

Abstract
The conversion of aryl halides to primary arylamines with a convenient and inexpensive source of ammonia has been a long-standing synthetic challenge. Aqueous ammonia would be the most convenient and least expensive form of ammonia, but such a palladium-catalyzed amination reaction with a high concentration of water faces challenges concerning catalyst stability and competing hydroxylation, and palladium-catalyzed reactions with this practical reagent are rare. Further, most reactions with ammonia to form primary amines are conducted with tert-butoxide base, but reactions with ammonium hydroxide would contain hydroxide as base. Thus, ammonia surrogates, ammonia in organic solvents, and ammonium salts have been used under anhydrous conditions instead with varying levels of selectivity for the primary amine. We report the palladium-catalyzed amination of aryl and heteroaryl chlorides and bromides with aqueous ammonia and a hydroxide base to form the primary arylamine with high selectivity. The palladium catalyst containing a new dialkyl biheteroaryl phosphine ligand (KPhos) suppresses both the formation of aryl alcohol and diarylamine side products. Mechanistic studies with a soluble hydroxide base revealed turnover-limiting reductive elimination of the arylamine and an equilibrium between arylpalladium amido and hydroxo complexes prior to the turnover-limiting step.
Graphical Abstract

INTRODUCTION
The coupling of nitrogen nucleophiles with aryl and heteroaryl halides with a transition-metal catalyst has become one of the most widely used reactions to synthesize arylamines in settings ranging from academic laboratories to agrochemical, pharmaceutical, and materials discovery and process groups.1 A wide range of nitrogen nucleophiles form C−N bonds with the carbon at the site of C−X bonds (X = halogen or pseudohalogen) by this reaction.2 While many reactions are conducted with complex nitrogen nucleophiles, the coupling with ammonia could be used to form arylamines on a large scale as intermediates to many amine, amide, sulfonamide, and heterocyclic end products. In this context, transition-metal-catalyzed C−N coupling with ammonia or its surrogates provides a route that is milder than classical sequences of nitration and reduction to form arylamines.
The synthesis of primary arylamines from aryl halides has been achieved by various research groups, including our own (Figure 1a),3 but these reactions have required toxic ammonia gas, ammonia solutions in organic solvents that are expensive and lose their ammonia over time, or ammonium salts that require excess base and generate an additional equivalent of halide (Figure 1b).4 Each process has required a tert-butoxide base. Protected or masked ammonia sources, such as benzophenone imine, bis(trimethylsilyl)amide, primary amides, tert-butyl carbamate, (di)allylamine, and benzylamine, have been used to form a single C−N bond with aryl halides, but these reagents require deprotection after the reaction is completed.
Figure 1.

Transition-metal-catalyzed amination of aryl halides with ammonia or its surrogates. (a) General scheme of the reaction. (b) Known sources of ammonia in Pd-catalyzed amination reactions and their limitations. (c) Challenge of employing aqueous ammonia in the Pd-catalyzed C−N coupling reactions: selective monoarylation of ammonia over diarylation and hydroxylation of aryl halides. (d) Development of the palladium catalyst for the selective amination of aryl halides with aqueous ammonia and a hydroxide base in this work.
These procedures were developed because no palladium system has been reported to catalyze the most preferable reaction: the coupling of aqueous ammonia with aryl halides and a simple hydroxide base (Figure 1c).5 Aqueous ammonia would be the ideal source because it is readily available, less expensive than organic solutions of ammonia, and easier to handle and store safely than anhydrous ammonia. However, the palladium-catalyzed amination of aryl halides with aqueous ammonia is difficult to achieve. Such a reaction must confront a series of potential side reactions, including the formation of aryl alcohol and diarylether, instead of the desired primary arylamine, potential catalyst decomposition through intermediate palladium hydroxo compounds,6 and the need for hydroxide as base that is rarely used for palladium-catalyzed aminations. Although several copper-catalyzed C−N bond-forming reactions occur with aqueous ammonia, the scope of aryl halide coupling partners is largely limited to aryl iodides and bromides, the reactions require high temperatures, and catalyst loadings are high, usually between 5 and 10 mol %.7 The narrow scope of aryl halide electrophiles and higher loadings of catalysts make the development of a more general and efficient amination method necessary.
We report the palladium-catalyzed amination of aryl and heteroaryl halides with aqueous ammonia and potassium hydroxide base enabled by the development of a new ligand (KPhos) based on a bipyrazole backbone (Figure 1d). The bipyrazole ligand suppresses formation of phenol, and appropriate substituents on the backbone and at phosphorus lead to the highest observed selectivities for formation of monoaryl- vs diarylamine products with a broad scope of aryl and heteroaryl halides. Mechanistic investigations, including kinetic analyses of the initial rates, show that reductive elimination of the arylamine is turnover-limiting and that an equilibrium between arylpalladium(II) amido and arylpalladium(II) hydroxo complexes occurs before this turnover-limiting step.
RESULTS AND DISCUSSION
Reaction Development.
At the outset of our study to develop the selective monoarylation of ammonium hydroxide, we evaluated phosphine ligands with the G3 palladacyclic dimer (Figure 2a). Reactions with the catalyst generated from representative dialkyl arylphosphines showed that 1-chloro-4-fluorobenzene 1a and aqueous ammonia form the desired aniline 2a as a major product, but with side products comprising the corresponding diarylamine (Ar2NH) from coupling of the product and aryl alcohol (ArOH) from coupling of the aryl halide with hydroxide. The biaryl substituent on phosphorus affected the selectivity for the formation of the phenol side product. Reactions with BrettPhos derivatives, which contain a sterically hindered biphenyl substituent, formed ArOH as the major side product,3g whereas reactions conducted with tBuBippyPhos as ligand, which contains a bipyrazole backbone, formed no observable ArOH.3f,8
Figure 2.
