaction development because it reacted in higher yield and with comparable selectivity than did 2c. In addition, the pyridyl fluoride in the product would enable further derivatization by nucleophilic aromatic substitution.
To improve the yield of the reaction between cis-4-octene and 6-fluoro-2-aminopyridine to form amine 3ab, we evaluated the effects of reaction time, temperature, solvent, and bisphosphine. However, the product formed in lower or similar yields (ca. 34%) in all cases (see the Supporting Information for details). Instead, the yield of the product increased to 89% when an excess of cis-4-octene (5 equiv) was used (Scheme 2A, entry 4). These results prompted us to assess whether the remote hydroamination was reversible and if a thermodynamic equilibrium between the isomeric alkenes and the 2-amine (3ab) affected the yield.
To test for reversible hydroamination, we subjected 2-aminoalkane 3ab to the catalytic conditions. Dehydroamination of 3ab occurred to form octene isomers as well as 2b, and 37% of 3ab remained. This result suggested that the low yield of 2-aminoalkene from a 1:1 ratio of cis-4-octene and 6-fluoro-2-aminopyridine resulted from unfavorable thermodynamics, rather than low activity or stability of the iridium catalyst.
The regioselectivity of the reaction between cis-4-octene (5 equiv) and 2b was further increased to 91% by conducting the reaction at 90 °C instead of at 120 °C (Scheme 2A, entry 5). However, prolonged reaction time (72 h) was required for the reaction to reach equilibrium at this temperature. Thus, an iridium complex that would catalyze the remote hydroamination with higher rates at the lower temperature was needed.
To do so, we sought to exploit the modularity of the ligand backbone and phosphine aryl groups by an approach that could survey the effects within a broad range of structures. Due to the challenge of synthesizing a library of bisphosphine ligands, particularly with biaryl backbones,55–59 we individually varied the components of the bisphosphines. We separately evaluated the effects of the electronic and steric properties of a series of backbones and a series of substituents on the aryl groups (Scheme 2B) of the bisphosphine ligands. After identifying the backbone and the aryl groups leading to the highest yield and selectivity, we combined these components to form a new ligand that would benefit from the properties of both components.
To evaluate the effect of the ligand backbone, we conducted the remote hydroamination with iridium catalysts of ligands possessing the same aryl group (DTBM group) but different backbones (L1–L7). The effect of the backbone of the ligand on the selectivity of the reaction was negligible, but the effect of the backbone on the rate was significant. The rate of the reaction was fastest with the ligand containing the electron-rich and bulky Ad-SEGPHOS backbone (L7).
The effect of the aryl group of the ligand on the reaction was assessed by conducting the remote hydroamination with iridium catalysts of ligands possessing the same backbone (SEGPHOS) but different aryl groups (L8–L13). This effect was determined with the SEGPHOS backbone because it is present in commonly used bisphosphines, including several commercially available versions, and because the racemic ligand is readily accessible by the homocoupling of benzodioxolyl diethyl phosphonate.57 The size of the meta substituents on the aryl rings significantly affected the selectivity of the reaction (L1, L8, L9), and the electronic properties of the meta and para substituents affected the rate (L8, L9, L12, L13). A midsized, slightly electron-donating, diisopropyl substituted aryl group (DIP, L11) was determined to possess the properties leading to high selectivity and high rate for the remote hydroamination.
Having identified the backbone (Ad-SEGPHOS) and aryl group (3,5-diisopropylphenyl, DIP) that individually led to the highest yield and selectivity, we prepared DIP-Ad-SEGPHOS (L14) that combined these two components and discovered that the reaction catalyzed by iridium and L14 yielded the product with selectivity equally high and with rates 6.5 and 1.8 times higher than of those of the reactions performed with the parent ligands L7 and L11, respectively (Scheme 2C). This new ligand, DIP-Ad-SEGPHOS (L14), was synthesized in a high 34% overall yield on gram scale. Moreover, the remote hydroamination of cis-4-octene required 4 h to reach equilibrium, whereas the reaction with widely used, commercially available DTBM-SEGPHOS (L11) required 72 h. This modular approach to the new ligand experimentally assessed a wide space of bisphosphine ligands with 13 representative examples and revealed a structure with enhanced catalytic performance in a rapid and rational manner.
