

Abstract
The Ni-catalyzed oxidation of unactivated alkanes, including the oxidation of polyethylenes, by meta-chloroperbenzoic acid (mCPBA) occur with high turnover numbers under mild conditions, but the mechanism of such transformations has been a subject of debate. Putative, high-valent nickel-oxo or nickel-oxyl intermediates have been proposed to cleave the C–H bond, but several studies on such complexes have not provided strong evidence to support such reactivity toward unactivated C(sp3)–H bonds. We report mechanistic investigations of Ni-catalyzed oxidations of unactivated C–H bonds by mCPBA. The lack of an effect of ligands, the formation of carbon-centered radicals with long lifetimes, and the decomposition of mCPBA in the presence of Ni complexes suggest that the reaction occurs through free alkyl radicals. Selectivity on model substrates and deuterium-labeling experiments imply that the m-chlorobenzoyloxy radical derived from mCPBA cleaves C–H bonds in the alkane to form an alkyl radical, which subsequently reacts with mCPBA to afford the alcohol product and regenerate the aroyloxy radical. This free-radical chain mechanism shows that Ni does not cleave the C(sp3)–H bonds as previously proposed; rather, it catalyzes the decomposition of mCPBA to form the aroyloxy radical.
Graphical Abstract
INTRODUCTION
Oxidation of aliphatic C–H bonds is an important chemical transformation that is widely practiced in nature and in synthetic chemistry. In most living organisms, cytochrome P450 enzymes catalyze the hydroxylation of saturated C–H bonds,1 and, in methanotrophic bacteria, methane monooxygenases (MMOs) catalyze the hydroxylation of methane to produce methanol.2 In synthetic chemistry, the oxidation of feedstock alkanes is a promising way to make valuable products, such as alcohols and ketones, from inexpensive starting materials. However, a high-yielding, selective oxidation of unactivated aliphatic C–H bonds is challenging because the large bond dissociation energies (BDEs) near 100 kcal/mol3,4 and high highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gaps make them inert to many chemical reactions and because the ubiquity of C(sp3)–H bonds in organic molecules makes it necessary to distinguish between similar, but inequivalent sites. In addition, oxidized products, such as alcohols and alkyl halides, are generally more reactive than the starting unfunctionalized alkanes, so overoxidation may occur, leading to undesired byproducts.5
Despite these challenges, many methods have been developed in the past century for the oxidation of aliphatic C–H bonds. Several organic reagents, such as ozone,6 dioxiranes,7,8 and aromatic peracids,9,10 were found to oxidize saturated hydrocarbons with high selectivity for tertiary C–H bonds. The oxidation of aliphatic C–H bonds catalyzed by transition-metal complexes also has been developed extensively. As early as the 1890s, Fenton reported the iron(II)-catalyzed oxidation of tartaric acid by hydrogen peroxide.11 Later, the Gif-Barton system,12 which utilizes iron catalysts and tBuOOH or H2O2 as oxidants, the Pt(II) catalytic system by Shilov and Periana,3,13,14 and the Ru porphyrin system by Groves and others15–17 were developed to achieve the oxidation of alkanes with high turnover numbers (TONs). More recently, white reported a selective oxidation of aliphatic C–H bonds in complex molecules with H2O2 and iron catalysts containing nitrogen-based ligands.18,19
Compared to these methods that involve group 8 transition-metal catalysts, Ni-catalyzed oxidations of alkanes are less explored but have been reported to occur with high TONs. Itoh and others have reported the hydroxylation of cyclohexane and adamantane with mCPBA and nickel catalysts containing tetradentate, nitrogen-based ligands, such as tris(2-pyridylmethyl)amine (TPA).20–22 Our group achieved selective hydroxylation of polyethylenes with mCPBA and Ni catalysts containing phenanthroline-based (Phen-based) ligands.23 This oxidation occurred with loadings of nickel as low as 0.1 mol %, required mild conditions (50 °C under air), and afforded the mono-oxidized product (alcohol rather than ketone or ester) with hundreds of TONs (Figure 1A).
Figure 1.

(A) Ni-Catalyzed hydroxylation of cyclohexane, adamantane, and polyethylenes by mCPBA. (B) Previously proposed pathway for Ni-catalyzed oxidation of aliphatic C–H bonds. (C) This work: free-radical chain mechanism of Ni-catalyzed oxidation of C(sp3)–H bonds by mCPBA.
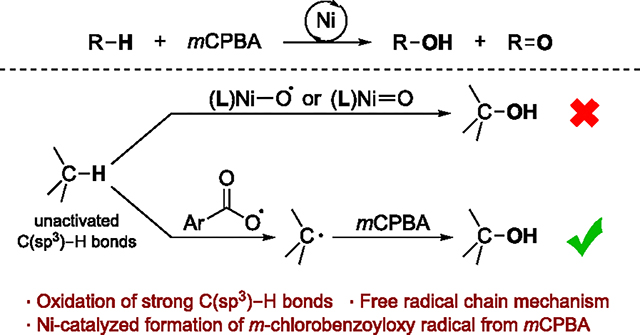
The Ni-catalyzed oxidation of saturated hydrocarbons by mCPBA has been proposed to occur by forming a transient Ni-oxyl or Ni-oxo intermediate24 (hereafter called a Ni-oxygen species) that cleaves the C–H bond (Figure 1B).20,25,26 Hikichi reported the isolation and characterization of a Ni complex of mCPBA containing a trispyrazolylborate-based (Tp-based) ligand.27 Several other Ni-oxygen species containing macrocyclic, nitrogen-based ligands also have been characterized spectroscopically.26,28–34 These nickel intermediates oxidize activated C–H bonds (BDE < 90 kcal/mol) in substrates such as 9,10-dihydroanthracene (DHA) and xanthene,27,28,33 but the reactivity of such intermediates with unactivated C–H bonds (BDE ≈ 100 kcal/mol) has not been convincingly established.35 This trend in reactivity is inconsistent with the results of the catalytic reactions, in which the strong C–H bonds in cyclohexane and polyethylenes undergo hydroxylation under mild conditions. Studies that address this issue of the reactivity of Ni-oxo or Ni-oxyl complexes with C–H bonds and mechanisms of Ni-catalyzed oxidation reactions are crucial to understanding the role of Ni in the catalytic system and to improving the efficiency and selectivity of these reactions further.
In this work, we report mechanistic investigations of the Ni-catalyzed oxidation of unactivated aliphatic C–H bonds. We provide evidence that, contrary to the previously proposed reaction pathways, Ni complexes are not involved in the cleavage of C–H bonds; rather, they catalyze the decomposition of mCPBA to generate an aroyloxy radical that cleaves the C–H bond via hydrogen atom abstraction (HAA). The resulting alkyl radical then reacts with mCPBA to form the hydroxylated product and regenerates the aroyloxy radical. Thus, the catalytic reaction proceeds via a free-radical chain mechanism (Figure 1C) in which the rate of initiation is determined by the concentration and identity of the nickel complex, but the nickel complex does not control the regioselectivity of the oxidations.