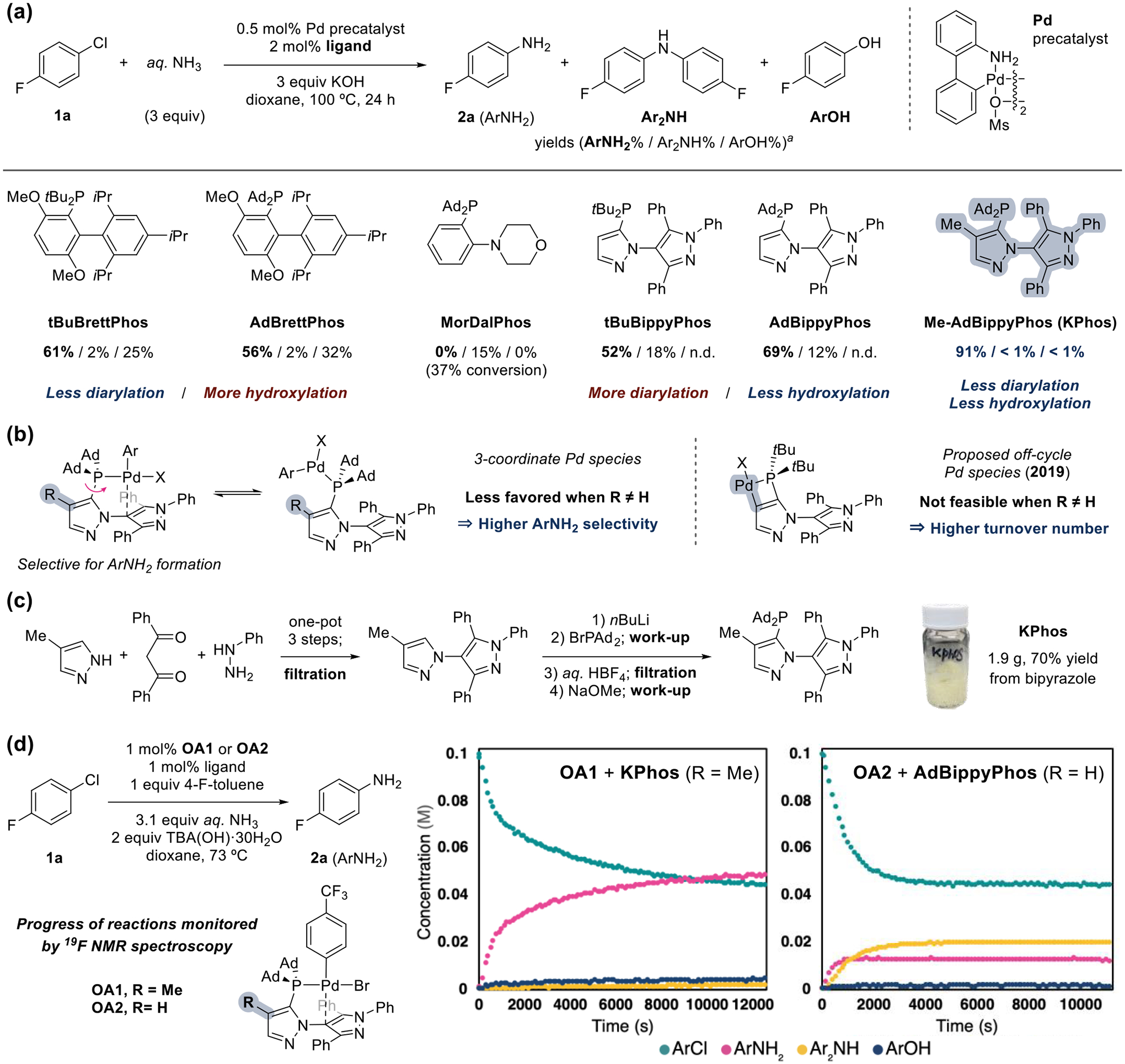
(a) Evaluation of representative dialkyl aryl phosphine ligands and KPhos. Standard reaction conditions: 1a (0.1 mmol), Pd-precatalyst (0.5 μmol), ligand (2.0 μmol), aq NH3 (0.3 mmol), and KOH (0.3 mmol) in 1,4-dioxane (1.0 mL) at 100 °C for 24 h. aConversion of 1a and yields of products were determined by 19F NMR spectroscopy with 1-fluoronaphthalene as an internal standard. (b) Working hypotheses of how the Kphos ligand achieves higher product selectivity and turnover number. (c) Gram scale synthesis of KPhos without purification by column chromatography. (d) Observation of the progress of amination reactions with KPhos or AdBippyPhos ligand recorded by 19F NMR spectroscopy.
With the result that the catalysts containing a bipyrazole ligand afford high selectivity for amination with ammonia over hydroxylation with water, we sought to further increase the formation of the desired monoarylamine by suppressing the formation of the diarylamine side product. The reaction with the parent BippyPhos formed Ar2NH as the major side product, generating a 2.9:1 ratio of ArNH2 to Ar2NH. Bulkier alkyl substituents on the phosphorus of BippyPhos led to a slight improvement in the selectivity to form 2a over Ar2NH with AdBippyPhos giving a 5.8:1 ratio of ArNH2 to Ar2NH.
To design an AdBippyPhos ligand that would lead to higher selectivity for the primary amine,9 we installed a methyl group on the C4 position adjacent to the C−P bond (Figure 2b). We followed the hypothesis that arylpalladium(II) complexes in the selectivity-determining step can adopt tetra- or tricoordinate isomers10 and proposed that the latter complex more readily coordinates the primary arylamine to provide the diarylamine side product. In this case, a new ligand with the methyl group in place of H could disfavor the tricoordinate complex, leading to a higher selectivity for the formation of primary arylamine. Furthermore, the methyl substituent should prevent metalation of the pyrazole C4 position that yields a palladacyclobutene complex, which would be an off-cycle species during the coupling reaction.11,12
The new bipyrazole ligand, 4-methyl AdBippyPhos (KPhos), was generated on a gram scale in up to 70% isolated yield over four steps from a bipyrazole without the need for chromato-graphic purification (Figure 2c). Fomepizole, 1,3-diphenyl-1,3-propanedione, and phenylhydrazine were assembled by a three-step process in one pot to afford the bipyrazole core.13 The C5−H bond of the bipyrazole was then lithiated in situ, and the resulting bipyrazolyl lithium was quenched with di(1-adamantyl)bromophosphine.14 Purification of KPhos from the crude mixture was accomplished by the formation of KPhos·HBF4, followed by filtration and neutralization.