Having identified a suitable N–H donor and ligand to realize the remote hydroamination, the scope of the reaction with varied alkenes was investigated (Scheme 3). Remote hydroamination occurred in high yields and with high regioselectivity with Z-alkenes, E-alkenes, and 1,1-disubstitued alkenes, including those requiring multiple isomerizations (3ab–3db); 2-amino product 3cb formed from six consecutive isomerizations prior to hydroamination. Selective remote hydroamination at the less hindered site was observed with an alkene (1db) possessing two potential reactive subterminal positions. The remote hydroamination also occurred with alkenes requiring contrathermodynamic isomerizations; the remote hydroamination of vinylarenes formed products 3eb and 3fb, without competing generation of products from hydroamination at the benzylic position, even though the isomerization to generate the reactive alkene requires deconjugation. The hydroamination of a β,γ-unsaturated ester (3gb) furnished the product of remote amination without the formation of the β-aminoalkyl product from conjugate addition of the aminopyridine into an α,β-unsaturated ester that formed from isomerization. Less than 20% of the alkene isomerized to the α,β-unsaturated ester during the remote hydroamination of alkene 1gb. In addition, minimal isomerization of the alkene (<10%) and no remote hydroamination occurred with the α,β-unsaturated ester as the substrate. These results indicated that isomerization toward the carbonyl unit to form the conjugated enoate is slower than isomerization to further deconjugated positions, and N–H addition to the more electron-rich alkene is faster than that to the conjugated alkene. The remote hydroamination also occurred with alkenes bearing a variety of suitably protected and free functional groups, including protected amines (3hb, 3nb), alcohols (3ib–3jb), and esters (3kb–3ob, 3qb–3rb) as well as arenes (3kb–3ob) and heteroarenes (3pb–3qb) bearing electron-donating and electron-withdrawing substituents. It also occurred with alkenes containing proximal, fully substituted β-carbons (3kb–3sb). The reaction even occurred with mixtures of alkenes to form a single product. Regioconvergent generation of 2-substituted amines from isomeric mixtures of alkenes occurred in high yield with excellent regioselectivity (3tb, 3ab). Finally, this reaction is compatible with the functionalization of alkenes derived from biologically relevant compounds (3ub–3xb). For certain substrates (1c, 1g, 1i, 1j), the reactions with DIP-MeOBIPHEP as ligand afforded products with higher regioselectivity than those obtained from the reaction with the (4-NMe2-3,5-DIP)-MeOBIPHEP ligand. For the reaction of β,γ-unsaturated ester (1g) and alkenes containing fully substituted β-carbons (1k–1q, 1s–1w), an increase in catalyst loading was required to achieve high yields.
Scheme 3. Scope of the Remote Hydroamination with Different Alkenesa.
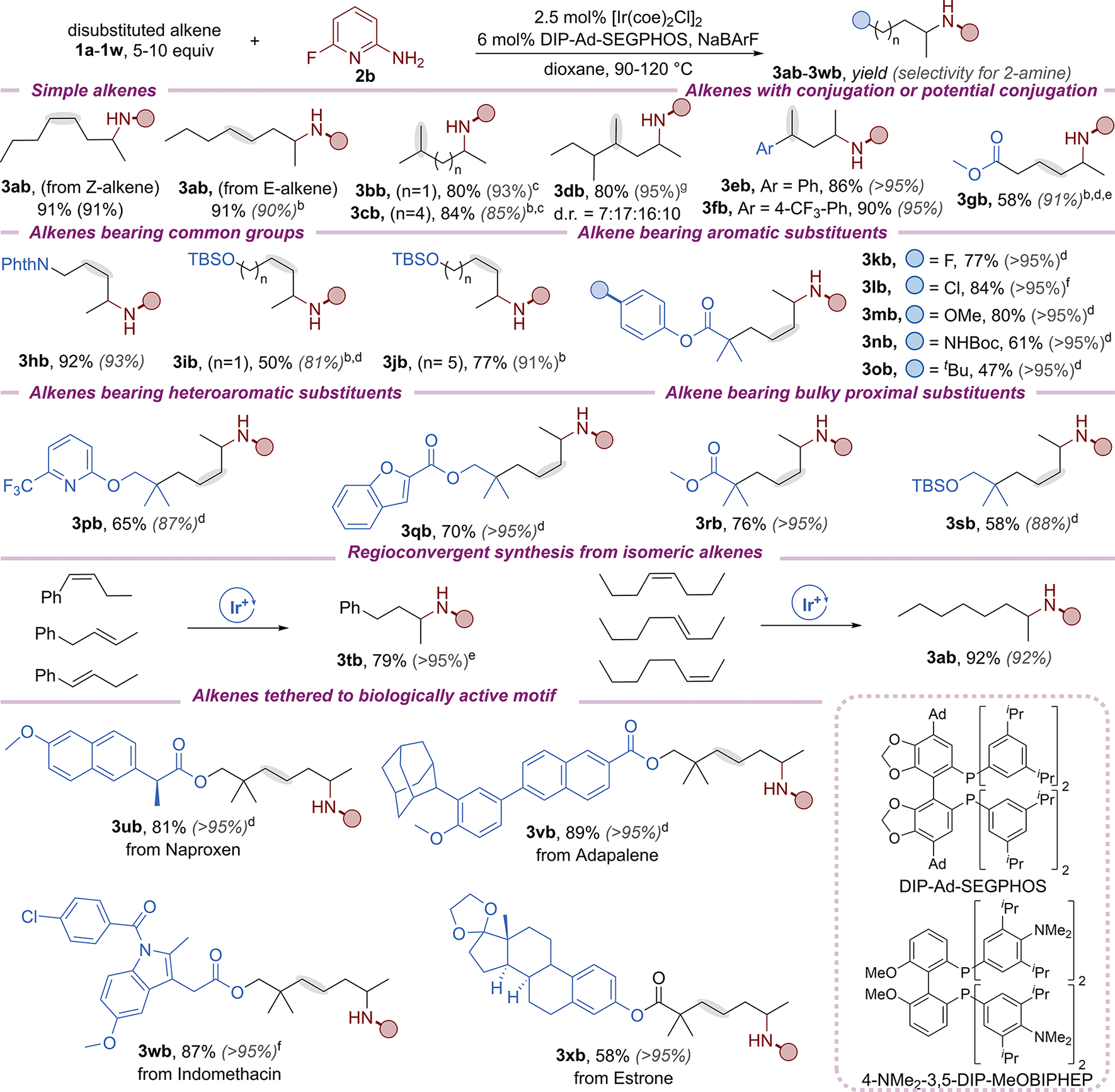
aProducts 3eb−3fb, 3ub, and 3xb were formed in 1:1 dr. b(4-NMe2-3,5-DIP)-MeOBIPHEP as the ligand. c7.5 equiv of alkene. d3.75 mol % [Ir(coe2)Cl]2, 9 mol % ligand and NaBArF. eTen equiv of alkene. fFive mol % [Ir(coe)2Cl]2, 12 mol % ligand and NaBArF. gTwenty equiv of alkene.