RESULTS AND DISCUSSIONS
1. Effect of Ligands on Selectivity.
To understand the role of Ni complexes in the catalytic reaction, we studied the effect of varying the ligands on the selectivity of oxidation. If a transient Ni-oxygen intermediate cleaved the C–H bonds in hydrocarbons, the identity of the ancillary ligands that possess varied electronic and steric properties should affect the selectivity of the catalytic reaction. Therefore, Ni complexes (1a–1h, Figure 2) that contain a series of nitrogen-based, bidentate or tetradentate ligands (L1–L8) were synthesized and used for the catalytic oxidation of cyclohexane (2) and adamantane (3).
Figure 2.

Structures of ligands L1−L8 and Ni catalysts 1a−1h.
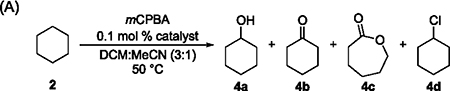
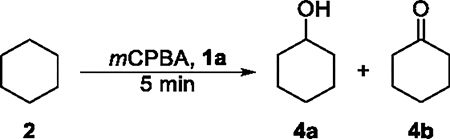
The results of the oxidation of cyclohexane (2) catalyzed by this series of nickel complexes are shown in Table 1A. Cyclohexanol (4a) was formed as the major product together with small amounts of cyclohexanone (4b), ε-caprolactone (4c), and chlorocyclohexane (4d). The formation of 4c can be attributed to the uncatalyzed Baeyer–Villiger oxidation of 4b. The ratio of alcohol to ketone and lactone, that is, A/(K + E), defined as the ratio of the yield of 4a to the combined yields of 4b and 4c, reflects the selectivity for mono-oxidation.
Table 1.
Results of the Oxidation of (A) Cyclohexane and (B) Adamantane in the Presence of Different Catalysts
| |||||||
|---|---|---|---|---|---|---|---|
| entry | catalyst | time (h)a | 4a (%) | 4b (%) | 4c (%) | 4d (%) | A/(K+E) |
|
| |||||||
| 1 | 1a | 1 | 33.8±6.0 | 10.9±0.3 | 0.0±0.0 | 12.8±3.2 | 3.1±0.6 |
| 2 | 1b | 1 | 38.1±2.5 | 10.3±1.0 | 1.1±1.0 | 11.4±2.9 | 3.3±0.6 |
| 3 | 1e | 1 | 39.1±2.1 | 10.4±0.6 | 1.0±1.0 | 12.4±3.0 | 3.4±0.5 |
| 4 | 1d | 1.5 | 31.1±1.7 | 6.3±0.5 | 3.3±0.2 | 8.3±0.8 | 3.2±0.3 |
| 5 | 1e | 1.5 | 32.9±2.3 | 8.3±1.4 | 2.1±1.3 | 13.0±3.8 | 3.2±0.9 |
| 6 | 1f | 2 | 34.6±1.9 | 5.2±0.7 | 4.8±0.7 | 9.5±1.5 | 3.5±0.5 |
| 7 | 1g | 3 | 33.7±2.1 | 4.7±1.4 | 5.4±0.8 | 10.6±0.5 | 3.3±0.7 |
| 8 | 1h | 4b | 10.0±2.3 | 2.3±0.4 | 5.1±0.3 | 5.8±1.1 | -c |
| 9 | NiCl2 | 4 | 30.7±1.0 | 8.3±1.6 | 4.4±1.8 | 9.7±0.8 | 2.4±0.6 |
| 10 | CoCl2 | 1.5 | 24.7±5.1 | 10.7±0.4 | 0.0±0.0 | 9.1±1.0 | 2.3±0.5 |
| 11 | none | 4b | 1.3±2.2 | 0.9±1.5 | 0.6±1.1 | 0.0±0.0 | -c |
| |||||||
| entry | catalyst | time (h)a | 5a (%) | 5b+5c (%) | 5d (%) | 5e (%) | 3°/2° |
|
| |||||||
| 1 | 1a | 1 | 36.7±6.4 | 3.1±0.7 | 2.7±0.4 | 3.9±3.6 | 5.6±3.1 |
| 2 | 1b | 1 | 30.6±3.8 | 2.6±0.2 | 2.8±0.5 | 2.7±2.3 | 6.3±2.8 |
| 3 | 1c | 1 | 30.6±5.7 | 2.4±0.3 | 2.4±0.2 | 2.4±2.1 | 6.9±3.3 |
| 4 | 1d | 1.5 | 24.9±2.1 | 2.5±0.7 | 4.0±0.7 | 4.0±0.6 | 4.4±0.7 |
| 5 | 1e | 1.5 | 30.0±1.2 | 2.5±0.1 | 4.8±0.1 | 4.9±0.6 | 4.7±0.4 |
| 6 | 1f | 2 | 38.5±6.0 | 3.0±0.3 | 3.5±0.9 | 4.1±0.4 | 5.9±0.9 |
| 7 | 1g | 3 | 29.0±1.2 | 2.6±0.5 | 5.2±0.6 | 5.0±0.7 | 4.5±0.5 |
| 8 | 1h | 4b | 20.7±2.1 | 1.4±0.2 | 4.0±0.4 | 2.6±0.3 | 6.2±0.8 |
| 9 | NiCl2 | 4 | 28.2±1.8 | 2.4±0.1 | 4.4±0.3 | 3.5±0.1 | 5.5±0.3 |
| 10 | CoCl2 | 1.5 | 27.9±0.7 | 2.8±0.1 | 2.0±0.5 | 2.1±1.8 | 6.1±2.3 |
| 11 | none | 4b | 8.8±5.0 | 0.6±1.1 | 0.4±0.7 | 0.2±0.3 | -c |
| 12 | 1a d | 1 | 6.5 | 5.0 | 53.8 | 13.4 | |
Conditions: 0.125 mmol mCPBA, 7.5 equiv of 2 or 2 equiv of 3, 0.1 mol % catalyst, DCM/MeCN (3:1, 0.125 M), 50 °C under air. All yields were based on mCPBA and reported as the arithmetic mean of three repeated experiments.
Time required to reach greater than 90% conversion of mCPBA unless noted otherwise.
Low conversion of mCPBA even after 4 h of reaction.
Conversion too low to determine.
In the presence of CCI4 (1 equiv).
Our data show that the identity of the ligand affects the rate of the oxidation process. Reactions in the presence of complexes 1a–1e, which contain sterically unhindered ligands L1–L5, reached full conversion of mCPBA within 1–1.5 h (Table 1A, entries 1–5), whereas those in the presence of 1f and 1g, which contain sterically encumbered ligands L6 and L7, required 2 and 3 h to reach greater than 90% conversion, respectively (Table 1A, entries 6 and 7). The conversion of the reaction with complex 1h, which contains the most sterically hindered ligand L8, in which two methyl groups flank the phenanthroline moiety, was low even after 4 h of heating (Table 1A, entry 8). Time courses further confirmed that the rates of the reactions in the presence of complex 1a were significantly higher than those in the presence of Ni complexes containing sterically hindered ligands, such as 1h (see the Supporting Information).