Reactions catalyzed by the G3 palladacyclic dimer and KPhos under the same conditions as those with other dialkyl (bi)arylphosphine ligands occurred with exceptionally high yield and selectivity for the primary amine (Figure 2a). Comparison of the selectivity to that with the nonmethylated analog (AdBippyPhos) showed that the additional methyl group present in KPhos increased the selectivity of monoarylation over diarylation dramatically, from 5.8:1 for AdBippyPhos to 178:1 for KPhos. This increase in selectivity for amination was evaluated further by tracking the progress of amination reactions with KPhos and AdBippyPhos (Figure 2d). Under homogeneous conditions with arylpalladium(II) bromide complex OA1 or OA2 as the catalyst and the soluble base TBA(OH), the time courses of the reactions were recorded by 19F NMR spectroscopy, and the results are shown in Figure 2d. The catalyst containing KPhos reacted with higher turnovers and selectivity for the formation of monoarylamine 2a over the corresponding diarylamine than did the catalyst containing AdBippyPhos. Although the KPhos-ligated palladium catalyst reacted more slowly than the AdBippyPhos-ligated analog, we used this new ligand for the rest of our studies because the selectivity for monoarylation and the turnover numbers were higher than with AdBippyPhos.15
To understand the origin of the high selectivity with the KPhos ligand, the geometry of arylpalladium(II) bromide complex OA1 was determined by single-crystal X-ray diffraction (Figure 3a). The geometry of OA1 at the palladium atom is distorted square planar (∠C26−Pd1−C14 = 168.95°), with the phosphine ligand coordinated in a bidentate fashion through phosphorus and C4 of the distal pyrazole ring. The distortion of the bond angles results from steric interactions between the adamantyl groups on phosphorus and the aryl group bound to the palladium. As shown in Figure 3b, the distances between one of the hydrogens in the methyl substituent and the hydrogens of the adamantyl groups are 2.156 and 2.124 Å respectively, implying that rotation of the C−P bond of OA1 to provide the three-coordinate intermediate in Figure 2b is restricted. In addition, the distances between the ortho-hydrogens of the aryl group to palladium and the hydrogens of the adamantyl groups (~2.3 Å) are shorter than the 1,3-diaxial distances between adamantyl hydrogens (~2.5 Å, Figure 3c). This feature limits the scope of the amination of ortho-substituted aryl halides and is discussed later in this paper.
Figure 3.

(a) Thermal ellipsoid plot of OA1 with 50% probability ellipsoids. (b) Selected distances between hydrogens of methyl at pyrazole and hydrogens of the adamantyl group at phosphorus. (c) Selected distances between ortho-hydrogens of the aryl group bound to palladium and hydrogens of the adamantyl group at phosphorus.
Scope of Aryl Halides that React with Aqueous Ammonia.
A diverse set of aryl chlorides and bromides undergo catalytic coupling with aqueous ammonia to give aniline products (Table 1). The reactions of aryl halides containing electron-donating para-substituents resulted in the formation of primary arylamines 2a−g in good yields. Electron deficient aryl halides bearing trifluoromethoxy (2h), trifluoromethyl (2i), acyl (2j−l), carbamoyl (2m), cyano (2p), nitro (2q), sulfonyl (2t), or sulfamoyl (2u) groups also underwent the reaction to furnish the corresponding aniline derivatives. We did not observe defluorinated side products derived from SNAr processes with aryl chlorides or bromides containing an aryl fluoride. Aminojulolidine product 2g was synthesized on 3 mmol scale from the corresponding aryl bromide, demonstrating the high activity of the palladium catalyst derived from KPhos and Pd-G3 dimer in >1.0 mmol scale reactions.
Table 1.
Scope of Aryl Halides for the Amination with Aqueous Ammoniaa

|
Standard reaction conditions: Aryl halide (1.00 mmol), Pd-precatalyst (5.00 μmol), KPhos (20.0 μmol), aq NH3 (3.00 mmol), and KOH (3.00 mmol) in 1,4-dioxane (10.0 mL) at 100 °C for 24 h.
Isolated yields.
Ratio was determined by 1H NMR spectroscopy of the crude mixture.
Isolated as N-Boc protected aniline.
Pd-precatalyst (10.0 μmol) and KPhos (40.0 μmol) were used.
1.5 equiv KOH was used. gPd-precatalyst (15.0 μmol) and KPhos (60.0 μmol) were used.
AdBippyPhos was used instead of KPhos. Five equiv aq NH3 was used.
Pd-precatalyst (25.0 μmol) and KPhos (100 μmol) were used.
1.2 equiv aq NH3 was used.
2,6-Diaminopyridine was formed in 18% yield. lReactions for 48 h.
Aryl halides derived from carbonyl derivatives, such as alkyl aryl ketones, a diaryl ketone, and an N,N-disubstituted benzamide, converted to the corresponding primary arylamines (2j−m) in good yields without side reactions, such as hydration, hydrolysis, or condensation. However, methyl and tert-butyl 4-chlorobenzoates underwent saponification under the reaction conditions, yielding the corresponding 4-amino- and 4-chloro-benzoic acids. An aryl chloride bearing a secondary benzylic alcohol underwent the catalytic amination, providing 2n without the formation of aryl alkyl ether or 4-aminoacetophenone that would arise from reaction of an arylpalladium(II) alkoxo complex. It is remarkable that 4-chloroaniline, which possesses a primary arylamine moiety, afforded 1,4-phenylenediamine 2o as the sole product, underscoring the high selectivity for monoarylation. Reactions involving functional groups on the aryl halide that have been detrimental to previously reported palladium catalyst systems gave the corresponding anilines in good yields (2p−s). Meta-substituted aryl halides and 2-chloronaphthalene participated in the amination reaction to afford primary arylamines 2l and 2v−y.
The reactions of 1-chloronaphthalene or aryl chlorides possessing ortho-substituents proceeded to lower conversion under standard reaction conditions than those lacking ortho-substituents, presumably due to the steric demand of the KPhos ligand from the increased size of the adamantyl substituents at phosphorus and the ortho-substituent of aryl halides caused by the methyl group on the ligand, as illustrated in the ORTEP diagram of OA1 (Figure 3c). However, reactions of such ortho-substituted aryl chlorides with aqueous ammonia conducted with AdBippyPhos in place of KPhos as the ligand formed the corresponding primary arylamines in good yields (2z−aa) with high selectivity for monoarylation that results from the steric properties of the substrate.