The scope of aminopyridines undergoing the remote hydroamination is shown in Scheme 4. Aminopyridines containing electron-withdrawing trifluoromethyl and difluoromethyl substituents at the 6-position underwent the remote hydroamination to afford the corresponding amines (2c–2d) in good yield with high regioselectivity. The remote hydroamination of cis-4-octene with aminopyridines containing synthetic handles for cross-coupling and SNAr reactions (2e−2h) also occurred in good yield. The remote hydroamination with aminopyridines containing a series of suitably protected or free functional groups, such as a protected alcohol (2i), protected amine (2j), ester (2k), alkyl (2l), aryl, and heteroaryl groups (2m–2s), also occurred.
Scheme 4. Scope of the Remote Hydroamination with Different Aminopyridines.
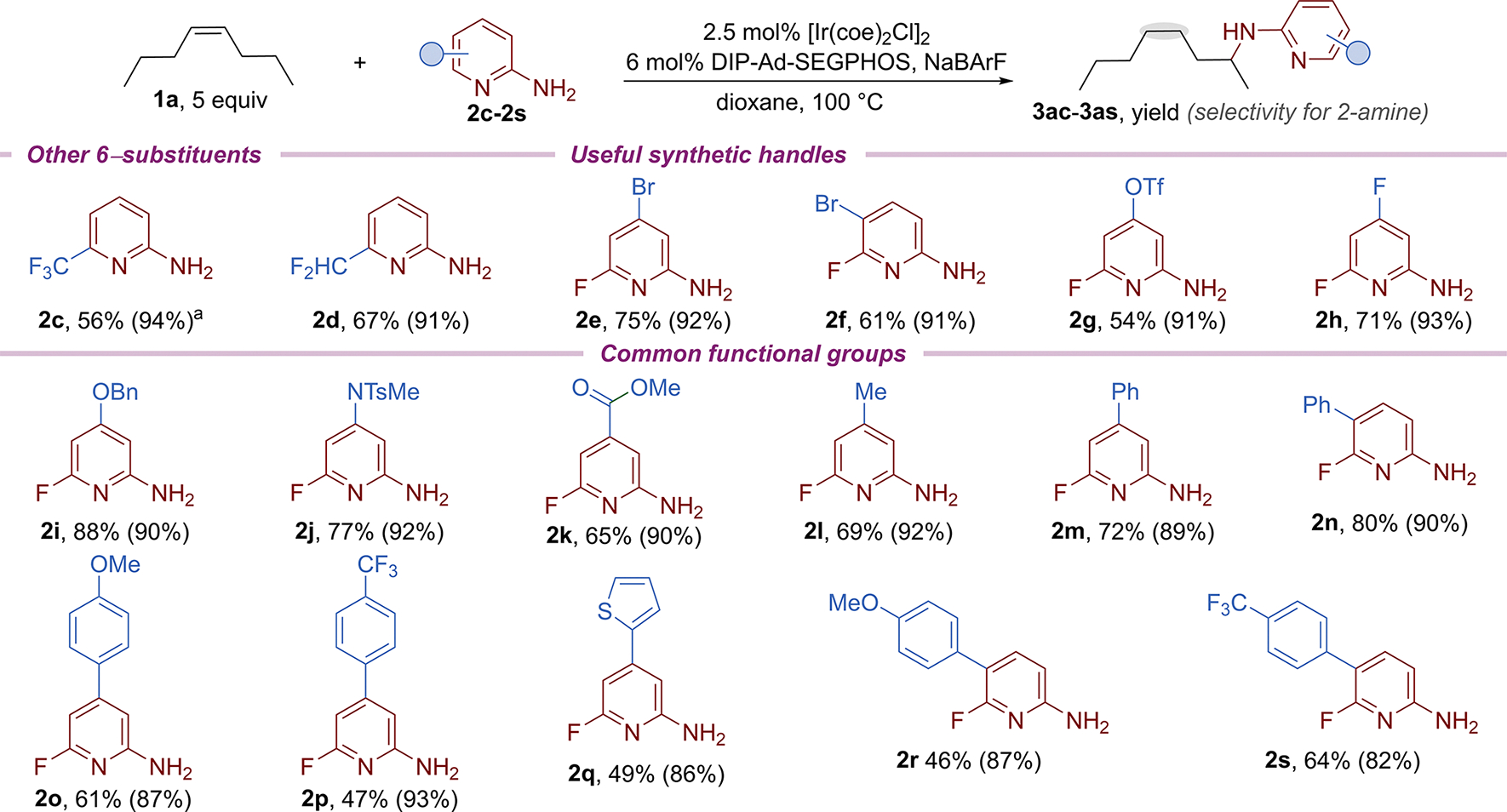
aFive mol % [Ir(coe)2Cl]2, 12 mol % ligand and NaBArF.