However, the data in Table 1 also show the lack of a pronounced effect of the ligand on the yield of oxidized products or on the A/(K + E) ratio. All reactions in the presence of complexes 1a–1g gave product 4a in 31%–39% yield and products 4b, 4c in 10%–11% combined yield; the A/(K + E) ratios of these reactions range from 3.1 to 3.5, which is well within experimental error. The oxidation of cyclohexane in the presence of ligandless NiCl2 (Table 1A, entry 9) formed product 4a in 31% yield and products 4b and 4c in 13% combined yield, corresponding to an A/(K + E) ratio of 2.4 ± 0.6. This ratio is comparable to those of reactions catalyzed by ligated Ni complexes 1a–1g. Similar results were observed for the oxidation of 2 catalyzed by CoCl2, although the cobalt-catalyzed reaction was faster than the nickel-catalyzed one and reached full conversion within 1.5 h (Table 1A, entry 10). The negligible effect of ligands on both product distribution and selectivity for mono-oxidation, along with the similar yield of products and selectivity of the catalytic reactions conducted without ligands, is inconsistent with the previously proposed pathway in which a Ni-oxygen species cleaves the C–H bond.
Although the chemoselectivity for the formation of alcohol, ketone, and lactone products 4a–4c provided preliminary evidence that a ligated nickel complex is not involved in the cleavage of C–H bonds, the ketone and ester are likely to be secondary products. Therefore, the distribution of these products could be envisioned to depend on several factors besides the identity of the species that cleaves the C–H bond. A more rigorous test would involve the measurement of the initial products. In addition, measuring the selectivity for tertiary versus secondary (or primary) C–H bonds in a model alkane is important to identify the species that cleaves the C–H bond and to understand the mechanism of Ni-catalyzed oxidations of hydrocarbons. Thus, we also measured the regioselectivity for the oxidation of adamantane.
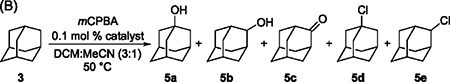
The results of the oxidations of adamantane (3) catalyzed by a series of nickel complexes are shown in Table 1B. 1-Adamantanol (5a) was the major product; 2-adamantanol (5b), 2-adamantanone (5c), and 1- and 2-chloroadamantane (5d and 5e) were the minor products. Like the reactions of cyclohexane, the rates of the reactions of adamantane depended on the ligand bound to nickel. Reactions catalyzed by Ni complexes 1a–1e reached greater than 90% conversion of mCPBA within 1–1.5 h (Table 1B, entries 1–5), whereas those catalyzed by 1f and 1g, which contain sterically encumbered ligands L6 and L7, required 2 and 3 h to reach similar levels of conversion (Table 1B, entries 6 and 7). The reaction in the presence of complex 1h, which contains the most sterically hindered ligand L8, did not reach 90% conversion, even after 4 h of heating (Table 1B, entry 8).
Despite the different rates of reactions catalyzed by these complexes, all of the reactions catalyzed by complexes 1a–1g furnished product 5a in 24.9%–38.5% yield, the combination of products 5b and 5c in 2.4%–3.1% yield, product 5d in 2.4%–5.2% yield, and product 5e in 2.4%–5.0% yield. The ratio of the combination of products from functionalization at the tertiary positions (5a and 5d) to the combination of those at the secondary position (5b, 5c, and 5e), which is denoted 3°/2°, reflects the regioselectivity of this reaction. These 3°/2° ratios were between 4.4 and 6.9 and well within the experimental error of each other. In addition, the oxidation of adamantane catalyzed by ligandless NiCl2 and CoCl2 furnished the combination of products 5a and 5d in 32.6% and 29.9% yield and the combination of products 5b, 5c and 5e in 5.9% and 4.9% yield, corresponding to 3°/2° ratios of 5.5 and 6.1, respectively, which are indistinguishable from those of reactions catalyzed by the ligated Ni complexes 1a–1g. The lack of dependence of the product distribution and regioselectivity of the oxidation of adamantane on this series of Ni complexes containing different ligands and on ligandless metal chlorides, again, is inconsistent with the hypothesis that a Ni-oxygen species cleaves unactivated C–H bonds in alkanes.
2. Formation of Carbon-Centered Radicals with Long Lifetimes.
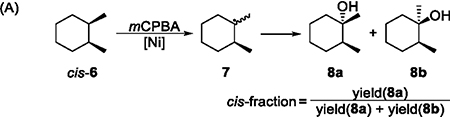
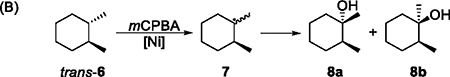
Determining whether carbon-centered radicals are formed and, if so, estimating the lifetime of these radicals is crucial to understanding the mechanism of oxidations of C–H bonds. The formation of chlorinated products 5d and 5e in high yields in a Ni-catalyzed oxidation of adamantane with 1 equiv of CCl4 (Table 1B, entry 12) strongly suggests the formation of carbon-centered radicals and subsequent trapping by CCl4. Substrates that react as radical clocks are often employed to study the lifetime of carbon-centered radicals and the rate of the formation of C–O bonds during the oxidation of C–H bonds.36 For example, several radical clocks based on cyclopropanes were used to study the mechanism of the hydroxylation of saturated C–H bonds catalyzed by P450 enzymes.1 In this work, we used the well-known cis- and trans-1,2-dimethylcyclohexane (cis- and trans-6) as radical clocks37 to investigate the lifetime of carbon-centered radicals formed in the Ni-catalyzed oxidation reaction.
The Ni-catalyzed oxidation of cis-6, which occurred predominantly at the tertiary position, generated the tertiary alkyl radical 7, which is known to epimerize rapidly (first-order rate constant k ≈ 109 s−1),36 and afforded tertiary alcohols 8a and 8b as major products (Table 2A). The fraction of the cis product (ratio of the yield of 8a to the combined yield of 8a and 8b) reflects the rate of the epimerization of 7 versus that of the formation of a C–O bond. If the formation of the C–O bond occurs much faster than the epimerization, then the cis product 8a would be formed predominantly. When the step that forms the C–O bond occurs more slowly than the epimerization of 7, the products 8a and 8b have been observed in similar amounts, corresponding to a cis-fraction close to 50%.36,38 As shown in Table 2A, the oxidation of cis-6 in the presence of a series of Ni complexes, including the ligandless NiCl2, formed the tertiary alcohols with a cis-fraction ranging from 42.6% to 48.9%. These results indicate that the lifetime of the carbon-centered radical formed in these reactions is long enough that complete epimerization occurs before the formation of the C–O bond.
Table 2.
Results of the Ni-Catalyzed Oxidationa of (A) cis-6 and (B) trans-6
| ||||
|---|---|---|---|---|
| Entry | [Ni] | Yield of 8a (%) | Yield of 8b (%) | cis-fraction (%) |
|
| ||||
| 1 | 1a | 22.9±5.4 | 30.7±6.8 | 42.6±0.6 |
| 2 | 1b | 20.3±2.7 | 26.2±3.5 | 43.7±0.5 |
| 3 | 1e | 17.8±1.9 | 23.5±2.5 | 43.1±0.2 |
| 4 | 1e | 17.9±2.1 | 18.8±3.0 | 48.9±1.8 |
| 5 | 1f | 23.8±4.1 | 26.8±2.4 | 46.8±2.3 |
| 6 | 1g | 17.5±4.1 | 19.9±6.5 | 47.3±2.5 |
| 7 | NiCl2 | 22.7±2.5 | 24.6±1.5 | 47.9±3.4 |
| ||||
| Entry | [Ni] | Yield of 8a (%) | Yield of 8b (%) | cis-fraction (%) |
|
| ||||
| 1 | 1a | 11.4±0.9 | 16.0±1.2 | 41.7±0.1 |
| 2 | 1b | 11.7±1.4 | 16.5±2.2 | 41.6±0.3 |
| 3 | 1e | 11.2±1.6 | 15.8±2.6 | 41.4±0.4 |
| 4 | 1e | 8.2±1.0 | 14.0±1.8 | 36.9±0.2 |
| 5 | 1f | 10.2±0.7 | 15.8±1.0 | 39.3±0.1 |
| 6 | 1g | 5.9±2.7 | 10.0±4.6 | 37.2±0.3 |
| 7 | NiCl2 | 8.1±3.3 | 12.8±5.5 | 39.1±1.0 |
Conditions: 0.125 mmol of mCPBA, 7.5 equiv of cis- or trans-6, 0.1 mol % [Ni], DCM/MeCN (3:1, 0.125 M), 50 °C under air, 1–4 h.