A wide range of heteroaryl chlorides and bromides also reacted to afford primary arylamines. Aryl halides containing 5-membered heterocycles such as 1,3-benzodioxole, indole, indazole, pyrrole, benzothiazole, and benzothiophene provided the corresponding products in good yields (2ab−ah). In addition, various aminopyridines (2ai−ak), quinolines (2al−ao), and quinoxaline (2ap) formed under the reaction conditions. The selectivity between the formation of mono- and diarylamine consistently exceeded 20:1 under the reaction conditions for all substrates included in Table 1 by the analysis of the crude reaction mixture by 1H NMR spectroscopy. While the vast majority of substrates tested reacted to form the arylamine product, a few examples of substrates that decomposed or were unreactive are provided in the Supporting Information.
We also investigated the effect of the ratio of ligand to catalyst and Pd(0) source on the yield of primary arylamine. The reaction to form 2ae with a 1:1 ratio of KPhos to Pd-G3 dimer instead of the standard 2:1 ratio formed 2ae in 83% isolated yield. Moreover, product 2ae was isolated in quantitative yield when 0.5 mol % Pd2(dba)3 was used instead of 0.5 mol % Pd-G3 dimer. These results demonstrate the high activity of the catalyst system generated in situ from KPhos and a Pd(0) source.
Investigation of the Reaction Mechanism.
The major phosphine-ligated palladium species during initial rate measurements was determined by 19F and 31P NMR spectroscopy to be the KPhos-ligated arylpalladium(II) hydroxo complex [(KPhos)Pd(Ar)(OH)] (Pd-(Ar)OH, Ar = 4-fluorophenyl) under conditions of the kinetic studies with the soluble base (Figure 4). This species was independently generated by combining (KPhos)Pd(4−F-C6H4)Cl (OA3) and TBA(OH)·30H2O in the presence of excess 1-chloro-4-fluorobenzene (see Supporting Information). We were unable to isolate Pd-(Ar)OH because it reacts to form the corresponding phenol and biaryl products in the absence of ammonia, and attempts to generate Pd-(Ar)OH from OA3 in the absence of the added aryl halide led to nonspecific decomposition and formation of palladium black. The resting state of the catalytic reaction with KOH as base is more complex than that with TBA(OH)·30H2O. We propose an arylpalladium complex lacking a phosphine ligand is the major species, but the lack of reactivity without ligand shows that the small amount of ligated palladium is the active catalyst (see Supporting Information).16
Figure 4.

Resting state of the catalyst under the conditions with TBA(OH) (top) or KOH (bottom) determined by (a) 19F and (b) 31P{1H} NMR spectroscopy, and comparison with [(KPhos)Pd(4−F-C6H4)OH] independently generated in situ (middle).
To reveal the effects of the atypical, monophasic, organic-aqueous solvent, and hydroxide base and the effect of the new structure of the phosphine on the mechanism of this process, we analyzed initial rates of the reactions depicted in Figure 5a with varied concentrations of the reagents, catalysts, and aniline product, and we monitored the evolution of the palladium catalyst by NMR spectroscopy. The initial rates were measured by 19F NMR spectroscopy with TBA(OH) base instead of KOH, so that the reaction is homogeneous, and with OA1 as the catalyst instead of Pd-G3, to minimize the induction period leading to the active catalyst.
Figure 5.

(a) Reaction conditions for the kinetic studies. (b) Dependence of the initial rates on varied concentrations of the reagents, product, or catalyst. (c) Linear free energy relationship between the para-substituents of aryl chloride and the relative initial rates. Hammett plot (Left) and Swain–Lupton plot (Right). (d) Proposed mechanism of the palladium-catalyzed amination of aryl halides with aqueous ammonia and a hydroxide base.
Kinetic measurements on the reaction with TBA(OH)·30H2O as base showed that the reaction with KPhos is zeroth-order in the concentrations of aryl chloride, added ligand, base, and added product aniline (Figure 5b).17 The reaction was first-order in the Pd/KPhos catalyst. Measurements of initial rates with varied concentrations of TBA(OH)·30H2O or aqueous ammonia without maintaining a constant [H2O] led to a partial negative order in the concentration of TBA(OH)·30H2O and to partial positive and negative orders in ammonia at lower and higher concentrations, respectively. These results implied that the amount of water affects the rate, and qualitative studies showed that additional water inhibits the reaction.18 Thus, we also measured the initial rates with varied concentrations of TBA(OH)·30H2O or aqueous ammonia at a constant concentration of water. Under these conditions, the initial rates as a function of ammonia concentration were first-order in the concentrations of ammonia up to 0.31 M, which is the concentration of the synthetic studies.19,20
The zeroth-order dependence of the initial rate on the concentration of ligand for reactions with TBA(OH)·30H2O suggests that the displacement by ammonia is effectively irreversible during these rate measurements, but the accumulation of primary amine selectively at long reaction times implies that a small concentration of phosphine-ligated species catalyzes the amination once the system reaches steady state. Consistent with this assertion, the reaction of 1a and aqueous ammonia with KOH as base catalyzed by a 1:1 ratio of KPhos/OA1 converted 43% of 1a to the corresponding monoarylamine 2a at 90 min, but the reaction catalyzed by a 3:1 ratio of KPhos/OA1 quantitatively converted 1a to 2a after the same 90 min reaction time.
To determine if the proton transfer step could be rate limiting, the kinetic isotope effect was measured for reactions with the combination of NH4·OH and KOH and the combination of ND4·OD and KOD. The two reactions in parallel under the standard conditions gave a KIE of 1.03 ± 0.16 (Scheme 1). This small value indicates that the proton transfer step is not turnover-limiting. However, this value is consistent with a potential equilibrium between Pd-(Ar)OH and the arylpalladium(II) amido complex [(KPhos)Pd(Ar)-(NH2)] (Pd-(Ar)NH2) prior to turnover-limiting reductive elimination, particularly because of the similarity in νN−H and νO−H values.