Subsequent functionalization of the remote hydroamination products containing pyridyl fluorides occurred by nucleophilic aromatic substitutions (Scheme 5). SNAr reactions of amine 3ab with sulfur-, oxygen-, and nitrogen-based nucleophiles proceeded smoothly to afford the substituted products (4ab–9ab), although deprotonation of the acidic N–H proton in 3ab under basic conditions significantly decreased its reactivity toward SNAr reactions.
Scheme 5.
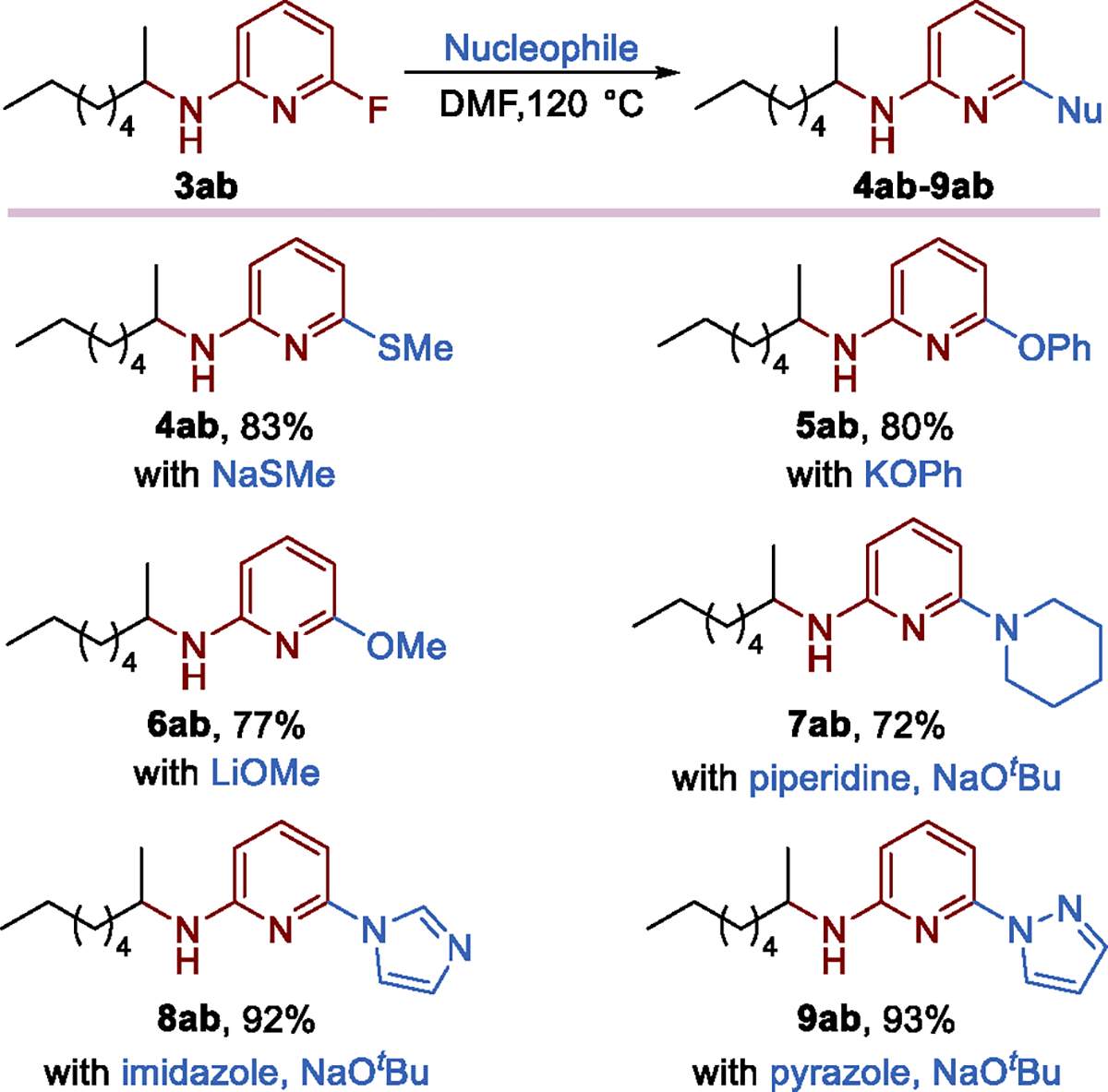
Diversification of the Product through Nucleophilic Aromatic Substitutions
In summary, we have discovered iridium-catalyzed remote hydroaminations of unactivated disubstituted alkenes with electron-poor 2-aminopyridines. The keys to this development are the use of aminopyridines bearing electron-withdrawing substituents at the 6-position to increase the selectivity for the remote amine product by changing the relative rates for hydroamination versus isomerization of the alkene and the creation of a new DIP-Ad-SEGPHOS ligand by evaluating the steric and electronic effects of ligand modules on reactivity and selectivity. Further work on elucidating the mechanism of the reaction is ongoing, and we anticipate that this work will inspire the development of other remote hydrofunctionalizations and the development of bisphosphines by experimental assessment of the components of such modular ligands.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Director, Office of Science of the US Department of Energy under contract number DE-AC02–05CH11231. We gratefully acknowledge Takasago for gifts of (S)-DTBM-SEGPHOS, Dr. Hasan Celik for assistance with NMR experiments (NIH S10OD024998), and John Brunn for HRMS. Yumeng Xi thanks Bristol-Myers Squibb for a graduate fellowship. Craig Day thanks European Union’s Horizon 2020 under the Marie Curie PREBIST grant agreement 754558.
ABBREVIATIONS
- Ad
1-adamantyl
- DMM
4-methoxy-3,5-dimethyl-phenyl
- DTBM
4-methoxy-3,5-ditertbutyl-phenyl
- DTMGM
4-methoxy-3,5-ditrimethylgermanyl-phenyl
- DM
3,5-dimethyl-phenyl
- DIP
3,5-diisopropyl-phenyl
- DIPO
3,5-diisopropoxyphenyl
- SNAr
nucleophilic aromatic substitution
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c13054.
Experimental procedures and characterization of new compounds (PDF)
Contributor Information
Senjie Ma, Department of Chemistry, University of California, Berkeley, California 94720, United States; Division of Chemical Sciences, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States.
Haoyu Fan, Department of Chemistry, University of California, Berkeley, California 94720, United States.
Craig S. Day, Department of Chemistry, University of California, Berkeley, California 94720, United States
Yumeng Xi, Department of Chemistry, University of California, Berkeley, California 94720, United States; Division of Chemical Sciences, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States.
John F. Hartwig, Department of Chemistry, University of California, Berkeley, California 94720, United States; Division of Chemical Sciences, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States
REFERENCES
- (1).Vasseur A; Bruffaerts J; Marek I Remote functionalization through alkene isomerization. Nat. Chem. 2016, 8 (3), 209–219. [DOI] [PubMed] [Google Scholar]