All yields were based on mCPBA and reported as the arithmetic mean of three repeated experiments.
The cis fraction from the Ni-catalyzed oxidation of trans-6, summarized in Table 2B, ranged from 36.9% to 41.7%. The lower fraction of the cis product from reactions catalyzed by 1e–1g and NiCl2, which are slower than the reactions catalyzed by 1a–1c (Table 2B, entries 4–7), is likely due to a competing background, uncatalyzed hydroxylation by mCPBA. Uncatalyzed oxidation of alkanes by mCPBA is known to occur with retention of configuration.9 These results are also consistent with the Ni-catalyzed reactions occuring through a long-lived alkyl radical before the step that forms the C–O bond.
3. Ni-Catalyzed Decomposition of mCPBA and a Callback to Fenton Chemistry.
Our attempts to isolate Ni-oxygen intermediates or Ni-mCPBA adducts ligated by phenanthrolines have simply led to the decomposition of mCPBA. In the absence of alkanes, mCPBA was consumed completely to form meta-chlorobenzoic acid (mCBA) in 72% yield and chlorobenzene (9) in 15% yield in the presence of Ni complex 1a under standard conditions (Figure 3A). Chlorobenzene, which was formed in ~51% yield in the catalytic oxidation of cyclohexane (Figure 3B), would arise from the decarboxylation of the meta-chlorobenzoyloxy radical (10). Moreover, our data show that the rate of decomposition of mCPBA is dependent on the identity of the ligand in the Ni catalyst. Time courses indicate that the rate of decomposition in the presence of complex 1a is significantly higher than that in the presence of complex 1h, which contains sterically hindered ligands (see the Supporting Information).
Figure 3.

(A) Results of Ni-catalyzed decomposition of mCPBA in the absence of alkanes. (B) Formation of chlorobenzene in Ni-catalyzed oxidation of 2 by mCPBA.
This Ni-catalyzed decomposition of an aromatic peracid that involves free-radical intermediates is reminiscent of the classic Fenton chemistry (Scheme 1),39,40 in which an iron(II) cation catalyzes the decomposition of hydrogen peroxide to generate the hydroxyl radical (11) (eq 1–1). If an alkane is present in the reaction system, the highly reactive radical 11 abstracts a hydrogen atom from a C–H bond to form water and an alkyl radical, which can subsequently form alcohol (eq 1–2). If no alkane is present, H2O2 will decompose to release O2 and water through either a radical chain (eqs 1–3 and 1–4) or a nonchain pathway (eqs 1–1, 1–3, and 1–5). The decomposition of mCPBA in the presence of Ni complexes, the lack of an effect of the ligands on the selectivity of catalytic reactions, and the formation of long-lived carbon-centered radicals led us to propose an alternative reaction pathway in which the Ni species does not cleave the C–H bond; instead, it reacts with mCPBA to generate free radicals that abstract a hydrogen atom from the C(sp3)–H bond. However, because the nickel-catalyzed reaction occurs with mCPBA instead of H2O2, the identity of the species that cleaves the C–H bond in such a scenario was unclear.
Scheme 1. Mechanism of Fenton Reactions of H2O2.

4. Identity of the Free Radical Responsible for C–H Bond Cleavage.
To identify the radical that cleaves the C–H bond during the reactions with mCPBA containing catalytic amounts of nickel complex, we conducted the reactions of mCPBA and Ni complexes with deuterium-labeled cyclohexane (2-d12) and adamantane (3) and compared the selectivity of these reactions to the known selectivities for the reactions involving abstraction of the hydrogen atom in a C–H bond by various radicals. Figure 4A shows four mCPBA-derived free radicals that might be formed in the reaction system containing nickel: the 3-chlorobenzoyloxy radical (10), the hydroxyl radical (11), the 3-chlorophenyl radical (12), which can be formed by decarboxylation of 10, and the 3-chlorobenzoylperoxy radical (13).
Figure 4.

(A) Free radicals derived from mCPBA that might cleave C–H bonds in the catalytic reaction. (B) Tertiary to secondary selectivity of free radicals for the oxidation of 3. (C) Photomediated formation of benzoylperoxy radical from benzil under an atmosphere of O2. (D) Results of Ni-catalyzed oxidation of 2-d12.
The selectivity of hydroxyl radical (11) for the oxidation of adamantane is low. The 3°/2° ratios from oxidations of 3 that involve cleavage of C–H bonds by radical 11 range from 0.4 to 1.3 (Figure 4B, entries 1–3).41 These values are much smaller than those of Ni-catalyzed oxidation of 3 by mCPBA, which range from 4.4 to 6.9. Thus, hydroxyl radical 11 is unlikely to be the species that cleaves the C–H bond in this reaction.
If 3-chlorophenyl radical (12) were the major species that cleaves the C–H bond in Ni-catalyzed oxidations of alkanes, then the oxidation of deuterated cyclohexane (2-d12) would afford deuterated cyclohexanol (4a-d11) and deuterated cyclohexanone (4b-d10) as the major products, along with a stoichiometric amount of deuterated chlorobenzene (9-d1). However, 2H NMR spectra of the reaction of 2-d12 with mCPBA and a catalytic amount of Ni complex 1a showed that this reaction formed 4a-d11 and 4b-d10 in 26% and 8% yields, respectively, but produced 9-d1 in only 2% yield (Figure 4D). This result indicates that aryl radical 12 cannot be the major intermediate that cleaves C–H bonds in hydrocarbons. The large amount of byproduct 9 observed in the catalytic oxidations of 2 and 3 (Figure 3B) was most likely formed by hydrogen atom transfer (HAT) from solvent molecules, such as dichloromethane (DCM), to 12. This hypothesis is supported by the detection of 1,1,2,2-tetrachloroethane in the reaction mixture, which is the homocoupling product of dichloromethyl radicals (CHCl2·) that would form by hydrogen atom transfer from DCM to 12. We also conducted the Ni-catalyzed oxidation of cyclohexane by mCPBA with DCM-d2 as the solvent and observed that 87% of the chlorobenzene that formed contained the deuterium label (see the Supporting Information). These results further support our hypothesis that chlorobenzene (9) is formed primarily by hydrogen atom transfer from solvent molecules to radical 12.