Scheme 1.

Kinetic Isotope Effect of 4-Chlorobenzotrifluoride from Reaction in Separate Vessels
To gain information on whether reductive elimination could be rate limiting, a linear free energy relationship between the initial rates of amination and para-substituents on the aryl chloride was investigated (Figure 5c).21 A Hammett plot of the initial rate versus σp was nonlinear with R2 = 0.77. A Swain−Lupton plot with σp values created by varying the field and resonance contributions of the substituents was linear (R2 = 0.97) with the optimized field and resonance parameters of σp = 0.37F + 0.63R−.22 The positive ρ value (+2.48) of the optimized Swain−Lupton plot and larger resonance than the field effect is consistent with values from prior studies varying substituents on the palladium-bound aryl group on reductive eliminations to form carbon−nitrogen bonds and, thereby, further supports turnover-limiting reductive elimination. Thus, our kinetic data are consistent with turnover-limiting reductive elimination of the primary arylamine from the parent arylpalladium(II) amido complex generated by the reversible reaction of ammonia with the arylpalladium hydroxo resting state.
To understand the origin of selectivity for the formation of aniline, we conducted density functional theory (DFT) calculations of putative reaction intermediates. However, the reaction conditions that include hydrated dioxane as solvent with dissolved NH3, varying ionicity from dissolved KOH and KCl, and need for explicit binding of solvent to the K+ and hydrogen bonding to the OH and NH2 ligands and anions, precluded accurate absolute energies. Yet, our calculations did offer some insight into the experimental selectivity.
The calculations were conducted with dioxane and water as solvents using the SMD model to assess differences in energies resulting from differences in the bulk solvent dielectric constants. These calculations were consistent with our experimental finding that Pd-(Ar′)OH (with Ar′ = Ph) is the resting state of the catalyst and that reaction of this species with ammonia to generate the reactive complex Pd-(Ar′)NH2 lies uphill (by approximately 4.3 kcal/mol in dioxane and 6.2 kcal/mol in water). These thermodynamic differences in energy corroborate the observation of Pd-(Ar)OH as the resting state and account for the lack of observation of Pd-(Ar)NH2 prior to turnover-limiting reductive elimination.
The computed barriers to reductive elimination to form the C−N bond in the aniline product (ΔGAniline‡) from Pd-(Ar′)NH2 without any explicit hydrogen bonding of the medium to the amido group were 10.8 kcal/mol in dioxane and 10.9 kcal/mol in water. The combination of these barriers and the 4.3 or 6.2 kcal/mol endothermicity for formation of Pd-(Ar′)NH2 from the resting state are lower than the values that would correspond to the experimentally observed initial rates for formation of 4-fluoroaniline of 9.3 × 10−5 M/s at 73 °C (ΔG‡ = ca. 27 kcal/mol). However, this computed barrier is lower in free energy than that computed for the formation of the C−O bond in the corresponding phenol by approximately 1.8 kcal/mol in dioxane. These relative energies of the transition states are consistent with the high selectivity for formation of aniline over the phenol.
Finally, the computed free energy of the arylpalladium(II) anilido complex Pd-(Ar′)NHPh and free ammonia was approximately 7.6 kcal/mol lower than that of the Pd-(Ar′)NH2 complex and free aniline. In addition, the computed barrier to reductive elimination to form diarylamine from Pd-(Ar′)NHPh was only about 12 kcal/mol, suggesting that the diarylamine product would readily form under the reaction conditions if the hydroxo or parent amido complex were to react with aniline to form the arylpalladium(II) anilido complex in the catalytic system. Because diarylamine is less than 1% of the product, we hypothesize that the barrier to formation of the arylpalladium(II) anilido complex is kinetically inaccessible from either of these two complexes. Consistent with this assertion and the assumption that the exchange of the hydroxo ligand with an arylamido ligand would be preceded by binding of aniline, the computed free energy for binding of aniline to Pd-(Ar′)OH is 5 kcal/mol higher than binding of ammonia to the free hydroxo complex Pd-(Ar′)OH (see Supporting Information for further discussion on the interpretation and limitations of computational methods).
The kinetic and spectroscopic studies are all consistent with the proposed catalytic cycle for the palladium-catalyzed amination of aryl chlorides with aqueous ammonia and a hydroxide base depicted in Figure 5d. The observation of the first-order dependence on the concentration of the palladium catalyst and the zeroth-order dependence on the concentration of aryl chloride, excess ligand, and base without the product inhibition supports turnover-limiting reductive elimination of the primary arylamine. The positive slope (ρ = +2.48) of the linear free energy relationship with the optimized Swain–Lupton parameters is further consistent with the proposed mechanism. The inhibitory effects of water on the initial rate, the first-order dependence on lower concentration of ammonia, and the KIE of 1.03 ± 0.16 imply that the arylpalladium(II) amido complex equilibrates with an arylpalladium(II) hydroxo complex and that reductive elimination of aniline is turnover-limiting. These conclusions are consistent with the arylpalladium(II) hydroxo complex being the sole or major resting state of the catalyst depending on the concentration of ammonia.
The lack of effect of added aniline on the rate or selectivity for monoarylamine product, together with DFT calculations indicating that reductive elimination to form the diarylamine product would occur with a barrier lower than that for reductive elimination to form the monoarylamine product, suggests that arylpalladium(II) anilido complexes Pd-(Ar)-NHPh do not form in the catalytic system.
This mechanism can be compared to that of a prior system with anhydrous ammonia and dimethoxyethane (DME) or dioxane solvent, NaOtBu as base, and the large, chelating bisphosphine ligand CyPFtBu.3a,c,23 The resting state and origin of selectivity of the two systems are clearly distinct and result from the difference in the reaction medium and ligand. Studies on this prior system showed that the arylpalladium(II) amido complex was the resting state, implying that reductive elimination of the primary arylamine was also the turnover-limiting step. However, in this prior system with the hindered alkoxide base NaOtBu, the resting state contained the Pd−N bond to an amido ligand instead of the Pd−O bond to the alkoxo or hydroxo ligand. For the prior system, reductive elimination of aniline was computed to be faster than reductive elimination of primary arylamine from the parent amido complex, so the selectivity to form the primary arylamine resulted from the thermodynamically favored formation of the parent amido complex over the anilido complex due to the steric properties of the bisphosphine. In our current system, the selectivity for primary amine over secondary amine appears to result from the kinetic selectivity for the formation of the parent amido complex. The selectivity for formation of arylamine over phenol must result from the low barrier to reductive elimination that forms the C−N bond, considering that Pd(Ar)NH2 is less thermodynamically stable than the hydroxo complex Pd(Ar)OH and that the hydroxo complex accumulates in the catalytic system.