- (2).Sommer H; Juliá-Hernández F; Martin R; Marek I Walking Metals for Remote Functionalization. ACS Cent. Sci. 2018, 4 (2), 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Janssen-Müller D.; Sahoo B; Sun S-Z; Martin R Tackling Remote sp3 C–H Functionalization via Ni-Catalyzed “chain-walking” Reactions. Isr. J. Chem. 2020, 60 (3–4), 195–206. [Google Scholar]
- (4).Ghosh S; Patel S; Chatterjee I Chain-walking reactions of transition metals for remote C–H bond functionalization of olefinic substrates. Chem. Commun. 2021, 57 (85), 11110–11130. [DOI] [PubMed] [Google Scholar]
- (5).Wang X-X; Lu X; Li Y; Wang J-W; Fu Y Recent advances in nickel-catalyzed reductive hydroalkylation and hydroarylation of electronically unbiased alkenes. Sci. China Chem. 2020, 63 (11), 1586–1600. [Google Scholar]
- (6).Buslov I; Becouse J; Mazza S; Montandon-Clerc M; Hu X Chemoselective Alkene Hydrosilylation Catalyzed by Nickel Pincer Complexes. Angew. Chem., Int. Ed. 2015, 54 (48), 14523–14526. [DOI] [PubMed] [Google Scholar]
- (7).Buslov I; Song F; Hu X An Easily Accessed Nickel Nanoparticle Catalyst for Alkene Hydrosilylation with Tertiary Silanes. Angew. Chem., Int. Ed. 2016, 55 (40), 12295–12299. [DOI] [PubMed] [Google Scholar]
- (8).Chen C; Hecht MB; Kavara A; Brennessel WW; Mercado BQ; Weix DJ; Holland PL Rapid, Regioconvergent, Solvent-Free Alkene Hydrosilylation with a Cobalt Catalyst. J. Am. Chem. Soc. 2015, 137 (41), 13244–13247. [DOI] [PubMed] [Google Scholar]
- (9).Jia X; Huang Z Conversion of alkanes to linear alkylsilanes using an iridium–iron-catalysed tandem dehydrogenation–isomerization–hydrosilylation. Nat. Chem. 2016, 8 (2), 157–161. [DOI] [PubMed] [Google Scholar]
- (10).Noda D; Tahara A; Sunada Y; Nagashima H Non-Precious-Metal Catalytic Systems Involving Iron or Cobalt Carboxylates and Alkyl Isocyanides for Hydrosilylation of Alkenes with Hydrosiloxanes. J. Am. Chem. Soc. 2016, 138 (8), 2480–2483. [DOI] [PubMed] [Google Scholar]
- (11).Hanna S; Butcher TW; Hartwig JF Contrathermodynamic Olefin Isomerization by Chain-Walking Hydrofunctionalization and Formal Retro-hydrofunctionalization. Org. Lett. 2019, 21 (17), 7129–7133. [DOI] [PubMed] [Google Scholar]
- (12).Ye W-T; Zhu R Dioxygen-promoted cobalt-catalyzed oxidative hydroamination using unactivated alkenes and free amines. Chem. Catal. 2022, 2 (2), 345–357. [Google Scholar]
- (13).Crudden CM; Edwards D Catalytic Asymmetric Hydroboration: Recent Advances and Applications in Carbon–Carbon Bond-Forming Reactions. Eur. J. Org. Chem. 2003, 2003 (24), 4695–4712. [Google Scholar]
- (14).Cipot J; Vogels CM; McDonald R; Westcott SA; Stradiotto M Catalytic Alkene Hydroboration Mediated by Cationic and Formally Zwitterionic Rhodium(I) and Iridium(I) Derivatives of a P,N-Substituted Indene. Organometallics 2006, 25 (25), 5965–5968. [Google Scholar]
- (15).Ghebreyessus KY; Angelici RJ Isomerizing-Hydroboration of the Monounsaturated Fatty Acid Ester Methyl Oleate. Organometallics 2006, 25 (12), 3040–3044. [Google Scholar]
- (16).Lata CJ; Crudden CM Dramatic Effect of Lewis Acids on the Rhodium-Catalyzed Hydroboration of Olefins. J. Am. Chem. Soc. 2010, 132 (1), 131–137. [DOI] [PubMed] [Google Scholar]