The benzoylperoxy radical (13′) can be generated by the photomediated homolysis of benzil under an atmosphere of O2 (Figure 4C).42,43 To measure the selectivity of radical 13′ for the oxidation of adamantane (3), we conducted the photochemical oxidation of 3 with benzil and O2 in 1,2-dichloroethane (DCE). The 3°/2° ratios derived from the products of these reactions range from 11.2 to 12.1 (Figure 4B, entry 4), which is higher than those from Ni-catalyzed oxidations of 3 conducted in the same solvent (Figure 4B, entry 5). Given the similar structures of radicals 13 and 13′, it is likely that the 3°/2° selectivity for the oxidation of 3 by these two radicals is similar. Therefore, the 3-chlorobenzoylperoxy radical (13) is unlikely to be the species that cleaves C–H bonds in Ni-catalyzed oxidations of alkanes.
Thus, the aroyloxy radical 10 is the most likely radical to cleave the C–H bond. The BDE of the O–H bond in mCBA was reported to be 107.3 kcal/mol,4 which is higher than those of unactivated C–H bonds in alkanes and makes HAT from alkanes to radical 10 thermodynamically favorable. The secondorder rate constant for the abstraction of a hydrogen atom from 2 by 4-methoxybenzoyloxy radical (14) was measured to be on the order of 105 M−1 s−1, whereas the first-order rate constant for decarboxylation of 14 was measured to be 104–105 s−1.44 Given the similarity between the structures of m-chloro- and p-methoxy-substituted benzoyloxy radicals 10 and 14, it is likely that the rate constants for abstraction of a hydrogen atom from an unactivated C(sp3)–H bond by these two radicals are similar and that the rate of abstraction of a hydrogen atom by 10 would be similar to that of the decarboxylation. This prediction based on the literature values fits with the observed oxidation process and formation of chlorobenzene.
However, reliable data on the selectivity of aroyloxy radical 10 for the abstraction of hydrogen atoms from alkanes were needed to assess more precisely whether this radical cleaves the C–H bonds in Ni-catalyzed oxidations. Thus, we generated radical 10 in situ via the thermal decomposition of bis(3-chlorobenzoyl)-peroxide (15), measured its selectivity for hydrogen-atom abstraction of a model alkane, 2,2,4,4-tetramethylpentane (16), and compared the selectivity of the metal-free bromination of 16 by peroxide 15 to that of Ni-catalyzed reactions (Figure 5). All reactions were conducted in the presence of CBr4, which is well-known to trap carbon-centered radicals rapidly. In this case, the distribution of products 17a and 17b from bromination at the secondary and primary C–H bonds accurately reflects the selectivity in the hydrogen-atom transfer step. The model substrate 16 was chosen because radicals derived from CBr4, such as CBr3·, do not cleave the strong primary and secondary C–H bonds in alkane 16 and, therefore, do not interfere with the measurement of the selectivity of hydrogen-atom abstraction by aroyloxy radical 10. The tertiary C–H bonds in adamantane, however, can be cleaved by these radicals derived from CBr4, so adamantane is not a suitable substrate for this experiment (see the Supporting Information).
Figure 5.

Bromination of alkane 16 by (A) peroxide 15, (B) mCPBA and Ni complex 1a, and (C) peracid 18 and Ni complex 1a. Conditions: (A) 0.0625 mmol of 15, 10 equiv of 16, 4 equiv of CBr4, DCM (0.0625 M), 80 °C under N2; (B) 0.125 mmol of mCPBA, 5 equiv of 16, 2 equiv of CBr4, 0.1 mol % 1a, DCM (0.125 M), 50 °C under N2. (C) 0.125 mmol of 18, 5 equiv of 16, 2 equiv of CBr4, 1 mol % 1a, DCM (0.125 M), 50 °C under N2. aAverage of eight repeated experiments. bAverage of three repeated experiments.
The reaction of peroxide 15 with alkane 16 in the presence of 2 equiv of CBr4 furnished 17a in 10.4% yield and 17b in 5.5% yield, corresponding to a 2°/1° ratio of 1.9 ± 0.4 (Figure 6A). The Ni-catalyzed bromination of 16 by mCPBA formed 17a in 35.2% yield and 17b in 17.9% yield, corresponding to a 2°/1° ratio of 2.0 ± 0.2 (Figure 6B). The similarity between the selectivity of the bromination reactions with diaroyl peroxide 15 and the process with the nickel complex and mCPBA strongly suggests that radical 10 is the species that cleaves the C–H bond in the catalytic system.
Figure 6.

Illustration of the GC experiment for the detection of alkyl hydroperoxide intermediates designed by Shul’pin.
To assess further our proposal that the aroyloxy radical derived from the aromatic peracid is the species that cleaves the C–H bond in the catalytic reaction, we conducted reactions with a different peracid. We envisioned that the selectivity for the Ni-catalyzed bromination of 16 would be different for reactions with aromatic peracids bearing different substituents than the chloride in mCPBA. Indeed, the Ni-catalyzed bromination of 16 by 3,5-bis(trifluoromethyl)perbenzoic acid (18) furnished 17a in 14.7% yield and 17b in 13.4% yield, corresponding to a 2°/1° ratio of 1.1 ± 0.1 (Figure 6C). This ratio is lower than those from reactions involving m-chlorobenzoyloxy radical 10 (Figure 6A,B) and further corroborates our hypothesis that aroyloxy radicals derived from aromatic peracids cleave the C–H bonds of alkanes in the catalytic system.
5. The C–O Bond Formation Step.
Having identified the free radical responsible for C–H bond cleavage, we investigated the step that forms the C–O bond. Three possible pathways can lead to the formation of a C–O bond from the carbon-centered radical generated by hydrogen-atom abstraction of the alkane by aroyloxy radical 10 (Scheme 2): (1) trapping by O2 to generate an alkylperoxy radical, which subsequently decomposes to form alcohol and ketone products (eq 2–1); (2) trapping by a putative nickel(III) hydroxide species, which would be generated by the oxidation of the Ni(II) complex by mCPBA, to form the alcohol product and a Ni(II) species (eq 2–2); (3) trapping by mCPBA to afford the alcohol product and radical 10 (eq 2–3).
Scheme 2. Three Possible Pathways for the Formation of C–O Bonds.

The decomposition of alkylperoxy radicals usually forms the corresponding alcohol and ketone products in an ~1:1 ratio,45 whereas the Ni-catalyzed oxidation of 2 gave A/(K + E) ratios between 3.1 and 3.5 (Table 1A). Furthermore, the yield and selectivity of the catalytic oxidations of 2 and 3 under an atmosphere of N2 were similar to those of the reactions under air. These straightforward observations suggest that the formation of the C–O bond does not involve O2 and that the steps in eq 2–1 are unlikely to be the major ones forming the C–O bond in the Ni-catalyzed oxidations. Nonetheless, we decided to assess more precisely whether trapping of alkyl radicals by O2 occurred and, if so, what percentage of the oxidized products were formed from O2.