CONCLUSIONS
We have developed a catalyst for highly selective and broadly applicable palladium-catalyzed amination of aryl chlorides and bromides with aqueous ammonia and a hydroxide base. The selective formation of primary arylamines was achieved by developing KPhos, a ligand that contains a methyl substituent on the AdBippyPhos ligand. This small modification to C4 of the bipyrazole adjacent to the C−P bond dramatically improved the selectivity for monoarylation over diarylation, and the palladium catalyst containing bipyrazole ligands are selective for amination over hydroxylation with aqueous ammonia. The reaction occurred with a broad range of aryl halides to provide the primary arylamines with at least 20:1 selectivity for the formation of the monoarylamine over the diarylamine. Mechanistic investigations under the reaction conditions with a soluble hydroxide base or KOH base have demonstrated that reductive elimination from the arylpalladium(II) amido complex, which is in equilibrium with the resting arylpalladium(II) hydroxo complex, is turnover-limiting. Studies exploring the applicability of the KPhos ligand for other catalytic cross-coupling reactions, in which nucleophiles may be delivered in aqueous solution and a hydroxide base may be used, are being conducted in our laboratory.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Drs. Hasan Celik, Raynald Giovine, and Pines Magnetic Resonance Center’s Core NMR Facility (PMRC Core) for spectroscopic assistance. The instrument used in this work is in part supported by NIH S10OD024998. The authors thank Isaac F. Yu for acquiring and solving single-crystal XRD data and Dr. Kyan A. D’Angelo, Isaac F. Yu, and Christina N. Pierson for discussions during the preparation of the paper. K.C. thanks Drs. Hasan Celik and Alicia Lund for kinetic experiments by NMR spectroscopy and Elena Kreimer for ICP-MS experiments.
Funding
The discovery of the ligand and catalyst system was funded by BASF under the California Research Alliance (CARA) program and the scope and mechanistic studies were supported by National Institutes of Health (NIH) under grant R35GM130387.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c05768.
Experimental procedures, characterization of compounds, and miscellaneous data (PDF)
Accession Codes
CCDC 2307394 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif or by emailing data_request@ccdc.cam.ac.uk or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.4c05768
The authors declare the following competing financial interest(s): A provisional patent based on this work has been filed.
Contributor Information
Kyoungmin Choi, Department of Chemistry, University of California, Berkeley, California 94720, United States; Present Address: Therapeutics & Biotechnology Division, Korea Research Institute of Chemical Technology, Daejeon 34114, Republic of Korea.
John N. Brunn, Department of Chemistry, University of California, Berkeley, California 94720, United States.
Kailaskumar Borate, BASF Chemicals India Pvt. Ltd., Navi Mumbai 400705, India.
Rahul Kaduskar, BASF Chemicals India Pvt. Ltd., Navi Mumbai 400705, India.
Carlos Lizandara Pueyo, BASF Corporation, Berkeley, California 94720, United States.
Harish Shinde, BASF Chemicals India Pvt. Ltd., Navi Mumbai 400705, India.
Roland Goetz, BASF SE, Ludwigshafen 67056, Germany.
John F. Hartwig, Department of Chemistry, University of California, Berkeley, California 94720, United States
REFERENCES
- (1).(a) Ruiz-Castillo P; Buchwald SL Applications of Palladium-Catalyzed C−N Cross-Coupling Reactions. Chem. Rev 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Devendar P; Qu R-Y; Kang W-M; He B; Yang G-F Palladium-Catalyzed Cross-Coupling Reactions: A Powerful Tool for the Synthesis of Agrochemicals. J. Agric. Food Chem 2018, 66, 8914–8934. [DOI] [PubMed] [Google Scholar]; (c) Lee BS; Park JG; Yoo JD; Jang JW; Lee YS; Mun SY; Lee IG; Baek HJ Compound for Organic Electronic Element, Organic Electronic Element Using the Same, and an Electronic Device Thereof U.S. Patent US2023/0118527 A1, 2023.; (d) Kim M; Suh SD; Lee SJ; Jeon HS; Hong SK Compound and Organic Light-Emitting Element Comprising Same U.S. Patent WO2024/005486 A1, 2024.; (e) Chung YS; Choi E; An E; Ahn H; Kang G; Kwon E; Lee Y; Jang D; Cho S; Jung Y; Kim HS; Choi H; Choi B Heterocyclic Compound, Organic Light-Emitting Device Including the Same, And Electronic Apparatus Including the Organic Light-Emitting Device US2024/0010636 A1, 2024.