- (17).Obligacion JV; Chirik PJ Bis(imino)pyridine CobaltCatalyzed Alkene Isomerization–Hydroboration: A Strategy for Remote Hydrofunctionalization with Terminal Selectivity. J. Am. Chem. Soc. 2013, 135 (51), 19107–19110. [DOI] [PubMed] [Google Scholar]
- (18).Palmer WN; Diao T; Pappas I; Chirik PJ High-Activity Cobalt Catalysts for Alkene Hydroboration with Electronically Responsive Terpyridine and α-Diimine Ligands. ACS Catal. 2015, 5 (2), 622–626. [Google Scholar]
- (19).Scheuermann ML; Johnson EJ; Chirik PJ Alkene Isomerization–Hydroboration Promoted by Phosphine-Ligated Cobalt Catalysts. Org. Lett. 2015, 17 (11), 2716–2719. [DOI] [PubMed] [Google Scholar]
- (20).Ogawa T; Ruddy AJ; Sydora OL; Stradiotto M; Turculet L Cobalt- and Iron-Catalyzed Isomerization–Hydroboration of Branched Alkenes: Terminal Hydroboration with Pinacolborane and 1,3,2-Diazaborolanes. Organometallics 2017, 36 (2), 417–423. [Google Scholar]
- (21).Obligacion JV; Chirik PJ Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroboration. Nat. Rev. Chem. 2018, 2 (5), 15–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hanna S; Bloomer B; Ciccia NR; Butcher TW; Conk RJ; Hartwig JF Contra-thermodynamic Olefin Isomerization by Chain-Walking Hydroboration and Dehydroboration. Org. Lett. 2022, 24 (4), 1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lim Y-G; Kang J-B; Kim YH Regioselective alkylation of 2-phenylpyridines with terminal alkenes via C–H bond activation by a rhodium catalyst. J. Chem. Soc., Perkin Trans. 1 1996, No. 17, 2201–2206. [Google Scholar]
- (24).Bair JS; Schramm Y; Sergeev AG; Clot E; Eisenstein O; Hartwig JF Linear-Selective Hydroarylation of Unactivated Terminal and Internal Olefins with Trifluoromethyl-Substituted Arenes. J. Am. Chem. Soc. 2014, 136 (38), 13098–13101. [DOI] [PubMed] [Google Scholar]
- (25).Borah AJ; Shi Z Rhodium-Catalyzed, Remote Terminal Hydroarylation of Activated Olefins through a Long-Range Deconjugative Isomerization. J. Am. Chem. Soc. 2018, 140 (19), 6062–6066. [DOI] [PubMed] [Google Scholar]
- (26).Zhang M; Hu L; Lang Y; Cao Y; Huang G Mechanism and Origins of Regio- and Enantioselectivities of Iridium-Catalyzed Hydroarylation of Alkenyl Ethers. J. Org. Chem. 2018, 83 (5), 2937–2947. [DOI] [PubMed] [Google Scholar]
- (27).Lee W-C; Wang C-H; Lin Y-H; Shih W-C; Ong T-G Tandem Isomerization and C–H Activation: Regioselective Hydroheteroarylation of Allylarenes. Org. Lett. 2013, 15 (20), 5358–5361. [DOI] [PubMed] [Google Scholar]
- (28).Yamakawa T; Yoshikai N Alkene Isomerization−Hydroarylation Tandem Catalysis: Indole C2-Alkylation with Aryl-Substituted Alkenes Leading to 1,1-Diarylalkanes. Chem.Asian J. 2014, 9 (5), 1242–1246. [DOI] [PubMed] [Google Scholar]
- (29).He Y; Cai Y; Zhu S Mild and Regioselective Benzylic C–H Functionalization: Ni-Catalyzed Reductive Arylation of Remote and Proximal Olefins. J. Am. Chem. Soc. 2017, 139 (3), 1061–1064. [DOI] [PubMed] [Google Scholar]
- (30).He Y; Liu C; Yu L; Zhu S Ligand-Enabled Nickel-Catalyzed Redox-Relay Migratory Hydroarylation of Alkenes with Arylborons. Angew. Chem., Int. Ed. 2020, 59 (23), 9186–9191. [DOI] [PubMed] [Google Scholar]