To assess this pathway further, we determined the amount of alkylperoxy intermediates in the product mixture that would form by the trapping of the alkyl radical with O2. Shul’pin et al. reported an experiment for this purpose (Figure 6).46,47 Alkylperoxy intermediates, such as cyclohexyl hydroperoxide (19), decompose by gas chromatography (GC) to generate the corresponding alcohol and ketone products 4a and 4b in a ~1:1 ratio. Treatment of a reaction mixture containing alcohol 4a, ketone 4b, and intermediate 19 with an excess of reductants, such as PPh3, reduces 19 to 4a in quantitative yield without affecting the amount of alcohol or ketone formed before the addition of PPh3. Thus, if intermediate 19 is present in a reaction mixture, the ratios of alcohol to ketone (A/K) derived from the GC trace of the mixture before and after the treatment with PPh3 will be different. Moreover, the percentage of 19 in the total amount of oxidized products can be calculated from these ratios.
The A/K ratio derived from GC traces of the mixture from the Ni-catalyzed oxidation of 2 under an atmosphere of N2 after 5 min before the addition of PPh3 was identical to that after the addition of PPh3 (Table 3, entries 1 and 2). This result indicates that intermediate 19 was not formed and that molecular O2 was not generated in situ during the reaction under N2. The A/K ratio derived from GC traces of the reaction conducted under air before the addition of PPh3 was 4.2 ± 0.7, and the ratio after the addition of PPh3 was 6.1 ± 0.8 (Table 3, entries 3 and 4). The small difference between these ratios suggests that some amount of intermediate 19 was formed under air by the pathway shown as eq 2–1 in Scheme 2. However, the percentage of 19 in the total amount of oxidized species (4a + 4b + 19) was estimated to be only 10% based on these two ratios (see the Supporting Information for details of the calculation). Thus, the trapping of alkyl radicals by O2, even under air, was not the major pathway for the formation of the C–O bond in the catalytic reactions.
Table 3.
GC Results of the Ni-Catalyzed Oxidationa of Cyclohexane under N2 and Air
| |||
|---|---|---|---|
| entry | N2/air | treatment with PPh3 | A/K b |
| 1 | N2 | yes | 4.6 ± 0.1 |
| 2 | no | 4.5 ± 0.0 | |
| 3 | air | yes | 6.1 ± 0.8 |
| 4 | no | 4.2 ± 0.7 | |
Conditions: 0.125 mmol mCPBA, 7.5 equiv 2, 0.1 mol % 1a, DCM/MeCN (3:1, 0.125 M), 50 °C, 5 min.
Arithmetic mean of three repeated experiments.
The lack of effect of the ligands on the yield and selectivity of the Ni-catalyzed oxidation of alkanes does not support the potential formation of alcohol by reaction of the alkyl radical with a nickel hydroxide species (Scheme 2, eq 2–2). If the C–O bonds were formed by this species, then the cis fraction of the alcohols formed from the reactions of 1,2-dimethylcyclohexanes would be expected to depend on the electronic and steric properties of the nickel species, which in turn would depend on the identity of the ligand.
Having ruled out the two pathways described above, we considered that the trapping of carbon-centered radicals by mCPBA (Scheme 2, eq 2–3) would be the major pathway for the formation of the C–O bond. Indeed, the reaction of an alkyl radical with an aliphatic peracid by a free-radical chain process has been well-established by many examples of decarboxylative formation of alcohols from aliphatic peracids.48
6. The Free-Radical Chain Mechanism.
Having investigated both the C–H bond-cleaving and the C–O bond-forming steps, we propose a free-radical chain mechanism for the Ni-catalyzed oxidation of unactivated C(sp3)–H bonds by mCPBA (Scheme 3). Contrary to the hypotheses in the literature20,25,26 that Ni-oxygen intermediates cleave C–H bonds, our data suggest that the Ni complex catalyzes the reaction of mCPBA to generate the 3-chlorobenzoyloxy radical (10) among other products (eq 3–1). Equation 3–1 likely consists of several redox reactions, but our data do not allow conclusions about the mechanism of this step. Radical 10 abstracts a hydrogen atom from the alkane to form the mCBA byproduct and a carbon-centered radical (eq 3–2), which then reacts with mCPBA to afford the alcohol product and regenerate radical 10 (eq 3–3), thus propagating the radical chain. As discussed previously, the trapping of alkyl radicals by O2 is a minor side reaction under air (eq 3–4). Decarboxylation of radical 10 to generate the 3-chlorophenyl radical 12, which captures a hydrogen atom from solvent molecules to form the chlorobenzene byproduct (eq 3–5), competes with the reaction of 10 with a C–H bond (eq 3–2).
Scheme 3. Free-Radical Chain Mechanism of Ni-Catalyzed Oxidation of Unactivated C(sp3)–H Bonds.

On the basis of this mechanism, the identity of the Ni complex influences the rate of decomposition of mCPBA and hence the rate of formation of radical 10, but it does not change the selectivity for reaction at one C–H bond over another. This conclusion is consistent with our observation that reactions catalyzed by Ni complexes with more sterically hindered ligands are slower, but the selectivities of these reactions are similar to those catalyzed by complexes with less hindered ligands. This radical chain mechanism, in which the C–H bond is not cleaved by a nickel complex, reconciles the combination of low reactivity of Ni-oxygen species toward strong C(sp3)–H bonds and the well-documented formation of oxidation products in high yield from Ni-catalyzed oxidations occurring at strong aliphatic C–H bonds.
Alexanian and co-workers achieved tunable, selective chlorination of C(sp3)–H bonds via free-radical pathways by modifying the substituents on the N-chloroamide reagents.49,50 The free-radical mechanism we propose for the Ni-catalyzed oxidation of unactivated alkanes suggests that novel, substituted aromatic peracids, likewise, could modify the selectivity of the oxidation of C(sp3)–H bonds in complex molecules and polymers.
CONCLUSION
In conclusion, we report detailed mechanistic investigations of the oxidation of unactivated C(sp3)–H bonds by mCPBA catalyzed by Ni complexes coordinated by nitrogen-based ligands. We show the absence of an effect of the ligand on the yield and selectivity of this reaction, as well as a clear correlation between reaction rates and the steric hindrance of the ligands. Experiments with radical clocks indicate that this reaction generates a carbon-centered radical that is sufficiently long-lived to epimerize before the formation of the C–O bond.
On the basis of these results and the observation of Ni-catalyzed decomposition of mCPBA, we propose an alternative pathway for oxidation that does not involve a nickel-oxo or -oxyl species. We conclude that the nickel facilitates the generation of the 3-chlorobenzoyloxy radical, which cleaves the C–H bonds in alkanes, as evidenced by a series of experiments on the selectivity for reactions of adamantane and 2,2,4,4-tetramethylpentane and on the oxidation of deuterium-labeled cyclohexane. We also conclude that trapping of alkyl radicals by mCPBA is the major pathway to form the C–O bond. Finally, we propose that the oxidation occurs by a free-radical chain mechanism. Our results and this proposed mechanism reconcile the lack of strong evidence for the reactivities of Ni-oxo or Ni-oxyl complexes toward strong C(sp3)–H bonds and the formation of oxidized products in good yield from Ni-catalyzed oxidations of unactivated alkanes with aromatic peracids. We envision that such mechanistic understanding can guide us to design more effective and selective organic oxidants, for example, substituted aromatic peracids, for the oxidation of unactivated C(sp3)–H bonds in complex molecules and polymers.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Director, Office of Science, of the U.S. Department of Energy under Contract No. DE-AC0205CH11231. We gratefully acknowledge Dr. H. Celik for assistance with the NMR experiments, Dr. N. Settineri for the X-ray crystallography, Dr. M. Zhang in the LBL Catalysis Facility for the IR and ESI HRMS experiments, Dr. E. Kreimer for the elemental analysis, and Dr. R. Chatterjee for the EPR analysis. Instruments in the CoC-NMR are supported in part by NIH S10OD024998. Y.Q. thanks Dr. A. Bunescu, Dr. L. Chen, and Dr. P.-f. Ji for helpful discussions.