- (2).(a) Dorel R; Grugel CP; Haydl AM The Buchwald–Hartwig Amination After 25 Years. Angew. Chem., Int. Ed 2019, 58, 17118–17129. [DOI] [PubMed] [Google Scholar]; (b) Beletskaya IP; Averin AD Metal-catalyzed reactions for the C(sp2)−N bond formation: achievements of recent years. Russ. Chem. Rev 2021, 90, 1359–1396. [Google Scholar]
- (3).(a) Shen Q; Hartwig JF Palladium-Catalyzed Coupling of Ammonia and Lithium Amide with Aryl Halides. J. Am. Chem. Soc 2006, 128, 10028–10029. [DOI] [PubMed] [Google Scholar]; (b) Surry DS; Buchwald SL Selective Palladium-Catalyzed Arylation of Ammonia: Synthesis of Anilines as Well as Symmetrical and Unsymmetrical Di- and Triarylamines. J. Am. Chem. Soc 2007, 129, 10354–10355. [DOI] [PubMed] [Google Scholar]; (c) Vo GD; Hartwig JF Palladium-Catalyzed Coupling of Ammonia with Aryl Chlorides, Bromides, Iodides, and Sulfonates: A General Method for the Preparation of Primary Arylamines. J. Am. Chem. Soc 2009, 131, 11049–11061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schulz T; Torborg C; Enthaler S; Schäffner B; Dumrath A; Spannenberg A; Neumann H; Börner A; Beller M A General Palladium-Catalyzed Amination of Aryl Halides with Ammonia. Chem. - Eur. J 2009, 15, 4528–4533. [DOI] [PubMed] [Google Scholar]; (e) Lundgren RJ; Peters BD; Alsabeh PG; Stradiotto M A P,N-Ligand for Palladium-Catalyzed Ammonia Arylation: Coupling of Deactivated Aryl Chlorides, Chemoselective Arylations, and Room Temperature Reactions. Angew. Chem., Int. Ed 2010, 49, 4071–4074. [DOI] [PubMed] [Google Scholar]; (f) Crawford SM; Lavery CB; Stradiotto M BippyPhos: A Single Ligand With Unprecedented Scope in the Buchwald–Hartwig Amination of (Hetero)aryl Chlorides. Chem. - Eur. J 2013, 19, 16760–16771. [DOI] [PubMed] [Google Scholar]; (g) Cheung CW; Surry DS; Buchwald SL Mild and Highly Selective Palladium-Catalyzed Monoarylation of Ammonia Enabled by the Use of Bulky Biarylphosphine Ligands and Palladacycle Precatalysts. Org. Lett 2013, 15, 3734–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Lombardi C; Day J; Chandrasoma N; Mitchell D; Rodriguez MJ; Framer JL; Organ MG Selective Cross-Coupling of (Hetero)aryl Halides with Ammonia To Produce Primary Arylamines using Pd-NHC Complexes. Organometallics 2017, 36, 251–254. [Google Scholar]; (i) Green RA; Hartwig JF Palladium-Catalyzed Amination of Aryl Chlorides and Bromides with Ammonium Salts. Org. Lett 2014, 16, 4388–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Green RA; Hartwig JF Nickel-Catalyzed Amination of Aryl Chlorides with Ammonia or Ammonium Salts. Angew. Chem., Int. Ed 2015, 54, 3768–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Borzenko A; Rotta-Loria NL; MacQueen PM; Lavoie CM; McDonald R; Stradiotto M Nickel-Catalyzed Monoarylation of Ammonia. Angew. Chem., Int. Ed 2015, 54, 3773–3777. [DOI] [PubMed] [Google Scholar]; (l) Lavoie CM; MacQueen PM; Rotta-Loria NL; Sawatzky RS; Borzenko A; Chisholm AJ; Hargreaves BKV; McDonald R; Ferguson MJ; Stradiotto M Challenging nickel-catalysed amine arylations enabled by tailoredancillary ligand design. Nat. Commun 2016, 7, No. 11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Enthaler S Ammonia: An Environmentally Friendly Nitrogen Source for Primary Aniline Synthesis. ChemSusChem 2010, 3, 1024–1029. [DOI] [PubMed] [Google Scholar]; (b) Klinkenberg JL; Hartwig JF Catalytic Organometallic Reactions of Ammonia. Angew. Chem., Int. Ed 2011, 50, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hartwig JF; Shaughnessy KH; Shekhar S; Green RA Palladium-Catalyzed Amination of Aryl Halides. In Organic Reactions; John Wiley & Sons, 2020; pp 853–958. [Google Scholar]; (d) Hartwig JF Evolution of a Fourth Generation Catalyst for the Amination and Thioetherification of Aryl Halides. Acc. Chem. Res 2008, 41, 1534–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Schranck J; Tlili A Transition-Metal-Catalyzed Monoarylation of Ammonia. ACS Catal 2018, 8, 405–418. [Google Scholar]
- (5).Naghipour, A. et al. reported that Pd(PPh3)2Cl2 catalyzes amination of aryl iodides and bromides in aqueous ammonia but the catalyst is not working for 1-chloro-4-fluorobenzene 1a with KOH base. For details, see:; Naghipour A; Ghorbani-Choghamarani A; Babaee H; Hashemi M; Notash B Crystal structure of a novel polymorph of trans-dichlorobis(triphenylphosphine) palladium (II) and its application as a novel, efficient and retrievable catalyst for the amination of aryl halides and stille cross-coupling reactions. J. Organomet. Chem 2017, 841, 31–38. [Google Scholar]
- (6).(a) Grushin VV; Alper H Alkali-Induced Disproportionation of Palladium(II) Tertiary Phosphine Complexes, [L2PdCl2], to LO and Palladium(0). Key Intermediates in the Biphasic Carbonylation of ArX Catalyzed by [L2PdCl2]. Organometallics 1993, 12, 1890–1901. [Google Scholar]; (b) Grushin VV Catalysis for Catalysis: Synthesis of Mixed Phosphine–Phosphine Oxide Ligands via Highly Selective, Pd-Catalyzed Monooxidation of Bidentate Phosphines. J. Am. Chem. Soc 1999, 121, 5831–5832. [Google Scholar]; (c) Grushin VV Synthesis of Hemilabile Phosphine–Phosphine Oxide Ligands via the Highly Selective Pd-Catalyzed Mono-oxidation of Bidentate Phosphines: Scope, Limitations, and Mechanism. Organometallics 2001, 20, 3950–3961. [Google Scholar]