- (31).He Y; Han B; Zhu S Terminal-Selective C(sp3)–H Arylation: NiH-Catalyzed Remote Hydroarylation of Unactivated Internal Olefins. Organometallics 2021, 40 (14), 2253–2264. [Google Scholar]
- (32).He Y; Ma J; Song H; Zhang Y; Liang Y; Wang Y; Zhu S Regio- and enantioselective remote hydroarylation using a ligand-relay strategy. Nat. Commun. 2022, 13 (1), 2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhang Y; Han B; Zhu S Rapid Access to Highly Functionalized Alkyl Boronates by NiH-Catalyzed Remote Hydroarylation of Boron-Containing Alkenes. Angew. Chem., Int. Ed. 2019, 58 (39), 13860–13864. [DOI] [PubMed] [Google Scholar]
- (34).Cheng Q; Liu W; Dang Y Insights into the mechanism and regioselectivity in Ni-catalysed redox-relay migratory hydroarylation of alkenes with arylborons. Chem. Commun. 2021, 57 (99), 13610–13613. [DOI] [PubMed] [Google Scholar]
- (35).Liu J; Gong H; Zhu S BH3 · Me2S: An Alternative Hydride Source for NiH-Catalyzed Reductive Migratory Hydroarylation and Hydroalkenylation of Alkenes. Eur. J. Org. Chem. 2021, 2021 (10), 1543–1546. [Google Scholar]
- (36).Zhang Y; Ma J; Chen J; Meng L; Liang Y; Zhu S A relay catalysis strategy for enantioselective nickel-catalyzed migratory hydroarylation forming chiral α-aryl alkylboronates. Chem. 2021, 7 (11), 3171–3188. [Google Scholar]
- (37).Werner EW; Mei T-S; Burckle AJ; Sigman MS Enantioselective Heck Arylations of Acyclic Alkenyl Alcohols Using a Redox-Relay Strategy. Science 2012, 338 (6113), 1455–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mei T-S; Werner EW; Burckle AJ; Sigman MS Enantioselective Redox-Relay Oxidative Heck Arylations of Acyclic Alkenyl Alcohols using Boronic Acids. J. Am. Chem. Soc. 2013, 135 (18), 6830–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Mei T-S; Patel HH; Sigman MS Enantioselective construction of remote quaternary stereocentres. Nature 2014, 508 (7496), 340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Qian D; Hu X Ligand-Controlled Regiodivergent Hydroalkylation of Pyrrolines. Angew. Chem., Int. Ed. 2019, 58 (51), 18519–18523. [DOI] [PubMed] [Google Scholar]
- (41).Zhou F; Zhang Y; Xu X; Zhu S NiH-Catalyzed Remote Asymmetric Hydroalkylation of Alkenes with Racemic α-Bromo Amides. Angew. Chem., Int. Ed. 2019, 58 (6), 1754–1758. [DOI] [PubMed] [Google Scholar]
- (42).Wang J-W; Liu D-G; Chang Z; Li Z; Fu Y; Lu X Nickel-Catalyzed Switchable Site-Selective Alkene Hydroalkylation by Temperature Regulation. Angew. Chem., Int. Ed. 2022, 61 (31), e202205537. [DOI] [PubMed] [Google Scholar]
- (43).Li P; Lee BC; Zhang X; Koh MJ Base-Mediated SiteSelective Hydroamination of Alkenes. Synthesis 2022, 54 (06), 1566–1576. [Google Scholar]
- (44).Miao H-Z; Liu Y; Chen Y-W; Lu H-Y; Li J; Lin G-Q; He Z-T Stereoselective Pd-Catalyzed Remote Hydroamination of Skipped Dienes with Azoles. Synlett 2022, 33, A–F. [Google Scholar]
- (45).Zhang Y; He J; Song P; Wang Y; Zhu S Ligand-Enabled NiH-Catalyzed Migratory Hydroamination: Chain Walking as a Strategy for Regiodivergent/Regioconvergent Remote sp3C–H Amination. CCS Chem. 2021, 3 (9), 2259–2268. [Google Scholar]