Footnotes
Notes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c09157
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c09157.
Experiment procedures, characterization of compounds (PDF)
X-ray crystallographic data for complex 1a (CIF)
X-ray crystallographic data for complex 1f (CIF)
X-ray crystallographic data for complex 1g (CIF)
X-ray crystallographic data for complex 1h (CIF)
Contributor Information
Yehao Qiu, Department of Chemistry, University of California, Berkeley, California 94720, United States.
John F. Hartwig, Department of Chemistry, University of California, Berkeley, California 94720, United States
REFERENCES
- (1).Meunier B; de Visser SP; Shaik S Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- (2).Merkx M; Kopp DA; Sazinsky MH; Blazyk JL; Muller J; Lippard SJ Dioxygen activation and methane hydroxylation by soluble methane monooxygenase: A tale of two irons and three proteins. Angew. Chem., Int. Ed. 2001, 40, 2782–2807. [DOI] [PubMed] [Google Scholar]
- (3).Shilov AE; Shul’pin GB Activation of C-H bonds by metal complexes. Chem. Rev. 1997, 97, 2879–2932. [DOI] [PubMed] [Google Scholar]
- (4).Luo Y-R; Luo Y-R Comprehensive handbook of chemical bond energies; CRC Press: Boca Raton, FL, 2007; p 1655. [Google Scholar]
- (5).Crabtree RH Alkane C-H activation and functionalization with homogeneous transition metal catalysts: a century of progress—a new millennium in prospect. J. Chem. Soc., Dalton Trans. 2001, 2437–2450. [Google Scholar]
- (6).Williamson DG; Cvetanovic RJ Rates of Ozone-Paraffin Reactions in Carbon Tetrachloride Solution. J. Am. Chem. Soc. 1970, 92, 2949–2952. [Google Scholar]
- (7).Bravo A; Fontana F; Fronza G; Minisci F; Zhao LH Molecule-induced homolysis versus “concerted oxenoid oxygen insertion” in the oxidation of organic compounds by dimethyldioxirane. J. Org. Chem. 1998, 63, 254–263. [Google Scholar]
- (8).Mello R; Fiorentino M; Fusco C; Curci R Oxidations by Methyl(Trifluoromethyl)Dioxirane. 2. Oxyfunctionalization of Saturated-Hydrocarbons. J. Am. Chem. Soc. 1989, 111, 6749–6757. [Google Scholar]
- (9).Bravo A; Bjorsvik HR; Fontana F; Minisci F; Serri A Radical versus “oxenoid” oxygen insertion mechanism in the oxidation of alkanes and alcohols by aromatic peracids. New synthetic developments. J. Org. Chem. 1996, 61, 9409–9416. [Google Scholar]
- (10).Schneider HJ; Mueller W Selective functionalization of hydrocarbons. 6. Mechanistic and preparative studies on the regio- and stereoselective paraffin hydroxylation with peracids. J. Org. Chem. 1985, 50, 4609–4615. [Google Scholar]
- (11).Fenton HJH Oxidation of tartaric acid in the presence of iron. J. Chem. Soc., Trans. 1894, 65, 899–910. [Google Scholar]
- (12).Barton DHR; Doller D The Selective Functionalization of Saturated-Hydrocarbons - Gif Chemistry. Acc. Chem. Res. 1992, 25, 504–512. [Google Scholar]
- (13).Periana RA; Taube DJ; Gamble S; Taube H; Satoh T; Fujii H Platinum catalysts for the high-yield oxidation of methane to a methanol derivative. Science 1998, 280, 560–564. [DOI] [PubMed] [Google Scholar]
- (14).Periana RA; Mironov O; Taube D; Bhalla G; Jones CJ Catalytic, oxidative condensation of CH4 to CH3COOH in one step via CH activation. Science 2003, 301, 814–818. [DOI] [PubMed] [Google Scholar]
- (15).Groves JT; Bonchio M; Carofiglio T; Shalyaev K Rapid catalytic oxygenation of hydrocarbons by ruthenium pentafluorophenylporphyrin complexes: Evidence for the involvement of a Ru(III) intermediate. J. Am. Chem. Soc. 1996, 118, 8961–8962. [Google Scholar]
- (16).Wang CQ; Shalyaev KV; Bonchio M; Carofiglio T; Groves JT Fast catalytic hydroxylation of hydrocarbons with ruthenium porphyrins. Inorg. Chem. 2006, 45, 4769–4782. [DOI] [PubMed] [Google Scholar]
- (17).Ohtake H; Higuchi T; Hirobe M Highly Efficient Oxidation of Alkanes and Alkyl Alcohols with Heteroaromatic N-Oxides Catalyzed by Ruthenium Porphyrins. J. Am. Chem. Soc. 1992, 114, 10660–10662. [Google Scholar]
- (18).Chen MS; White MC Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. [DOI] [PubMed] [Google Scholar]
- (19).Chen MS; White MC A predictably selective aliphatic C-H oxidation reaction for complex molecule synthesis. Science 2007, 318, 783–787. [DOI] [PubMed] [Google Scholar]
- (20).Nagataki T; Ishii K; Tachi Y; Itoh S Ligand effects on Ni(II)-catalysed alkane-hydroxylation with m-CPBA. Dalton Trans. 2007, 1120–1128. [DOI] [PubMed] [Google Scholar]
- (21).Nagataki T; Tachi Y; Itoh S NiII(TPA) as an efficient catalyst for alkane hydroxylation with m-CPBA. Chem. Commun. 2006, 4016–4018. [DOI] [PubMed] [Google Scholar]
- (22).Balamurugan M; Mayilmurugan R; Suresh E; Palaniandavar M Nickel(II) complexes of tripodal 4N ligands as catalysts for alkane oxidation using m-CPBA as oxidant: ligand stereoelectronic effects on catalysis. Dalton Trans. 2011, 40, 9413–9424. [DOI] [PubMed] [Google Scholar]
- (23).Bunescu A; Lee SW; Li Q; Hartwig JF Catalytic Hydroxylation of Polyethylenes. ACS Cent. Sci. 2017, 3, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ni-oxyl, commonly written as Ni–O·, refers to a nickel-oxygen complex that contains a formal Ni–O single bond in which the oxygen atom bears an unpaired electron. Ni-oxo, commonly written as Ni═O, refers to a nickel-oxygen complex that contains a formal Ni═O double bond in which the oxygen atom bears no unpaired electrons. For more details, see ref 34.