- (7).(a) Lang F; Zewge D; Houpis IN; Volante RP Amination of aryl halides using copper catalysis. Tetrahedron Lett 2001, 42, 3251–3254. [Google Scholar]; (b) Kim J; Chang S Ammonium salts as an inexpensive and convenient nitrogen source in the Cu-catalyzed amination of aryl halides at room temperature. Chem. Commun 2008, 3052–3054. [DOI] [PubMed] [Google Scholar]; (c) Xia N; Taillefer M A Very Simple Copper-Catalyzed Synthesis of Anilines by Employing Aqueous Ammonia. Angew. Chem., Int. Ed 2009, 48, 337–339. [DOI] [PubMed] [Google Scholar]; (d) Quan Z; Xia H; Zhang Z; Da Y; Wang X Copper-Catalyzed Amination of Aryl Halides with Aqueous Ammonia under Mild Conditions. Chin. J. Chem 2013, 31, 501–506. [Google Scholar]; (e) Fan M; Zhou W; Jiang Y; Ma D Assembly of Primary (Hetero)Arylamines via CuI/Oxalic Diamide-Catalyzed Coupling of Aryl Chlorides and Ammonia. Org. Lett 2015, 17, 5934–5937. [DOI] [PubMed] [Google Scholar]; (f) Gao J; Bhunia S; Wang K; Gan L; Xia S; Ma D Discovery of N-(Naphthalen-1-yl)-N′-alkyl Oxalamide Ligands Enables Cu-Catalyzed Aryl Amination with High Turnovers. Org. Lett 2017, 19, 2809–2812. [DOI] [PubMed] [Google Scholar]
- (8).Singer RA BippyPhos: A Highly Versatile Ligand for Pd-Catalyzed C−N, C−O and C−C Couplings. Isr. J. Chem 2020, 60, 294–302. [Google Scholar]
- (9).(a) Stradiotto M; Lundgren RJ Application of Sterically Demanding Phosphine Ligands in Palladium-Catalyzed Cross-Coupling leading to C(sp2)−E Bond Formation (E = − NH2, − OH, and − F). In Ligand Design in Metal Chemistry: Reactivity and Catalysis; John Wiley & Sons, 2016; pp 104–133. [Google Scholar]; (b) Kim S-T; Kim S; Baik M-H How bulky ligands control the chemoselectivity of Pd-catalyzed N-arylation of ammonia. Chem. Sci 2020, 11, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For experimental and computational results on preferable coordination of primary or secondary amines to tricoordinate (dialkyl biarylphosphine)palladium oxidative addition complexes, see:; (a) Barder TE; Buchwald SL Insights into Amine Binding to Biaryl Phosphine Palladium Oxidative Addition Complexes and Reductive Elimination from Biaryl Phosphine Arylpalladium Amido Complexes via Density Functional Theory. J. Am. Chem. Soc 2007, 129, 12003–12010. [DOI] [PubMed] [Google Scholar]; (b) Biscoe MR; Barder TE; Buchwald SL Electronic Effects on the Selectivity of Pd-Catalyzed C−N Bond-Forming Reactions Using Biarylphosphine Ligands: The Competitive Roles of Amine Binding and Acidity. Angew. Chem., Int. Ed 2007, 46, 7232–7235. [DOI] [PubMed] [Google Scholar]; (c) For an example of the development of dialkyl biarylphosphine ligands for amination of aryl halides under amine binding hypothesis, see: McCann SD; Reichert EC; Arrechea PL; Buchwald SL Development of an Aryl Amination Catalyst with Broad Scope Guided by Consideration of Catalyst Stability. J. Am. Chem. Soc 2020, 142, 15027–15037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Strotman NA; Soumeillant MC; Zhu K; Markwalter CE; Wei CS; Hsiao Y; Eastgate MD Effects of Multiple Catalyst Deactivation Pathways and Continuous Ligand Recycling on the Kinetics of Pd-Catalyzed C−N Coupling Reactions. J. Org. Chem 2019, 84, 4653–4660. [DOI] [PubMed] [Google Scholar]
- (12). We did not observe any traces of palladacyclobutene complexes in 31P NMR spectra during and after the reactions with AdBippyPhos.
- (13).Withbroe GJ; Singer RA; Sieser JE Streamlined Synthesis of the Bippyphos Family of Ligands and Cross-Coupling Applications. Org. Process Res. Dev 2008, 12, 480–489. [Google Scholar]
- (14). Bipyrazolyl lithium did not undergo phosphinylation reaction to provide KPhos when commercially available di(1-adamantyl) chlorophosphine was used.
- (15). We did not observe any traces of palladacyclopentene complexes in 31P NMR spectra during and after the reactions with KPhos.
- (16). We were unable to determine the major phosphine-ligated palladium complex in the reactions with AdBippyPhos and AdBrettPhos as ligand in place of KPhos.
- (17). We assessed qualitatively the effect of added aniline on the rate and selectivity of the reactions with AdBrettPhos and AdBippyPhos as ligand. When 1 equiv. of exogeneous aniline was added to the reaction of AdBrettPhos, the rate of consumption of aryl chloride was indistinguishable from that without added aniline and the ratio of primary to secondary amine decreased to 1:1.36. When exogeneous aniline was added to the reaction of AdBippyPhos, the rate was higher but the diarylamine became the major product with a ratio of 1:12.7. See Supporting Information for details.
- (18). For details, see Supporting Information.
- (19). At higher concentrations, the rate saturated, and the curve of rate vs [NH3] fit the values Vmax = 6.8 × 10−5 M·s−1, KM = 1.22 M. For details, see Supporting Information.
- (20). Kinetic studies under the standard reaction conditions with KOH did not provide consistent results due to the heterogeneity of the reaction. For the preliminary studies including the quantitative ICP-MS analysis of the palladium in the aqueous phase, see Supporting Information for details.
- (21).Hansch C; Leo A; Taft RW A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev 1991, 91, 165–195. [Google Scholar]
- (22).For examples of using R− instead of R to generate an optimized Swain-Lupton parameter σp in carbon–heteroatom bond-forming reductive elimination reactions, see references:; (a) Driver MS; Hartwig JF Carbon–Nitrogen-Bond-Forming Reductive Elimination of Arylamines from Palladium(II) Phosphine Complexes. J. Am. Chem. Soc 1997, 119, 8232–8245. [Google Scholar]; (b) Mann G; Baranano D; Hartwig JF; Rheingold AL; Guzei IA Carbon–Sulfur Bond-Forming Reductive Elimination Involving sp-, sp2-, and sp3-Hybridized Carbon. Mechanism, Steric Effects, and Electronic Effects on Sulfide Formation. J. Am. Chem. Soc 1998, 120, 9205–9219. [Google Scholar]
- (23).Klinkenberg JL; Hartwig JF Slow Reductive Elimination from Arylpalladium parent Amido Complexes. J. Am. Chem. Soc 2010, 132, 11830–11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.