- (46).Lee C; Seo H; Jeon J; Hong S γ-Selective C(sp3)–H amination via controlled migratory hydroamination. Nat. Commun. 2021, 12 (1), 5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Du B; Ouyang Y; Chen Q; Yu W-Y Thioether-Directed NiH-Catalyzed Remote γ-C(sp3)–H Hydroamidation of Alkenes by 1,4,2-Dioxazol-5-ones. J. Am. Chem. Soc. 2021, 143 (37), 14962–14968. [DOI] [PubMed] [Google Scholar]
- (48).Wagner-Carlberg N; Rovis T Rhodium(III)-Catalyzed Anti-Markovnikov Hydroamidation of Unactivated Alkenes Using Dioxazolones as Amidating Reagents. J. Am. Chem. Soc. 2022, 144 (49), 22426–22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Rosenfeld DC; Shekhar S; Takemiya A; Utsunomiya M; Hartwig JF Hydroamination and Hydroalkoxylation Catalyzed by Triflic Acid. Parallels to Reactions Initiated with Metal Triflates. Org. Lett. 2006, 8 (19), 4179–4182. [DOI] [PubMed] [Google Scholar]
- (50).Xi Y; Ma S; Hartwig JF Catalytic asymmetric addition of an amine N–H bond across internal alkenes. Nature 2020, 588 (7837), 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ma S; Xi Y; Fan H; Roediger S; Hartwig JF Enantioselective hydroamination of unactivated terminal alkenes. Chem. 2022, 8 (2), 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Ma S; Hill CK; Olen CL; Hartwig JF Ruthenium-Catalyzed Hydroamination of Unactivated Terminal Alkenes with Stoichiometric Amounts of Alkene and an Ammonia Surrogate by Sequential Oxidation and Reduction. J. Am. Chem. Soc. 2021, 143 (1), 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hanley PS; Hartwig JF Intermolecular Migratory Insertion of Unactivated Olefins into Palladium–Nitrogen Bonds. Steric and Electronic Effects on the Rate of Migratory Insertion. J. Am. Chem. Soc. 2011, 133 (39), 15661–15673. [DOI] [PubMed] [Google Scholar]
- (54).Hanley PS; Hartwig JF Migratory Insertion of Alkenes into Metal–Oxygen and Metal–Nitrogen Bonds. Angew. Chem., Int. Ed. 2013, 52 (33), 8510–8525. [DOI] [PubMed] [Google Scholar]
- (55).Miyashita A; Yasuda A; Takaya H; Toriumi K; Ito T; Souchi T; Noyori R Synthesis of 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), an atropisomeric chiral bis(triaryl)phosphine, and its use in the rhodium(I)-catalyzed asymmetric hydrogenation of. alpha.-(acylamino)acrylic acids. J. Am. Chem. Soc. 1980, 102 (27), 7932–7934. [Google Scholar]
- (56).Müller D; Umbricht G; Weber B; Pfaltz A C2-Symmetric 4,4′,5,5′-Tetrahydrobi(oxazoles) and 4,4′,5,5′-Tetrahydro-2,2′-methylenebis[oxazoles] as Chiral Ligands for Enantioselective Catalysis Preliminary Communication. Helv. Chim. Acta 1991, 74 (1), 232–240. [Google Scholar]
- (57).Saito T; Yokozawa T; Ishizaki T; Moroi T; Sayo N; Miura T; Kumobayashi H New Chiral Diphosphine Ligands Designed to have a Narrow Dihedral Angle in the Biaryl Backbone. Adv. Synth. Catal. 2001, 343 (3), 264–267. [Google Scholar]
- (58).Pai C-C; Li Y-M; Zhou Z-Y; Chan ASC Synthesis of new chiral diphosphine ligand (BisbenzodioxanPhos) and its application in asymmetric catalytic hydrogenation. Tetrahedron Lett. 2002, 43 (15), 2789–2792. [Google Scholar]
- (59).Duprat de Paule S; Jeulin S; Ratovelomanana-Vidal V; Genêt J-P; Champion N; Deschaux G; Dellis P SYNPHOS: a New Atropisomeric Diphosphine Ligand. From Laboratory-scale Synthesis to Scale-up Development. Org. Process Res. Dev. 2003, 7 (3), 399–406. (60) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.