- (25).Sankaralingam M; Vadivelu P; Suresh E; Palaniandavar M Mixed ligand nickel(II) complexes as catalysts for alkane hydroxylation using m-chloroperbenzoic acid as oxidant. Inorg. Chim. Acta 2013, 407, 98–107. [Google Scholar]
- (26).Hikichi S; Hanaue K; Fujimura T; Okuda H; Nakazawa J; Ohzu Y; Kobayashi C; Akita M Characterization of nickel(II)-acylperoxo species relevant to catalytic alkane hydroxylation by nickel complex with mCPBA. Dalton Trans. 2013, 42, 3346–3356. [DOI] [PubMed] [Google Scholar]
- (27).Nakazawa J; Terada S; Yamada M; Hikichi S Structural characterization and oxidation reactivity of a nickel(II) acylperoxo complex. J. Am. Chem. Soc. 2013, 135, 6010–6013. [DOI] [PubMed] [Google Scholar]
- (28).Pirovano P; Farquhar ER; Swart M; McDonald AR Tuning the Reactivity of Terminal Nickel(III)-Oxygen Adducts for C-H Bond Activation. J. Am. Chem. Soc. 2016, 138, 14362–14370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pfaff FF; Heims F; Kundu S; Mebs S; Ray K Spectroscopic capture and reactivity of S = 1/2 nickel(III)-oxygen intermediates in the reaction of a Ni(II)-salt with mCPBA. Chem. Commun. 2012, 48, 3730–3732. [DOI] [PubMed] [Google Scholar]
- (30).Corona T; Draksharapu A; Padamati SK; Gamba I; Martin-Diaconescu V; Acuna-Pares F; Browne WR; Company A Rapid Hydrogen and Oxygen Atom Transfer by a High-Valent Nickel-Oxygen Species. J. Am. Chem. Soc. 2016, 138, 12987–12996. [DOI] [PubMed] [Google Scholar]
- (31).Cho J; Kang HY; Liu LV; Sarangi R; Solomon EI; Nam W Mononuclear nickel(II)-superoxo and nickel(III)-peroxo complexes bearing a common macrocyclic TMC ligand. Chem. Sci. 2013, 4, 1502–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Company A; Yao S; Ray K; Driess M Dioxygenase-like reactivity of an isolable superoxo-nickel(II) complex. Chem. - Eur. J. 2010, 16, 9669–9675. [DOI] [PubMed] [Google Scholar]
- (33).Corona T; Pfaff FF; Acuna-Pares F; Draksharapu A; Whiteoak CJ; Martin-Diaconescu V; Lloret-Fillol J; Browne WR; Ray K; Company A Reactivity of a Nickel(II) Bis(amidate) Complex with meta-Chloroperbenzoic Acid: Formation of a Potent Oxidizing Species. Chem. - Eur. J. 2015, 21, 15029–15038. [DOI] [PubMed] [Google Scholar]
- (34).Corona T; Company A Spectroscopically Characterized Synthetic Mononuclear Nickel-Oxygen Species. Chem. - Eur. J. 2016, 22, 13422–13429. [DOI] [PubMed] [Google Scholar]
- (35).Oxidation of strong C–H bonds in cyclohexane by a nickel-acylperoxy and a nickel-oxyl species was reported in refs 26 and 32, respectively. However, in ref 26, the authors commented that the oxidations of cyclohexane and substrates containing strong C–H bonds may occur through different mechanisms and that a pathway that involves cleavage of the O–O bond in the Ni-acylperoxy complex, which would generate free radicals, may be dominant in the oxidation of cyclohexane. In ref 32, the Ni-oxyl species was generated by oxidation of a nickel precursor by 3 equiv of mCPBA. Therefore, background catalytic oxidation of substrates by excessive mCPBA cannot be ruled out. Indeed, the authors commented that “background catalytic reaction (of mCPBA) significantly interferes with the kinetic trace (of the reaction between cyclohexane and the Ni-oxyl species)”.
- (36).Costas M; Chen K; Que L Biomimetic nonheme iron catalysts for alkane hydroxylation. Coord. Chem. Rev. 2000, 200, 517–544. [Google Scholar]
- (37).Chen LZ; Su XJ; Jurss JW Selective Alkane C-H Bond Oxidation Catalyzed by a Non-Heme Iron Complex Featuring a Robust Tetradentate Ligand. Organometallics 2018, 37, 4535–4539. [Google Scholar]
- (38).Miyajima S; Simamura O The Stereochemistry of Autoxidation of Methylcyclohexanes. Bull. Chem. Soc. Jpn. 1975, 48, 526–530. [Google Scholar]
- (39).Walling C Fenton’s Reagent Revisited. Acc. Chem. Res. 1975, 8, 125–131. [Google Scholar]
- (40).Haber FWJJ The catalytic decomposition of hydrogen peroxide by iron salts. Proc. R. Soc. London, Ser. A 1934, 147, 332–351. [Google Scholar]
- (41).Shul’pin GB; Kozlov YN; Nizova GV; Suss-Fink G; Stanislas S; Kitaygorodskiy A; Kulikova VS Oxidations by the reagent “O2-H2O2-vanadium derivative-pyrazine-2-carboxylic acid”. Part 12. Main features, kinetics and mechanism of alkane hydroperoxidation. J. Chem. Soc., Perkin Trans. 2001, 2, 1351–1371. [Google Scholar]
- (42).Shimizu N; Bartlett PD Photooxidation of Olefins Sensitized by Alpha-Diketones and by Benzophenone - Practical Epoxidation Method with Biacetyl. J. Am. Chem. Soc. 1976, 98, 4193–4200. [Google Scholar]
- (43).Sawaki Y; Ogata Y Reactivities of Acylperoxy Radicals in the Photoreaction of Alpha-Diketones and Oxygen. J. Org. Chem. 1984, 49, 3344–3349. [Google Scholar]
- (44).Chateauneuf J; Lusztyk J; Ingold KU Spectroscopic and kinetic characteristics of aroyloxyl radicals. 1. The 4-methoxybenzoyloxyl radical. J. Am. Chem. Soc. 1988, 110, 2877–2885. [Google Scholar]
- (45).Russell GA Deuterium-Isotope Effects in the Autoxidation of Aralkyl Hydrocarbons - Mechanism of the Interaction of Peroxy Radicals. J. Am. Chem. Soc. 1957, 79, 3871–3877. [Google Scholar]
- (46).Shul’pin GB Alkane Oxidation: Estimation of Alkyl Hydroperoxide Content by GC Analysis of the Reaction Solution Samples Before and after Reduction with Triphenylphosphine. Chemistry Preprint Archive 2001, 2001, 21–26. [Google Scholar]
- (47).Shul’pin GB; Nizova GV; Kozlov YN; Gonzalez Cuervo L; Suss-Fink G Hydrogen peroxide oxygenation of alkanes including methane and ethane catalyzed by iron complexes in acetonitrile. Adv. Synth. Catal. 2004, 346, 317–332. [Google Scholar]
- (48).Fossey J; Lefort D; Sorba J Peracids and Free-Radicals - a Theoretical and Experimental Approach. Top. Curr. Chem. 1993, 164, 99–113. [Google Scholar]
- (49).Quinn RK; Konst ZA; Michalak SE; Schmidt Y; Szklarski AR; Flores AR; Nam S; Horne DA; Vanderwal CD; Alexanian EJ Site-Selective Aliphatic C-H Chlorination Using N-Chloroamides Enables a Synthesis of Chlorolissoclimide. J. Am. Chem. Soc. 2016, 138, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Carestia AM; Ravelli D; Alexanian EJ Reagent-dictated site selectivity in intermolecular aliphatic C-H functionalizations using nitrogencentered radicals. Chem. Sci. 2018, 9, 5360–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.